
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cement retarding mechanism of phosphonates and their interaction with aluminium†
Daniel
Axthammer
 and
Joachim
Dengler
*
and
Joachim
Dengler
*
BASF Construction Additives GmbH, Global Research Advanced Materials & Systems, Dispersions & Colloidal Materials, Trostberg, Germany. E-mail: joachim.dengler@basf.com
First published on 18th June 2024
Abstract
Phosphonates are widely used components of retarder and dispersant formulations in concrete and mortar. In this study, 11 different phosphonate retarders were analyzed for their retarding performance in ordinary Portland cement (OPC) and alite. The interaction of the phosphonates with clinker phases was investigated using calorimetry and pore solution analysis in both the OPC and alite systems. Calorimetry, coupled with saturation indices calculated from the pore solution analysis, enables a better understanding of the retarding mechanism for phosphonate retarders in OPC and alite. The objective of this research is to provide improved design principles for retarding admixtures, allowing for the achievement of specific performance profiles.
Introduction
Ordinary Portland cement (OPC) is a crucial ingredient in the construction materials concrete and mortar.1 It comprises several compounds, including silicate, aluminate, sulfates, and iron phases, which interact and affect the hydration process. The silicate phases, alite (an impure form of tricalcium silicate (C3S‡)) and belite (an impure form of dicalcium silicate (C2S)), are primarily responsible for the setting and hardening of the cement,2,3 while the aluminate reaction determines the early rheology.4,5 To control these properties, dispersants can be added to adjust rheology,6,7 and retarders8–11 or accelerators12 can be used to influence the setting time based on specific application requirements. The adjustment of setting time is important for various applications. Long workability times are necessary when concrete must be transported for extended periods, such as offshore job sites or when concrete must be pumped over long distances or heights. Roller-compacted concretes also require long setting times, as fast setting can create cold joints. The most significant application for retarders is to compensate for high temperatures. Increased temperatures reduce the open times, which can be counteracted by the addition of retarding molecules. Retarding additives, such as hydroxycarboxylic acids13,14 (tartrates,15–17 gluconate,18 or citrates19), saccharides,20,21 and phosphorous-based retarders, are commonly used in modern concrete or mortar admixtures. Among these, phosphonate groups containing retarders have gained importance due to their high dosage efficiency and linear dosage-performance profile over a wide range.22,23 The high dosage efficiency, however, requires proper dosage and mixing at the plant. Dosage fluctuations or local overdoses due to insufficient mixing can lead to over-retardation, which may cause strength issues. Other important aspects are not only the setting of the system but also how fast the construction material builds strength after the loss of workability. In many applications, we observe the need for a fast increase in compressive strength after the end of the workable time. Others need a slow increase in strength as, for example, surface treatment is necessary. Such performance criteria decide on which retarding molecules are best suited for different applications. Although being an important formulation additive in the admixture industry, only limited research has been conducted on the use of phosphonic retarders in cementitious systems.13,23–31 Several mechanistic studies have been performed, yielding different outcomes. Ramachandran et al. found a correlation between the number of phosphonate groups and retarding performance and explained the retarding effect by poisoning the nuclei of Ca(OH)2 and C–S–H.24 Gu also traces the retardation back to the poisoning and stabilization of C–S–H nuclei, which is probably related to the chelating ability of the phosphonates. Rickert and Pang proposed an insoluble calcium phosphate layer inhibiting the dissolution of clinker phases resulting in the desired retardation.30,31 Barron showed some mechanistic studies of nitrilotris(methylene)triphosphonate (ATP), including the influence of the aluminate reaction on the performance of the additive. They conclude in a mixed effect on nucleation and the formation of insoluble clusters on anhydrous surfaces, preventing further hydration.22 Daake also discussed the effect of the aluminate reaction on the adsorption and performance of EDTMP by adding the additives at different times.32 Coveney and Billingham propose that phosphonates prevent the recrystallization of amorphous ettringite resulting in an extended surface blocking.26,29,33,34 Lupu also interprets the retardation as a result of surface blockage caused by complexes of calcium and retarders, with a subsequent restart of hydration through the rearrangement of these organometallic structures. However, these studies offer somewhat fragmented explanations, and some interpretations rely on outdated models of cement hydration.35 In this context, we will analyze the retarding performance of different organic phosphonate-based retarders and investigate their interaction with different clinker phases to gain insights into the mechanism. This will be done using a combined analysis of calorimetry and pore solution composition.Materials
Additives
The retarding additives used in this study are presented in Table 1. They were purchased from Sigma Aldrich Chemie GmbH, Munich, Germany or synthesized according to literature protocols.
|
Cement
The cement used in this experimental setup was a CEM I 42.5R. The chemical composition was determined by XRF and the mineralogical analysis was conducted using Rietveld refinement of XRD data. The data are summarized in Table 2. Alite was synthesized according to standard procedures.36 The chemical composition is summarized in Table 3.| Chemical composition [% (w/w)] | Mineralogical composition [% (w/w)] | ||
|---|---|---|---|
| CaO | 61.76 | C3S alite | 56.65 |
| SiO2 | 19.52 | C2S belite | 14.85 |
| Al2O3 | 4.96 | C3A aluminate | 6.90 |
| Fe2O3 | 3.01 | C4AF ferrite | 8.15 |
| MgO | 3.01 | CaSO4 anhydrite | 2.75 |
| Na2O | 0.238 | Hemihydrate | 1.25 |
| K2O | 0.900 | K2SO4 arcanite | 0.60 |
| K3Na(SO4)2 aphthitalite | 0.40 | ||
| CaO calcium oxide | 0.65 | ||
| MgO perikcase | 2.50 | ||
| CaCO3 calcite | 3.80 | ||
| CaMg(CO3)2 dolomite | 0.55 | ||
| SiO2 quartz | 0.70 | ||
| Ca(OH)2 portlandite | 0.20 | ||
| Sum | 93.40 | Sum | 99.9 |
| Chemical composition [% (w/w)] | |
|---|---|
| CaO | 72.0 |
| SiO2 | 25.4 |
| Al2O3 | 1.0 |
| Fe2O3 | 0.3 |
| MgO | 1.0 |
| Sum | 100.2 |
Methods
Definition of the retardation
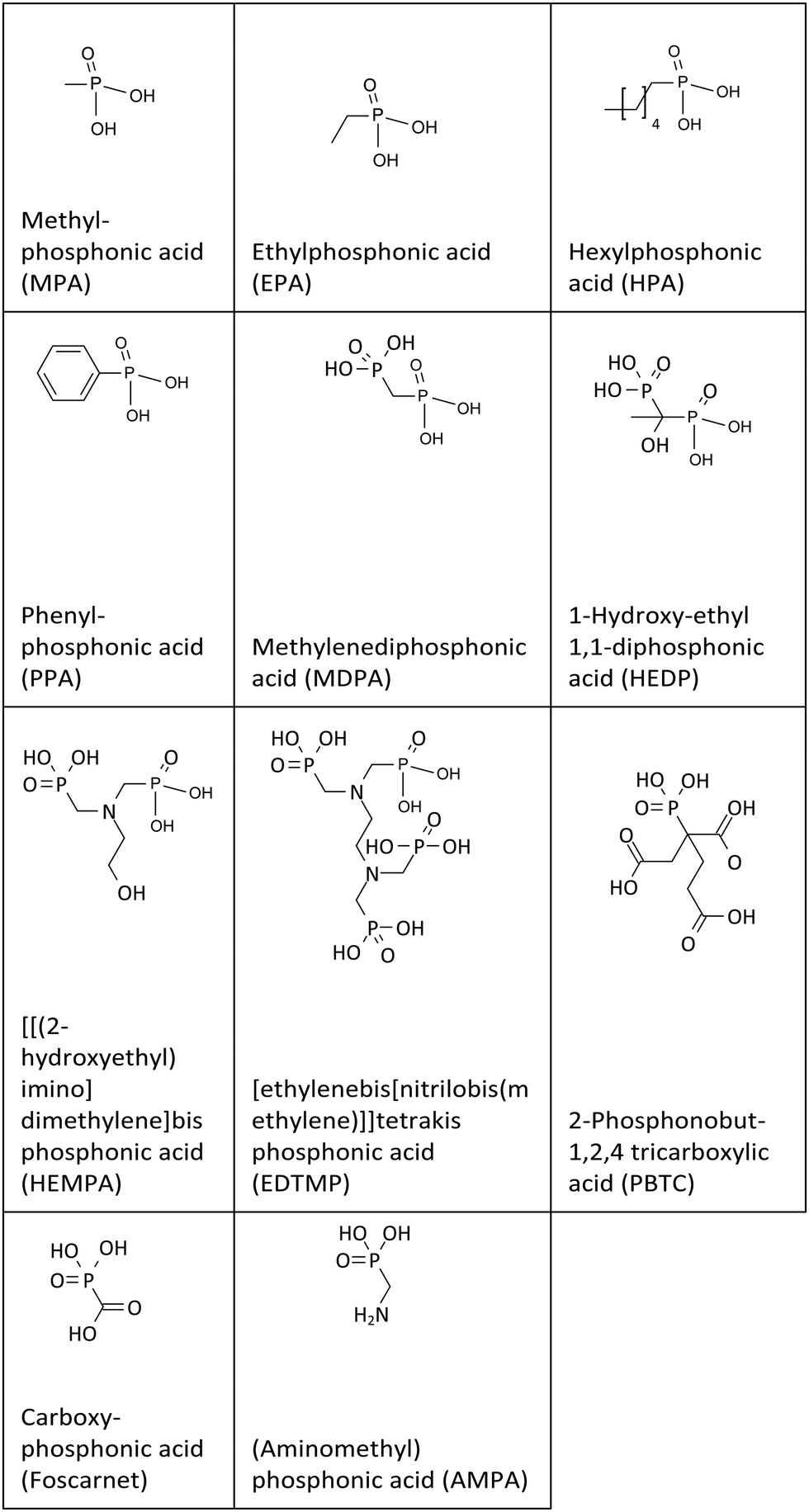
The quantification of the effectiveness of additives in retarding the hydration process, based on calorimetry measurements, was adapted from Nalet et al.37 In Fig. 1 and Fig. S3–S5 (ESI†), the typical calorimetric curves obtained from Portland cement are shown, both with and without a retarder. These curves illustrate the heat flow, which represents the reaction heat generated during the dissolution of the anhydrate phase and the subsequent precipitation of hydrates over time.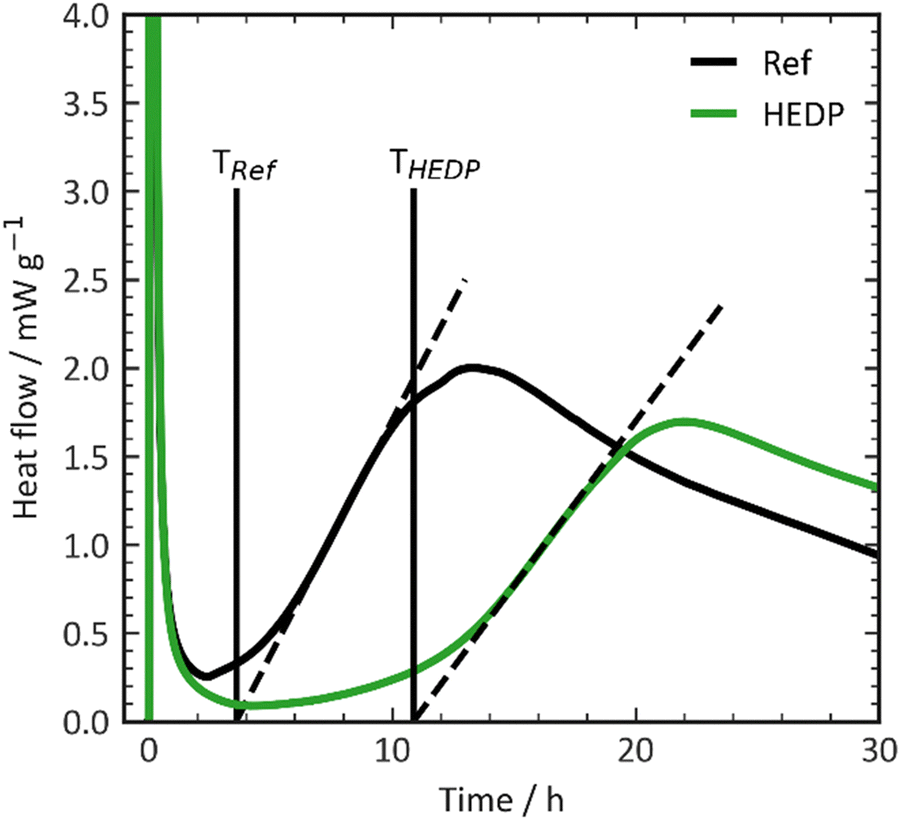 | ||
| Fig. 1 Heat flow curves from calorimetry without and with 5 mmol L−1 HEDP displaying dormant period and slope of the silicate reaction. | ||
In this study, we utilize two different parameters to describe the impact of retarders on the hydration kinetics of cement. The first parameter is the time at which the induction period of cement ends. This time is defined as the point where the slope of the calorimetric curve intersects the x-axis during the acceleration period. By using different concentrations of the retarder (0.03, 0.06, and 0.09% by weight of cement), three distinct ending times for the dormant period are received (refer to Fig. 1). At these relatively low concentrations, there is a linear correlation between the end of the dormant period and the concentration of the retarder (in mmol L−1). The slope of this correlation is considered as the efficiency of the retarder.
The second parameter describes the effect of the retarder on the kinetics of silicate hydration. This value is obtained by measuring the slope of the calorimetric curve at the inflection point of the silicate reaction during the acceleration period, which represents the steepness of the highest acceleration. Similarly, these values are plotted on a graph using the steepness/concentration (in mmol L−1) function, with the slope defining the impact of the retarder on the hydration kinetics of the silicate reaction.
Results and discussion
Impact of phosphonate molecules on the hydration of Portland cement
The effectiveness of the retarders listed in Table 1 was assessed using calorimetry, and the findings are presented in Fig. 2. The graph depicts the end of the dormant period, as previously explained, on the y-axis, and the molar concentration of the added additives on the x-axis. To ensure consistency and account for the substantial variance in molecular weights and functionalities of the retarder structures, the additive concentrations are presented in mmol per liter of mixing water. This method guarantees that all experiments were conducted with an equivalent number of retarder molecules, enabling a more accurate correlation between chemical structure and effect.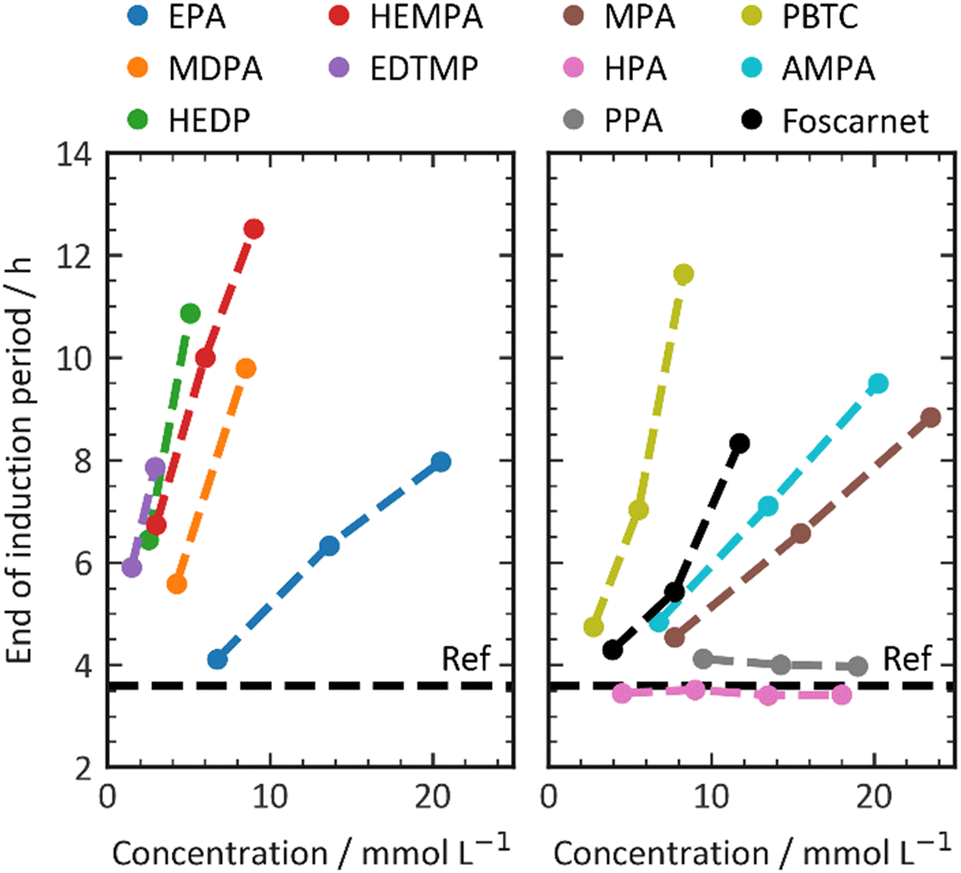 | ||
| Fig. 2 Retarder dose efficiency. | ||
In Portland cement with phosphonates, an increase in the concentration of retarders results in a shift towards later times for the end of the dormant period. Within this range of dosages, there is a linear relationship between the dosage and the end of the induction period. To ensure better comparability and reduce potential errors associated with calorimetry measurements, the slope of the straight line is used as a means of comparing the effectiveness of different retarder structures.
The phosphonates used in this study demonstrate varying levels of retarder efficiency. For example, EDTMP shows a steep slope in the straight line, indicating a high efficiency of 1.4 h mmol−1 L−1. It only requires less than 4 mmol L−1 to retard the cement by 4 hours. In contrast, HPA does not exhibit any retarding effect. As discussed previously in the literature, the structure of the retarder plays a crucial role in determining its effectiveness.24,26
Ramachandran et al. suggested that the retarding efficiency of a retarder molecule is influenced by the number of phosphonate groups it possesses.24,25 However, their experiments had a somewhat limited range of structural diversity. In our own experiments, we generally observed a limited correlation among the investigated additives (Fig. 3). This correlation was established by plotting the retardation efficiency against the number of charges present in the retarder molecule. Carboxylate and phosphonate groups were assumed to be fully dissociated, while hydroxy and amine groups were considered to be undissociated. A few data points in the plot deviate from the trend. HEDP and HEMPA exhibit a higher retarding efficiency compared to the others. These hydroxy groups appear to significantly enhance the retarding performance of phosphonates, potentially by increasing the solubility of calcium or aluminium complexes compared to their non-hydroxylated counterparts. In summary, while there is a general correlation between the number of charges in the retarder molecule and its retarding efficiency, certain factors such as hydroxy groups and hydrophobicity can have a more complex impact on the performance.
 | ||
| Fig. 3 Impact of charges of retarders on the (a) retardation efficiency and (b) C3S kinetics. | ||
PPA and HPA, which are the most hydrophobic additives in our experimental set, show nearly no retarding performance. This observation suggests a potential influence of complexation with aluminum or calcium salts, as well as the solubility of the additive complexes in the pore solution.38 For a retarding effect to occur, the organic additive must interact with inorganic surfaces such as clinker phases, hydration products, or nuclei through processes like adsorption or complexation. This interaction enables the desired retardation effect. Higher charge densities enhance complexation energies through a chelate effect, thereby facilitating complex formation. However, it is important to note that an increase in charge density does not necessarily result in improved adsorption, as depicted in Fig. 4. Initially, the retarding performance improves as the additive adsorption increases up to 100%. However, as the retarding performance continues to increase, the adsorptivity of the phosphonates begins to decrease. This finding further confirms the complexity of the organic–inorganic interaction, which underlies the observed retardation performance.
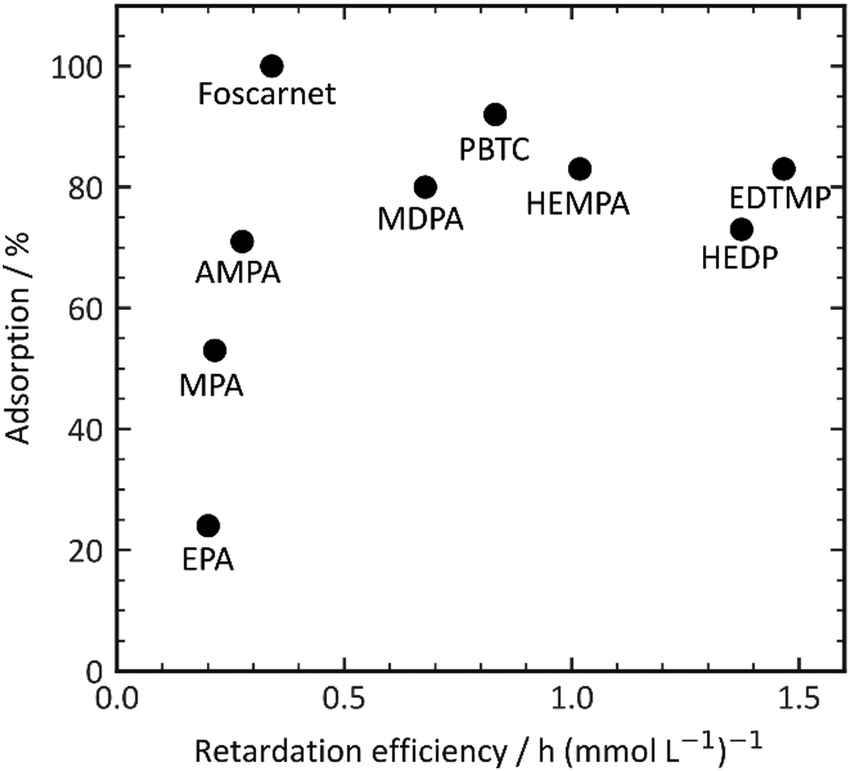 | ||
| Fig. 4 Correlation of retarder adsorption and efficiency. | ||
The maximum slope, as shown in Fig. 5, is another factor that can be extracted from the calorimetric curves. The slope of the calorimetric curve directly correlates with the reaction kinetics of the C3S hydration process, which involves a series of dissolution, nucleation, and crystal growth. Unlike the effect on the dormant period, where all phosphonates exhibited a retarding effect by prolonging the duration of the dormant period, the addition of phosphonate retarders resulted in diverse effects on the hydration kinetics of C3S. The influence of the retarder on C3S dissolution depends on the dosage of the retarder and increases with higher concentrations. The slope values of the regression lines vary, ranging from +0.1 × 10−8 mW g−1 mol−1 (AMPA) to −4.5 × 10−8 mW g−1 mol−1 (MDPA). In this study, the additives used serve two distinct purposes. Firstly, they aim to extend the dormant period, which in turn enhances the workability and setting times of the cement composition. Secondly, they either increase or decrease the C3S reaction, which is responsible for the material's structural build-up and hardening after setting. Fig. 2 illustrates how the additive affects the C3S dissolution kinetics and the retarder's ability to prolong the workability time of the cement. It is important to note that these two factors are not directly correlated. Additives that significantly delay the onset of the C3S reaction do not necessarily retard the C3S reaction kinetics, and vice versa.
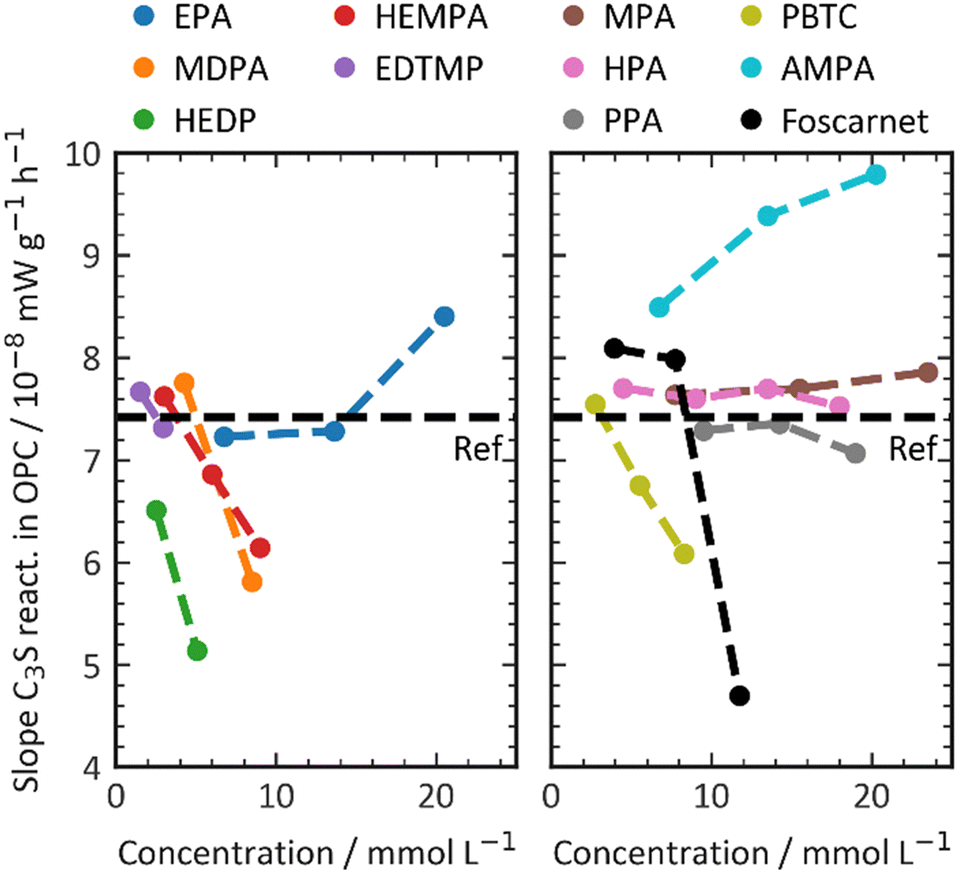 | ||
| Fig. 5 Impact of the retarder on the C3S kinetics at the accelerating period of the silicate reaction. | ||
The direct explanation of the effect on C3S kinetics using simple molecular descriptors proved to be challenging. Factors such as charge density, number of charges, and functional groups did not show clear correlations. Fig. 5 demonstrates that only molecules with a low number of charges have an accelerating effect on the C3S reaction kinetics. However, it should be noted that a highly charged molecule does not necessarily imply a strong decelerating effect. To categorize the retarders into three distinct clusters, the relationship between the slope of the silicate reaction and the retarder's impact on the length of the dormant period can be visualized (Fig. 6). The first cluster, indicated by a broken line, comprises monophosphonates. Foscarnet, which is another monophosphonate has a different action due to the presence of a carboxylic group. These additives exhibit low retarding performance and do have a positive effect on the kinetics of the silicate reaction. They all share the common feature of having a single phosphonate group and no carboxylate groups. The second cluster, indicated by a continuous line, includes PBTC, HEMPA, and EDTMP. These additives have a strong retarding effect on the open time but only a moderate impact on the kinetics of the silicate reaction. The additives within this cluster contain multiple charged groups, such as one phosphonate and several carboxylates, or two or more phosphonates. The third cluster, denoted by a dotted line, encompasses Foscarnet, MDPA, HEDP. These additives exhibit a significant retardation effect on the formation of C–S–H. The impact on the dormant period varies from low to high retardation. In this cluster, the common characteristic is the presence of multiple charge carriers, such as phosphonates or a combination of phosphonate and carboxylate groups (as seen in Foscarnet). In general, the last two types of additives share similarities. One key structural feature is the distance between the charge carriers. In the case of the additives in the third group, the phosphonates are α,α-phosphonates, meaning they are attached to the same carbon atom. In the case of Foscarnet, the second charged functional group, the carboxylate, is directly linked to the phosphonate group. The additives in the second cluster have functional groups (carboxylates and phosphonates) that are connected in a less rigid manner, with the spacing between phosphonates or charged functional groups consisting of at least three covalent bonds. However, to gain further insight into complexation, molecular modeling and single crystal analysis would be needed, which is beyond the scope of this paper.
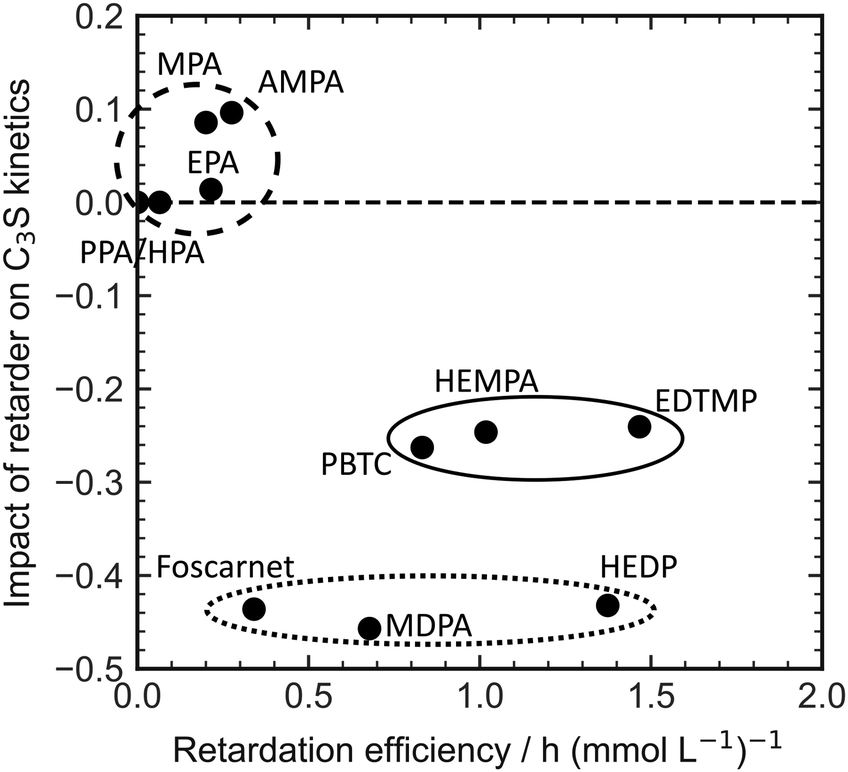 | ||
| Fig. 6 Correlation of the retardation efficiency and the impact of different retarders on the slope of the silicate reaction in OPC. | ||
Several mechanisms have been proposed to elucidate the retarding effects of phosphonates in cement. Some researchers suggested a mechanism where the precipitation of Ca-phosphonate blocks the dissolution of anhydrous phases.15,22,30,31,39 Others proposed a mechanism that involves the poisoning of the nuclei of C–S–H (calcium silicate hydrate) and Ca(OH)2 (calcium hydroxide).24,25
The hydration processes in cement encompass a sequence of successive reactions, including dissolution, nucleation, and growth. Despite the long history of C–S–H, there are still fundamental questions that remain unanswered. Moreover, our comprehension of C–S–H particle growth is still relatively constrained. Prior to growth, nucleation serves as the initial step. As the C–S–H crystals remain relatively small, it seems that secondary nucleation becomes more prominent compared to crystal growth once a certain size is attained.8,40 This emphasizes the significance of nucleation in the precipitation process, particularly in relation to the early mechanical properties of C–S–H.
Our study delves deeper into the mechanisms of organic additives on cement hydration by examining the hydration kinetics. Each kinetic parameter, including dissolution, nucleation, growth, or secondary nucleation, contributes to the overall kinetics of the reaction. Similar consecutive reactions are frequently observed in chemical reactions and can be described mathematically. In such cases, the rate of a chain reaction is typically not controlled by a single step.41 By decreasing the reactivity of one step in this consecutive reaction chain, the overall reaction kinetics is reduced. This change in reaction kinetics can be observed in the development of the educt (anhydrous phases), product (C–S–H crystals), and intermediate species (ions in pore solution and nuclei/growing C–S–H).
The initial stage of the process entails the dissolution of anhydrous phases, which can be further subdivided into separate kinetic steps, including etch/kink formation and dissolution.42
When the dissolution rate is reduced, a lower concentration of ions in the pore solution compared to the system without any additive can be expected. This assumption holds true as long as the subsequent processes of nucleation and growth are not affected by the additive. To gain a deeper understanding of this phenomenon, we analyzed the pore solution in cement paste and calculated the saturation levels of clinker phases and hydration products. To introduce retardation in a comparable manner, we extended the induction time to 7 hours by the retarders. The dosages necessary are depicted in Table 4. Therefore, pore solution was obtained for each retarder at the same level of retardation. Saturation of the pore solution was calculated on this basis (Fig. 7).
| Phosphonate | Dosage for Tind = 7 h in Portland cement [mmol L−1] | Duration of dormant period in alite per h | Slope of the C3S reaction during Alite hydration [10−8 mW g−1 h−1] |
|---|---|---|---|
| Reference | — | 6.70 | 6.35 |
| MPA | 16.44 | 34.36 | 1.52 |
| EPA | 16.58 | 27.84 | 4.44 |
| AMPA | 13.20 | 33.22 | 1.30 |
| Foscarnet | 9.91 | 10.30 | 4.99 |
| MDPA | 5.68 | 28.17 | 4.83 |
| PBTC | 5.49 | 38.19 | 2.03 |
| HEMPA | 3.24 | 35.95 | 1.89 |
| HEDP | 2.85 | 60 | 0.63 |
| EDTMP | 2.31 | 60 | 0.31 |
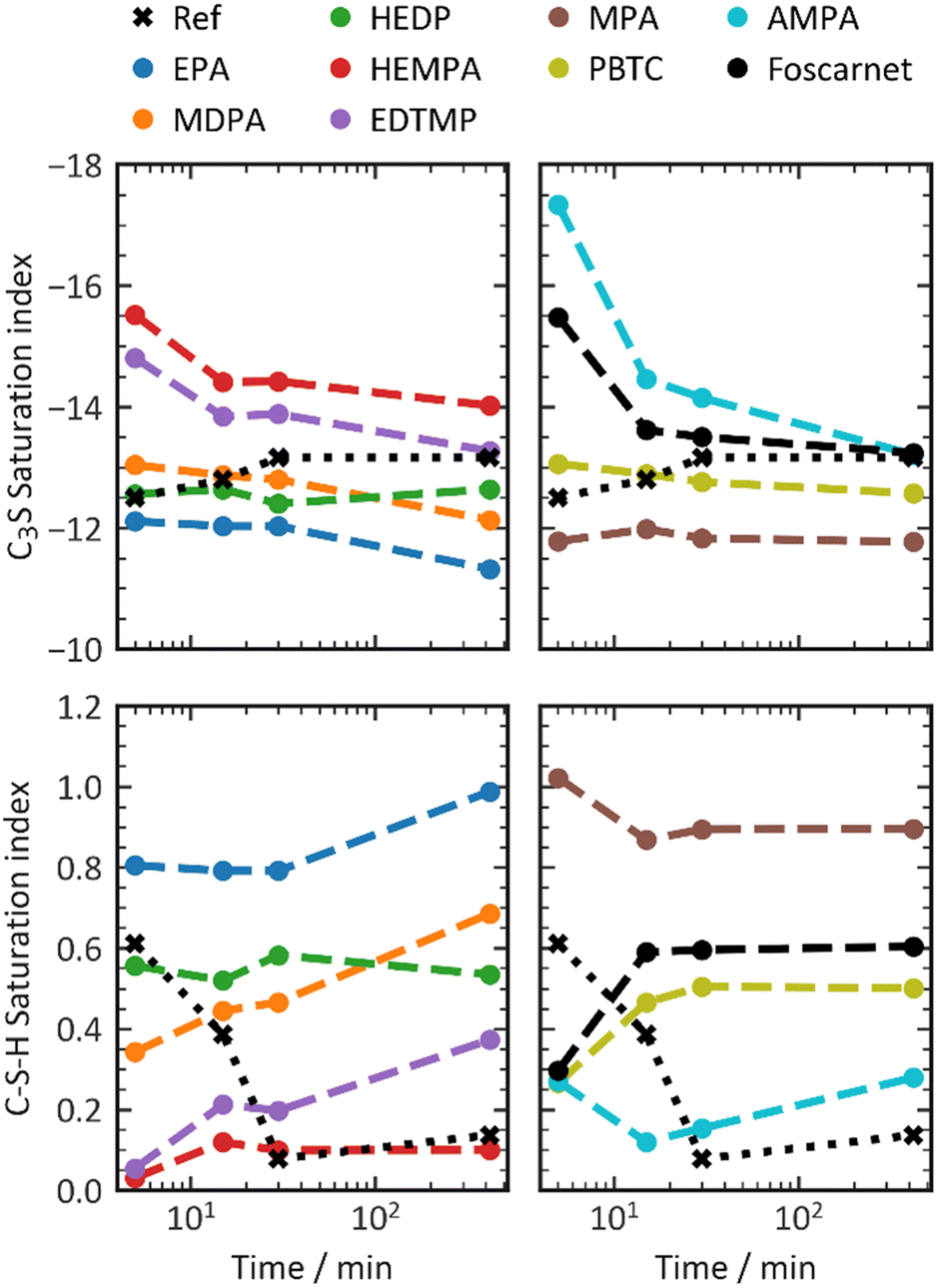 | ||
| Fig. 7 C–S–H saturation indices in presence of phosphonate retarders in an amount necessary to achieve a dormant period of 7 h. | ||
In general, the presence of retarders in the cement paste leads to a higher undersaturation of the pore solution with respect to C3S compared to the reference value at the 5–15 minute mark. However, methyl- and ethylphosphonic acid have minimal impact on retardation and result in slightly lower C3S undersaturation. This suggests a reduced dissolution rate of C3S when combined with phosphonate retarders. Over the subsequent 415 minutes, the undersaturation of C3S decreases, while the undersaturation of C3S in pastes without additives slightly increases. By the end of the 7-hour induction period, the supersaturation index falls within the range of ±1 of the reference system. AMPA, HEMPA, EDTMP, and Foscarnet exhibit higher undersaturation of C3S, while PBTC, HEDP, MDPA, MPA, and EPA result in lower undersaturation. This coincides with a relative increase in the oversaturation of C–S–H, indicating a potential inhibition of nucleation or growth of C–S–H. Only the addition of HEMPA and AMPA results in a supersaturation comparable to the reference value between 30 and 420 minutes. When discussing the effect of retarders on the supersaturation of C3S and C–S–H, it is crucial to distinguish between the initial 15 minutes and later time periods. During the early stages of the reaction, C3S saturation is more negative, while C–S–H is less oversaturated. This can be attributed to the reduced dissolution kinetics of C3S, which aligns with the high adsorption rates of phosphonates. However, as time progresses, a high C–S–H supersaturation combined with C3S undersaturation, similar to non-retarded systems, clearly indicates a significant influence on the nucleation or growth of C–S–H for most phosphonates.
An alternative explanation for an increased ion concentration in the pore solution could be a faster dissolution kinetics of C3S. However, this possibility can be ruled out for most additives based on calorimetry results, except for EPA and MPA. These two phosphonates appear to enhance the dissolution kinetics of C3S, leading to increased C–S–H supersaturation and decreased C3S undersaturation. Therefore, it can be concluded that for most phosphonates, their action is primarily through nucleation or growth inhibition after 15 minutes of aging, although this does not fully explain the observed variations in performance.
Influence of aluminium on the performance of phosphonate containing molecules in cement
Several research teams have been discussing the impact of the aluminate reaction on the effectiveness of retarders and dispersants.11,15,17,43–49 The water-reducing performance of dispersants, such as poly-naphthalene sulfonate, strongly depends on the aluminate content. It has been proven that the adsorption and intercalation of these additives in hydrate phases like ettringite is one factor responsible for the loss of the dispersing effect over time.6,50,51 Nalet and Nonat conducted an evaluation of the influence of adsorption on ettringite on the effectiveness of retarders.37 They found that the complexation of ions in the pore solution and adsorption onto ettringite varies depending on the stereochemistry of sugar alcohols. The aluminate content has a strong influence on the retarding performance, with a higher C3A content resulting in a decrease in the retarding action of the additives.The complexation of aluminium and adsorption on the surface of ettringite leads to a reduced efficiency as the organic retarder is no longer available to retard the silicate phase. The complexation of aluminium ions was measured in a very diluted system at L/S = 100, resulting in a significant increase in aluminium concentration in the solvent. Other researchers explain the aluminate dependence of the retarder by phase selective adsorption.52,53 Other research groups have demonstrated that a high concentration of aluminium can retard the silicate reaction. Nehring et al. showed this influence in ternary systems consisting of calcium aluminate cement, Portland cement, and calcium sulfate.54 Their hypothesis was that there was a hindered C–S–H seeding. This was further elaborated for OPC by Bergold et al., who showed the effect of AFm precipitation on the C–S–H formation kinetics.55 The reduced C–S–H formation was related to a higher aluminium concentration in the pore solution. This relationship between aluminium concentration and the dissolution kinetics of C3S was also proven by Nicoleau et al.56 They described the poisoning effect of aluminium on the formation of C–S–H. Aluminium ions covalently bind to surface silicate monomers and inhibit the C3S dissolution. The cleavage of these bonds is then dependent on the OH− and Ca2+ concentration. Pustovgar et al. also demonstrated, using MD simulation, strong ionic interactions between aluminium and calcium on the surface of silicate.57
Calorimetry measurements were conducted to test the influence of aluminate phases, primarily C3A and C4(A,F), on additives with alite. In this experimental setup, the dosages of the additives were adjusted to achieve an induction period ending time of t = 7 hours in Portland cement. The dosages required to achieve this target increased with a decreasing efficiency of the retarder. These admixture dosages are summarized in Table 4. The end of the induction period and the slope of the C3S reactions in alite were measured at the same dosage applied for the OPC reaction and summarized in Table 4. All retarders still exhibited retarding effects, but they had varying, mostly stronger effects on alite. Once again, there was no clear relationship between molecular structure and performance. Factors such as charge density, phosphorus or carboxylate content, number of functional groups, or molecular weight did not provide a direct explanation for the retarding efficiency of these additives on alite. The performance of different additives appears to be influenced by multiple factors and cannot be easily explained by simple observations such as adsorption.
The impact of retarders on the duration of the dormant period in alite is much more significant compared to Portland cement. This aligns with the findings of Nalet et al., where the interaction of additives with formed ettringite reduces the concentration of retarder available for retarding the formation of C–S–H in the pore solution. In most cases, the end of the induction period increases from 7 hours in cement to 32 hours ±20%, primarily due to the presence of aluminium and particle size effects. A higher heat flow during the initial reaction of cement corroborates a strong interaction of cement the aluminate phases with the retarding agents (Fig. S1, ESI†).
The effect of the least dosed additives, HEDP and EDTMP, on the dormant period in alite is even stronger, with a delay of 60 hours before C–S–H formation recommences. This indicates a stronger interaction of the additive with aluminium, which compromises the retarding effect. This is likely due to reduced adsorption of the additive on the clinker grains, possibly caused by clusters of ettringite that are not retained by the filter and consume the additive.58 On the other hand, Foscarnet results in minor retardation at the given dosage. Its retarding mechanism, acting similarly to other additives as a nucleation and dissolution inhibitor, is believed to be influenced by its interaction with aluminate ions, rather than solely its chemical compound with the silicate phases.
These variations in retardation caused by different structures suggest that the interaction between organic and inorganic components with aluminate phases in Portland cement plays a role in retarding C–S–H formation. The interaction and adsorption of additives with aluminium compounds, such as ettringite, C3A, C4(A,F), or complexation with aluminium ions in the pore solution, are significant for the performance of additives in OPC.
The presence of aluminate phases in Portland cement has a significant impact on both the dormant period and dissolution kinetics. The interaction between aluminium and retarders plays a crucial role in this process. When phosphonate-based retarders are present, the rate of the silicate reaction in alite is consistently reduced compared to the reference value without retardants. As a result, the onset of the silicate reaction is delayed, and there is a decrease in the formation kinetics of C–S–H in alite. To better understand this, Fig. 8 provides a clear visualization of the dormant period and reaction kinetics of the silicate reaction.
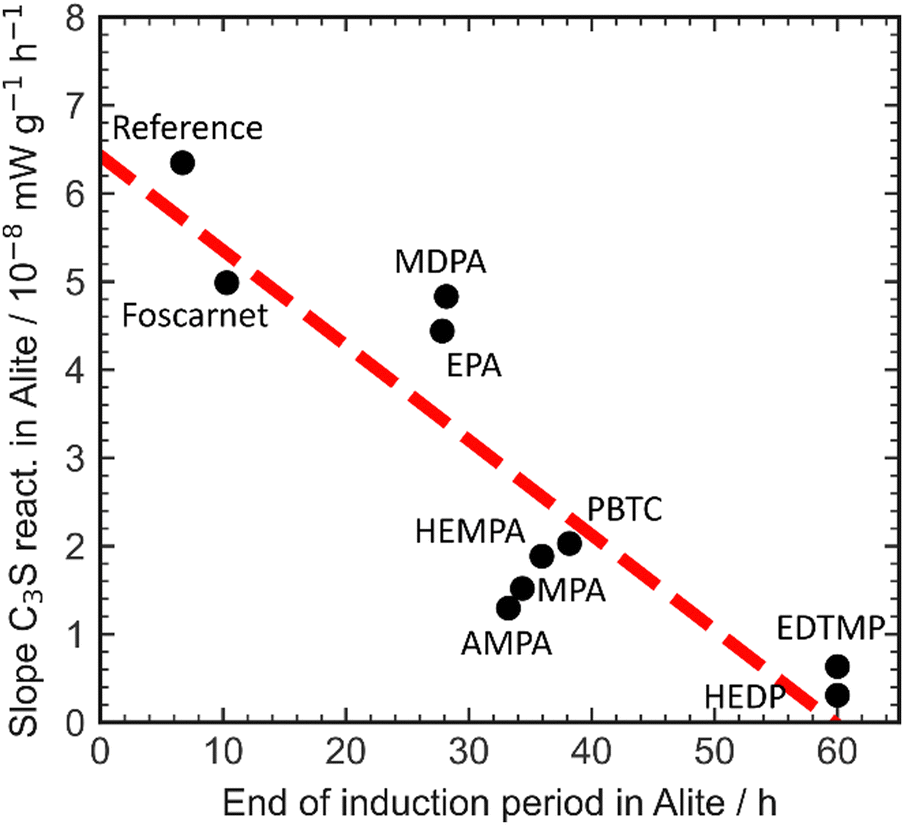 | ||
| Fig. 8 Correlation of the retardation efficiency and the impact of different retarders on the slope of the silicate reaction in alite. | ||
The retardation of the C–S–H reaction in alite ultimately leads to a reduction of the silicate reaction, establishing a correlation between the dormant period and dissolution kinetics of the accelerating silicate reaction. However, the increase in aluminate phases from alite to Portland cement disrupts this correlation, emphasizing the importance of aluminate content and aluminate reaction in Portland cement when assessing the impact of additives on the dormant period and reaction kinetics during the acceleration phase of the silicate reaction.
The interaction between organics and aluminium phases and ions does not follow a consistent performance relationship. For instance, the interaction of AMPA with aluminium enhances the formation kinetics of C–S–H, whereas EDTMP decreases it.
The debate surrounding the hydration kinetics and its origin during the main hydration peak after the dormant period has been intense. According to the most recent model, the calorimetric curve of the main hydration peak can be described by the nucleation rate and the length of the C–S–H needles. The needle length determines the slowdown of the hydration reaction when the cement grain is fully covered by C–S–H needles. The acceleration phase is influenced by the nucleation rate on surfaces, which leads to the prevalence of secondary nucleation,8,40,59 as the size of C–S–H crystallites are limited to the nanometer range. This leads to the assumption that the rate of the silicate reaction in cement is connected to the number of nuclei. In the case of alite, regardless of the retarding agent used, the open time and the number of nuclei depend on each other. This means that the nucleation inhibition and subsequent saturation of the pore solution are the reasons for the enhanced dormant period. The dormant period occurs as a result of the dissolution of clinker phases, where the kinetics depends on the saturation of ions in the pore solution.42 However, this does not hold true for additives in cement. The dissolution of clinker phases is relatively independent of the aluminium content, despite the influence of adsorption and particle size distribution. The crucial parameter here is the influence of aluminium on the nucleation of C–S–H. Previous research has shown that the influence of additives on aluminium dissolution and their complexation changes the nucleation of C–S–H. This has been observed with zinc, where the incorporation of zinc ions in C3A increased the kinetics and size of C–S–H needles, not due to larger crystallites but rather the higher agglomeration of C–S–H crystallites forming a larger mesocrystal.60,61 The retarding influence of magnesium was explained by the formation of fewer nuclei. SEM analysis of different cement pastes with AMPA, EDTMP, and HEDP, all at the same hydration turnover but exhibiting different nucleation kinetics, did not show any significant influence on the size of C–S–H mesocrystals compared to the reference (Fig. 9). Therefore, it is highly likely that the aluminium complexes inhibit the nucleation of C–S–H with less impact on the growth of the crystals.
 | ||
| Fig. 9 Morphologies of the hydrated cement pates at maximum heat flow of the C3S reaction. (a) EDTMP, (b) HEDP, (c) reference (d) AMPA. | ||
Retarding mechanism and retarder design principles
Understanding how molecular structures influence retarding performance is crucial for the further development of additives. To design an efficient retarder, various design principles can be employed. Charge density remains the major factor impacting the performance of additives. The higher the charges, the better the inhibiting effect on the nucleation of C–S–H. However, the molecule's solubility and complexing behavior in the pore solution are also crucial. Compared to other non-phosphorous containing molecules,13 retarders have relatively high adsorption, leading to a strong retardation of the dissolution of the clinker phases in the beginning. Later, the retardation mechanism switches and prevents nucleation. With higher performance, the additives reduce adsorptivity and act more in the pore solution. This explains the additional benefit of aminic or hydroxylic functionalities, which increase the solubility of calcium or aluminum complexes with the retarding agents. Nevertheless, it is unclear whether multiple hydroxylic or primary aminic groups will further improve the dose efficiency.In cases where fast strength gain is necessary after a long workable time, additional functionalities become necessary. The interaction of additives with aluminate phases determines the speed of strength increase. The open time and speed of subsequent strength gain are correlated with low aluminum in the clinker. In Portland cement, where the aluminum phases are present at higher concentrations, the molecular structure of the retarder influences the kinetics of the dissolution of the silicate phases responsible for the increase in strength. It is evident that charged functional groups linked flexibly are more effective in achieving this effect. Longer alkyl links between charged functionalities can create flexible molecules that interact beneficially with aluminum phases. However, it is still unclear why such behavior occurs or how the additives are complexing the ions in the pore solution. Molecular modeling could provide further insights into this phenomenon.
Conclusions
This study examines the performance of phosphonate additives in cement and their influence on aluminium. Phosphonate retarders are an important type of retarders that offer a high degree of structural variability, allowing for a wide range of applications due to different mechanisms at play. In Portland cement, retarders have two distinct effects on the overall OPC reaction kinetics: they prolong the dormant period and affect the speed of the consecutive C–S–H reaction. The mechanism by which additives impact the dormant period involves a combination of dissolution and nucleation inhibition. In alite, which contains low levels of aluminium, the dormant period and the slope of the silicate are correlated. However, increasing the amount of aluminium in cement reduces the effectiveness of the additives due to their interaction with aluminium phases. These aluminium complexes significantly impact the nucleation of C–S–H, making it independent of the dormant time or the dissolution of clinker phases. The structure of the additives and their ability to form complexes become crucial factors. These complexes play a decisive role in the inhibition of C–S–H nucleation.To design an efficient retarder, multiple charged functional groups are beneficial. Solubilizers such as alcohols and amines can also be used to enhance th performance of the retarder. To improve the C–S–H reaction, the functional groups should be connected in a flexible manner.
Author contributions
Joachim E. Dengler: conceptualization, methodology, resources, supervision, writing – original draft, writing – review & editing. Daniel Axthammer: investigation, visualization, data curation, formal analysis, writing – review & editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts of interest.Acknowledgements
D. A. would like to thank Prof. Konstantin Karaghiosoff for supervising this Master's Thesis. We thank Dr Xuerun Li and Cornelia Kronast for their help with the SEM.Notes and references
- D. Herfort and D. E. Macphee, in Lea's Chemistry of Cement and Concrete, ed. P. C. Hewlett and M. Liska, Butterworth-Heinemann, 5th edn, 2019, pp. 57–86 DOI:10.1016/B978-0-08-100773-0.00003-4.
- Y. Chen and I. Odler, Cem. Concr. Res., 1992, 22, 1130–1140 CrossRef CAS.
- R. H. Bogue and W. Lerch, Ind. Eng. Chem., 1934, 26, 837–847 CrossRef CAS.
- D. Nicia, C. Jakob, D. Jansen, D. Ivanov, O. Mazanec, J. Dengler, J. Neubauer and D. Lowke, Cem. Concr. Res., 2023, 173, 107289 CrossRef CAS.
- C. Jakob, D. Jansen, J. Dengler and J. Neubauer, Cem. Concr. Res., 2023, 166, 107095 CrossRef CAS.
- R. J. Flatt and Y. F. Houst, Cem. Concr. Res., 2001, 31, 1169–1176 CrossRef CAS.
- P. C. Nkinamubanzi, S. Mantellato and R. J. Flatt, in Science and Technology of Concrete Admixtures, ed. P.-C. Aïtcin and R. J. Flatt, Woodhead Publishing, 2016, pp. 353–377 DOI:10.1016/B978-0-08-100693-1.00016-3.
- L. Nicoleau, T. Gadt, L. Chitu, G. Maier and O. Paris, Soft Matter, 2013, 9, 4864–4874 RSC.
- P. C. Aïtcin, in Science and Technology of Concrete Admixtures, ed. P.-C. Aïtcin and R. J. Flatt, Woodhead Publishing, 2016, pp. 405–413 DOI:10.1016/B978-0-08-100693-1.00019-9.
- D. Marchon and R. J. Flatt, in Science and Technology of Concrete Admixtures, ed. P.-C. Aïtcin and R. J. Flatt, Woodhead Publishing, 2016, pp. 279–304 DOI:10.1016/B978-0-08-100693-1.00012-6.
- J. Cheung, A. Jeknavorian, L. Roberts and D. Silva, Cem. Concr. Res., 2011, 41, 1289–1309 CrossRef CAS.
- P. C. Aïtcin, in Science and Technology of Concrete Admixtures, ed. P.-C. Aïtcin and R. J. Flatt, Woodhead Publishing, 2016, pp. 395–404 DOI:10.1016/B978-0-08-100693-1.00018-7.
- J. E. Dengler, H. Grassl, J. Bizzozero and T. Gädt, Mater. Today Commun., 2024, 39, 109095 CrossRef CAS.
- X. Li, H. Grassl, C. Hesse and J. Dengler, Commun. Mater., 2024, 5, 1 CrossRef CAS.
- M. Bishop and A. R. Barron, Ind. Eng. Chem. Res., 2006, 45, 7042–7049 CrossRef CAS.
- X. Guan, F. Wang, Q. Ren, Y. Zheng, K. Y. Mei, C. Zhang and X. Cheng, Constr. Build. Mater., 2023, 399, 132580 CrossRef CAS.
- I. Kirchberger, F. Goetz-Neunhoeffer and J. Neubauer, Cem. Concr. Res., 2023, 170, 107188 CrossRef CAS.
- N. B. Singh, Cem. Concr. Res., 1976, 6, 455–460 CrossRef CAS.
- V. S. Ramachandran and M. S. Lowery, Thermochim. Acta, 1992, 195, 373–387 CrossRef CAS.
- N. L. Thomas and J. D. Birchall, Cem. Concr. Res., 1983, 13, 830–842 CrossRef CAS.
- L. Zhang, L. J. J. Catalan, R. J. Balec, A. C. Larsen, H. H. Esmaeili and S. D. Kinrade, J. Am. Ceram. Soc., 2010, 93, 279–287 CrossRef CAS.
- M. Bishop, S. G. Bott and A. R. Barron, Chem. Mater., 2003, 15, 3074–3088 CrossRef CAS.
- L. Coppola, S. Lorenzi, P. Kara and S. Garlati, Buildings, 2017, 7, 62 CrossRef.
- V. S. Ramachandran, M. S. Lowery, T. Wise and G. M. Polomark, Mater. Struct., 1993, 26, 425–432 CrossRef CAS.
- P. Gu, V. S. Ramachandran, J. J. Beaudoin and E. Quinn, Adv. Cem. Based Mater., 1995, 2, 182–188 CrossRef CAS.
- P. V. Coveney and W. Humphries, J. Chem. Soc., Faraday Trans., 1996, 92, 831–841 RSC.
- X. Lv and J. Li, J. Mater. Civ. Eng., 2023, 35, 130684 CrossRef.
- B. Li, X. Lv, Y. Dong, S. Zhou and J. Zhang, Constr. Build. Mater., 2018, 168, 958–965 CrossRef CAS.
- P. V. Coveney, R. J. Davey, J. L. W. Griffin and A. Whiting, Chem. Commun., 1998, 1467–1468, 10.1039/a802371i.
- J. Rickert and G. Thielen, Cem., Concr., Aggregates, 2004, 26, 1–10 CrossRef.
- X. Pang, P. Boontheung and P. J. Boul, Cem. Concr. Res., 2014, 63, 20–28 CrossRef CAS.
- H. von Daake and D. Stephan, Cem. Concr. Res., 2017, 102, 119–126 CrossRef CAS.
- J. Billingham and P. V. Coveney, J. Chem. Soc., Faraday Trans., 1993, 89, 3021–3028 RSC.
- J. L. W. Griffin, P. V. Coveney, A. Whiting and R. Davey, J. Chem. Soc., Perkin Trans. 2, 1999, 1973–1981, 10.1039/A902760B.
- K. L. Scrivener, P. Juilland and P. J. M. Monteiro, Cem. Concr. Res., 2015, 78, 38–56 CrossRef CAS.
- X. Li, A. Ouzia and K. Scrivener, Cem. Concr. Res., 2018, 108, 201–207 CrossRef CAS.
- C. Nalet and A. Nonat, Cem. Concr. Res., 2016, 90, 137–143 CrossRef CAS.
- W. Duan, H. Oota and K. Sawada, J. Chem. Soc., Dalton Trans., 1999, 3075–3080, 10.1039/a904461b.
- C. Lupu, R. S. Arvidson, A. Lüttge and A. R. Barron, Chem. Commun., 2005, 2354–2356, 10.1039/B500192G.
- C. Labbez, B. Jönsson, C. Woodward, A. Nonat and M. Delhorme, Phys. Chem. Chem. Phys., 2014, 16, 23800–23808 RSC.
- K. J. Laidler, Chemical kinetics, Harper and Row, 3rd edn, 1987 Search PubMed.
- L. Nicoleau and M. A. Bertolim, J. Am. Ceram. Soc., 2016, 99, 773–786 CrossRef CAS.
- S. Hanehara and K. Yamada, Cem. Concr. Res., 2008, 38, 175–195 CrossRef CAS.
- L. M. Meyer and W. F. Perenchio, Concr. Int., 1979, 1, 36–43 CAS.
- K. Burek, J. Dengler, F. Emmerling, I. Feldmann, M. U. Kumke and J. Stroh, ChemistryOpen, 2019, 8, 1441–1452 CrossRef CAS PubMed.
- T. Hirsch, T. Matschei and D. Stephan, Cem. Concr. Res., 2023, 168, 107150 CrossRef CAS.
- D. Wagner, F. Bellmann and J. Neubauer, Cem. Concr. Res., 2020, 137, 106198 CrossRef CAS.
- F. A. Hartmann and J. Plank, Constr. Build. Mater., 2021, 274, 122204 CrossRef.
- R. Costa, T. Cardoso, M. Degen, L. Silvestro, E. Rodríguez and A. P. Kirchheim, Cement, 2023, 11, 100057 CrossRef CAS.
- J. Plank, H. Keller, P. R. Andres and Z. Dai, Inorg. Chim. Acta, 2006, 359, 4901–4908 CrossRef CAS.
- H. Uchikawa, Function of organic admixture supporting high performance concrete, in Proceedings of the international RILEM conference on the role of admixtures in high performance concrete, ed. J. G. Cabrera and R. Rivera-Villareal, 1999 Search PubMed.
- B. J. Smith, L. R. Roberts, G. P. Funkhouser, V. Gupta and B. F. Chmelka, Langmuir, 2012, 28, 14202–14217 CrossRef CAS.
- B. J. Smith, A. Rawal, G. P. Funkhouser, L. R. Roberts, V. Gupta, J. N. Israelachvili and B. F. Chmelka, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 8949–8954 CrossRef CAS PubMed.
- J. Nehring, J. Neubauer, S. Berger and F. Goetz-Neunhoeffer, Cem. Concr. Res., 2018, 107, 264–274 CrossRef CAS.
- S. T. Bergold, F. Goetz-Neunhoeffer and J. Neubauer, Cem. Concr. Res., 2017, 93, 32–44 CrossRef CAS.
- L. Nicoleau, E. Schreiner and A. Nonat, Cem. Concr. Res., 2014, 59, 118–138 CrossRef CAS.
- E. Pustovgar, R. K. Mishra, M. Palacios, J.-B. d’Espinose de Lacaillerie, T. Matschei, A. S. Andreev, H. Heinz, R. Verel and R. J. Flatt, Cem. Concr. Res., 2017, 100, 245–262 CrossRef CAS.
- F. Caruso, S. Mantellato, M. Palacios and R. J. Flatt, Cem. Concr. Res., 2017, 91, 52–60 CrossRef CAS.
- A. Picker, L. Nicoleau, A. Nonat, C. Labbez and H. Cölfen, Cem. Concr. Res., 2023, 174, 107329 CrossRef CAS.
- X. Li and K. L. Scrivener, Cem. Concr. Res., 2022, 154, 106734 CrossRef CAS.
- A. Bazzoni, S. H. Ma, Q. Q. Wang, X. D. Shen, M. Cantoni and K. L. Scrivener, J. Am. Ceram. Soc., 2014, 97, 3684–3693 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma00299g |
| ‡ Cement nomenclature: C = CaO, S = SiO2, A = Al2O3, and H = H2O; C3S refers to 3CaO·SiO2, namely, tricalcium silicate. |
| This journal is © The Royal Society of Chemistry 2024 |