
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A comprehensive study on rheological properties of photocrosslinkable gallol-metal complexed hyaluronic acid-based biomaterial inks†
Hatai
Jongprasitkul
 a,
Sanna
Turunen
ab,
Minna
Kellomäki
a and
Vijay Singh
Parihar
*a
a,
Sanna
Turunen
ab,
Minna
Kellomäki
a and
Vijay Singh
Parihar
*a
aBiomaterials and Tissue Engineering Group, Faculty of Medicine and Health Technology, Tampere University, 33720 Tampere, Finland. E-mail: vijay.parihar@tuni.fi
bNew Materials and Processes Group, Faculty of Engineering and Business, Turku University of Applied Sciences, 20520 Turku, Finland
First published on 10th June 2024
Abstract
This study describes the development of bioadhesive hydrogels as biomaterial inks, which combine catechol chemistry and metal coordination using gallic acid and hyaluronic acid. By controlling the phase of gelation, the properties of hydrogels can be modulated through pH and Fe3+ ion concentrations, thus allowing for diverse coordination states within the polymeric network. The dual-crosslinking mechanism based on catechol–metal coordination and methacrylation improves the hydrogels' printability and facilitates adaptive swelling behaviour, which is crucial for in situ bioprinting. The developed hydrogels address the challenge of effective adhesion in moist environments such as human tissues, exhibiting rapid self-healing and wet adhesion properties. Our findings indicate that mono- and bis-complex hydrogels are optimal for printing, while bis- and tris-complex hydrogels offer higher stability, which is suitable for injection. However, tris-coordinated hydrogels have limited self-healing and adhesive properties due to excessive oxidative crosslinking over time. Overall, this work advances the potential application of hyaluronan-based hydrogels in biomaterial inks, stimuli-responsive hydrogels, and bioadhesives inspired by mussel byssus cuticle chemistry.
Introduction
Hydrogels possess adhesive properties, yet their efficacy is compromised by the significant water content within their polymer network. The high water content creates a weak boundary layer that prevents the hydrogels from directly contacting substrates, leading to decreased surface energy and weakened adhesion.1 Additionally, hydrogen bonding interactions between water molecules and the hydrogel's adhesive groups decrease the reactivity of the hydrogel with solid materials at the interface.2 This issue becomes more complex when hydrogels are used in biomedical engineering, where most surfaces, such as tissues and wound surfaces, are wet and moist.3 The development of hydrogels with tissue adhesion is crucial for advanced wound dressing applications, particularly those involving in situ crosslinking.4 However, many available hydrogels lack tissue adhesion, resulting in inadequate fixation of wound edges or dressing in wound healing applications.5The use of bioadhesive hydrogels has been an emerging approach due to their unique wet adhesive properties and potential for a wide range of applications, including wound sealing, drug delivery, and mimicking tissue-like constructs.6 Many bioadhesives have been developed by incorporating catechol-related groups, drawing inspiration from bacterial adhesions or molecular pairing in organisms.2 Recent studies have shown that hydrogels containing catechol have been utilized for interfacial engineering, leading to the development of tissue-adhesive hydrogels with enhanced properties. For example, there is an injectable, self-healing hydrogel with thiol and catechol groups, a mussel-inspired hydrogel with hyaluronic acid and catechol, and a bioinspired adhesive hydrogel with quinone and catechol groups.7–9 Metal ligand coordination in catechol-based polymers offers high tunability in bonding strength and bonding dynamics by varying the stoichiometric ratios of catechol:Fe3+ ions and the system's pH, leading to adjustable network formation. Metal complexation is extensively used in natural materials to modulate the mechanical properties of bulk and interfacial structures.10 Metal–catechol complexes in catechol groups like dihydroxyphenylalanine (DOPA), found in mussel adhesive proteins, penetrate water layers and form strong bonds with various substrates.11,12 Holten-Anderson et al. utilised metal salts such as VCl3, FeCl3 and AlCl3 to form catechol–metal complexes on DOPA-polyethylene glycol (PEG) hydrogels.13–15 The mechanical properties of the polymeric network, such as shear modulus and relaxation time, can be precisely adjusted by varying the type of metal ion and pH. On the other hand, the oxidative polymerisation in a DOPA-rich polymer solution is relatively slow due to the limited number of free hydroxyl groups. The oxidative crosslinking can be enhanced by the presence of an additional hydroxyl group in the aromatic compound, which is found in gallol-based molecules.16
Gallol is a well-known phenolic moiety that shares a molecular backbone with catecholic polyphenol but contains three hydroxyls attached to a benzene ring.17,18 Due to the additional hydroxyl groups (–OH), gallol-based polymers exhibit rapid self-assembly, which reduces the gelation time compared to other phenolic building blocks, such as phenol and catechol.16,17 In addition, the chemical interactions within the molecules are highly reversible due to the formation of dynamic crosslinks via hydrogen bonds, hydrophobic interactions, intermolecular bonding motifs and metal–ligand complexation at basic pH, resulting in self-healing and bioadhesive properties.13,19 Networks with reversible bonds have several advantages over non-reversible systems; they possess inherent shear thinning and self-healing properties without adding crosslinking or thickening agents to facilitate those innate properties.20 Ink rheology can also be tuned by adding fibers to the hydrogel, as has been reported by Choi et al. in their study about infusing gelatin fibers into a gelatin and alginate hydrogel matrix.21
However, only a few reports have exploited gallol-modified hyaluronic acid polymers as all-in-one hydrogels with various unique properties, including rapid self-healing, printability and tissue adhesion.18,22–26 Previously, Shin et al. have explored the formulation of gallol-functionalized HA and gelatin as a bioink for direct extrusion printing. However, the stability of hydrogels lasted only six days.27 In another study, Mörö et al. employed hydrazone and catechol chemistry to create dynamic covalent crosslinking to stabilise the hydrogel construct. However, the combination of oxidative and dynamic crosslinking resulted in rapid gelation of the hydrogels. The premature hydrogel formation has caused challenges for its use in printing or injecting applications.28
This study designed bioadhesive hydrogels as printable biomaterial inks, using gallic acid and hyaluronic acid to exploit the synergistic effects of catechol and metal coordination. Gelation is controlled via pH and Fe3+ ions. A dual crosslinking approach, combining catechol–metal chelation and photocrosslinking with methacrylate groups to create a complementary network, enhancing tissue adhesion and long-term stability, as shown in Fig. 1. We investigated the rheological properties, printability, and gelation kinetics of HAMA–GA formulations induced by catechol–metal chelation and oxidative crosslinking. We hypothesized that the ink formulations were able to improve tissue adhesion and achieve shear-thinning, self-healing properties, suitable for biomaterial inks and injectable hydrogels.
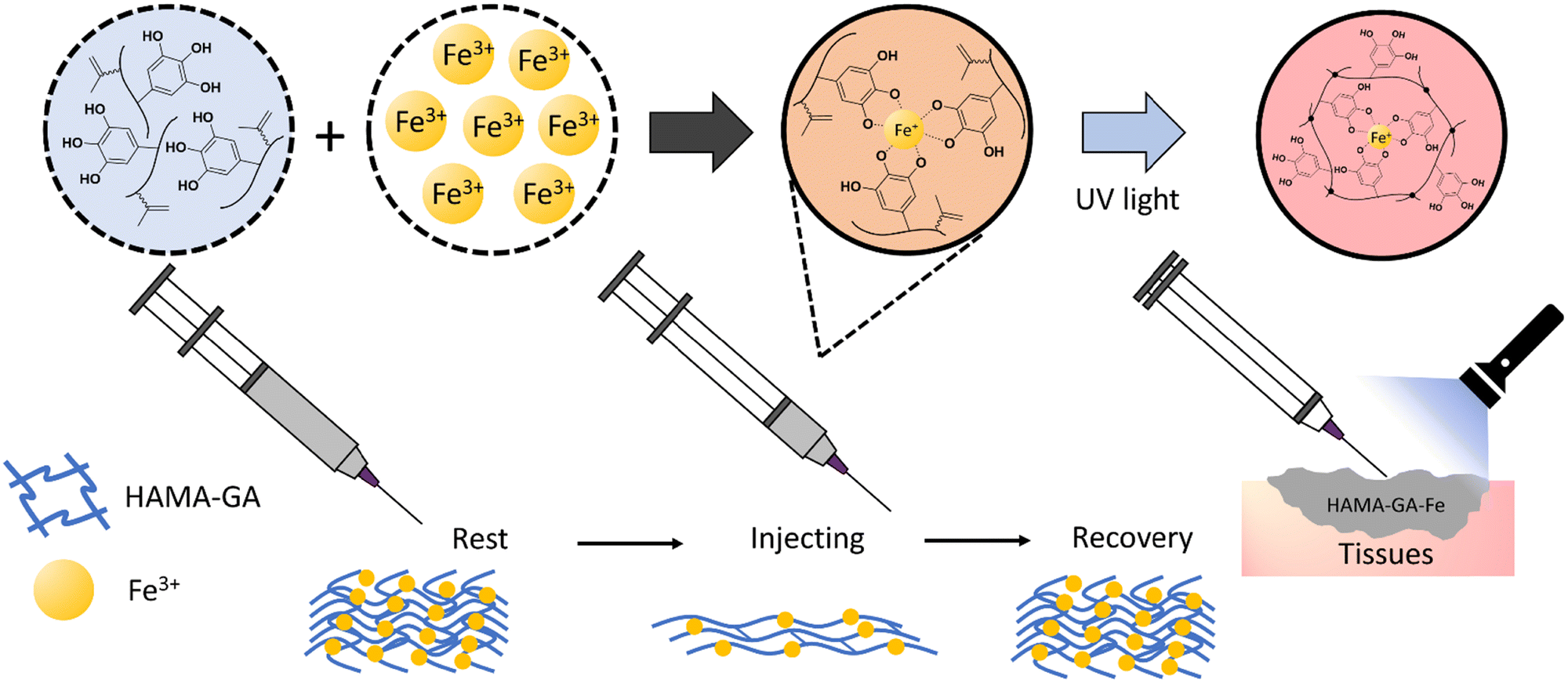 | ||
| Fig. 1 The process of HAMA–GA gel formation under the influence of Fe3+ provides extrudability and self-healing properties during extrusion. After extrusion at the target site, the gel is photocrosslinked, resulting in a stable matrix through metalcoordination bonds and covalent linkages. | ||
Experimental
Materials
Hyaluronic acid (HA, MW 100 kDa) was purchased from Lifecore Biomedical (Chaska, USA). Methacrylic anhydride (MA), gallic acid (GA, 3,4,5-trihydroxy benzoic acid), hydrazine hydrate, 1-ethyl-3-(3-dimethyl aminopropyl)-carbodiimide hydrochloride (EDC), 1-hydroxy benzotriazole hydrate (HOBt), dimethyl sulfoxide (DMSO), and Irgacure 2959 (I2959) were purchased from Merck KGaA, Darmstadt, Germany. Dialysis membranes used for purification were purchased from Spectra Por-6 (MWCO 3500). DI water (deionised water, Miele Aqua Purificator G 7795, Siemens) was used. Dulbecco's phosphate-buffered saline (DPBS) was prepared in the lab. All solvents were of analytical quality. 1H-NMR analysis was conducted on an NMR spectrometer (SCZ500R, JEOL Resonance, Japan).Synthesis of catechol-modified hyaluronic acid methacrylate (HAMA)
Hyaluronic acid methacrylate with a 30% degree of modification (HAMA30) and gallic acid hydrazide (GH) were prepared according to the previous studies,29,30 as described in ESI† (Fig. S1 and S2).29,30 HAMA–GA was synthesised by GA conjugation to the HAMA backbone. In brief, 400 mg of HAMA (1 mmol of HAMA, in equivalent) was dissolved in 75 mL DI water, followed by the addition of 1 mmol HOBt (153 mg, 1 equivalent). The GH (184 mg, 1 equivalent) was separately dissolved in 25 mL of DMSO and added dropwise to the reaction mixture. The pH of the reaction solution was maintained at 4.75 using 1 M HCl and 1 M NaOH. GA modification was set at 10% by adjusting the ratio of EDC (29 mg, 0.15 mmol) and left to stir overnight. After that, the reaction mixture was transferred into a dialysis bag (Spectra Por-6, MWCO 3500 g mol−1) and dialysed against dilute HCl (pH = 3.5) containing 100 mM NaCl (6 × 2 L, 48 h) and then dialysed against deionised water (4 × 2 L, 24 h). The solution was freeze-dried, obtaining a white fluffy material. The conjugation of GA and the degree of modification of GA in HAMA was further confirmed by the presence of a distinctive aromatic peak at 6.99 ppm of GA in the 1H NMR spectrum. All 1H-NMR spectra were recorded using a JEOL-500 MHz instrument with D2O as solvent.Preparation of catechol–metal coordination complexes hydrogel and pH-dependent HAMA–GA hydrogel
The HAMA–GA with 30% modification of methacrylate and 10% modification of GA was prepared at a concentration of 5% w/v for all characterisation processes. The photoinitiator, I2959, was added to the solution at a concentration of 0.5% w/v in DPBS for dual-crosslinking hydrogel. To analyse the phase transition of HAMA–GA hydrogel influenced by pH and Fe3+, the pH of all hydrogel formulations was adjusted using 0.5 M NaOH and varied into acidic (pH = 4 and 5), neutral (pH = 7), and basic (pH = 8 and 9) pH. The Fe3+ solution was prepared by varying the concentration of iron(III) chloride (FeCl3). In this study, each hydrogel formulation was termed according to a concentration of FeCl3 (0.125, 0.25 and 0.5% w/v), i.e., HAMA–GA 1.25Fe, HAMA–GA-2.5Fe and HAMA–GA-5Fe. The photocrosslinked hydrogels or dual-crosslinked hydrogels were prepared similarly to the samples without photocrosslinking. The dual-crosslinking was performed by irradiating the Fe3+ -induced hydrogels with UV light at 365 nm in wavelength (BlueWave® 50 UV curing spot lamp, DYMAX Corp., USA) at the intensity of 25 mW cm−2 for 120s. The photocrosslinked samples were then characterised by oscillatory measurements. The hydrogel sample (without photocrosslinking) was prepared by casting 250 μL of hydrogel solution into a cut 4 mL syringe used as a mould and left to cure for 2 h. After curing, the hydrogel samples were subjected to rheological characterisation to observe the flow behaviours. The hydrogel samples with proper gelation (solid-like phase) were selected for the rheological characterisations.Gelation kinetics of Fe3+ and pH-induced hydrogels
A UV-vis spectrophotometer (Shimadzu UV-3600 plus UV-VIS-NIR spectrophotometer) was used to analyse the different crosslinking types in HAMA–GA hydrogels influenced by Fe3+ and pH. The experiment was carried out by varying the Fe3+ concentration (0.125, 0.25 and 0.5% w/v) and pH conditions (3, 7 and 9) in DI water. The sample was prepared by combining 150 μL of 5% w/v HAMA–GA solution with 10 μL of the Fe3+ solution in 1 mL of DI water, then loaded into a 1 cm quartz cuvette. The zeta potential of HAMA–GA was measured with the Zetasizer Nano ZS measurement system (Malvern Ltd., UK) to determine the surface charge at various pH values. The sample was prepared at 1 mg mL−1 concentration in DI water at 25 °C using disposable folded capillary DTS1070 cells.The flow behaviour of HAMA–GA with various Fe3+ concentrations
The flow behaviour of different hydrogel formulations was evaluated by a rotational rheometer (Discovery HR-2, TA Instruments Inc., USA) with a plate-plate geometry (12 mm in diameter, gap size of 2.5 mm) at RT. The performed tests were flow sweep (shear viscosity-shear rate), stress ramp (viscosity-shear stress) and thixotropic behaviour (viscosity as a function of repeating cycles of low/high shear rate). The flow sweep was utilised to determine the shear-thinning properties of the hydrogel by varying the shear rate from 0.01 to 100 s−1. The shear-thinning behaviour of each sample was confirmed again via oscillatory measurements with frequency sweep (complex modulus as a function of frequency, 0.1–500 rad s−1, at 1% of strain). The stress ramp test was carried out to obtain the yield stress values, ranging from 0.1 Pa to 1000 Pa for all samples (the shear rate was kept constant at 0.1 s−1). The yield point was determined by determining the intersection point between the linear and viscosity-drop regions from the viscosity-shear stress plot. The yield point indicates the hydrogel's flow initiation at the applied shear stress level. The measurements of recovery behaviour were performed by repeating cycles of low (0.1 s−1) and high shear rates (100 s−1) to simulate the actual conditions during the injection.The flow model of catechol–metal complex hydrogels
To further analyse the effect of Fe3+ on the physical properties of hydrogels, the Carreau model was applied to the viscosity curves of HAMA–GA–Fe hydrogels. The parameters of shear-thinning coefficients (n), zero-shear viscosity (η0) and time constant (λ) are presented in eqn (1).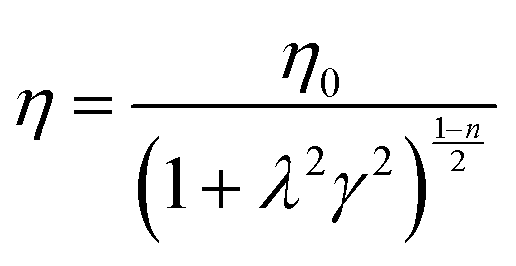 | (1) |
3D printing and printability assessment of catechol–metal complex hydrogels
For the bioprinting setup, we used an extrusion-based 3D bioprinter (BRINTER® ONE 3D Bioprinter, Brinter Ltd., Finland). 5% w/v of HAMA–GA 1.25Fe hydrogel at pH 5 was selected as a biomaterial ink, which was immediately loaded into the printing cartridge after mixing to prevent extensive crosslinking over time, then incubated under the shaker for 10 min to remove air bubbles. To pre-screen the printability, the biomaterial ink was extruded using an air pressure-controlled print head through the nozzle (250 μm, SmoothFlow™, Nordson EFD, USA) to produce a filament in the air. The lattice CAD model (20 × 20 × 1 mm3) was selected for 4-layered grid printing. Printing speed and print head temperature were fixed at 6 mm s−1 and RT. The first layer z height was set to 250 μm to increase the ink's adhesion to the substrate. The second layer height was selected as the same as the nozzle diameter, i.e. 250 μm. Grids were printed using a pressure of 3000 mbar and extruded as a continuous filament to avoid any unnecessary cut-offs in the printing pressure. Microscope glass slides were used as substrates, and the printing bed temperature was set at RT.Rheological and self-healing properties of dual-crosslinked hydrogel
To characterise the viscoelastic properties of dual-crosslinked hydrogels, oscillatory measurements were performed on a rotational rheometer with time, frequency, and amplitude sweep. The effect of photocrosslinking on the hydrogel was determined via a time sweep of oscillatory mode via in situ polymerisation with an external UV lamp at 365 nm in wavelength and 25 mW cm−2 in UV intensity. In oscillatory time sweep measurement (photocrosslinking kinetics of hydrogels), storage and loss moduli, G′ and G′′ were measured as a function of time (500 s, UV lamp was activated at 100 s) while strain and frequency were kept constant at 1% and 1 Hz, respectively. A frequency sweep was performed between 0.1–100 Hz under 1% of constant strain, while an amplitude sweep was conducted in the range of 0.1–1000% strain to observe the critical strain and the frequency was set at 1 Hz. The strain recovery or self-healing behaviour tests were conducted under the repeating seven cycles of low (1%) and high oscillation strain (500%) conditions, and the oscillation frequency was set at 1 Hz. The holding period of each cycle was set at 60 s.The self-healing properties of GA-based hydrogels were evaluated via a cutting-healing method.30 The hydrogels were first cut into two separate pieces, after which the cut edges were faced together until they merged. The stress relaxation of hydrogel samples was also determined by rheology as a function of time.31 The strain (5%) was constantly applied to the hydrogel samples for 1000 s, and relaxation of stress, relaxation time and the initial elastic modulus were recorded simultaneously. The relaxation stress values were normalised and plotted on the graph. The experiments were performed at RT.
pH-dependent stability of photocrosslinked hydrogels
All dual-crosslinked hydrogels were immersed in 0.1 M phosphate-buffered saline (PBS) solution with different pH levels (3, 5, 7, 8, and 9) to examine their stability. The pH of the PBS was re-adjusted by 1 M HCl or 1 M NaOH to reach the target pH. Each pH represents a different physiological pH level of skin in normal and infected conditions. The hydrogels were maintained at 37 °C ± 0.5 °C in a shaking incubator at 90 rpm until various time points (0, 1, 2, 3, 5, 7 and 15 days). The swelling ratio was calculated as Ws/W0. At the zero-time point, the samples were defined with a weight of W0. At a specific time, the hydrogel samples were removed and gently blotted with filter paper to remove the residual solution and weighted as Ws.Bioadhesion of hydrogels
A tack test was performed for HAMA and HAMA–GA–Fe hydrogels with and without photocrosslinking using a rotational rheometer. The bioadhesion of hydrogels (n = 5, each parameter) was examined through in situ photocrosslinking and detachment capability in the axial direction. The protocol has been reported in a previous study.30,32 Briefly, porcine muscles were carefully cut into circular sheets with a 12 mm diameter and attached to the geometry and the bottom plate. Next, the sample was loaded into a 2.5 mm gap between the two tissue sheets (upper and bottom plates) and left to oxidise for 30 min. The sample was irradiated with UV light for 2 min to crosslink the sample (for dual-crosslinking samples). After that, the geometry was lifted in axial motion at a constant rate of 20 μm s−1. The change in axial force was recorded at the point of detachment. A graph was then plotted to observe the influence of GA–Fe3+ and photocrosslinking on the adhesive properties of the hydrogel compared to the plain HAMA.Antioxidant properties
The antioxidant capacity of HAMA–GA was tested by assessing its ability to scavenge free radicals using the DPPH (2,2,1-diphenyl-1-picrylhydrazyl) method, as described in a previous study.30,32 HAMA–GA was dissolved in 1 mL of distilled water at a concentration of 30 μg mL−1 and then mixed with 1 mL of DPPH solution (1 mg/12 mL in methanol). The mixture was allowed to incubate at 25 °C for 30 minutes, after which the absorbance was measured at 517 nm using a UV-vis spectrophotometer.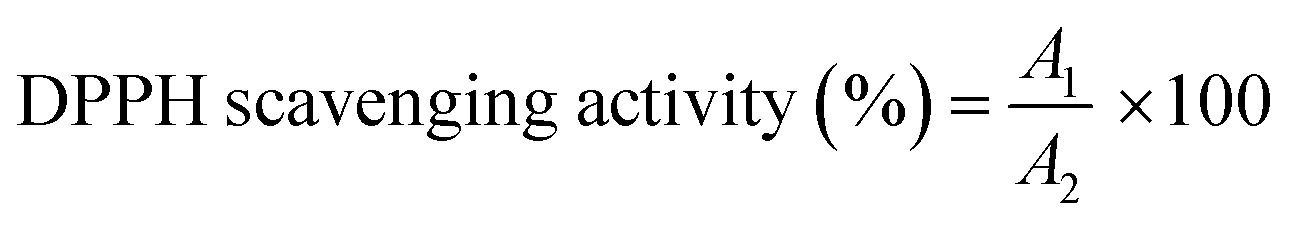 | (2) |
Statistical analysis
All experiments were repeated five times. The quantities are presented as mean ± standard deviation (SD). The analysis was performed using a one-way analysis of variance to determine the differences between groups, and p < 0.05 was considered significant.Results and discussion
According to the studies, bioinspired hydrogels based on catechol chemistry can form reversible metal-coordinated bonds, which are able to undergo autocondensation to form a covalent network in the presence of oxidative agents or basic pH. The nomenclature mono, bis, or tris denotes the number of reactive groups on molecules essential to the gelation process, where higher functionality correlates with the development of more resilient and interconnected gel structures.33 Furthermore, the strategic application of phase-controlled gelation in these hydrogels presents potential advantages in regulating their swelling behaviour in response to varying pH conditions. The hydrogels, characterised by diverse coordination degrees (mono, bis, and tris) within the polymer network, exhibit the ability to dissolve, shrink, and swell at specific pH levels. This property becomes particularly significant in the context of wound healing, where variations in pH levels occur during the process. Healthy skin typically maintains an acidic pH range of 4–6, while infected skin often manifests a basic pH between 7.3 and 9.8.34 Leveraging the swelling behaviour of these hydrogels, they can be effectively employed for wound sealing in infected areas. Notably, the hydrogels demonstrate stability at basic pH levels, gradually dissolving as the wound heals and pH decreases. This system has been extensively studied as a dynamic, self-healing network such as PEG-DOPA polymers mixed with metal ions (Al3+, Fe3+ or V3+).19 Our previous studies have utilised gallol-modified gelatine methacrylate (GelMA–GA) or gallol-modified hyaluronic acid (HAGA) to create an effective bioinspired biomaterial ink for in situ wound sealing applications.30,32 However, HAGA alone suffered from poor stability in the cell culture medium, and it also lacked strong bioadhesion due to the absence of covalent bonding with the target tissue.26,27 It has been previously reported that HAGA–HAMA blends show high stability, pH responsiveness and tissue adhesion.30 To improve the overall properties of the catechol-based hydrogel, we functionalised HA with gallic acid and methacrylol groups (HAMA–GA) to enable non-covalent and covalent interactions. In addition, the presence of Fe3+ further enhances the mechanical properties of hydrogels via metal coordination complexes.Synthesis of catechol-modified HAMA
Unlike the previous investigation involving HAMA–HAGA and HAGA–GelGA blends,27,30 this finding emphasized that both GA and MA were grafted onto the same HA backbone. Functionalizing HAMA and GA on the same polymer backbone offers several advantages over blending different polymers such as uniform and cohesive network structures. In addition, consistent gelation behaviour can be precisely controlled through pH and metal ion concentration, leading to more reliable hydrogel formation.HAMA–GA was synthesised, as shown in Fig. 2 and Fig. S3 (ESI†). To conjugate GA to the HAMA backbone, GA carboxylate was firstly modified to GH, then conjugated with HAMA using carbodiimide chemistry. The GH-modified HAMA was carried out by an EDC/HOBt coupling reaction in acidic conditions (∼pH 4.75). The results of GH synthesis and its expansion can be observed in Fig. S4–S8 (ESI†), shown in the 1H-NMR spectra. The conjugation of GH in HAMA was confirmed by the presence of distinctive peaks at 6.99 ppm due to aromatic protons of GA in the 1H NMR spectrum (Fig. S9A and B, ESI†). The degree of GA modification was ascertained to be ∼10 mol% with respect to the repeated disaccharide units of HA. In addition, HAMA was synthesised at a modification degree of 30% of the disaccharide repeating units, which was also determined by 1H-NMR (Fig. S10A and B, ESI†). The modification was determined by integrating the olefinic peaks of the methacrylate group at 6.13 and 5.70 ppm against the methyl peak of the N-acetyl of HA at 1.95 ppm.
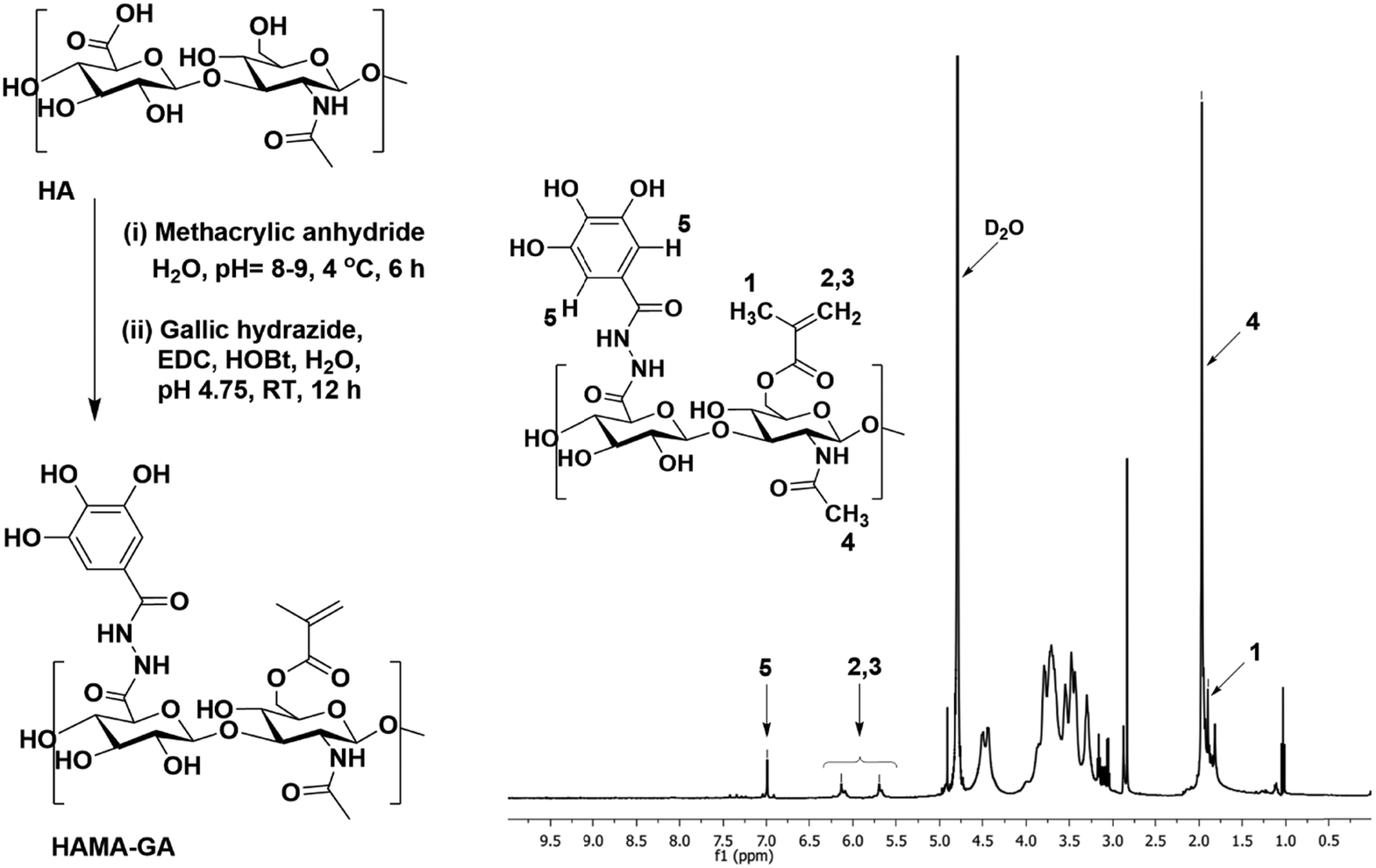 | ||
| Fig. 2 The synthesis pathway and 1H-NMR spectra of catechol-modified HAMA. | ||
Phase transition of HAMA–GA hydrogel under the influence of pH and Fe3+
The gelation condition for HAMA–GA hydrogels for phase transition was optimised by varying the pH levels and the concentration of Fe3+. To observe the effect of pH in HAMA–GA hydrogel in the absence of Fe3+, the sol–gel phase transition was investigated from pH 3 to 9 using rheological measurements for shear viscosity and zeta-potential for anionicity indication.15 The sol–gel phase transition was apparent through visual analysis. The gelation was observed as the HAMA–GA solution, initially at pH 3, shifted to pH 9 (Fig. 3). Fig. 3A shows that the transparent colour of the HAMA–GA solution at acidic conditions turned to a translucent pink colour at the neural conditions, and the initial solution changed to a yellow colour at the basic conditions due to the highest oxidation of catechol. According to the literature, catechol-conjugated polymers can form hydrogels in basic pH conditions with oxidising agents such as sodium periodate (NaIO4) or horse-radish peroxidase, exhibiting colour shifting from a clear to dark, deep red colour.33 On the other hand, the results showed that HAMA–GA mixtures undergo the sol–gel transition under basic conditions without using an oxidising agent through the catechol oxidation because of the formation of quinone species in gallol moieties.26 This quinone species has remarkable properties for engaging in hydrogen bonding interactions with other gallol moieties present in the system.35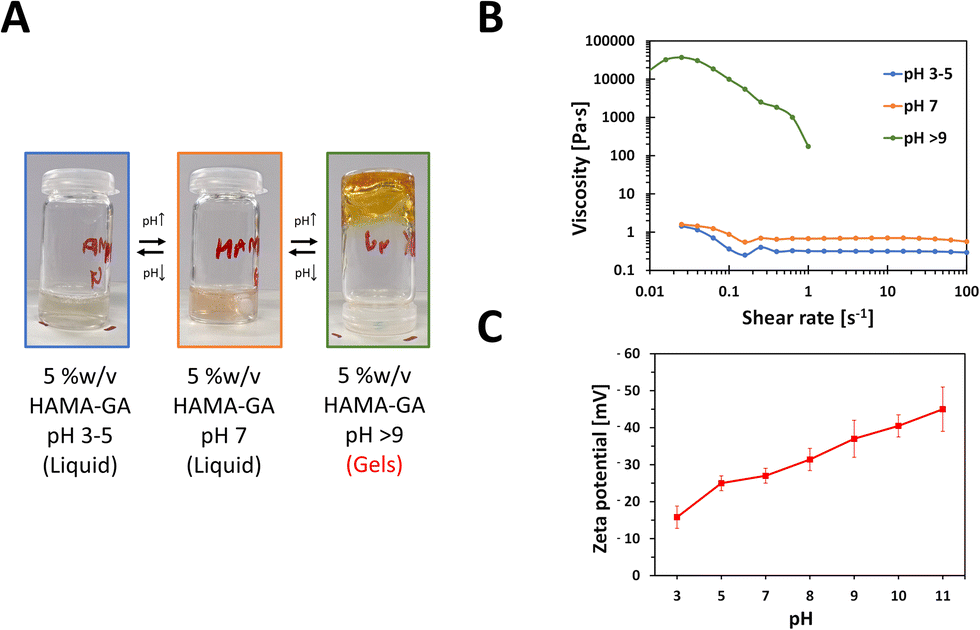 | ||
| Fig. 3 Gelation kinetics of HAMA–GA are influenced by a range of pH conditions, from acidic to basic. (A) Visual inspection of HAMA–GA gelation in vials. (B) Flow tests of HAMA–GA with progressively increasing shear rates to assess shear viscosity changes. (C) Zeta potential values of HAMA–GA with varying pH levels. | ||
To further characterise the phase transition of HAMA–GA gel, the HAMA–GA formulations at pH 3–5, 7 and > 9 were measured via a rotational rheometer with flow mode to determine the pH-dependent viscosity of the gels. Fig. 3B shows that the viscosity of the mixtures at acidic (pH 3–5) and neutral (pH 7) pH was less than 1 Pa s, exhibiting Newtonian behaviour. In contrast, the gels at basic condition (pH > 9) displayed higher viscosity (more than 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Pa s), and the sample was slipped out of geometry due to the gel-like properties, which were confirmed via frequency sweep of oscillatory measurement (G′ > G′′).
000 Pa s), and the sample was slipped out of geometry due to the gel-like properties, which were confirmed via frequency sweep of oscillatory measurement (G′ > G′′).
The gelation kinetics were also confirmed by determining the attractive interaction between HAMA–GA molecules. In general, the strong anionic attribute of HA is primarily from the carboxylate group with its backbone.10 The catechol interaction has strongly influenced the gelation kinetics due to the electrostatic interaction. The increasingly negative values of zeta potential observed in the HAMA–GA system indicate a greater extent of anionic behaviour as the pH increases (Fig. 3C).
The stoichiometry of catechol–metal complexes is explained as mono-, bis- and tris-complexes, and it is controlled by pH levels and Fe3+ concentrations.10 The specific pH ranges in catechol–metal complexed hydrogels are required to surpass the sol–gel point: mono-(acidic), bis-(neutral) and tris-(basic) complexes. The concentration of Fe3+ in the system ranged from 0.125, 0.25 to 0.5% w/v and was controlled to ensure that it did not exceed 0.5% w/v to prevent Fe3+ precipitation. The optimisation of the Fe3+ concentration was essential to maintain the stability and homogeneity of the hydrogels. The phase transition mechanism of catechol–Fe3+ complexes was verified by diluting HAMA–GA solution with FeCl3 solution with an increasing pH. The initial characterisation of the samples was to analyse the effect of Fe3+ in the system at a constant pH of 3–5. The previous studies reported that catechol-modified polysaccharides could form into a gel at acidic pH with sufficient curing time to initiate the coupling reaction.27,33 Similarly, Fe3+-catechol coordination of HAMA–GA (HAMA–GA 2.5Fe) also gelated at acidic conditions with optimum Fe3+ concentration (0.25% w/v of FeCl3) and curing time; however, HAMA–GA 5Fe started to precipitate at 0.5% w/v of FeCl3. The colour variation of HAMA–GA–Fe mixtures was influenced by the Fe3+ concentrations, as it turned from a clear to a darker colour corresponding to higher metal coordination (Fig. 4A). The stoichiometry of catechol–metal complexes was verified by UV-vis spectroscopy. The absorption peak of the mono-complexes appeared around 615 nm in all HAMA–GA–Fe mixtures (Fig. 4B), but it became more distinctive at higher concentrations of Fe3+.
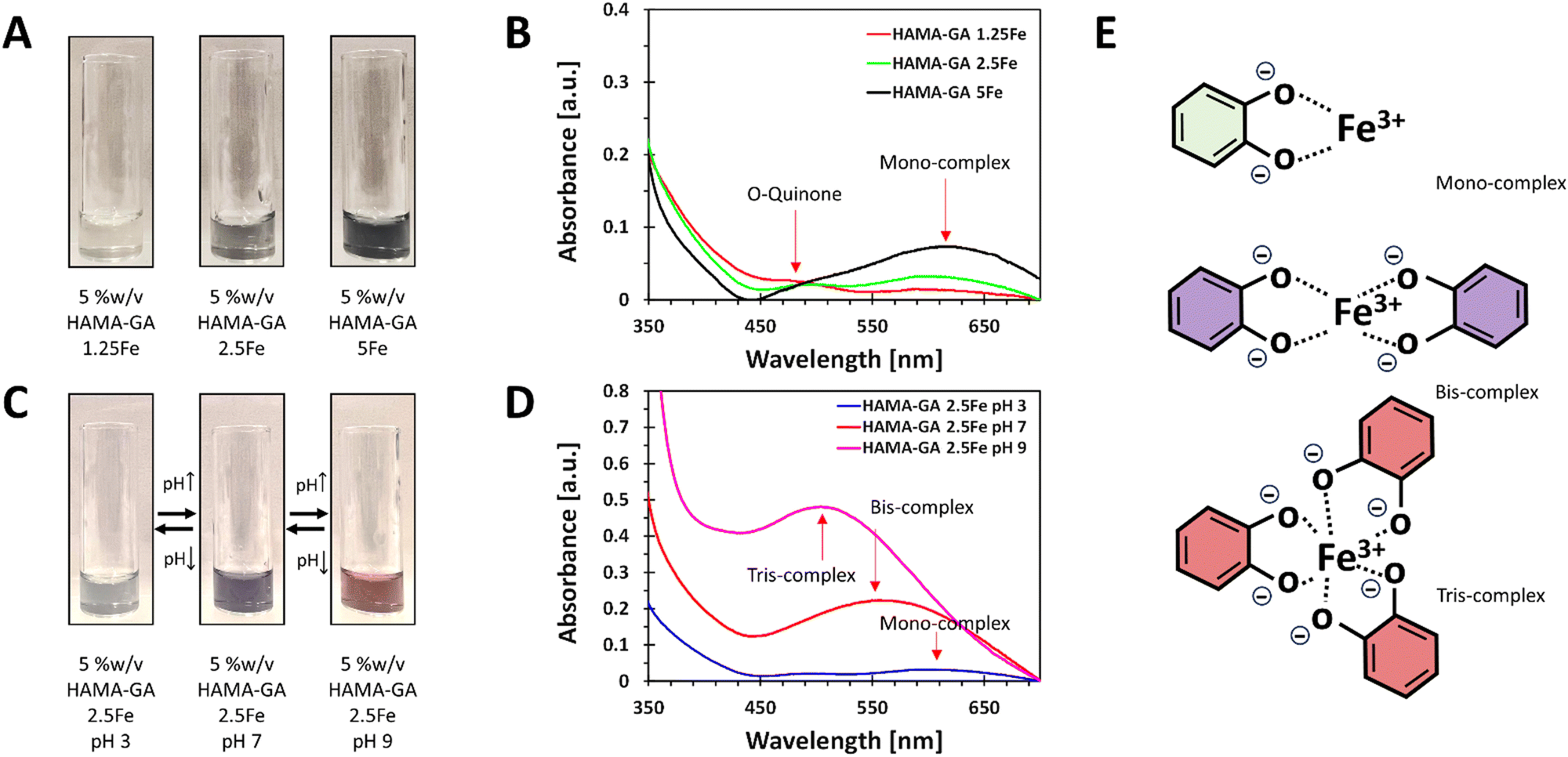 | ||
| Fig. 4 The quantitative analysis of catechol-induced gelation in HAMA–GA–Fe3+. (A) and (B) UV-vis spectra and the corresponding colour of HAMA–GA samples in vials at different Fe3+ concentrations (0.125, 0.25 and 0.5% w/v) under pH 3 (acidic). (C) and (D) UV-vis spectra and the colour of HAMA–GA 2.5Fe samples at various pH conditions (3, 7 and 9). (E) HAMA–GA hydrogels at acidic, neutral, and basic conditions, induced by different Fe3+ concentrations to form mono- (green), bis- (violet) and tris-complex (red) structures. | ||
The assessment of the phase transition of HAMA–GA–Fe mixtures (0.125, 0.25 and 0.5% w/v of FeCl3) was performed by progressing the pH levels (pH 3, 7 and > 9) in the diluted solutions. The initial colour at pH 3 was observed as a clear grey colour and slowly turned dark purple upon increasing pH to 7, and finally, it became a deep red colour when it reached pH 9 (Fig. 4C). This phenomenon has been previously reported, demonstrating a change in stoichiometry from mono to bis to tris as the pH increases.36 UV-vis spectroscopy confirmed the dominance of mono-, bis-, and tris-catechol–Fe3+ complexes, at pH ∼ 3, ∼7 and > 9. The shift in absorbance has been reported elsewhere.37Fig. 4D shows the hypsochromic shift of the absorption spectra from mono- (615 nm) to bis- (550 nm) and tris- (490 nm) under increasing pH.38 However, Fig. 4B and D show that the o-quinone absorption peak was clearly observed in mono-complex because the catechol-group formed bis- and tris-complexes under basic pH, reducing the free phenolic hydroxy groups to form o-quinone.13
To characterise further the phase transition of catechol–Fe3+ hydrogels, the HAMA–GA 2.5Fe hydrogels were prepared without incubating them at acidic, neutral, and basic pH to form mono-, bis- and tris-complex, respectively (Fig. 5A). The fresh sample mixtures were then subjected to oscillatory measurement via frequency sweep. HAMA–GA 2.5Fe at pH ∼ 3 and ∼7 displayed gel-like properties (G′ > G′′). On the other hand, the mixtures at pH ∼ 7 showed viscous properties (G′′ > G′) at low frequency, which then reached sol–gel transition (1 rad s−1) and exhibited viscoelastic solid (G′ > G′′) as the frequency increased, which behaved according to the Maxwell viscoelastic model.14 The rapid formation of mono-complex hydrogels at acidic pH can be attributed to the immediate complexation driven by the abundant presence of aromatic hydroxyl groups, in contrast to DOPA-functionalized hydrogels.33,39 Even though the tris-complex hydrogels at basic pH formed a true hydrogel (G′ > G′′) rapidly at low frequency, the instant cohesive interaction of tris-coordination caused inhomogeneity of the gel structure. Compared to mono- and bis-coordination, those gels displayed slower gelation, providing the time to form the gels homogeneously. The storage modulus of the samples (starting from 1 rad s−1 frequency) increased with increasing pH from 47.2 Pa, 750.7 Pa to 987.1 Pa at pH 3, 7 and 9, respectively, due to the strong hydrogen bonding with multiple-metal catechol complexes.
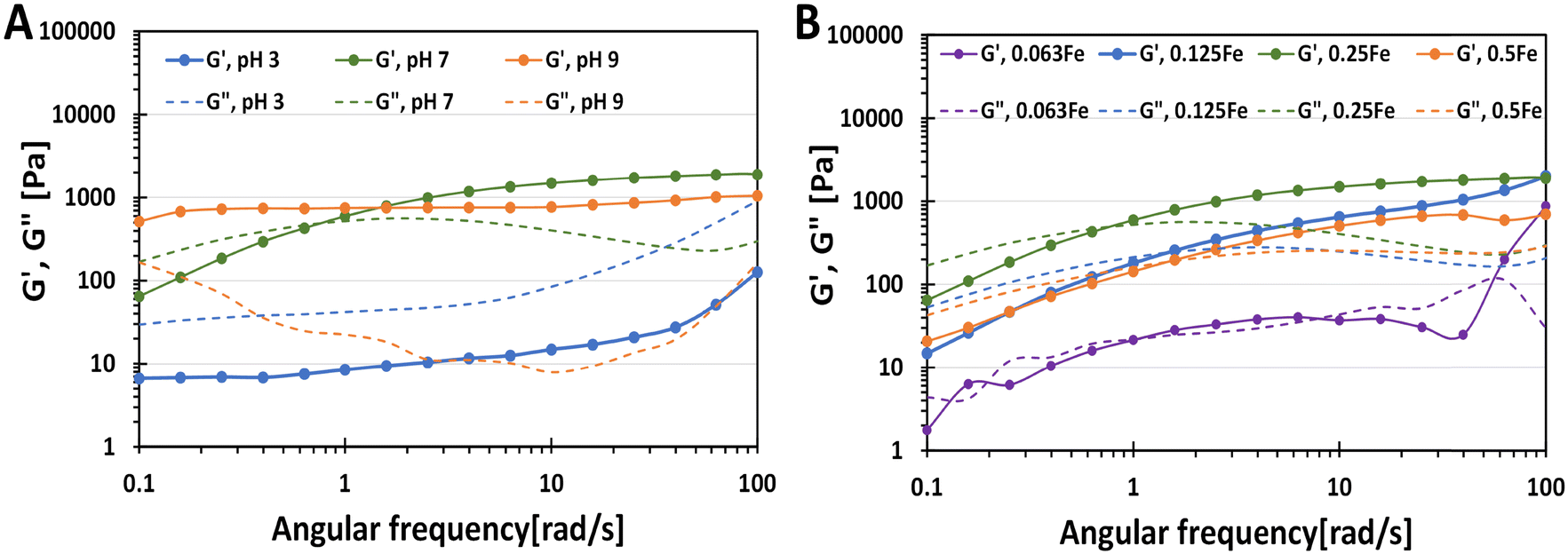 | ||
| Fig. 5 The rheological properties of sol–gel transition of HAMA–GA at different pH (A) and Fe3+ concentration (B). (A) HAMA–GA hydrogel with pH 3, 7 and 9 at 0.25% w/v of FeCl3, (B) HAMA–GA hydrogel with 0.063, 0.125, 0.25 and 0.5% w/v of FeCl3 at pH 7. | ||
To study the phase controllable hydrogels via varying Fe3+ ion concentrations, Fe3+ induced hydrogels were prepared at different concentrations of FeCl3 at pH 7. The characterisation was also performed via oscillatory measurement (frequency sweep). The sample mixtures were also prepared without incubation to prevent excessive crosslinking in the hydrogel structure. As shown in Fig. 5B, the gel samples obeyed the Maxwell viscoelastic model, exhibiting viscoelastic fluid (G′′ > G′) at low frequency and viscoelastic solid at high frequency (G′ > G′′) because bis-complex formed slower than mono-complex. The storage modulus of bis-complex hydrogel was enhanced by increasing FeCl3 concentration due to oxidative crosslinking with Fe3+. The gelation time was reduced by utilising a higher concentration of FeCl3. Moreover, the additional catechol modification in HA could lead to higher crosslinking in the hydrogel, reducing the gelation time.26
The flow behaviour of HAMA–GA with various Fe3+ concentrations
HAMA–GA hydrogels were pre-screened under three conditions: acidic, neutral, and basic. These conditions were induced by varying concentrations of Fe3+ to facilitate the formation of mono-, bis-, and tris-complex structures. 5% w/v of HAMA–GA mixture was prepared at pH ∼ 3, ∼7 and ∼9 with various concentrations of FeCl3 (0.125, 0.25 and 0.5% w/v), termed as HAMA–GA 1.25Fe, HAMA–GA 2.5Fe and HAMA–GA 5Fe, respectively (Fig. S11, ESI†). The HAMA–GA 2.5Fe mixture remained dark viscous fluid at pH ∼ 3 and later gelated after curing for 30 min. The HAMA–GA 1.25Fe mixture at pH ∼ 3 remained in a sol state, indicating that the metal coordination bonding was insufficient to establish a hydrogel network. Before curing, the mixtures formed highly elastic hydrogels with a dark purple colour when the pH reached 7. A stiff gel immediately formed when the pH reached 9.To characterise the injectability of catechol–Fe3+ hydrogels via rheological measurement (flow mode), the samples at gel state, including mono-complex (HAMA–GA 2.5Fe), bis-complex (HAMA–GA 1.25, 2.5 and 5Fe) and tris-complex (HAMA–GA 5Fe), were selected. In general, non-covalent interactions have been used for self-healing injectable hydrogels due to their reversibility and can reform after breaking.21 The results showed that pH level directly affected gels' viscosity due to hydrogen bonding, which turned them into true gels at basic pH (tris-complex). The study emphasised the relationship between the coordination degree (mono, bis and tris), the viscosity and the injectability. The Carreau model and Cox–Merz rule were used to evaluate the influence of Fe3+ concentration on the shear-thinning properties. The Cox–Merz rule was applied to the small amplitude oscillatory shear test, effectively reducing material escape from the plate. The measurements in flow mode yielded an edge failure, resulting in the material spilling from the rheometer plates.40,41Fig. 6A and B show that the shear thinning observed in flow experiments aligned with the results predicted by the Cox–Merz rule, indicating the model's applicability. In general, zero shear viscosity is used to characterise the viscosity of complex fluid.19 The flow behaviour results show that higher values of zero shear viscosity correlated with lower relaxation time and extended the Newtonian plateau in all tested samples, associated with coordination degree and enhancement in polymeric chains.40 The highest zero shear viscosity was observed in tris-complex hydrogels (HAMA–GA 2.5Fe at basic pH). While zero shear viscosity between two bis-complex hydrogels (HAMA–GA 2.5Fe and HAMA–GA 5Fe at neutral pH) displayed comparable values in the Newtonian plateau, but HAMA–GA 5Fe was slightly lower. On the other hand, HAMA–GA 0.63Fe hydrogels at pH 7 exhibited slight shear-thickening, in which the viscosity increased sharply and then decreased slowly in a shear-thinning fashion. In general, shear-thickening behaviour occurs in colloidal solution, which is affected by phase volume, dispersity of particles, particle distribution and particle shape.42 In this case, the degree of coordination in metal complex hydrogels varied with the applied shear force, resulting in the abruption of crosslinking density, which leads to shear-thickening, particularly at the yield point. According to the literature, the viscosity is influenced by the ligand exchange rate constant, in which the faster exchange rate occurs at basic pH.16 Due to three hydroxyl groups in gallic acid, the availability of hydroxyl groups is higher for ligand exchange, leading to higher viscosity.16,43 As shown in Fig. 6A and Table S1 (ESI†), the mono-complex hydrogels exhibited shear-thinning behaviour, showing a decrease in viscosity with an increase in shear rate (n = 0.4). The mono-complex HAMA–GA 2.5Fe also displayed a clear yield point (∼10 Pa), indicating the high injectability in this group. Furthermore, the relaxation time in hydrogels changed with the degree of coordination and FeCl3 concentration, which allowed the macromolecule to relax more in mono-complexes and at lower FeCl3 concentrations. The shear-thinning behaviour in bis-complex hydrogels (Fig. 6B) was observed at a shear rate above 1 s−1. On the other hand, the higher concentration of FeCl3 resulted in an increase in the range of shear rate to above 10 s−1, revealing a more extended Newtonian plateau regime. Consequently, the catechol–metal coordination complexes initiated the formation of a strong crosslinked network, which could not break after applied high stress and showed less relaxation, leading to poor injectability of hydrogels. Moreover, the results in Fig. 6C, F, and I show that tris-complex hydrogels formed true hydrogels and lost the injectability as observed in Fig. 6B (shear-thickening) because the samples underwent rapid oxidation after the pH reached the basic conditions and did not have free hydroxyl group available for metal coordination.
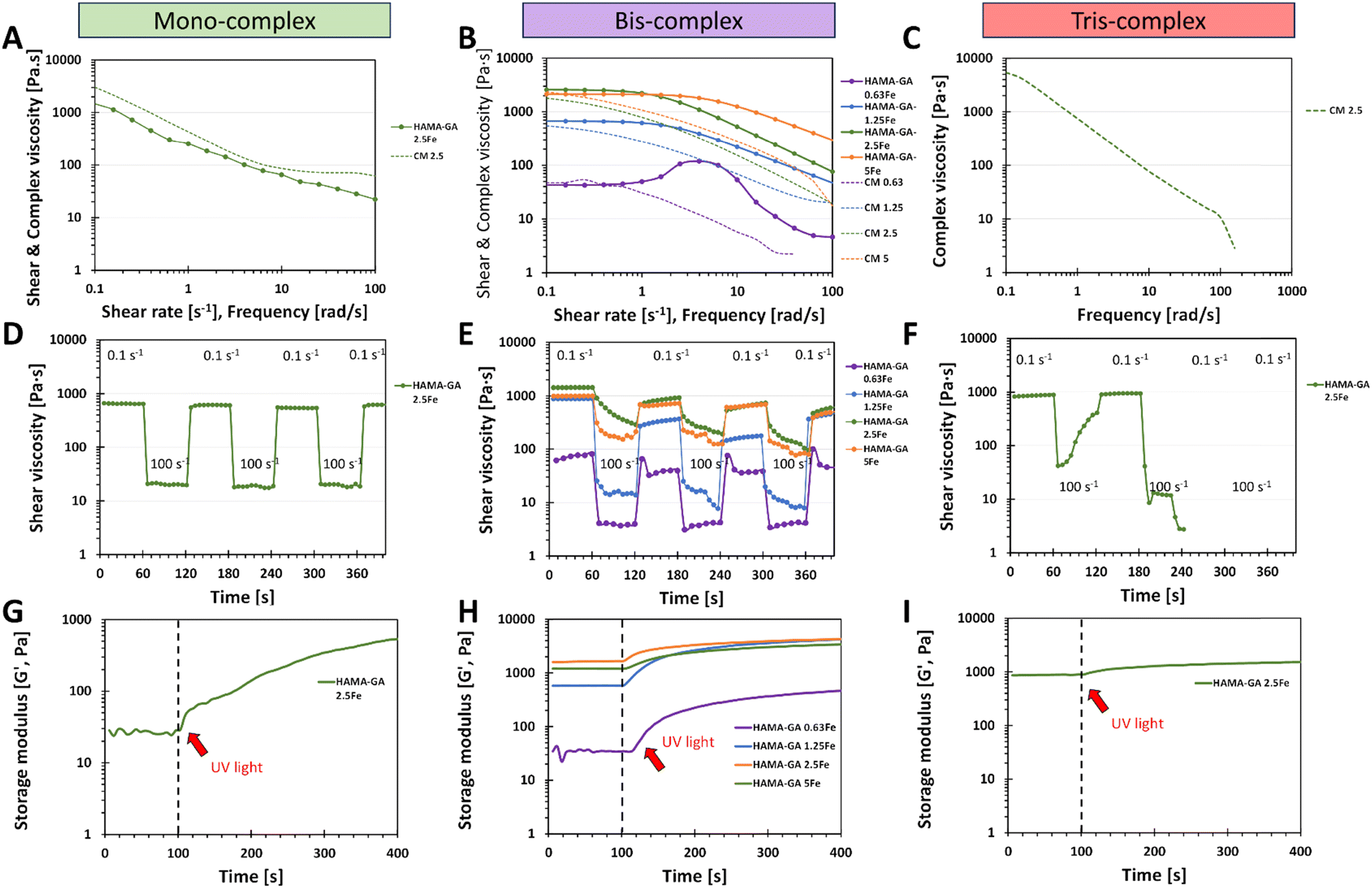 | ||
| Fig. 6 The overview results of rheological properties: shear-thinning, shear viscosity (A–C), recovery behaviour (D–F) and gelation time (G–I), to determine the injectability of HAMA–GA hydrogels (before photocrosslinking) under the influence of pH and FeCl3 denoted as mono-, bis- and tris-complexes. CM indicates the complex viscosity and frequency plot obtained from the Cox–Merz rules. | ||
Recovery behaviour testing was used to assess the injectability of tested samples after they were extruded from the needle. Each sample was subjected to the shear rate ramp test, which contained seven cycles of low (0.1 s−1) and high shear rate (100 s−1). The low shear rate mimicked the hydrogels' state while resting in the syringe, whereas the high shear rate represented the conditions experienced during injection. After that, another low shear rate mimicked the hydrogels' state at the injected site to measure the recovery of the hydrogels. Fig. 6F shows that it was impossible to measure the recovery behaviour of tris-complex hydrogels because this composition formed true hydrogels, and they slipped out from the geometry due to high rotational movement. Similar results occurred in bis-complex hydrogels (Fig. 6E); the samples formed into hydrogels, but the viscosity did not decrease due to the strong cohesion force and chain entanglements of hydrogels. However, the mono-complex hydrogels (Fig. 6D) showed excellent recovery behaviour and survived all seven repeating cycles and the viscosity of samples recovered to the initial values. These results could also be considered in the initial material design screening phases, especially in choosing crosslinking strategies.
The results of in situ photo rheology (Fig. 6G–I) revealed that these injectable hydrogels can be cured with UV light to form covalent bonds utilising a dual-crosslinking (photocrosslinking due to hyaluronic acid methacrylate and catechol–metal coordination complexes due to gallic acid–Fe3+ interaction). However, Fig. 6I shows the incompatibility of catechol motifs and photocrosslinking in a dual-crosslinking system. The photocrosslinking was disrupted in tris-complex samples because the high oxidative state in catechol–metal hydrogel interfered with the free-radical polymerisation of methacrylate groups, which eventually reduced the crosslinking density of photocrosslinked hydrogels and hindered photopolymerization, leading to a reduction in photocrosslinking in situ.6
3D printing of HAMA–GA hydrogels
The shear-thinning behaviour of HAMA–GA hydrogels was induced by suitable pH level and FeCl3 concentration. Based on pre-screening results (Fig. S11, ESI†) and flow behaviours (Fig. 6), shear-thinning properties and printed shape fidelity varied based on the degree of coordination in hydrogels and were influenced by pH level and FeCl3 concentrations (Fig. 7A and B). Specifically, the lattice model was chosen to observe the printing quality of different ink formulations, and its usage has been reported previously.30,32,44 The printability results were categorised into three groups and coloured red, blue, and green. The red indicates ‘unextrudable’, meaning the ink turned into true hydrogel inside the cartridge and could not be extruded through the nozzle. The green label represents ‘printable condition’, meaning that the deposited filaments were uniform and could stack into grid structures. However, the circularity of the pores inside the grids was not evaluated. The blue colour means ‘too liquid’, where the extruded filament could not maintain its shape after deposition on the substrate, leading to a pool of material instead of distinct filaments. When ink formulations do not reach the printability criteria due to under or over-gelation, the shape of the pores inside the grids deviates from the original CAD model, whereas high printability of the inks can maintain uniform filament during and after printing.45 As shown in Fig. 7B, the initial printable 5% w/v HAMA–GA inks were prepared at pH 5 and with 0.063% w/v of FeCl3, named HAMA–GA 0.63Fe, providing sufficient shape fidelity during extrusion in the air (Fig. S12, ESI†) and deposition on the substrate. In detail, the circular pores inside the printed grid structures were observed in the following ink formulations: HAMA–GA 0.63Fe at pH 7 and HAMA–GA 1.25Fe at pH 5. The ink prepared at pH 5 and low FeCl3 concentration (>0.063% w/v) exhibited Newtonian behaviour, leading to poor shape fidelity and spread after printing. On the other hand, inks prepared at high pH (<9) and high concentration of FeCl3 (<0.125% w/v) underwent excessive crosslinking and transformed into true hydrogels, which were unable to extrude from the nozzle. To achieve good printability, HAMA–GA was prepared at pH 7 with 0.125% w/v of FeCl3. The uniform flow was observed throughout the printing process (Fig. S12, ESI†), resulting in high shape fidelity, which could be maintained in 3D construct. In contrast, HAMA–GA 0.63Fe at pH 7 collapsed after printing at higher layers (Fig. S12, ESI†). It is important to note that three hydroxyl groups on the benzene ring of gallol interact with metal ions better than DOPA (two hydroxyl groups), leading to a stronger cohesive force between a polymeric network of hydrogels due to hydrogen bond formation and a metal complex.46 Due to their self-healing properties, the irregular filaments produced by HAMA–GA 0.125Fe returned to their original moduli after printing, ultimately forming a smooth printed tubular structure, as also shown in Fig. S12 (ESI†).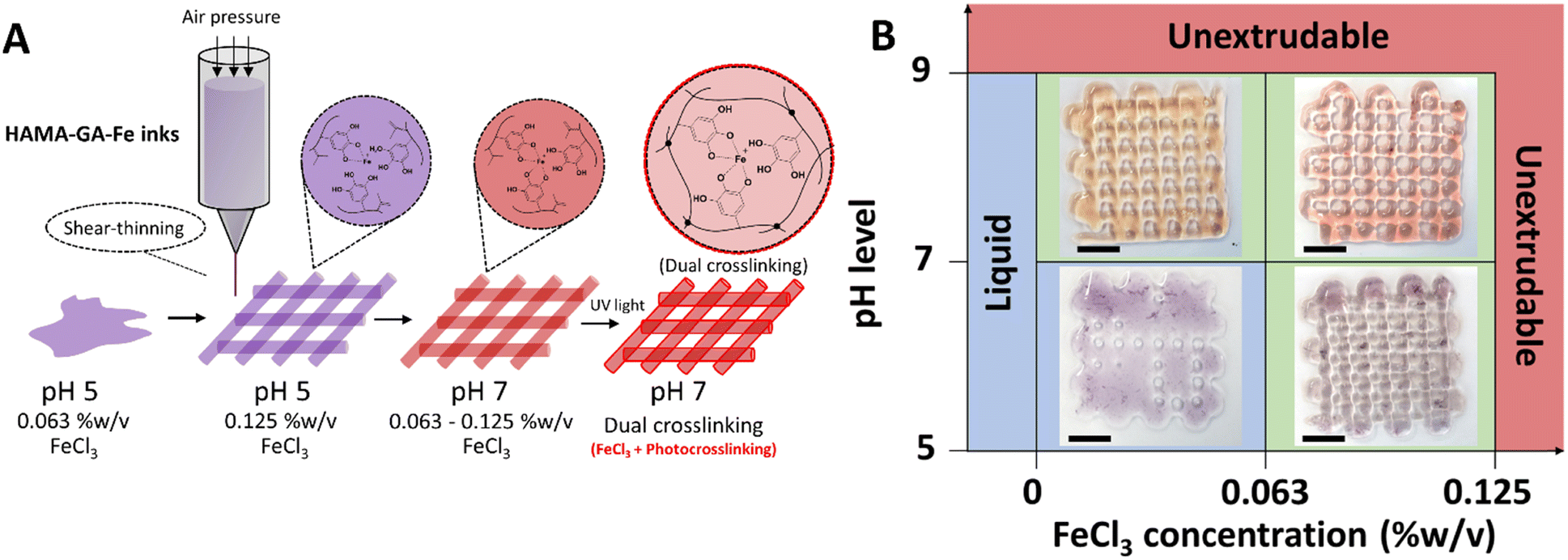 | ||
| Fig. 7 3D printing of HAMA–GA–Fe biomaterial inks. (A) The illustration of 3D printed structures, which the printability of inks depends on the sol–gel transition caused by pH level (5 and 4) and FeCl3 concentrations (0.063 and 0.125% w/v). The stability of the printed structures was enhanced by photocrosslinking after printing. (B) Printability of HAMA–GA by varying pH level and FeCl3 concentrations. Scale bar = 1 cm. | ||
Rheological properties of dual-crosslinked hydrogels with photocrosslinking
The viscoelastic properties of dual-crosslinked HAMA–GA–Fe hydrogels (catechol–metal complexes and photocrosslinking) were characterised by oscillatory measurement with amplitude sweep. The large amplitude oscillatory shear (LAOS) measurement was used to determine the behaviour of dual-crosslinking hydrogels because the microstructure of the complex hydrogel network behaves differently under large deformation.47Fig. 8 shows that all hydrogel samples display the constant moduli and distinct crossover point between G′ and G′′ between 1–1000% of strain. Mono-complex hydrogels (Fig. 8A) exhibit strain overshoot phenomenon, as G′ decreases but G′′ increases and then decreases after 100% of strain.48 According to the literature, the strain overshoot occurs in highly extendable structure hydrogels formed by the electrostatic repulsion from the charged group on the side chains (hydrogen bonding).49 When the hydrogel is exposed to a high strain, the complex structure resists against the deformation, where the overshoot of G′′ occurs.47 Although mono-complex hydrogel formed a structural complex due to the presence of metal ions, mono-complex hydrogels inherit a weaker network than bis-complex hydrogels (Fig. 8B), which results in lower critical strain and storage moduli. According to the literature, the extended linear viscoelastic region (LVR) region of bis-complex hydrogels corresponds to chain structure,13,19,33 which is related to inter- and intramolecular interactions between catechol–metal ions and vicinal catechol groups. These results highlight the importance of the additional gelation process induced by pH conditions; the pH level plays a vital role in catechol and HA chain arrangement, in which unreacted catechol in HA at acidic pH forms a weaker hydrogel network. An increase in storage modulus in mono- and bis-complex hydrogels corresponded to an increase in the coordination state. On the other hand, tris-complex hydrogels (Fig. 8C) also showed the strain overshoot phenomena. These hydrogels display the weakest critical strain and storage moduli compared to other gels because they underwent a high oxidation process and formed hydrogen bonding (o-quinone), constructing a solid hydrogel network at basic pH.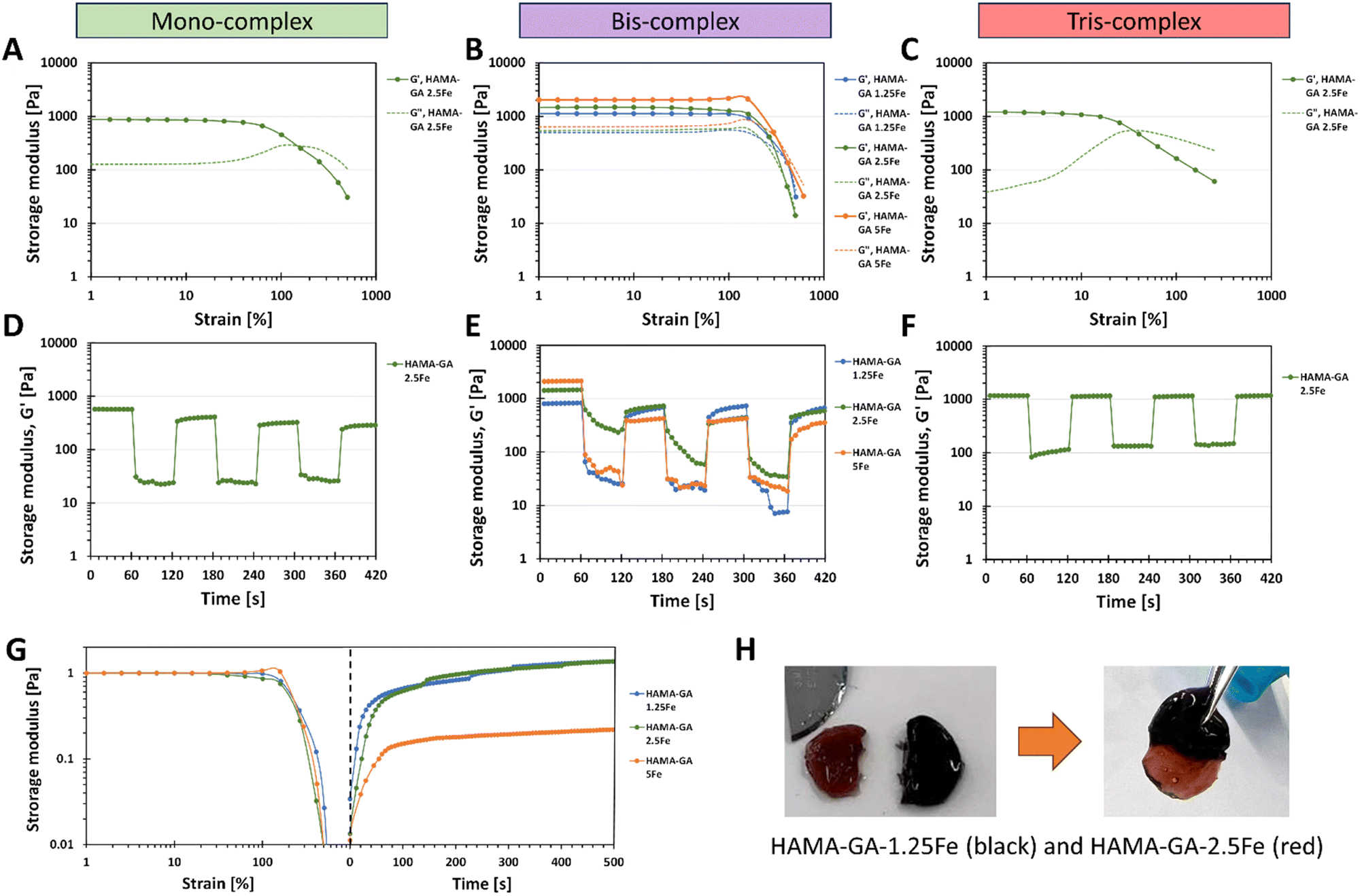 | ||
| Fig. 8 The viscoelasticity measurement of HAMA–HA with FeCl3 after photocrosslinking is denoted as mono-, bis-, and tris-complexes to verify the self-healing properties. (A) and (D) mono-complex HAMA–GA 2.5Fe at pH 3, (B) and (E) bis-complex HAMA–GA 1.25, 2.5 and 5Fe at pH 7, (C) and (F) tris-complex HAMA–GA 2.5Fe at pH 9. (G) Recovery of stiffness after structural failure at critical strain (500%). (H) The image represents the cut and healing methods to verify rapid self-healing properties between HAMA–GA 1.25Fe (black) and HAMA–GA 2.5Fe (red). | ||
Consequently, this strong network formation limits the availability for further bonding with metal ions. This is due to three hydroxy groups on the phenolic building block of gallol, which has a high reactivity in forming non-covalent bonds (hydrogen bonds). Furthermore, the additional photocrosslinking in tris-complex hydrogels increased the crosslinking density of hydrogel, which might accelerate the oxidation process.
Self-healing properties of dual-crosslinked hydrogels with photocrosslinking
We explored the combination of non-covalent and covalent crosslinking in HAMA–GA–Fe, which can influence the self-healing properties of hydrogels (reformation after breaking). The self-healing properties of HAMA–GA–Fe hydrogels were investigated by strain ramp test. Fig. 8D–F show the seven low and high strain cycles. The high strain was chosen according to the critical strain of the hydrogel samples, which induced the rupture of hydrogels. However, some hydrogels were splashed, causing the loss of sample volume due to the large oscillatory movement and incomplete storage moduli after high strain was observed.50 The results show that the storage moduli in all samples were restored. Still, bis-complex hydrogels could not recover their initial storage moduli entirely due to the loss of hydrogel volume during measurement. Despite the incomplete recovery after the first cycle in Fig. 8E, the moduli were restored progressively until the end of seven cycles, indicating the potential for self-healing properties. To confirm the rapid self-healing ability of hydrogels (Fig. 8H), the two hydrogel samples were cut into two pieces (dark gel indicates HAMA–GA 1.25Fe, and red gel indicates HAMA–GA 2.5Fe). Then, the two separate hydrogels were attached to each other. The two gels were merged instantly, which is attributed to the formation of the junctions between cut surfaces.16,45 Compared to other catechol-based hydrogels such as DOPA, the self-healing time was longer due to the lower availability of the hydroxyl group, reducing bond strength and reactivity. Another measurement was demonstrated by a two-step test using the amplitude sweep and time sweep. In Fig. 8G, the strain sweep was applied to the hydrogel sample in the range of 1 to 1000%, after which the samples were allowed to restore their initial storage moduli under 0% strain for 500 s. Only bis-complex hydrogels were selected in this test because weak hydrogels completely formed into true hydrogels with a stable network after photocrosslinking. In contrast, mono-complex was too soft, and tris-complex was too stiff. The results also showed that the storage modulus of HAMA–GA 5Fe was partially recovered to some extent after the high strain was removed due to the loss of sample during measurement. HAMA–GA 5Fe underwent higher crosslinking density due to the higher concentration of metal ions, resulting in stiffer hydrogels. However, all the gels exhibited rapid recovery after 50 s of observation. The reversible coordinate bonds recognised the gel network, leading to the rapid recovery of hydrogels.20 We assume that the rapid recovery of bis-complex hydrogels after high strain depends on the degree of coordination and metal–ligand association (pH conditions and FeCl3 concentration), which induced reformation at the molecular scale.Stress relaxation of dual-crosslinked hydrogels
Fig. 9 shows the normalised stress when the 5% strain was applied. This strain value was selected because the damage of phase-separated structure was anticipated to occur at higher strain values.51Fig. 9A–C show that hydrogels formed with less concentration of FeCl3 and at acidic pH exhibited faster relaxation. Our findings are consistent with the outcomes of a prior study, which suggests that stress relaxation in hydrogels is based on the crosslinking density polymers, which in this case was influenced by variations in metal ion concentrations and pH levels.52 The decreases in the stress are due to the breaking of non-covalent interactions, and the more extended stress relaxation comes from the high elasticity in the hydrogel network formed by catechol–metal coordination and photocrosslinking. Furthermore, the metal ion concentrations affected the initial elastic moduli of hydrogel samples, in which the storage moduli values of HAMA–GA-5Fe at neutral pH and HAMA–GA 2.5Fe at basic pH were equal. The relaxation time was determined when the stress value was relaxed to half (τ1/2). Specifically, HAMA–GA 1.5Fe (bis-complex) and HAMA–GA 2.5Fe (mono-complex) relaxed fastest compared to all hydrogel samples (∼1 s), followed by bis-complex of HAMA–GA 2.5Fe, HAMA–GA 5Fe, and tris-complex of HAMA–GA 2.5Fe (∼3, ∼10 and ∼20 s respectively). The previous studies show the importance of stiffness and stress relaxation of hydrogels.31 Studies show that highly crosslinked gels have limited stress relaxation, glycosaminoglycans and collagen production, cartilage matrix distribution, and chondrocyte proliferation.53,54 Considering matrix stress relaxation as a fundamental signal is crucial in understanding the basics of cell–ECM interactions and the underlying biophysics of mechanotransduction since most physiological extracellular matrices show some degree of stress relaxation. These findings suggest using stress relaxation as a design parameter for materials in tissue engineering, particularly in regulating cell proliferation and promoting bone regeneration.31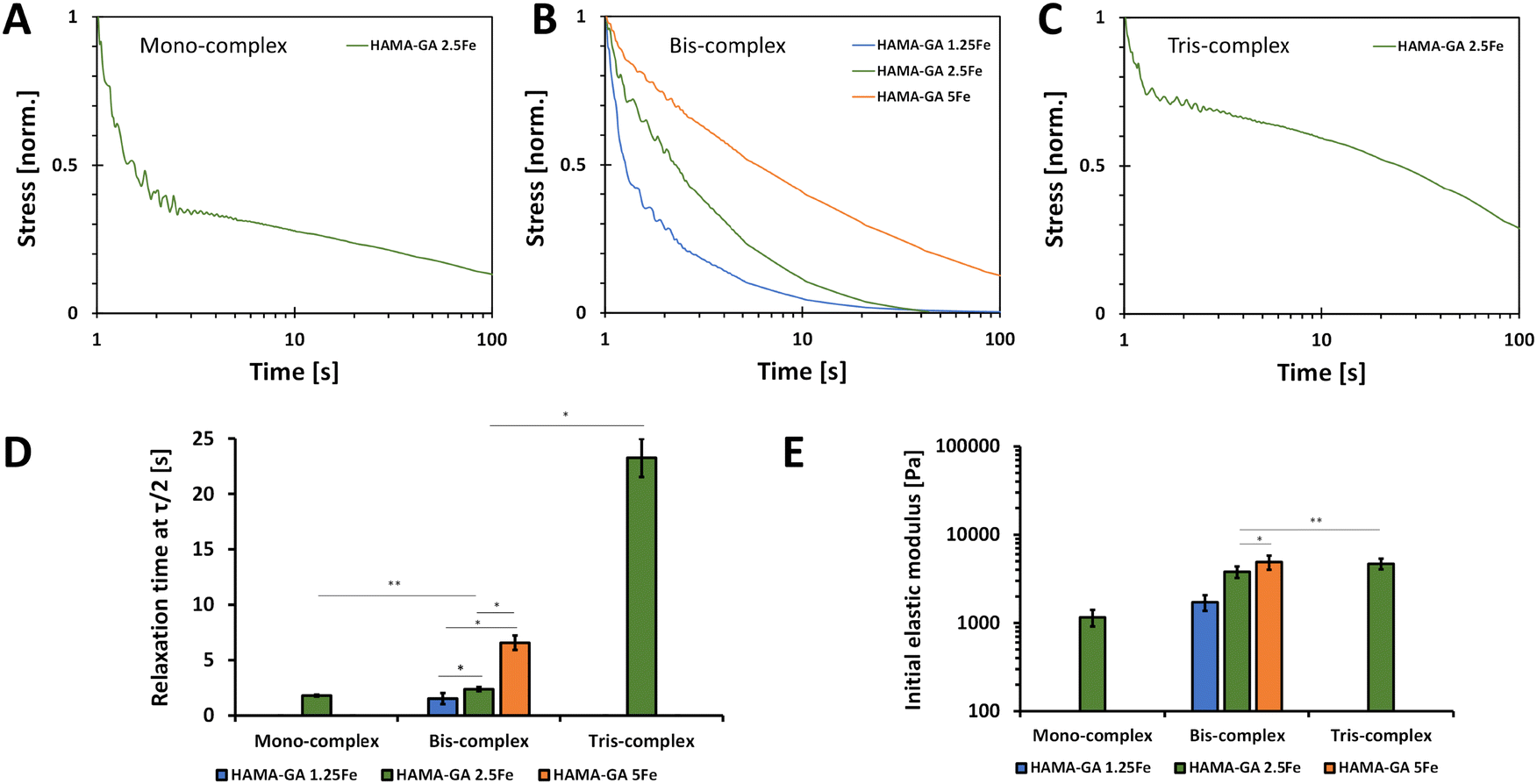 | ||
| Fig. 9 The stress relaxation profiles of mono-, bis- and tris-complex hydrogels with different concentrations of FeCl3. (A)–(C) The time scale stress relaxation, (D) relaxation time at τ1/2, (E) initial storage modulus of mono-, bis- and tris-complex hydrogels (n = 4). | ||
pH-dependent stability of photocrosslinked hydrogels
The swelling kinetics of hydrogels were analysed by determining the change in weight over time. As shown in Fig. 10, hydrogel samples were immersed in a DPBS solution within pH ranges 3, 5, 7, 8, and 9, representing the skin in different conditions. Specifically, pH 3–5 designates healthy skin, pH 7 designates acute wound infection, and pH 8–9 means infectious skin.34 We hypothesised that FeCl3-induced HAMA–GA at acidic pH (pH 3–5) would weaken the properties of metal–catechol complexes in the hydrogel network, resulting in high water uptake and loose hydrogel network. Whereas the hydrogel samples at neutral and basic pH would be more stable because catechol–metal complex hydrogels generally form coordinate bonds at pH 8 or above, similar to natural wet adhesion in mussel byssus cuticle at seawater pH ∼ 8.13 HAMA30 hydrogels (Fig. 10A) were used as a control, exhibiting less swelling (swelling ratio of 1.5) and being more stabilised at all pH ranges throughout observation time. HAMA30 at pH 9 showed the highest rate of swelling (swelling ratio of 3) compared to other pH levels. Fig. 10B–D show that the swelling ratio of HAMA–GA–Fe hydrogels significantly increased as the pH of the medium increased, and the hydrogel disintegrated at pH 9, which contrasted with the hypothesis. On the other hand, the hydrogels at acidic pH tended to swell during the first three days and then dissolved slowly over time until 15 d (Fig. 10B–D). The data indicates that the swelling behaviour of hydrogels responded to the environmental pH. The HAMA–GA 2.5Fe at pH 8 showed the highest swelling rate, followed by HAMA–GA 1.5Fe and HAMA–GA 5Fe. It is assumed that HAMA–GA–Fe at pH 9 had the highest swelling rate, disrupting the polymeric network. The catechol–metal complex hydrogels exhibited increased water absorption under neutral and basic conditions because the catechol chemistry within the hydrogels was stable at that pH range. Hydrogels formed from catechol–metal complexes are assumed to be highly porous, exhibiting a high swelling ratio.53 Based on the results, the HAMA–GA–Fe hydrogels provided pH-responsive behaviour. In addition, Fig. S13 (ESI†) illustrates the surface swelling of HAMA–GA under basic conditions. We expect that the increased swelling of the hydrogels in basic pH conditions will enhance the release of drugs or nanoparticles for infectious wounds (pH ∼ 8).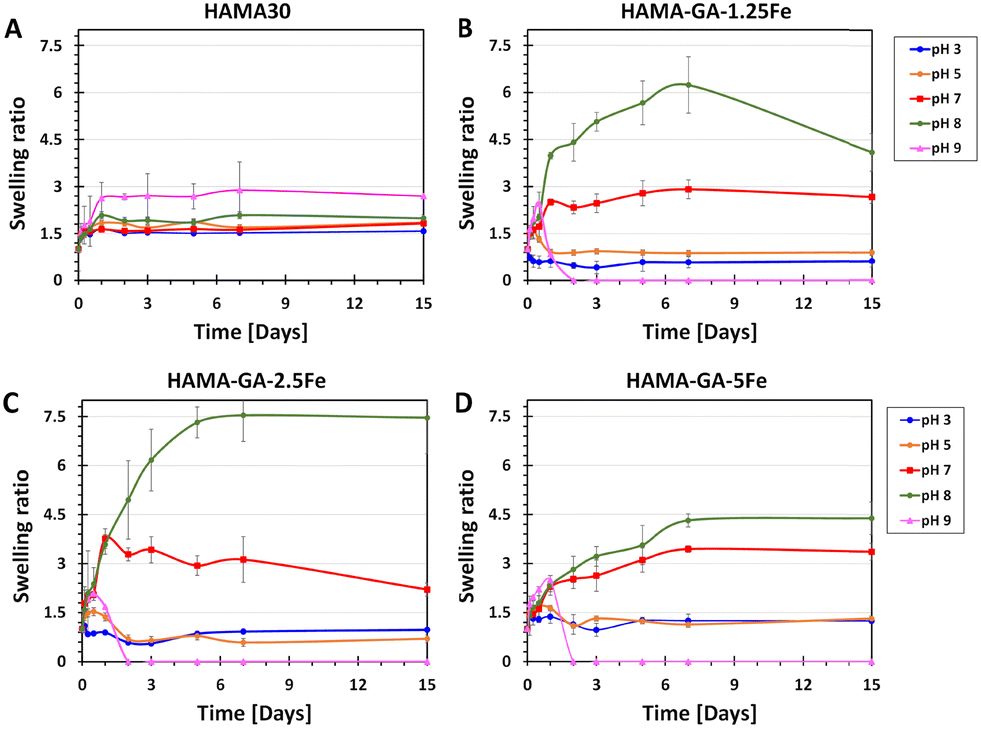 | ||
| Fig. 10 Time-dependent swelling behaviour of (A) HAMA30, (B) HAMA–GA 1.25Fe, (C) 2.5Fe and (D) 5Fe hydrogels as a response to pH 3–9 (n = 10, the error bars indicate the SD). | ||
Bioadhesion of hydrogels
The synergy between non-covalent (catechol–metal coordination complexes) and covalent (photocrosslinking) not only improves the stiffness and self-healing ability of hydrogels but also provides the bioadhesion of hydrogels to biological tissues. Catechol groups and FeCl3 have generally been used as adhesive hydrogels under wet conditions.55,56 In Fig. 11A, a tack test was performed on fresh porcine muscles. The hydrogel samples (HAMA, HAMA–GA 1.25, HAMA–GA 2.5 and HAMA–GA 5Fe) were characterised without (Fig. 11B) and with photocrosslinking (Fig. 11C) to prove the effect of synergy between two types of crosslinking (dual-crosslinking). Fig. 11B shows the tack test results of the samples before photocrosslinking, indicating that FeCl3-induced catechol hydrogels (HAMA–GA 1.25Fe, HAMA–GA 2.5Fe and HAMA–GA 5Fe) increased the tissue adhesion. In contrast, the HAMA solution did not adhere to the porcine tissues and escaped from the tissues. Fig. 11C shows the tack test of in situ photocrosslinked hydrogel samples. Despite improving the tissue adhesion after photocrosslinking, HAMA's adhesive strength exhibited higher tissue binding strength (burst). On the contrary, catechol–metal hydrogels possessed lower tissue binding strength but displayed more elasticity in the axial direction. We conclude that tissue adhesion force from covalent interaction alone is stronger but provides less elasticity than dual-crosslinking in catechol–metal hydrogels. Although catechol-conjugated hydrogel exhibits poorer tissue adhesion after photocrosslinking, its adhesive properties before photocrosslinking remain crucial for wound closure or fixation. These results highlight the role of catechol and Fe3+ in this dual-crosslinking hydrogel system. The concentration of FeCl3 also influenced the coordination degree in catechol–metal hydrogels, which affected the viscoelasticity and tissue adhesion of hydrogels.2 The trend in axial force of catechol–metal hydrogels (HAMA–GA 2.5Fe > HAMA–GA 5Fe > HAMA–GA 1.25Fe) follows the order of crosslinking kinetics of hydrogels, indicating more hydrophobicity as the G′ values increase.29 On the other hand, HAMA–GA 1.25Fe was not sufficiently crosslinked by the metal ions because the free catechol motifs in hydrogels are oxidised into o-quinone, leading to the lowest adhesion force.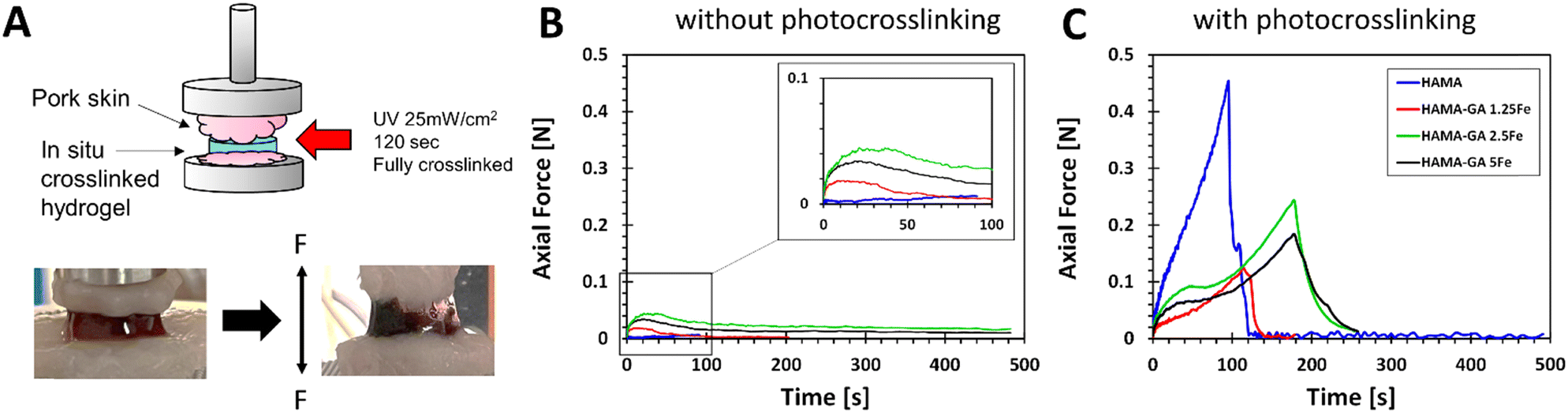 | ||
| Fig. 11 Measurement of tissue adhesion force of hydrogel samples: HAMA30, HAMA–GA 1.25Fe, HAMA–GA 2.5Fe and HAMA–GA 5Fe (n = 4). (A) The tack test setup using porcine muscles. (B) Hydrogel samples without photocrosslinking. (C) Hydrogel samples with photocrosslinking. | ||
Conclusions
This approach based on the chemistry of mussel byssus cuticle and photocrosslinking allows precise tuning of sol–gel transition and enhanced viscoelastic properties for hydrogels. The dual-crosslinking mechanism in catechol–metal coordination and methacrylated hydrogels offers versatility with pH-controllable sol–gel transition, enhanced viscoelasticity, self-healing, printability, and improved tissue adhesion. These properties are significantly influenced by the concentration of FeCl3 and pH conditions, which dictate the strength of the crosslinking bonds and coordination degree of hydrogels. The introduction of dual crosslinking through catechol chemistry and photocrosslinking further augments tissue adhesion, making it promising for application in bioadhesive, stimuli-responsive hydrogels, and biomaterial inks.Author contributions
Hatai Jongprasitkul: conceptualization, data curation, formal analysis, investigation, methodology, project administration, validation, visualization, writing – original draft, and writing – review & editing. Vijay Singh Parihar: conceptualization, data curation, formal analysis, investigation, methodology, project administration, validation, visualization, and writing – review & editing. Sanna Turunen: writing – review & editing. Minna Kellomäki: funding acquisition, resources, supervision, and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to The Centre of Excellence in Body-on-Chip Research (CoEBoC) by the Academy of Finland for financial support (decisions #312409, #326587, #336663 and #353173) and to the Tampere University funding for the CoEBoC consortium.References
- K. Lei, M. Chen, P. Guo, J. Fang, J. Zhang, X. Liu, W. Wang, Y. Li, Z. Hu, Y. Ma, H. Jiang, J. Cui, J. Li, K. Lei, M. Chen, P. Guo, J. Fang, J. Zhang, X. Liu, W. Wang, Y. Li, Z. Hu, J. Li, Y. Ma and H. Jiang, Adv. Funct. Mater., 2023, 33, 2303511 CrossRef CAS.
- W. Zhang, R. Wang, Z. M. Sun, X. Zhu, Q. Zhao, T. Zhang, A. Cholewinski, F. Yang, B. Zhao, R. Pinnaratip, P. K. Forooshani and B. P. Lee, Chem. Soc. Rev., 2020, 49, 433 RSC.
- Y. Hong, F. Zhou, Y. Hua, X. Zhang, C. Ni, D. Pan, Y. Zhang, D. Jiang, L. Yang, Q. Lin, Y. Zou, D. Yu, D. E. Arnot, X. Zou, L. Zhu, S. Zhang and H. Ouyang, Nat. Commun., 2019, 10(1), 1–11 CrossRef CAS PubMed.
- A. Assmann, A. Vegh, M. Ghasemi-Rad, S. Bagherifard, G. Cheng, E. S. Sani, G. U. Ruiz-Esparza, I. Noshadi, A. D. Lassaletta, S. Gangadharan, A. Tamayol, A. Khademhosseini and N. Annabi, Biomaterials, 2017, 140, 115–127 CrossRef CAS PubMed.
- L. Han, L. Yan, K. Wang, L. Fang, H. Zhang, Y. Tang, Y. Ding, L. T. Weng, J. Xu, J. Weng, Y. Liu, F. Ren and X. Lu, NPG Asia Mater., 2017, 9, e372–e372 CrossRef CAS.
- H. Montazerian, S. Mitra, A. Hassani Najafabadi, R. Seyedmahmoud, Y. Zheng, M. R. Dokmeci, N. Annabi, A. Khademhosseini and P. S. Weiss, ACS Mater. Lett., 2023, 5, 1672–1683 CrossRef CAS.
- J. Zhang, B. Li, J. Zuo, R. Gu, B. Liu, C. Ma, J. Li and K. Liu, Adv. Healthcare Mater., 2021, 10, 2100109 CrossRef CAS PubMed.
- H. J. Park, Y. Jin, J. Shin, K. Yang, C. Lee, H. S. Yang and S. W. Cho, Biomacromolecules, 2016, 17, 1939–1948 CrossRef CAS PubMed.
- Z. Jia, M. Wen, Y. Cheng and Y. Zheng, Adv. Funct. Mater., 2021, 31, 2008821 CrossRef CAS.
- S. Y. Zheng, H. Ding, J. Qian, J. Yin, Z. L. Wu, Y. Song and Q. Zheng, Macromolecules, 2016, 49, 9637–9646 CrossRef CAS.
- A. Andersen, M. Krogsgaard and H. Birkedal, Biomacromolecules, 2018, 19, 1402–1409 CrossRef CAS PubMed.
- G. Dai, L. Sun, J. Xu, G. Zhao, Z. Tan, C. Wang, X. Sun, K. Xu and W. Zhong, Acta Biomater., 2021, 129, 84–95 CrossRef CAS PubMed.
- N. Holten-Andersen, M. J. Harrington, H. Birkedal, B. P. Lee, P. B. Messersmith, K. Y. C. Lee and J. H. Waite, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2651–2655 CrossRef CAS PubMed.
- D. G. Barrett, D. E. Fullenkamp, L. He, N. Holten-Andersen, K. Y. C. Lee and P. B. Messersmith, Adv. Funct. Mater., 2013, 23, 1111–1119 CrossRef CAS PubMed.
- N. Holten-Andersen, A. Jaishankar, M. J. Harrington, D. E. Fullenkamp, G. Dimarco, L. He, G. H. McKinley, P. B. Messersmith and K. Y. C. Lee, J. Mater. Chem. B, 2014, 2, 2467–2472 RSC.
- H. Choi and K. Lee, Appl. Sci., 2022, 12, 11626 CrossRef CAS.
- S. Samanta, V. K. Rangasami, N. A. Murugan, V. S. Parihar, O. P. Varghese and O. P. Oommen, Polym. Chem., 2021, 12, 2987–2991 RSC.
- S. Samanta, V. K. Rangasami, H. Sarlus, J. R. K. Samal, A. D. Evans, V. S. Parihar, O. P. Varghese, R. A. Harris and O. P. Oommen, Acta Biomater., 2022, 142, 36–48 CrossRef CAS PubMed.
- M. K. Włodarczyk-Biegun, J. I. Paez, M. Villiou, J. Feng and A. Del Campo, Biofabrication, 2020, 12, 035009 CrossRef PubMed.
- P. Bertsch, M. Diba, D. J. Mooney and S. C. G. Leeuwenburgh, Chem. Rev., 2023, 123, 834–873 CrossRef CAS PubMed.
- S. Choi, K. Y. Lee, S. L. Kim, L. A. Macqueen, H. Chang, J. F. Zimmerman, Q. Jin, M. M. Peters, H. A. M. Ardoña, X. Liu, A.-C. Heiler, R. Gabardi, C. Richardson, W. T. Pu, A. R. Bausch and K. K. Parker, Nat. Mater., 2023, 22, 1039 CrossRef CAS PubMed.
- J. Y. Lai and L. J. Luo, Biomacromolecules, 2015, 16, 2950–2963 CrossRef CAS PubMed.
- M. M. Fernandes-Negreiros, L. A. N. C. Batista, R. L. S. Viana, D. A. Sabry, A. A. O. Paiva, W. S. Paiva, R. I. A. Machado, F. L. de Sousa Junior, D. de L. Pontes, J. de O. Vitoriano, C. Alves Junior, G. L. Sassaki and H. A. O. Rocha, Antioxidants, 2020, 9, 1–24 CrossRef PubMed.
- S. A. Monteiro e Silva, G. M. F. Calixto, J. Cajado, P. C. A. de Carvalho, C. F. Rodero, M. Chorilli and G. R. Leonardi, Polymers, 2017, 9, 391 CrossRef PubMed.
- B. Kang, T. P. Vales, B. K. Cho, J. K. Kim and H. J. Kim, Molecules, 2017, 22, 1976 CrossRef PubMed.
- M. Shin and H. Lee, Chem. Mater., 2017, 29, 8211–8220 CrossRef CAS.
- M. Shin, J. H. Galarraga, M. Y. Kwon, H. Lee and J. A. Burdick, Acta Biomater., 2019, 95, 165–175 CrossRef CAS PubMed.
- A. Mörö, S. Samanta, L. Honkamäki, V. K. Rangasami, P. Puistola, M. Kauppila, S. Narkilahti, S. Miettinen, O. Oommen and H. Skottman, Biofabrication, 2023, 15, 015020 CrossRef PubMed.
- H. Jongprasitkul, S. Turunen, V. S. Parihar, S. Annurakshita and M. Kellomaki, Biomacromolecules, 2021, 22, 481–493 CrossRef CAS PubMed.
- H. Jongprasitkul, V. S. Parihar, S. Turunen and M. Kellomäki, ACS Appl. Mater. Interfaces, 2023, 15(28), 33972–33984 CrossRef CAS PubMed.
- O. Chaudhuri, L. Gu, D. Klumpers, M. Darnell, S. A. Bencherif, J. C. Weaver, N. Huebsch, H. P. Lee, E. Lippens, G. N. Duda and D. J. Mooney, Nat. Mater., 2016, 15, 326–334 CrossRef CAS PubMed.
- H. Jongprasitkul, S. Turunen, V. S. Parihar and M. Kellomäki, Biomacromolecules, 2023, 24, 502–514 CrossRef CAS PubMed.
- J. Lee, K. Chang, S. Kim, V. Gite, H. Chung and D. Sohn, Macromolecules, 2016, 49, 7450–7459 CrossRef CAS.
- H. Haidari, Z. Kopecki, A. T. Sutton, S. Garg, A. J. Cowin and K. Vasilev, Antibiotics, 2021, 10, 1–15 CrossRef PubMed.
- N. Gamboa-Valero, P. D. Astudillo, M. A. González-Fuentes, M. A. Leyva, M. D. J. Rosales-Hoz and F. J. González, Electrochim. Acta, 2016, 188, 602–610 CrossRef CAS.
- M. J. Sever and J. J. Wilker, Dalton Trans., 2006, 813–822 RSC.
- Z. An, J. Sun, Q. Mei, B. Wei, M. Li, J. Xie, M. He and Q. Wang, J. Environ. Sci., 2022, 115, 392–402 CrossRef CAS PubMed.
- M. J. Sever and J. J. Wilker, Dalton Trans., 2004, 1061–1072 RSC.
- K. Kaniewska, P. Kościelniak and M. Karbarz, Gels, 2023, 9, 447 CrossRef CAS PubMed.
- M. K. Włodarczyk-Biegun and A. del Campo, Biomaterials, 2017, 134, 180–201 CrossRef PubMed.
- S. Kimura, M. Yokoya and M. Yamanaka, Chem. Lett., 2021, 50, 459–466 CrossRef CAS.
- N. J. Wagner and J. F. Brady, Phys. Today, 2009, 62, 27–32 CrossRef CAS.
- Y. Wang, J. P. Park, S. Hyeon Hong, H. Lee, Y. Wang, S. H. Hong, H. Lee and J. P. Park, Adv. Mater., 2016, 28, 9961–9968 CrossRef CAS PubMed.
- H. Jongprasitkul, S. Turunen, V. S. Parihar and M. Kellomäki, Bioprinting, 2022, 25, e00185 CrossRef.
- N. Paxton, W. Smolan, T. Böck, F. Melchels, J. Groll and T. Jungst, Biofabrication, 2017, 9, 44107 CrossRef PubMed.
- S. Lee and D. S. Hwang, Macromol. Rapid Commun., 2023, 44, 2200845 CrossRef CAS PubMed.
- K. Hyun, S. H. Kim, K. H. Ahn and S. J. Lee, J. Non-Newtonian Fluid Mech., 2002, 107, 51–65 CrossRef CAS.
- X. Fan, H. Xu, Q. Zhang, D. Xiao, Y. Song and Q. Zheng, Polymer, 2019, 167, 109–117 CrossRef CAS.
- W. E. Rochefort and S. Middleman, J. Rheol., 1987, 31, 337–369 CrossRef CAS.
- W. Yu, W. Sun, H. Chen, J. Wang, B. Xue and Y. Cao, Int. J. Mol. Sci., 2022, 23, 5170 CrossRef CAS PubMed.
- M. Tavafoghi, A. Sheikhi, R. Tutar, J. Jahangiry, A. Baidya, R. Haghniaz and A. Khademhosseini, Adv. Healthcare Mater., 2020, 9, 1901722 CrossRef CAS PubMed.
- X. Meng, Y. Qiao, C. Do, W. Bras, C. He, Y. Ke, T. P. Russell and D. Qiu, Adv. Mater., 2022, 34, 2108243 CrossRef CAS PubMed.
- H. P. Lee, L. Gu, D. J. Mooney, M. E. Levenston and O. Chaudhuri, Nat. Mater., 2017, 16, 1243–1251 CrossRef CAS PubMed.
- C. Lee, J. Shin, J. S. Lee, E. Byun, J. H. Ryu, S. H. Um, D. I. Kim, H. Lee and S. W. Cho, Biomacromolecules, 2013, 14, 2004–2013 CrossRef CAS PubMed.
- M. S. Akram Bhuiyan, J. D. Roland, B. Liu, M. Reaume, Z. Zhang, J. D. Kelley and B. P. Lee, J. Am. Chem. Soc., 2020, 142, 4631–4638 CrossRef CAS PubMed.
- A. R. Narkar, B. Barker, M. Clisch, J. Jiang and B. P. Lee, Chem. Mater., 2016, 28, 5432–5439 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma00273c |
| This journal is © The Royal Society of Chemistry 2024 |