
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In situ formation of robust nanostructured cobalt oxyhydroxide/cobalt oxide oxygen evolution reaction electrocatalysts†
Yupeng
Zhao
ab,
Dandan
Gao
 ab,
Johannes
Biskupek
c,
Ute
Kaiser
c,
Rongji
Liu
*abd and
Carsten
Streb
*abd
ab,
Johannes
Biskupek
c,
Ute
Kaiser
c,
Rongji
Liu
*abd and
Carsten
Streb
*abd
aDepartment of Chemistry, Johannes Gutenberg University Mainz, Duesbergweg 10-14, 55131 Mainz, Germany. E-mail: rongji.liu@uni-mainz.de; carsten.streb@uni-mainz.de
bInstitute of Inorganic Chemistry I, Ulm University, Albert-Einstein-Allee 11, 89081 Ulm, Germany
cCentral Facility of Electron Microscopy for Materials Science, Ulm University, Albert-Einstein-Allee 11, 89081 Ulm, Germany
dHelmholtz-Institute Ulm, Electrochemical Energy Conversion, Helmholtzstr. 11, 89081 Ulm, Germany
First published on 22nd April 2024
Abstract
The design of efficient and stable oxygen evolution reaction (OER) catalyst-based earth-abundant metal precursors is crucial for large-scale energy conversion and storage. To-date, many catalyst materials are limited by poor stability in harsh oxidative conditions. Thus, much research is targeted at developing materials that can operate under demanding OER conditions. One promising approach is the in situ formation of catalysts which are inherently stable under the oxidizing, alkaline conditions often used in OER studies. Here, we report how mixed metal sulfide precursors (i.e. CoMo2S4 and FeS2) which give the low overpotentials (307 mV at j = 10 mA cm−2) at the beginning of catalysis, are converted in situ to give a highly stable composite OER catalyst under alkaline OER conditions (1 M aqueous KOH solution, pH = 13.8). Mechanistic studies reveal that under operation, the precursor materials are converted to γ-CoOOH nanofibers and Co2O3 nanoparticles, both well-known prototype OER catalysts. The report demonstrates that the presence of crystalline mix metal sulfide precursors is critical for the simultaneous in situ formation of the active catalysts, highlighting that use of these earth-abundant minerals might offer an economically and chemically viable route for scalable catalyst development.
1. Introduction
Electrocatalytic splitting of water into hydrogen and oxygen plays a crucial role in converting and storing electrical energy.1–3 However, specifically the anodic oxygen evolution reaction (OER) is severely hindered by a challenging proton-coupled four-electron transfer, together with operation at harsh oxidative conditions under highly alkaline or highly acidic conditions.4,5 Thus, the reaction often shows slow kinetics and is considered the bottleneck of electrochemical water splitting.6 Developing efficient and stable oxygen evolution catalysts (OECs) therefore is critical to enable large-scale conversion of electricity from intermittent sources, e.g. wind or solar energy. Early studies were primarily focused on noble-metal oxide catalysts, such as RuO2 and IrO2,7–10 but high cost, scarcity, and poor long-term stability limit their large-scale deployment.11 Thus, recent research has been centered on developing efficient catalysts based on economically viable earth-abundant metals to enable scale-up and industrial use.12,13One challenge in OER catalyst design is the stable electrical wiring of the reactive site to electrically conductive support, e.g. high surface-area carbon. One facile route to this end is the use of zeolitic imidazolate frameworks (ZIFs) as precursors, which combine tunable composition, facile incorporation of metals, and easy thermal conversion to conductive carbon matrices.14,15 Of specific interest is the cobalt-containing ZIF-67 which can easily be synthesized at large scale and features tunable chemical composition and reproducible particle morphology.16 Various methods have been developed to convert ZIF-67 into the corresponding carbon-anchored metal oxide,17,18 metal phosphide,19 metal carbide,20 and metal sulfide.21 For example, Li et al. synthesized a ZIF-67 derived Co3O4@Z67-N700@CeO2, which exhibited a low overpotential (η = 350 mV) at a current density (j) of 10 mA cm−2.18 Similarly, highly-dispersed Co3O4/N-doped porous carbons were synthesized by calcination of ZIF-67/COF, which also featured good catalytic activities (η = 330 mV at j = 10 mA cm−2, a Tafel slope of 79 mV dec−1).17 Additionally, Wang et al. also prepared a cobalt sulfide electrocatalyst (h-CoxSy) which exhibited outstanding electrocatalytic OER performance. Despite this progress, many challenges still exist. One major issue is that many ZIF-67-derived composites are unstable under harsh oxidative and/or alkaline conditions. This is specifically challenging for ZIF-67-derived metal sulfides which can be oxidized to sulfur-oxygen compounds (e.g., sulfite or sulfate) under typical OER conditions. Often, this conversion occurs during operation and results in catalyst degradation and loss of reactivity.22 Here, we build on these studies and propose the use of ZIF-67-based metal sulfide as catalyst precursors which are converted into highly stable and highly active metal oxide/oxyhydroxide phases under oxidative, alkaline OER conditions. This conversion and activation route could provide a facile path in catalyst design to enable the self-selection of the most stable catalysts formed under the relevant catalytic conditions, resulting in high stability and high catalytic performance.
Here, we exemplify this approach by developing mixed metal sulfide catalyst precursors (i.e., CoMo2S4 and FeS2) using a facile synthetic route. We show that under OER conditions, the metal sulfides convert into Co2O3 nanoparticles and γ-CoOOH nanofibers, which act as high-performance active sites in the following OER catalysis.
2. Results and discussion
Briefly, pristine ZIF-67 polyhedrons were prepared based on a modified literature route.23,24 The samples were subsequently treated with the etching agent/Mo-precursor [H3PMo12O40]·xH2O (phosphomolybdic acid, PMA) and the Fe-precursor FeCl2·4H2O in ethanol solution, resulting in the formation of PMA@ZIF-CoFe (Scheme 1, for details, see ESI†). After that, the original purple ZIF-67 became brown because of the partial replacement of the Co ions in ZIF-67 by Fe ions and the introduction of [PMo12O40]3− (Fig. S1a, ESI†), as shown by Fourier transform infrared spectroscopy (FT-IR, see ESI,† Fig. S1b).24,25 Subsequently, PMA@ZIF-CoFe was converted to the metal sulfide-based precursor (hereafter referred to as Composite 1) by a solvothermal reaction with thioacetamide (TAA) at ethanol (90 °C for 3 h, followed by 200 °C for 3 h). To explore the temperature effect on the catalyst, Composite 2 was prepared by a similar synthetic route, where the solvothermal reaction was carried out at 90 °C for 6 h.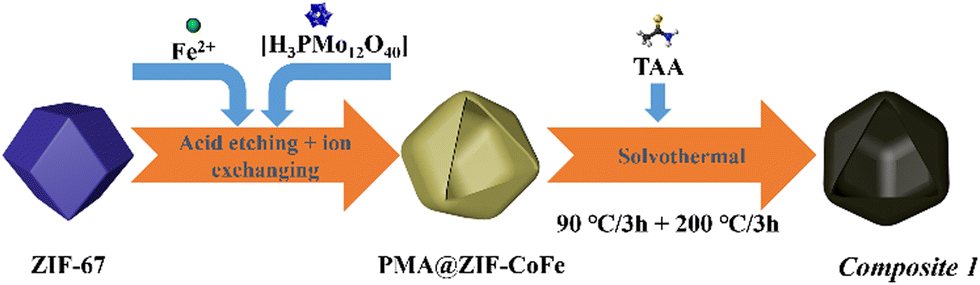 | ||
| Scheme 1 Schematic illustration of the preparation of Composite 1. | ||
Powder X-ray diffraction (pXRD) and FT-IR spectroscopy were used to examine the composition of Composite 1 and Composite 2. As shown in Fig. 1a, the pXRD data shows the presence of crystalline FeS2 and CoMo2S4 in Composite 1. The presence of these metal sulfides is further supported by the FT-IR spectrum of Composite 1 (Fig. 1b) which indicates the presence of S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S, and metal (M)–S bonds.26–29 In comparison, the pXRD and FT-IR data of Composite 2 (ESI,† Fig. S2) indicate the presence of amorphous metal sulfides. Notably, the presence of S–O bonds was also indicated by FT-IR spectroscopy, which suggests partial (surface) oxidation of the metal sulfides.23,30 This highlights that the elevated solvothermal temperature (200 °C for 3 h) is required to access crystalline metal sulfide phases as observed in Composite 1. Aberration-corrected high-resolution transmission electron microscopy (HRTEM), scanning electron microscopy (SEM), and scanning transmission electron microscopy (STEM) together with energy dispersive X-ray spectroscopy (EDX) were employed to examine the structure and morphologic structure of Composite 1 and Composite 2. As shown in Fig. 1c and Fig. S4a (ESI†), Composite 1 exhibits hollow polyhedral structures with an average diameter of around 800 nm, which is slightly smaller than that in the pristine ZIF-67 (Fig. S3a, ESI†). We assign this size reduction to the acid etching process, as similar changes have been observed previously.31 The surface of the polyhedral shows some roughening which is not present in the pristine ZIF-67. The same morphological feature can be seen in the case of Composite 2 as well (Fig. S3b and c, ESI†) and is also assigned to the etching procedure. For Composite 1, the HRTEM image (Fig. 1d and f) indicates the presence of nanocrystalline regions (blue circles) on the particle surface and particle edges. The nanocrystals are identified as FeS2 (JCPDS 42-1340) and CoMo2S4 (JCPDS 24-0332) by the corresponding selective area electron diffraction (SAED) analysis (Fig. 1e). The HRTEM image (Fig. 1f) of Composite 1 shows lattice and fringes with interplanar spacing of 0.26 nm and 0.19 nm, corresponding to the (
S, and metal (M)–S bonds.26–29 In comparison, the pXRD and FT-IR data of Composite 2 (ESI,† Fig. S2) indicate the presence of amorphous metal sulfides. Notably, the presence of S–O bonds was also indicated by FT-IR spectroscopy, which suggests partial (surface) oxidation of the metal sulfides.23,30 This highlights that the elevated solvothermal temperature (200 °C for 3 h) is required to access crystalline metal sulfide phases as observed in Composite 1. Aberration-corrected high-resolution transmission electron microscopy (HRTEM), scanning electron microscopy (SEM), and scanning transmission electron microscopy (STEM) together with energy dispersive X-ray spectroscopy (EDX) were employed to examine the structure and morphologic structure of Composite 1 and Composite 2. As shown in Fig. 1c and Fig. S4a (ESI†), Composite 1 exhibits hollow polyhedral structures with an average diameter of around 800 nm, which is slightly smaller than that in the pristine ZIF-67 (Fig. S3a, ESI†). We assign this size reduction to the acid etching process, as similar changes have been observed previously.31 The surface of the polyhedral shows some roughening which is not present in the pristine ZIF-67. The same morphological feature can be seen in the case of Composite 2 as well (Fig. S3b and c, ESI†) and is also assigned to the etching procedure. For Composite 1, the HRTEM image (Fig. 1d and f) indicates the presence of nanocrystalline regions (blue circles) on the particle surface and particle edges. The nanocrystals are identified as FeS2 (JCPDS 42-1340) and CoMo2S4 (JCPDS 24-0332) by the corresponding selective area electron diffraction (SAED) analysis (Fig. 1e). The HRTEM image (Fig. 1f) of Composite 1 shows lattice and fringes with interplanar spacing of 0.26 nm and 0.19 nm, corresponding to the (![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 04) plane of CoMo2S4 and (220) plane of FeS2. However, no obvious nanocrystals can be observed in HRTEM images (Fig. S3d, ESI†) of Composite 2, and the broad diffraction rings rather than thin ones or single reflections in the corresponding SAED (Fig. S3d insert, ESI†) indicates its amorphous feature. The results agree with the powder XRD measurement. In addition, STEM-energy-dispersive X-ray spectroscopy (EDX) elemental mappings verify the presence of homogeneously dispersed Co, Fe, Mo, and S within Composite 1 (Fig. S4b, ESI†) and Composite 2 (Fig. S3f, ESI†). All these results indicate that the synthesized PMA@ZIF-CoFe was successfully converted to metal sulfides (i.e. CoMo2S4 and FeS2) and led to a homogeneous distribution of these metal ions. In addition, the atomic ratio of Co, Fe, Mo, and S (2.63
04) plane of CoMo2S4 and (220) plane of FeS2. However, no obvious nanocrystals can be observed in HRTEM images (Fig. S3d, ESI†) of Composite 2, and the broad diffraction rings rather than thin ones or single reflections in the corresponding SAED (Fig. S3d insert, ESI†) indicates its amorphous feature. The results agree with the powder XRD measurement. In addition, STEM-energy-dispersive X-ray spectroscopy (EDX) elemental mappings verify the presence of homogeneously dispersed Co, Fe, Mo, and S within Composite 1 (Fig. S4b, ESI†) and Composite 2 (Fig. S3f, ESI†). All these results indicate that the synthesized PMA@ZIF-CoFe was successfully converted to metal sulfides (i.e. CoMo2S4 and FeS2) and led to a homogeneous distribution of these metal ions. In addition, the atomic ratio of Co, Fe, Mo, and S (2.63![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1.4:6.5) in Composite 1 was determined by inductively coupled plasma optical emission spectroscopy (ICP-OES). The result indicates that in addition to the formation of crystalline CoMo2S4 and FeS2, some amorphous cobalt sulfide might be present in Composite 1.
:1:1.4:6.5) in Composite 1 was determined by inductively coupled plasma optical emission spectroscopy (ICP-OES). The result indicates that in addition to the formation of crystalline CoMo2S4 and FeS2, some amorphous cobalt sulfide might be present in Composite 1.
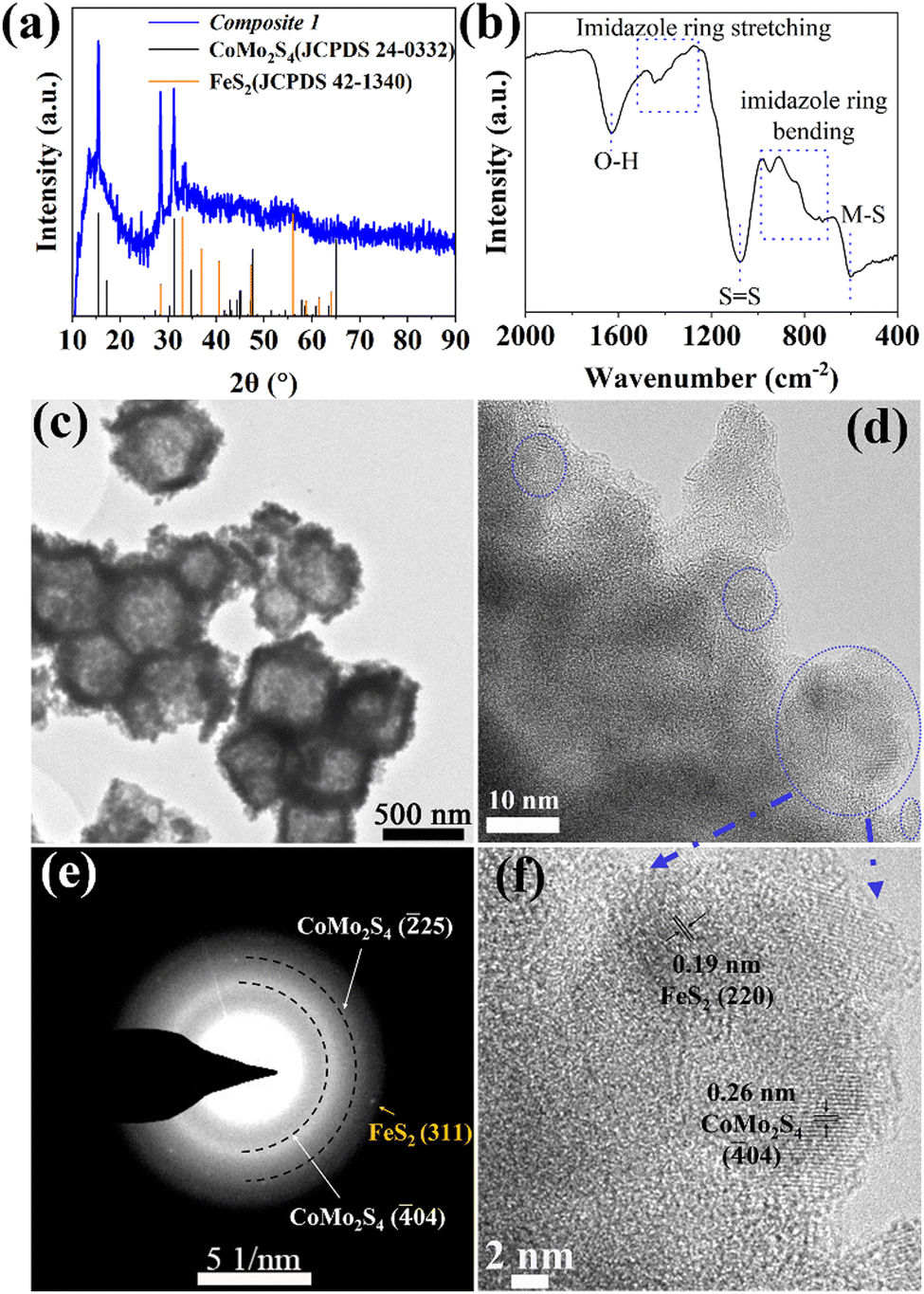 | ||
| Fig. 1 (a) pXRD pattern, (b) FT-IR spectrum, (c) TEM, and (d) and (f) HTREM images with (e) corresponding selected area electron diffraction pattern of Composite 1. | ||
The elemental composition and oxidation states in Composite 1 and Composite 2 were studied by X-ray photoelectron spectroscopy (XPS). The survey XPS spectra indicate the presence of Co, Fe, Mo, and S, in both Composite 1 (Fig. S5a, ESI†) and Composite 2 (Fig. S5b, ESI†). The deconvoluted high-resolution Fe 2p XPS spectra in Composite 1 (Fig. 2a) and Composite 2 (Fig. S5c, ESI†) show characteristic peaks at ∼712 eV, which are indicative of Fe2+ species.32 Satellite features (∼786 eV) in the deconvoluted Co 2p XPS spectra for Composite 1 (Fig. 2b) and Composite 2 (Fig. S5d, ESI†) suggest the presence of Co2+. This is further supported by peaks at ∼781.5 eV which are characteristic of Co2+ 2p3/2 in both Composite 1 and Composite 2.33 It should be noted that a strong and sharp signal appeared at 779 eV in the Co 2p3/2 spectrum of Composite 1 which can be assigned to Co–S interactions. Given that the binding energies of Co are close to metallic Co, this observation aligns with recently reported data on CoMo2S4 catalysts.34–36 The metal-like properties of Co in CoMo2S4 may facilitate electron delocalization, thereby enhancing conductivity. In contrast, for Composite 2, the Co 2p3/2 spectrum shows a signal at 780.4 eV which is assigned to Co3+ (Fig. S5c, ESI†). As for Mo 3d XPS spectrum of Composite 1 (Fig. 2c), the main binding energies located at 229 eV (Mo 3d5/2) and 232.2 eV (Mo 3d3/2) for Mo4+, belong to the Mo–S bond.34 In addition, the peaks at 232.5 eV and 235.6 eV are assigned to Mo6+. We assign it to the partial oxidation of metal sulfides. The predominant valence state of Mo4+ in CoMo2S4 provides additional confirmation of the metallic properties exhibited by Co within the CoMo2S4 structure. Notably, the S 2s signal also appeared at the same region of Mo 3d and was deconvoluted into three peaks at 233.9 eV, 226.4 eV, and 226 eV, corresponding to the three chemical states of the S species bonding with O (SO42−), Mo, and Co ions.35 However, only Mo6+ is observed in Composite 2 (ESI,† Fig. S5e). This is expected, as CoMo2S4 is only formed at an elevated reaction temperature (200 °C for Composite 1), while Composite 2 is not exposed to this temperature.23 The deconvoluted XPS spectrum for S 2p (Fig. 2d) indicates the existence of metal sulfides in Composite 1 (161.7 eV for S2− 2p3/2 and 162.8 eV for S22− 2p3/2).37 Similarly, signals for S2− 2p3/2 and S22− 2p3/2 were observed in Composite 2. It is worth noting that the XPS results of previously reported binding energies of Co 2p3/2 (778.9 eV), Mo 3d5/2 (228.7 eV), and S 2p3/2 (161.9 eV) in CoMo2S4 are in line with our observations, providing further support for the presence of CoMo2S4.36 Also, the signals belonging to SO42− were also observed at 168.7 eV (2p3/2) and 169.9 eV (2p1/2), which also indicates the partial oxidation of the metal sulfides.38
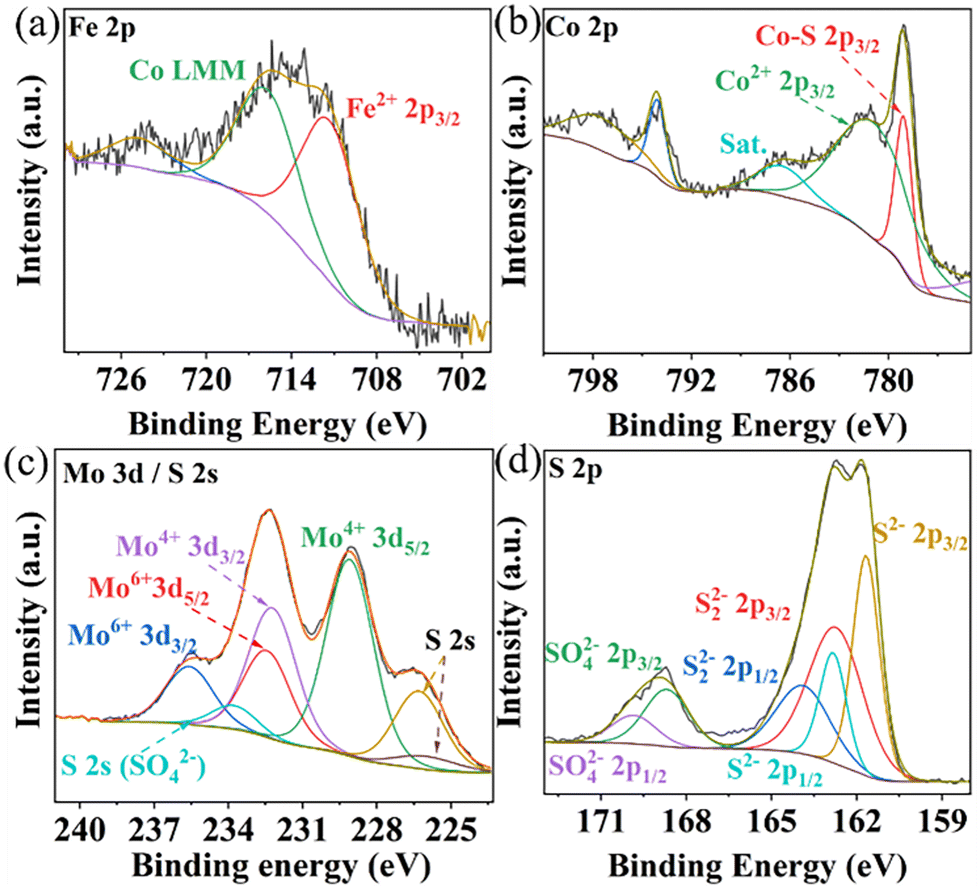 | ||
| Fig. 2 Deconvoluted high-resolution XPS spectra of Fe 2p (a), Co 2p (b), Mo 3d (c), and S 2p (d) of Composite 1. | ||
Next, we studied the in situ conversion of Composite 1 into the real active OER catalyst under oxidative electrochemical conditions. To this end, we studied the changes of Composite 1 in alkaline aqueous solution (1.0 M aqueous KOH) using a conventional three-electrode electrochemical setup. The catalyst was deposited on glassy carbon rotating disk electrode (RDE) working electrodes as follows: catalyst inks were prepared by sonicating a dispersion of the respective catalyst, 5 wt% Nafion solution, deionized (DI) water, and ethanol (for details, see ESI†). Subsequently, the respective ink was drop-cast onto the glassy carbon RDE working electrode. A Hg/HgO electrode was used as reference electrode, while a graphite rod was used as counter electrode. All potentials in this study have been converted to reversible hydrogen electrode potentials (RHE, see details in the ESI†). To assess synergistic effects of multiple metal reaction sites on OER, reference catalysts were prepared for comparison, labeled as Composite 3 (without Mo), Composite 4 (without Fe), and Composite 5 (without Fe and Mo), (see ESI† for synthetic and analytical details).
Linear sweep voltammetry (LSV) at a scan rate of 5 mV s−1 was conducted to evaluate the OER electrocatalytic performance. As illustrated in Fig. 3a and b, Composite 1 showed the lowest overpotential of η = 307 mV at a current density of j = 10 mA cm−2 (Fig. 3a and b). Composite 2 exhibited a slightly higher overpotential (η = 315 mV). However, in the absence of Mo (Composite 3), Fe (Composite 4), or both Mo and Fe (Composite 5), significantly higher overpotentials at the same current density were observed (Fig. 3a), indicating that the combined presence of Mo, Fe, and Co improve the OER activities, e.g., by synergistic effects on the composite electronic properties. Furthermore, Tafel slopes (η versus log(j)) of 74 mV dec−1 and 82 mV dec−1 were obtained for Composite 1 and Composite 2 (Fig. S6a, ESI†), while the other composites exhibited higher Tafel slopes. The significantly enhanced catalytic properties and OER kinetics of Composite 1 and Composite 2 underscore the vital role played by the concurrent presence of both Fe and Mo in the mixed metal sulfide-based catalysts.
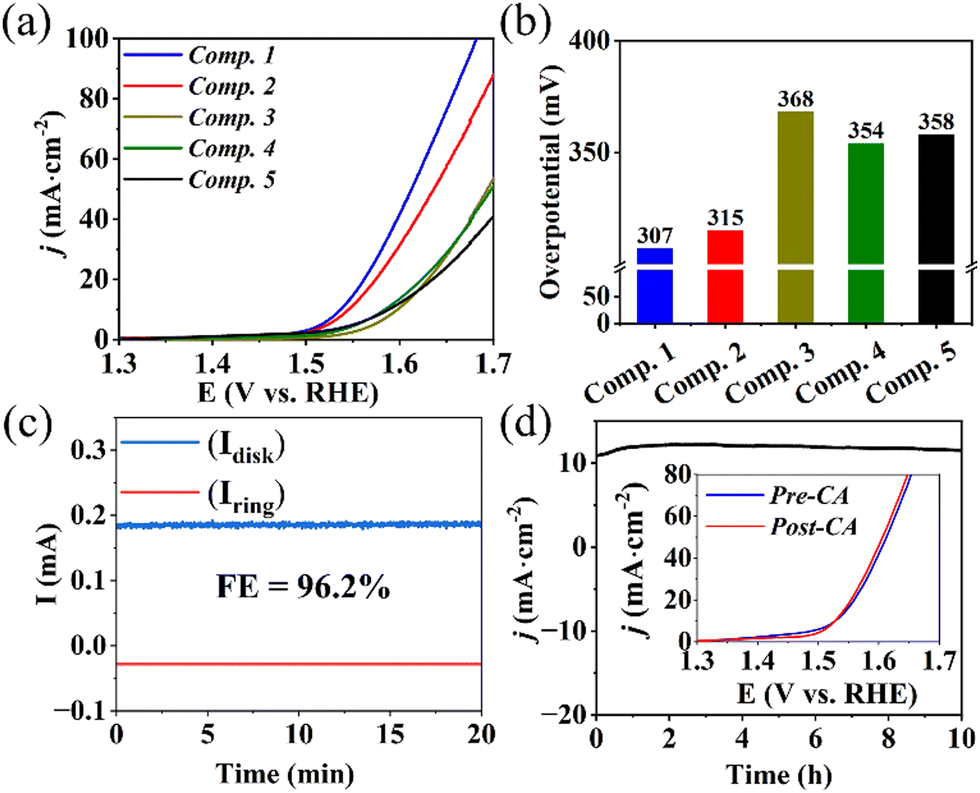 | ||
| Fig. 3 Electrocatalytic OER performance of Composite 1, Composite 2, and reference composites in 1 M aqueous KOH: (a) iR-corrected OER polarization curves, (b) Overpotentials at j = 10 mA cm−2, (c) RRDE analysis to determine faradaic efficiency, and (d) Chronoamperometry curves at η = 330 mV without iR compensation (insert: LSV polarization curves comparison before and after 10 h stability test) of Composite 1. | ||
Rotating ring-disk electrode (RRDE) voltammetry was employed to gain a deeper understanding of the OER reaction mechanism of Composite 1. To this end, an LSV measurement between 1.3 V and 1.7 V was performed on the disk electrode, where the OER reaction takes place, while the Pt ring electrode (set at a fixed potential of 1.5 V) was used to detect any partially oxidized intermediates formed during OER, such as H2O2 (for analytical details see ESI†). Under the given conditions, a negligible ring current was detected, and the electron transfer number was calculated to be close to 4, indicating that H2O2 intermediates were scarcely generated, confirming a desirable 4-electron pathway for OER (ESI,† Fig. S6b and c). In addition, the faradaic efficiency (FE) was measured using RRDE voltammetry at potentials of 0.4 V (ring) and 1.5 V (disk) at a rotation rate of 1600 rpm, so that oxygen generated on the disk surface could be reductively collected at the ring electrode (current collection efficiency: 15.7%). Based on the detected ring current (∼28 μA) and disk current (∼185 μA), a FE of 96.2% was calculated for Composite 1 (Fig. 3c, also refer to ESI† for detailed calculations).
Next, electrochemical impedance spectroscopy (EIS) was used to better understand the origins of the electrocatalytic performance. The results (ESI,† Fig. S7) indicate that Composite 1 features the lowest charge-transfer resistance (Rct = 9 Ω) among the catalyst samples studied, suggesting efficient interfacial electron transfer. Also, the double-layer capacitance (Cdl), proportional to the electrochemically active surface area (ECSA), was determined to compare the amount of reactive sites in the composites. As shown in Fig. S8 (ESI†), all composites tested show comparable ECSAs. Therefore, we conclude that the superior OER performance of Composite 1 is attributed to its low internal electron transfer resistance as well as intrinsic reactivity of the reactive sites.
Subsequently, chronoamperometry (CA) measurements were utilized to evaluate the stability and study the morphological and compositional changes of Composite 1 at 1.56 V in 1 M aqueous KOH. As illustrated in Fig. 3d, the current density j remained essentially unchanged with only a minor increase in the initial phase (0–3 h), and a marginal decrease in the second phase (3–10 h) of CA. This finding is supported by LSV analysis before and after CA, which are essentially identical (Fig. 3d and ESI,† Fig. S6d and S9).
To explore our hypothesis of structural conversion of the metal sulfide precursors under OER conditions, a morphological and chemical investigation was carried out after the stability test using AC-HRTEM and XPS. As shown in Fig. 4a, after 3 h of electrocatalysis, the hollow structure of Composite 1 was converted into a mixture of 2-dimensional (2D) nanoplates and 1D nanofibers, which formed on the particle surface and particle edges (Fig. 4b and c and ESI,† Fig. S10a). These nanofibers were identified as γ-CoOOH, based on a 0.44 nm interplanar spacing in the HRTEM image (Fig. 4c) and the matching SAED pattern (ESI,† Fig. S10b, match with γ-CoOOH JCPDS 14-0673). Elemental mapping by STEM-EDX further confirmed the presence of Co and O (ESI† Fig. S11). Based on these findings, the improved catalytic activities were attributed to the formation of γ-CoOOH, a recognized active OER catalyst.1,39,40 This result aligns with the XPS measurement (ESI,† Fig. S12), which revealed the absence of Co2+ and the presence of Co3+, as no satellite features (≈786 eV) for Co2+ were found. Also, XPS analysis indicated the absence of Mo and Fe signals. In addition, S2−/S22− was oxidized to SO42−, which is indicated by S 2p XPS data in Fig. S12 (ESI†). The findings indicate the decomposition of FeS2 and CoMo2S4, which can be attributed to the extremely harsh conditions encountered during OER. Moreover, after 10 h of electrocatalysis, nanoparticles (indicated by a white box in Fig. 4f and ESI,† Fig. S10d), were observed and identified as Co2O3 based on the corresponding SAED pattern (ESI,† Fig. S10e, JCPDS 02-0770). The oxidation of Co2+ was also witnessed by cyclic voltammetry (CV), a clear peak at 1.13 V can be observed, which are assigned to Co3+/2+ (ESI,† Fig. S10f).41 In addition, aggregation of γ-CoOOH nanofibers was observed after 10 h of electrocatalysis, as shown in Fig. 4b and e, and Fig. S13 (ESI†). The average length of γ-CoOOH nanofibers increased from ∼40 nm to ∼110 nm after 3 and 10 h OER catalysis. This aggregation of γ-CoOOH nanofibers might be linked to the marginal loss of catalytic reactivity observed during the 10 h CA experiments (Fig. 3f). As a comparison, we also investigated structural and chemical changes of Composite 2. Notably, Composite 2 exhibited poorer stability compared to Composite 1 (ESI,† Fig. S14). Composite 2 showed a CA behavior similar to Composite 1, featuring an initial slight current density increase followed by a subsequent drop in current density. Notably, the loss of reactivity was more pronounced than observed for Composite 1. Morphological and chemical investigations on Composite 2 were carried out after the stability test to explore the origins of the stability difference between Composite 1 and Composite 2. As shown in the ESI,† Fig. S13–S17, similar morphological and chemical changes were observed after catalysis in Composite 2 compared to Composite 1, including the formation of γ-CoOOH nanofibers and Co2O3 nanoparticles. However, there were fewer crystalline nanofibers and nanoparticles observed in Composite 2 after electrocatalysis for 10 hours compared to Composite 1. Consequently, the lower number of γ-CoOOH nanofibers and Co2O3 nanoparticles provided fewer active sites for OER, resulting in its poorer stability. Thus, we propose that the crystalline metal sulfides could effectively prevent Co leakage during OER and facilitate the oxidation of Co2+ to Co3+ and the formation of Co2O3 and γ-CoOOH, contributing to high stability. In contrast, amorphous metal sulfides might show higher solubility or faster degradation than the crystalline systems, resulting in faster degradation and leaching.41,42
 | ||
| Fig. 4 TEM and HRTEM images of Composite 1 after electrocatalysis for 3 h (a)–(c) and 10 h (d)–(f) | ||
3. Conclusion
In summary, we report the in situ conversion of a CoMo2S4/FeS2 composite heterojunction was prepared by a facile synthetic method. The synergistic effects of its structural and compositional characteristics contribute to the remarkable electrocatalytic performance, displaying a low overpotential (307 mV at j = 10 mA cm−2). As the electrocatalysis progresses, the crystalline metal sulfides are converted in situ into Co2O3 nanoparticles and γ-CoOOH nanofibers. These species act as the true active sites in OER electrocatalysis, resulting in high activity and high stability. We also observed the aggregation of γ-CoOOH nanofibers during prolonged electrocatalysis, resulting in a slight degradation of its electrocatalytic properties. This synthetic approach and structural/compositional evolution hold significant promise for pre-catalyst design and stability enhancement in metal sulfide-based catalysts, providing insights into the genuine active sites for metal sulfide-based OER electrocatalysts.4. Experimental section
4.1 Chemicals and solvents
Cobalt dinitrate hexahydrate (Co(NO3)2·6H20, ≥ 99%, CAS no. 10026-22-9), 2-methylimidazole (2-MIM, C4H6N2, 99%, CAS no. 693-98-1, 2-MIM), and iron (II) dichloride tetrahydrate (FeCl2·4H2O, 99%, CAS no. 13478-10-9) were purchased from Sigma-Aldrich. Ethanol (CH3CH2OH, absolute, CAS no. 64-17-5, EtOH) and potassium hydroxide (KOH, reagent grade, CAS no. 1310-58-3) were purchased from Fisher Scientific. Thioacetamide (CH3CSNH2, analysis, CAS no. 62-55-5, TAA) and 12-Molybdophosphoric acid hydrate (PMA, [H3PMo12O40]·xH2O, analysis, CAS no. 51429-74-4, PMA) were purchased from Merck. Methanol (CH3OH, reagent grade, CAS no. 67-56-1, MeOH) was purchased from VWR.4.2 Materials synthesis
In addition, Composite 2 was prepared by the same procedures, except the reaction temperature was set at 90 °C for 6 hours in the sulfidation. Composite 3, Composite 4, and Composite 5 were also prepared with the same procedures, but without the addition of PMA (Composite 3), FeCl2·4H2O (Composite 4), PMA, and 40 mg of FeCl2·4H2O (Composite 5), respectively during the acid etching and ion exchange.
Author contributions
Y. Z., R. L., and C. S. conceived the project. Y. Z. carried out the material fabrication and characterization. Y. Z. designed and performed electrocatalytic studies. R. L., Y. Z., and C. S. performed data analysis. Y. Z. and D. G. performed SEM/EDX analysis. J. B. and U. K. performed (S)TEM analyses. All authors co-wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge financial support by the Deutsche Forschungsgemeinschaft DFG (Cluster of Excellence EXC2154, POLiS, project number: 390874152 and TRR 234 CataLight, project number: 364549901 and project no 389183496). R. L. gratefully acknowledges financial support by the Alexander von Humboldt Foundation. D. G. acknowledges the Deutsche Forschungsgemeinschaft (DFG) for a Walter Benjamin Fellowship (project no. 510966757). R. L., D. G., and C. S. gratefully acknowledge financial support by Johannes Gutenberg University Mainz, the Top-Level Research Initiative SusInnoScience and the Gutenberg Research College. Dr Shuai Chen is acknowledged for XRD characterization support.References
- Z. P. Ifkovits, J. M. Evans, M. C. Meier, K. M. Papadantonakis and N. S. Lewis, Decoupled Electrochemical Water-Splitting Systems: A Review and Perspective, Energy Environ. Sci., 2021, 14(9), 4740–4759 RSC.
- X. Zou and Y. Zhang, Noble Metal-Free Hydrogen Evolution Catalysts for Water Splitting, Chem. Soc. Rev., 2015, 44(15), 5148–5180 RSC.
- B. Zhang, Y. Zheng, T. Ma, C. Yang, Y. Peng, Z. Zhou, M. Zhou, S. Li, Y. Wang and C. Cheng, Designing MOF Nanoarchitectures for Electrochemical Water Splitting, Adv. Mater., 2021, 33(17), 2006042 CrossRef CAS PubMed.
- J. Song, C. Wei, Z. F. Huang, C. Liu, L. Zeng, X. Wang and Z. J. Xu, A Review on Fundamentals for Designing Oxygen Evolution Electrocatalysts, Chem. Soc. Rev., 2020, 49(7), 2196–2214 RSC.
- L. Tian, X. Zhai, X. Wang, J. Li and Z. Li, Advances in Manganese-Based Oxides for Oxygen Evolution Reaction, J. Mater. Chem. A, 2020, 8(29), 14400–14414 RSC.
- N. T. Suen, S. F. Hung, Q. Quan, N. Zhang, Y. J. Xu and H. M. Chen, Electrocatalysis for the Oxygen Evolution Reaction: Recent Development and Future Perspectives, Chem. Soc. Rev., 2017, 46(2), 337–365 RSC.
- C. Spöri, J. T. H. Kwan, A. Bonakdarpour, D. P. Wilkinson and P. Strasser, The Stability Challenges of Oxygen Evolving Catalysts: Towards a Common Fundamental Understanding and Mitigation of Catalyst Degradation, Angew. Chem., Int. Ed., 2017, 56(22), 5994–6021 CrossRef PubMed.
- H. Liu, X.-H. Zhang, Y.-X. Li, X. Li, C.-K. Dong, D.-Y. Wu, C.-C. Tang, S.-L. Chou, F. Fang, X.-W. Du, H. Liu, C. Dong, D. Wu, X. Du, X. Zhang, Y. Li, X. Li, C. Tang, S. Chou and F. Fang, Conductive Boron Nitride as Promising Catalyst Support for the Oxygen Evolution Reaction, Adv. Energy Mater., 2020, 10(25), 1902521 CrossRef CAS.
- J. Wang, L. Han, B. Huang, Q. Shao, H. L. Xin and X. Huang, Amorphization Activated Ruthenium-Tellurium Nanorods for Efficient Water Splitting, Nat. Commun., 2019, 10(1), 1–11 CrossRef PubMed.
- T. Reier, Z. Pawolek, S. Cherevko, M. Bruns, T. Jones, D. Teschner, S. Selve, A. Bergmann, H. N. Nong, R. Schlögl, K. J. J. Mayrhofer and P. Strasser, Molecular Insight in Structure and Activity of Highly Efficient, Low-Ir Ir-Ni Oxide Catalysts for Electrochemical Water Splitting (OER), J. Am. Chem. Soc., 2015, 137(40), 13031–13040 CrossRef CAS PubMed.
- T. X. Nguyen, Y. H. Su, C. C. Lin and J. M. Ting, Self-Reconstruction of Sulfate-Containing High Entropy Sulfide for Exceptionally High-Performance Oxygen Evolution Reaction Electrocatalyst, Adv. Funct. Mater., 2021, 31(48), 2106229 CrossRef CAS.
- Z.-P. Wu, F. Lu, S.-Q. Zang, X. Wen, D. Lou, Z.-P. Wu, X. F. Lu, X. W. Lou and S.-Q. Zang, Non-Noble-Metal-Based Electrocatalysts toward the Oxygen Evolution Reaction, Adv. Funct. Mater., 2020, 30(15), 1910274 CrossRef CAS.
- W. Xiong, H. Yin, T. Wu and H. Li, Challenges and Opportunities of Transition Metal Oxides as Electrocatalysts, Chem. – Eur. J., 2023, 29(5), e202202872 CrossRef CAS PubMed.
- H. Wen, S. Zhang, T. Yu, Z. Yi and R. Guo, ZIF-67-Based Catalysts for Oxygen Evolution Reaction, Nanoscale, 2021, 13(28), 12058–12087 RSC.
- Y. Wang, Y. Wang, L. Zhang, C.-S. Liu and H. Pang, Core–Shell-Type ZIF-8@ZIF-67@POM Hybrids as Efficient Electrocatalysts for the Oxygen Evolution Reaction, Inorg. Chem. Front., 2019, 6(9), 2514–2520 RSC.
- C. Cui, X. Lai, R. Guo, E. Ren, W. Qin, L. Liu, M. Zhou and H. Xiao, Waste Paper-Based Carbon Aerogel Supported ZIF-67 Derived Hollow NiCo Phosphate Nanocages for Electrocatalytic Oxygen Evolution Reaction, Electrochim. Acta, 2021, 393, 139076 CrossRef CAS.
- G. L. Zhuang, Y. F. Gao, X. Zhou, X. Y. Tao, J. M. Luo, Y. J. Gao, Y. L. Yan, P. Y. Gao, X. Zhong and J. G. Wang, ZIF-67/COF-Derived Highly Dispersed Co3O4/N-Doped Porous Carbon with Excellent Performance for Oxygen Evolution Reaction and Li-Ion Batteries, Chem. Eng. J., 2017, 330, 1255–1264 CrossRef CAS.
- X. Li, S. You, J. Du, Y. Dai, H. Chen, Z. Cai, N. Ren and J. Zou, ZIF-67-Derived Co3O4@carbon Protected by Oxygen-Buffering CeO2 as an Efficient Catalyst for Boosting Oxygen Reduction/Evolution Reactions, J. Mater. Chem. A, 2019, 7(45), 25853–25864 RSC.
- B. Liu, S. Xue, S. Tan, D. Zhang, Z. Deng, H. Pan, W. Tu, R. Zhang, H. Zhang and Y. Wang, Hollow-Co3O4@CoP/NS-RGO Heterojunction Structure Derived from ZIF-67 Promotes Hydrogen Evolution Reaction and Oxygen Evolution Reaction Bifunctional Catalysis, Energy Fuels, 2022, 36(8), 4532–4540 CrossRef CAS.
- Y. Li, Z. Yin, M. Cui, S. Chen and T. Ma, Bimetallic Cobalt Molybdenum Carbide–Cobalt Composites as Superior Bifunctional Oxygen Electrocatalysts for Zn–Air Batteries. Mater Today, Energy, 2020, 18, 100565 CAS.
- Z. Zhang, S. Li, X. Bu, Y. Dai, J. Wang, X. Bao and T. Wang, Hollow ZIF-67 Derived Porous Cobalt Sulfide as an Efficient Bifunctional Electrocatalyst for Overall Water Splitting, New J. Chem., 2021, 45(37), 17313–17319 RSC.
- M. Cui, C. Yang, B. Li, Q. Dong, M. Wu, S. Hwang, H. Xie, X. Wang, G. Wang, L. Hu, M. Cui, C. Yang, Q. Dong, M. Wu, H. Xie, X. Wang, L. Hu, B. Li, G. Wang and S. Hwang, High-Entropy Metal Sulfide Nanoparticles Promise High-Performance Oxygen Evolution Reaction, Adv. Energy Mater., 2021, 11(3), 2002887 CrossRef CAS.
- Y. Guo, J. Tang, H. Qian, Z. Wang and Y. Yamauchi, One-Pot Synthesis of Zeolitic Imidazolate Framework 67-Derived Hollow Co3S4@MoS2 Heterostructures as Efficient Bifunctional Catalysts, Chem. Mater., 2017, 29(13), 5566–5573 CrossRef CAS.
- X. Wang, L. Yu, Y. Guan, S. Song, X. Wen, D. Lou, X. Wang, L. Yu, B. Y. Guan, X. W. Lou and S. Song, Metal–Organic Framework Hybrid-Assisted Formation of Co3O4/Co-Fe Oxide Double-Shelled Nanoboxes for Enhanced Oxygen Evolution, Adv. Mater., 2018, 30(29), 1801211 CrossRef PubMed.
- S. Anantharaj, S. R. Ede, K. Sakthikumar, K. Karthick, S. Mishra and S. Kundu, Recent Trends and Perspectives in Electrochemical Water Splitting with an Emphasis on Sulfide, Selenide, and Phosphide Catalysts of Fe, Co, and Ni: A Review, ACS Catal., 2016, 6(12), 8069–8097 CrossRef CAS.
- T. Abza, D. G. Dadi, F. G. Hone, T. C. Meharu, G. Tekle, E. B. Abebe and K. S. Ahmed, Characterization of Cobalt Sulfide Thin Films Synthesized from Acidic Chemical Baths, Adv. Mater. Sci. Eng., 2020, 2628706 CAS.
- W. Feng, L. Chen, M. Qin, X. Zhou, Q. Zhang, Y. Miao, K. Qiu, Y. Zhang and C. He, Flower-like PEGylated MoS2 Nanoflakes for near-Infrared Photothermal Cancer Therapy, Sci. Rep., 2015, 5(1), 1–13 Search PubMed.
- F. Sang, Z. Yin, W. Wang, E. Almatrafi, Y. Wang, B. Zhao, J. Gong, C. Zhou, C. Zhang, G. Zeng and B. Song, Degradation of Ciprofloxacin Using Heterogeneous Fenton Catalysts Derived from Natural Pyrite and Rice Straw Biochar, J. Cleaner Prod., 2022, 378, 134459 CrossRef CAS.
- Y. Sun, D. Lv, J. Zhou, X. Zhou, Z. Lou, S. A. Baig and X. Xu, Adsorption of Mercury (II) from Aqueous Solutions Using FeS and Pyrite: A Comparative Study, Chemosphere, 2017, 185, 452–461 CrossRef CAS PubMed.
- L. Yu, J. F. Yang, B. Y. Guan, Y. Lu and X. W. Lou, Hierarchical Hollow Nanoprisms Based on Ultrathin Ni-Fe Layered Double Hydroxide Nanosheets with Enhanced Electrocatalytic Activity towards Oxygen Evolution, Angew. Chem., 2018, 130(1), 178–182 CrossRef.
- Y. Zhao, D. Gao, J. Biskupek, U. Kaiser, R. Liu and C. Streb, Polyoxometalate-Assisted Synthesis of Amorphous Zeolitic Imidazolate for Efficient Electrocatalytic Oxygen Evolution, Results Chem., 2022, 4, 100568 CrossRef CAS.
- B. Fei, Z. Chen, J. Liu, H. Xu, X. Yan, H. Qing, M. Chen, R. Wu, B. Fei, Z. Chen, J. Liu, H. Xu, X. Yan, H. Qing, M. Chen and R. Wu, Ultrathinning Nickel Sulfide with Modulated Electron Density for Efficient Water Splitting, Adv. Energy Mater., 2020, 10(41), 2001963 CrossRef CAS.
- S. Dou, C.-L. Dong, Z. Hu, Y.-C. Huang, J. Chen, L. Tao, D. Yan, D. Chen, S. Shen, S. Chou, S. Wang, S. Dou, L. Tao, D. Yan, D. Chen, S. Wang, C. Dong, Y. Huang, J. Chen, Z. Hu, S. Chou and S. Shen, Atomic-Scale CoOx Species in Metal–Organic Frameworks for Oxygen Evolution Reaction, Adv. Funct. Mater., 2017, 27(36), 1702546 CrossRef.
- H. Cheng, Q. Liu, Y. Diao, L. Wei, J. Chen, F. Wang, H. Cheng, Q. Liu, Y. Diao, L. Wei, J. Chen and F. Wang, CoMo2S4 with Superior Conductivity for Electrocatalytic Hydrogen Evolution: Elucidating the Key Role of Co, Adv. Funct. Mater., 2021, 31(37), 2103732 CrossRef CAS.
- Y. Guo, J. Tang, J. Henzie, B. Jiang, W. Xia, T. Chen, Y. Bando, Y. M. Kang, M. S. A. Hossain, Y. Sugahara and Y. Yamauchi, Mesoporous Iron-Doped MoS2/CoMo2S4 Heterostructures through Organic-Metal Cooperative Interactions on Spherical Micelles for Electrochemical Water Splitting, ACS Nano, 2020, 14(4), 4141–4152 CrossRef CAS PubMed.
- I. Alstrup, I. Chorkendorff, R. Candia, B. S. Clausen and H. Topsøe, A Combined X-Ray Photoelectron and Mössbauer Emission Spectroscopy Study of the State of Cobalt in Sulfided, Supported, and Unsupported Co-Mo Catalysts, J. Catal., 1982, 77(2), 397–409 CrossRef CAS.
- D. Ma, B. Hu, W. Wu, X. Liu, J. Zai, C. Shu, T. Tadesse Tsega, L. Chen, X. Qian and T. L. Liu, Highly Active Nanostructured CoS2/CoS Heterojunction Electrocatalysts for Aqueous Polysulfide/Iodide Redox Flow Batteries, Nat. Commun., 2019, 10(1), 1–8 CrossRef CAS PubMed.
- A. M. De Jong, H. J. Borg, L. J. Van Ijzendoorn, V. G. F. M. Soudant, V. H. J. De Beer, J. A. R. Van Veen and J. W. Niemantsverdriet, Sulfidation Mechanism of Molybdenum Catalysts Supported on a SiO2/Si(100) Model Support Studied by Surface Spectroscopy, J. Phys. Chem., 1993, 97, 6477–6483 CrossRef CAS.
- D. Zhang, X. Kong, Y. Zhao, M. Jiang and X. Lei, CoOOH Ultrathin Nanoflake Arrays Aligned on Nickel Foam: Fabrication and Use in High-Performance Supercapacitor Devices, J. Mater. Chem. A, 2016, 4(33), 12833–12840 RSC.
- B. Zhang, Y. Zheng, T. Ma, C. Yang, Y. Peng, Z. Zhou, M. Zhou, S. Li, Y. Wang and C. Cheng, Designing MOF Nanoarchitectures for Electrochemical Water Splitting, Adv. Mater., 2021, 33(17), 2006042 CrossRef CAS PubMed.
- Y. Ou, L. P. Twight, B. Samanta, L. Liu, S. Biswas, J. L. Fehrs, N. A. Sagui, J. Villalobos, J. Morales-Santelices, D. Antipin, M. Risch, M. C. Toroker and S. W. Boettcher, Cooperative Fe sites on transition metal (oxy)hydroxides drive high oxygen evolution activity in base, Nat. Commun., 2023, 14, 1–12 Search PubMed.
- S. Anantharaj, P. N. Reddy and S. Kundu, Core-Oxidized Amorphous Cobalt Phosphide Nanostructures: An Advanced and Highly Efficient Oxygen Evolution Catalyst, Inorg. Chem., 2017, 56(3), 1742–1756 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma00382a |
| This journal is © The Royal Society of Chemistry 2024 |