
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Room temperature chemical transformation of SnSe to Ag2Se nanocrystals via cation exchange†‡
Yiqiao
Huang
and
Pierre F. P.
Poudeu
 *
*
Laboratory for Emerging Energy and Electronic Materials, Department of Materials Science and Engineering, University of Michigan, Ann Arbor, Michigan 48109, USA. E-mail: ppoudeup@umich.edu
First published on 26th April 2024
Abstract
Atomic-scale control of the chemical composition of semiconductor nanocrystals through a cation exchange reaction affords greater tunability in the design of multifunctional semiconductor composite nanocrystals. Here, we report a facile route to SnSe–Ag2Se composite nanocrystals using cation exchange at room temperature. Starting from freshly synthesized SnSe nanorods, we leverage the strong distortion of the Sn2+ octahedral coordination in SnSe and the hard–soft acid–base (HSAB) principle, to promote the exchange of an Sn2+ ion with two Ag+ ions in methanol leading to Ag2Se/SnSe nanocomposites. The morphology and chemistry of the nanocrystals evolve from nanorods with SnSe@Ag2Se (core@shell) structures for SnSe-rich composites to nanorods with a random distribution of Sn2+ and Ag+ ions for nearly equimolar composites, and finally to irregular fragmented nanocrystals for Ag2Se-rich composites. A mechanistic understanding of the observed morphology evolution is discussed using the change in the cation coordination from octahedral (Sn2+) to tetrahedral (Ag+) geometry and the accompanying expansion of the hcp Se2− sublattice. Interestingly, the synthesized composite nanocrystals exhibit an optical band gap value tunable within a wide energy range by increasing the Ag2Se/SnSe ratio. This work provides a useful and facile strategy to modify the optical behavior of semiconductor nanomaterials, which can be leveraged for the design of better optical and/or photovoltaic devices.
Introduction
Semiconductor nanocomposites have received considerable research attention in recent years due to their potential in multiple applications including thermoelectrics, photovoltaics, optoelectronics, photocatalysis, electrochemistry, and medical technology.1–11 Achieving semiconductor nanocomposites with targeted properties necessitates the utilization of synthesis methods that enable control over the materials' chemistry and structure, which are critical parameters for performance optimization. Cation exchange has emerged as an elegant approach for the modification of the chemistry and microstructure of pre-synthesized semiconductor nanocrystals, which enables greater tunability of functional behavior and paves the way for the design of multifunctional semiconductor composite nanocrystals.1,12–14 In a typical cation exchange reaction, the mobile cations in the parent nanocrystals tend to be replaced by the guest cations in solution, while the anion sublattice usually remains rigid.13,15 The cation exchange process generally uses solvents and soluble salts to provide guest cations. The cation exchange reaction that results in the retention of the anion sublattice requires some degrees of similarity in the atomic structure of the parent and the targeted phases. Typically, both the outgoing and the incoming cations adopt a similar coordination geometry within the anion sublattice and the exchange process results in a marginal change in the unit cell volume, which facilitates the conservation of the shape of the original nanocrystal.16,17 Such a cation exchange process has been widely explored for the synthesis of metastable phases.16 The cation exchange reaction that results in changes in the anion sublattice has also been reported.18–20 This is typically observed when the ion exchange product either has a different crystal structure from the parent phase,19 or has a similar structure with a significant change in the unit cell volume.18 In such ion-exchange reactions, the shape of the original nanocrystal is not conserved due to the rearrangement of the anion sublattice.One common structural feature of many cation exchange reactions reported to date, regardless of whether the cation exchange process changes or maintains the anion sublattice, is that the outgoing ion in the parent phase and the incoming cation in the targeted phase essentially adopt similar coordination geometry albeit marginal distortions. There are a few examples of cation exchange reactions that result in a change in the coordination of the exchanged cations. For instance, a reversible transformation between CdQ zinc blende nanoparticles and PbQ rock-salt nanoparticles (Q = S, Se, Te) has been demonstrated.21–26 However, these reactions require thermal activation as well as complex organic solvents/ligands such as octadecene (ODE), 1-dodecanethiol (DDT), trioctylphosphine oxide (TOPO), trioctylphosphine (TOP), and oleylamine (OLA)27–31 to facilitate the change in the coordination of Cd2+ and Pb2+ ions between the tetrahedral and octahedral sites in the face-centered cubic Q2− sublattice. Moreover, these reactions are typically conducted under oxygen-free conditions, which poses a constraint on their scalability for industrial applications.
Here, we report a facile cation exchange between the octahedrally coordinated Sn2+ ion in SnSe and the tetrahedrally coordinated Ag+ ion in Ag2Se, which enables the fabrication of a range of Ag2Se/SnSe nanocomposites starting from the SnSe nanorod (NR) precursor. It is remarkable that the transformation of SnSe to Ag2Se proceeds at room temperature and does not use complex organic solvents, unlike other cation exchange reactions that result in a change in the coordination of the exchanged cations.27–31 The Ag2Se/SnSe ratio in the final nanocomposites can be tuned by controlling the Ag/Sn molar ratio in the starting mixture. Interestingly, such control of chemical composition also enables the modification of the morphology as well as the elemental distribution within the final nanoparticles. We discuss the underlying atomic-scale mechanism by using X-ray powder diffraction (XRD), transmission electron microscopy (TEM), scanning transmission electron microscopy (STEM), and X-ray photoelectron spectroscopy (XPS) data on various SnSe/Ag2Se composites to systematically track the chemical transformation process. The optical band gap of various composites extracted from UV-vis-NIR diffuse reflectance spectroscopy data is also reported.
Results and discussion
Synthesis and structure
SnSe nanocrystals were synthesized following a modified surfactant-free method32 by mixing freshly prepared solutions of NaHSe and Na2SnO2 at room temperature followed by 2 h magnetic stirring. The phase purity and crystal structure of the synthesized SnSe precursor were confirmed by X-ray diffraction (XRD) and transmission electron microscopy. The experimental XRD pattern (Fig. 1(a)) of the synthesized SnSe nanoparticles can be indexed to the orthorhombic crystal structure (space group Pnma (#62), a = 11.502 Å, b = 4.153 Å, and c = 4.45 Å) of SnSe (ICSD# 50542). Transmission electron microscopy (TEM) revealed that the SnSe precursor adopts a nanorod-like morphology with dimensions ranging from 80 to 200 nm in length and 10 to 20 nm in diameter that are agglomerated into clusters of various sizes (Fig. 2(a)). The crystallinity of the SnSe nanorod was confirmed by high resolution TEM (HRTEM), which reveals the 0.36 nm interplanar distance of the (201) plane of the SnSe crystal (Fig. 2(f)).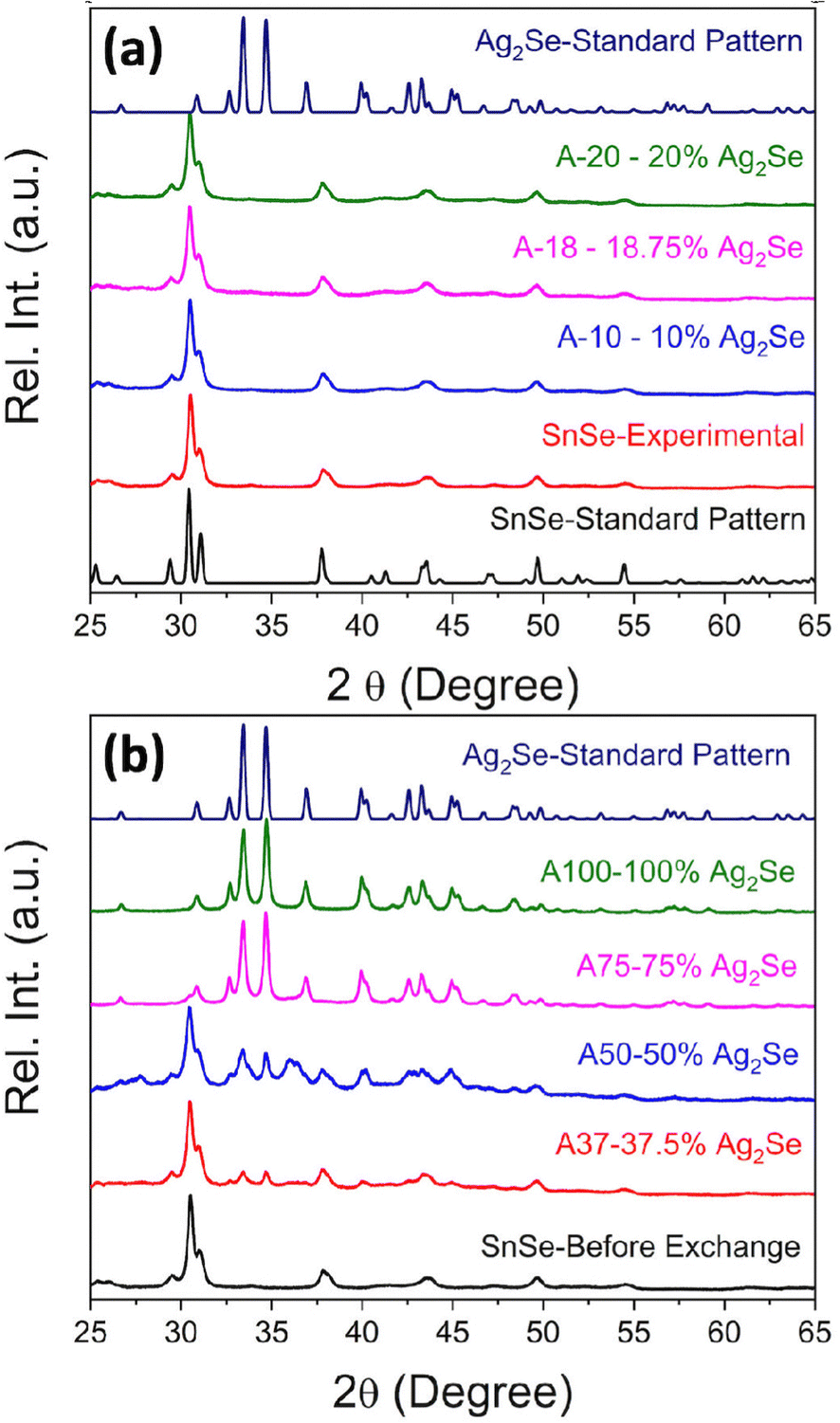 | ||
| Fig. 1 (a) XRD pattern of the synthesized SnSe precursor compared with the standard XRD pattern of SnSe (ISCD#50542), and XRD patterns of Ag2Se/SnSe nanocomposites with low Ag/Sn ratio showing only peaks from the SnSe phase; (b) XRD patterns of Ag2Se/SnSe nanocomposites with high Ag/Sn ratio displaying diffraction peaks of both SnSe and Ag2Se phases. | ||
 | ||
| Fig. 2 STEM images and the distribution of Ag and Sn in selected Ag2Se/SnSe samples: (a) and (f) SnSe precursor; (b) and (g) 37.5% Ag2Se (A-37); (c) and (h) 50% Ag2Se (A-50); (d) and (i) 75% Ag2Se (A-75); and (e) and (j) Ag2Se nanocrystals. (k)–(n) Schematic illustration of the chemical transformation from the SnSe nanorod precursor to the Ag2Se nanoparticles. | ||
Cation exchange reactions towards the synthesis of various Ag2Se/SnSe composite nanocrystals were performed by mixing the SnSe nanorods dispersed in methanol with varying amounts of Ag+ solutions prepared by dissolution of AgNO3 crystals in methanol. The mixture underwent ultrasonic processing for 4 hours followed by 2 days of magnetic stirring at room temperature. The resulting cation exchange reaction can be described using the following chemical eqn (1).
| SnSe + 2Ag+ → Ag2Se + Sn2+ | (1) |
The nominal composition of the synthesized Ag2Se/SnSe composites was controlled by adjusting the Ag/Sn molar ratio in the starting mixture (Table 1).
| Sample code | Ag/Sn ratio | Ag2Se mole% | SnSe mole% |
|---|---|---|---|
| SnSe | 0 | 0 | 100 |
| A-10 | 0.2/1 | 10 | 90 |
| A-18 | 0.375/1 | 18.75 | 81.25 |
| A-20 | 0.4/1 | 20 | 80 |
| A-37 | 0.75/1 | 37.5 | 62.5 |
| A-50 | 1/1 | 50 | 50 |
| A-75 | 1.5/1 | 75 | 25 |
| A-100 | 2/1 | 100 | 0 |
Fig. 1(a) and (b) show the XRD patterns of the Ag2Se/SnSe nanocomposites synthesized through cation exchange reactions. For nanocomposites with a low Ag/Sn ratio (Ag/Sn ≤ 0.4/1), only diffraction peaks from the SnSe phase are observed on the XRD patterns. This suggests either the formation of interstitial solid solution through the incorporation of Ag+ ions within the crystal lattice of SnSe nanorods, or that the Ag2Se phase formed in these samples are likely amorphous with no long-range ordered structure. Upon increasing the Ag/Sn ratio to above 0.75/1 (62.5% SnSe/37.5% Ag2Se), diffraction peaks associated with the Ag2Se phase can be observed on the XRD patterns along with peaks from the SnSe phase. Interestingly, the relative intensity of diffraction peaks from the SnSe phase decreases with the increasing Ag/Sn ratio whereas that of diffraction peaks from the Ag2Se phase increases suggesting a gradual chemical transformation of the SnSe nanocrystals into Ag2Se. Astonishingly, only diffraction peaks corresponding to the Ag2Se phase are observed on the XRD patterns of nanocomposites with the Ag/Sn ratio exceeding 1.5/1 (25% SnSe/75% Ag2Se). This suggests that the exchange of a large fraction of Sn2+ ions within the SnSe nanocrystals by Ag+ ions resulted in the destruction of the original crystal into amorphous smaller particles.
The diffraction peaks from the experimental XRD patterns of samples with 75% Ag2Se and 100% Ag2Se can be indexed to the orthorhombic crystal structure (P212121 (#19) a = 4.336 Å, b = 7.0774 Å, and c = 7.774 Å) of Ag2Se. This indicates a successful chemical transformation from the orthorhombic SnSe (Pnma (#62)) to the lower symmetry orthorhombic structure of Ag2Se (P212121 (#19)) (Fig. 3). A careful comparison of both crystal structures reveals that the cation exchange reaction results in a change in the coordination geometry of the exchanged ions. Indeed, the removal of one Sn2+ ion from the severely distorted [1 + 2 + 2 + 1] octahedral coordination (Fig. 3d) in the SnSe structure is electronically compensated by two Ag+ ions, which are accommodated within the two tetrahedral voids (Ag(1) and Ag(2)), adjacent to the octahedral void, in the deformed hexagonal close packed (hcp) Se2− sublattice of SnSe (Fig. 3a and b). This substitution reaction results in the formation, by the Ag+ ions, of edge-sharing tetrahedral clusters (Fig. 3d) in the orthorhombic structure of Ag2Se. Interestingly, the chemical transformation from SnSe to Ag2Se retains the hcp Se2− sublattice but is accompanied by an ∼12% increase in the unit cell volume from 212 Å3 (SnSe) to 238 Å3 (Ag2Se). This implies that the cation exchange between Sn2+ and Ag+ requires a local expansion of the Se2− framework in SnSe. Such expansion of the Se2− hcp sublattice is likely necessary not only to facilitate the diffusion of Ag+ ions within the tetrahedral voids, but also to accommodate the repulsion between Ag+(1) and Ag+(2) ions in the edge-sharing tetrahedral cluster. Interestingly, the lattice parameters (Table 2 and Table S2, ESI‡) of the Ag2Se phase in the SnSe/Ag2Se composites with 75% Ag2Se (A-75) as well as in the fully transformed sample with 100% Ag2Se (A-100) are highly consistent with the reported values for pristine Ag2Se demonstrating the effectiveness of the cation exchange process for the synthesis of the highly crystalline Ag2Se phase at 300 K.
 | ||
| Fig. 3 Schematic illustration of the atomic-scale cation exchange process leading to the transformation from SnSe to Ag2Se orthorhombic structure. (a) Crystal structure of SnSe highlighting the hcp Se2− sublattice with Sn2+ in octahedral void; the two empty tetrahedral voids (triangles) adjacent to the octahedrally coordinated Sn2+ are occupied by Ag+ ions in the crystal structure of Ag2Se (b). (c) distorted octahedral coordination geometry of Sn2+; (d) edge-sharing tetrahedral coordination of Ag+ ions in Ag2Se. | ||
| Lattice parameters | Ag2Se (standard pattern) | A-75 | A-100 |
|---|---|---|---|
| a (Å) | 4.336 | 4.338 | 4.331 |
| b (Å) | 7.070 | 7.074 | 7.064 |
| c (Å) | 7.774 | 7.789 | 7.777 |
| V (Å3) | 238.32 | 239.01 | 237.95 |
The chemical composition of the SnSe precursor and the synthesized Ag2Se/SnSe nanocomposites was determined using EDX analysis. Table S1 and Fig. S1(a)–(c) (ESI‡) show the nominal atomic percentages (at%) of Se, Sn, and Ag as well as the experimentally determined values for all samples. The Ag content in all samples is very close to the nominal value. Interestingly, the experimental atomic percentages of Se are always higher than the theoretical value especially in the nanocomposites with high Ag/Sn molar ratios, while the experimental values for the Sn element are always lower than the theoretical value. This result suggests the possibility of partial hydrolysis and/or oxidation of Sn2+ upon extraction from the SnSe crystal lattice.
Driving force for cation exchange
Given the large volume change (ΔV/V ∼ 12%) of the Se2− hcp sublattice and the change in the cation coordination, it is remarkable that the chemical transformation from SnSe to Ag2Se occurs at room temperature, which prompts the question, what is the driving force for the cation exchange process? Compared to the solid state synthesis of Ag2Se from the elements, which typically requires high temperatures and long annealing, the relatively fast cation exchange at room temperature between SnSe and Ag+ ion in methanol suggests that the formation of Ag2Se is thermodynamically favorable under these conditions. This implies a relatively low activation energy for the removal of Sn2+ from the distorted octahedral position within the hcp Se2− sublattice of SnSe and the incorporation of Ag+ ions at the tetrahedral positions. Indeed, a close examination of the cation coordination geometry in SnSe and Ag2Se revealed a severe distortion of Sn–Se bonding (3 short and 3 long) within the octahedral coordination with bond distances ranging from 2.735 Å to 3.474 Å (Δx/x ∼ 37%), whereas the tetrahedral coordination of Ag+ ions in Ag2Se shows weaker distortions, Δx/x ∼ 21% for Ag(2) and Δx/x ∼ 8.5% for Ag(1). Such severe distortion of the Sn2+ coordination in SnSe is a manifestation of the structural instability of SnSe, presumably due to the relatively small size of the Sn2+ ion (ionic radius (IR): 81 pm33 to 93 pm34 for CN = 6) for the large octahedral void. Conversely, the weaker distortion of the Ag+ (IR = 100 pm35,36 for CN = 4) tetrahedral coordination in Ag2Se points to a relatively more stable structure compared to SnSe.The relative stability of Ag2Se over SnSe can also be assessed by comparing their solubility product constant (KSP) in methanol/water. For instance, the KSP of Ag2Se in water ranges from 2 × 10−64 to 3 × 10−54, which is much lower than that of SnSe (5 × 10−34)13,37,38 indicating that Ag2Se is more stable in water than SnSe. Following a similar trend, one can anticipate that Ag2Se would also possess a relatively lower KSP than SnSe in polar solvents like methanol, indicating that Ag2Se is more thermodynamically stable than SnSe in the cation exchange environment, which is consistent with the observed relatively easy chemical transformation of SnSe to Ag2Se through exchange of Sn2+ by Ag+ at 300 K.
The relatively facile transformation of SnSe to Ag2Se can also be rationalized by considering the close spatial proximity of the octahedral (O) and tetrahedral (T+, T−) voids within the hcp Se2− sublattice in the SnSe structure as well as their 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio that enables the accommodation of both Ag+ ions used to electronically compensate for the outgoing Sn2+ ions. Upon removal of Sn2+ ions, the large octahedral voids left behind likely serve as the point of entry of Ag+ (IR ∼ 100 pm35,36) ions, which are slightly larger (Δ(IR)/(IR) ∼23%) than the outgoing Sn2+ ion, but still quite small for the large octahedral coordination within the Se2− hcp sublattice in SnSe. This results in an unstable intermediate structure as the octahedral voids are too large for the Ag+ ion. Such structural instability is likely resolved by the subsequent diffusion of Ag+ ions into the much smaller tetrahedral voids that are located ∼4 Å to ∼5 Å away from the octahedral site (Fig. 3b).
:2 ratio that enables the accommodation of both Ag+ ions used to electronically compensate for the outgoing Sn2+ ions. Upon removal of Sn2+ ions, the large octahedral voids left behind likely serve as the point of entry of Ag+ (IR ∼ 100 pm35,36) ions, which are slightly larger (Δ(IR)/(IR) ∼23%) than the outgoing Sn2+ ion, but still quite small for the large octahedral coordination within the Se2− hcp sublattice in SnSe. This results in an unstable intermediate structure as the octahedral voids are too large for the Ag+ ion. Such structural instability is likely resolved by the subsequent diffusion of Ag+ ions into the much smaller tetrahedral voids that are located ∼4 Å to ∼5 Å away from the octahedral site (Fig. 3b).
The driving force for the cation exchange reaction was also likely improved by the choice of a solvent that can both dissolve AgNO3 solids to provide free Ag+ cations and promote the cation exchange process by extracting Sn2+ from the original SnSe lattice. In this work, we leverage the Pearson's hard and soft acids and bases (HSAB) theory, which can be used to establish a qualitative affinity between metal ions and ligands/solvents based on the concept of absolute hardness (η),13 for the selection of methanol as the suitable solvent for the cation exchange process. According to the HSAB theory, a hard base tends to bind with a hard acid while a soft base prefers to bind with a soft acid.13,39,40 Therefore, one can anticipate a successful cation exchange reaction between SnSe and Ag+ ions using a hard base such as methanol or water as the solvent, since the guest cation (Ag+) is softer (η = 6.96) than the host cation Sn2+ (η = 7.94). Because of the relatively easy hydrolysis of Sn2+ ions in water, methanol was selected as the most appropriate solvent for the ion exchange process. Under this condition, the hard base molecule (methanol) tends to bind with the hard acid cation (Sn2+), which should facilitate its removal from the SnSe nanorods, creating empty cation sites (octahedral voids) for Ag+ ions to hop into. This is presumably the mechanism crucial for the successful fabrication of SnSe/Ag2Se composites through the cation exchange process at 300 K.
Mechanism of cation exchange
To probe the mechanism of the chemical transformation of SnSe nanocrystals into Ag2Se, the evolution of the composition and morphology of nanocrystals in selected Ag2Se/SnSe nanocomposites was investigated by transmission electron microscopy (TEM) and scanning transmission electron microscopy (STEM). A careful examination of the dark field (DF) and bright field (BF) images of various samples and the associated EDX elemental mappings revealed that both the nanocrystal morphology and the distribution of Ag and Sn within the composites change with the increasing Ag/Sn ratio (Fig. 2(b) to (e)). For instance, the TEM BF image of sample A-37 (37.5% Ag2Se) showed the formation of a core–shell structure (Fig. 2(b)). The corresponding elemental mappings revealed the concentration of Ag atoms on the shell part of the nanorod while Sn atoms are mainly located within the core (Fig. 2(g) and (l)). This suggests that for samples with low Ag/Sn ratio, the cation exchange proceeds from the extraction of Sn2+ on the surface of the initial SnSe nanorod to form a solid solution or a thin amorphous Ag2Se shell, which could explain the lack of diffraction peaks for the Ag2Se phase on the XRD patterns (Fig. 1a). Upon increasing the Ag/Sn ratio to 1/1(50% Ag2Se), the TEM-BF image (Fig. 2(c)) along with the corresponding elemental mapping (Fig. 2(h)) of the resulting sample (A-50) revealed a random distribution of nano-domains of the Ag2Se and SnSe phases throughout the nanorod. Such a change in the distribution of Ag and Sn indicates that as the Ag/Sn molar ratio increases, the initial core–shell heterostructure becomes unstable, presumably due to the large lattice mismatch at the Ag2Se/SnSe interface and the large change in the unit cell volume (ΔV/V ∼ 12%) arising from the expansion of the Se2− hcp sublattice (Fig. 3). During the exchange process, one Sn2+ must be replaced by two Ag+ ions to maintain the charge balance. Considering that the ionic radius of Sn2+ (∼81 pm33 to 93 pm34 for CN = 6) is slightly smaller than that of Ag+ (100 pm35,36 for CN = 4), the insertion of two Ag+ ions in replacement for one Sn2+ ion is anticipated to introduce intense internal stress to the original hcp Se2− sublattice in the SnSe structure. The internal stress is alleviated in the Ag2Se structure by the expansion of the Se2− framework leading to the formation of incoherent SnSe/Ag2Se interfaces. Such a high-energy interface likely facilitates the separation of the thin Ag2Se shell from the SnSe nanorod enabling additional diffusion of Ag+ ions into the remaining SnSe crystal. This process presumably continues until depletion of Ag+ ions from the solution, which results in the fragmentation of the initial SnSe nanorod into small clusters of nanocrystals with SnSe and Ag2Se compositions as observed in Fig. 2(c) and (h). As the Ag2Se molar percentage further increases to 75% (A-75), the initial nanorods are broken into multiple smaller Ag-rich nanoparticles connected by Sn-rich bridges, as shown in Fig. 2(d) and (i), and illustrated in Fig. 2(m). This morphological evolution from a core–shell structure to random distribution of Ag2Se and SnSe nanodomains and eventually to complete Ag-rich nanoparticles connected by Sn-rich bridges as schematically illustrated in Fig. 2(k) to (n) can be rationalized by considering that the incoherent SnSe/Ag2Se high energy interfaces lead to the separation of Ag2Se domains from the SnSe nanorod, which in turn facilitates the diffusion of Ag+ ions into the freshly exposed surface of the SnSe nanorod. The continuation of this cation-exchange process results in random distribution of Ag2Se and SnSe nano domains in the remaining nanorods for equimolar ratio and finally to the formation of small nanocrystals with Ag2Se composition and structure (Fig. 2(e), (j) and (n)).To elucidate the crystallinity near the interface between the SnSe and Ag2Se phases, additional high-resolution TEM (HRTEM) images were acquired for sample A-75 (Fig. 4). It can be seen from Fig. 4(a) and (b) that the sample A-75 features agglomerated nano-sized particles with irregular shapes. The higher magnification TEM image (Fig. 4(c)) shows multiple small nanoparticles on the surface of the larger piece. The corresponding STEM-BF image (Fig. 4(g)) and elemental mapping (Fig. 4(h) and (i)) reveal that the small nanoparticles are Ag2Se phase, while the larger piece is mainly the original SnSe phase with some degree of intermixing with Ag2Se. The high resolution TEM (HRTEM) image of the interface between the SnSe-rich particles and the Ag2Se nanoparticles (Fig. 4(d)) depicts the crystallinity of the SnSe phase (large piece) as well as that of the Ag2Se phase (small particle). As shown in Fig. 4(e), the local area at the interface between SnSe and Ag2Se shows a clear lattice pattern, on the SnSe side, with an interplanar spacing of 0.34 nm corresponding to the (210) plane of the SnSe crystal. The interface area shows no long-range ordered structure, and it is difficult to acquire the lattice information. Such a local amorphous region may be caused by the process of Sn2+ extraction and Ag+ insertion at the interface. For each Sn2+ extracted, there are 2 Ag+ ions that must enter the original Se2− sublattice to maintain the charge balance. This non-isovalent exchange process likely causes internal stress to the original lattice leading to severe deformation. As a result, the original long-range ordered SnSe structure is broken to form the Sn–Ag intermixing interface with poor crystallinity. Interestingly, the HRTEM image of the small Ag2Se nanoparticle region (Fig. 4(f)) revealed a clear lattice pattern with an interplanar spacing of 0.289 nm corresponding to the (102) plane of the Ag2Se phase. This suggests that a long-range ordered structure is formed upon complete exchange of Sn2+ by Ag+ from the original SnSe crystal.
 | ||
| Fig. 4 (a)–(c) TEM images of the nanocomposites A-75 under various magnifications, showing the agglomeration feature and interface between the small Ag2Se nanoparticles and large piece SnSe matrix; (d) HRTEM image of the interface between SnSe precursor and Ag2Se nanoparticles. The two highlighted areas show the lattice feature of the interface area (e) and the small Ag2Se nanoparticle area (f); (e) zoomed-in HRTEM image of the interface area, showing the lattice feature of SnSe (210) plane at the left side of the interface; (f) zoomed-in HRTEM image of the small nanoparticles, showing the lattice feature of the Ag2Se (102) plane; (g) STEM-BF image of the same SnSe/Ag2Se interface shown in (d) with corresponding elemental mapping results ((h), and (i)), confirming that the large matrix is SnSe-rich while the small nanoparticle is Ag2Se-rich. | ||
Further analysis of the SnSe/Ag2Se interface at a different area of the sample A-75 (Fig. 5(a)) also revealed a well-ordered structure at both the SnSe-rich and Ag2Se-rich ends whereas the phase boundary showed a disordered structure. As can be seen from Fig. 5(d) and (f), the HRTEM and the corresponding fast-Fourier transformed (FFT) pattern for the SnSe-rich (Fig. 5(e)) and Ag2Se-rich (Fig. 5(g)) areas show a highly periodic lattice structure, whereas the phase boundary (interface) looks amorphous without any long-range ordered structure (Fig. 5(b)) and the FFT pattern only shows circular rings feature (Fig. 5(c)), instead of patterned points feature, which also indicates that the interface area contains no long-range ordered structure. Fig. 5(d) shows the crystal lattice feature on the SnSe-rich side with an interplanar distance of 0.304 nm, which corresponds to the (011) plane of SnSe. The lattice feature on the small Ag2Se nanoparticle side (Fig. 5(f)) with an interplanar distance of 0.290 nm can be ascribed to the (102) plane of Ag2Se.
 | ||
| Fig. 5 (a) HRTEM image of the lower side of the small nanoparticle shown in Fig. 4(c). The 3 highlighted areas were selected to explore the lattice features at the interface (b), on the SnSe side of the interface (d), and on the Ag2Se side of the interface (f); (b) and (c) zoomed-in HRTEM image of the two-phase interface with the corresponding FFT pattern, showing an amorphous (no long-range-ordered) structure; (d) and (e) zoomed-in HRTEM image of the SnSe-rich side of the interface with the corresponding FFT pattern, showing the (011) plane of SnSe; (f) and (g) zoomed-in HRTEM image of the Ag2Se-rich side of the interface with the corresponding FFT pattern, showing the (102) plane of Ag2Se. | ||
Another noticeable feature of the synthesized nanocomposites is the morphology change of the SnSe nanorod along with various compositions. Fig. 6 displays the morphology of nanoparticles in A-37 (a), A-50 (b), and A-75 (c) SnSe/Ag2Se nanocomposites. The original shape of the SnSe nanorod precursor is preserved in sample A-37 (Fig. 6(a)). However, when it comes to sample A-50 (Fig. 6(b)), the nanorod begins to break into smaller nanoparticles. Furthermore, the nanorod almost completely breaks into smaller nanoparticles in samples A-75 (Fig. 6(c)). There is a clear trend that with a higher Ag2Se percentage in the nanocomposites (from 37.5% to 75%), the degree of cation exchange is higher, and as a result, the disintegration of the original SnSe nanorod into smaller particles becomes more prominent. This can be rationalized by the local expansion of the Se2− sublattice that results from the replacement of a relatively smaller Sn2+ (81–93 pm) ion by two relatively larger Ag+ (100 pm) ions during the cation exchange process to maintain the charge neutrality. When the degree of cation exchange is low (for example, in A-37), this tension stress is not strong enough to undermine the integrity of the original nanorod. However, as the degree of cation exchange increases (for example, A-50 and A-75), the build-up tension stress can reach a considerable level, which can damage the original lattice and break the nanorod into smaller nanoparticles.
 | ||
| Fig. 6 HRTEM images of samples A-37 (a), A-50 (b), and A-75 (c), showing the morphology change from nanorods to smaller nanoparticles due to the cracking of the initial nanorod. | ||
To further probe the mechanism of the ion exchange process, we have examined the oxidation state of Sn, Ag and Se atoms in various samples using X-ray photoelectron spectroscopy (XPS). Fig. 7(a) shows the Sn-3d XPS spectra of the SnSe precursor and various SnSe/Ag2Se nanocomposites. For the SnSe precursor, the Sn-3d spectrum shows two peaks at 485.4 eV and 493.8 eV, corresponding to the binding energy of Sn-3d5/2 and Sn-3d3/2 states in divalent tin (Sn2+), respectively.41 Upon partial exchange of Sn2+ by Ag+ in SnSe nanorods, broadening of both Sn-3d5/2 and Sn-3d3/2 peaks with a shift of the central position to higher binding energy is observed on the Sn-3d XPS spectra of various composites. A careful analysis of the peak shape indicates the appearance of a shoulder peak at 487.2 eV and 495.6 eV, which are respectively associated with the Sn-3d5/2 and Sn-3d3/2 states in tetravalent tin (Sn4+) in SnO2.42,43 This result suggests the oxidation of a small fraction of Sn2+ ions upon removal from the SnSe crystal lattice.44,45
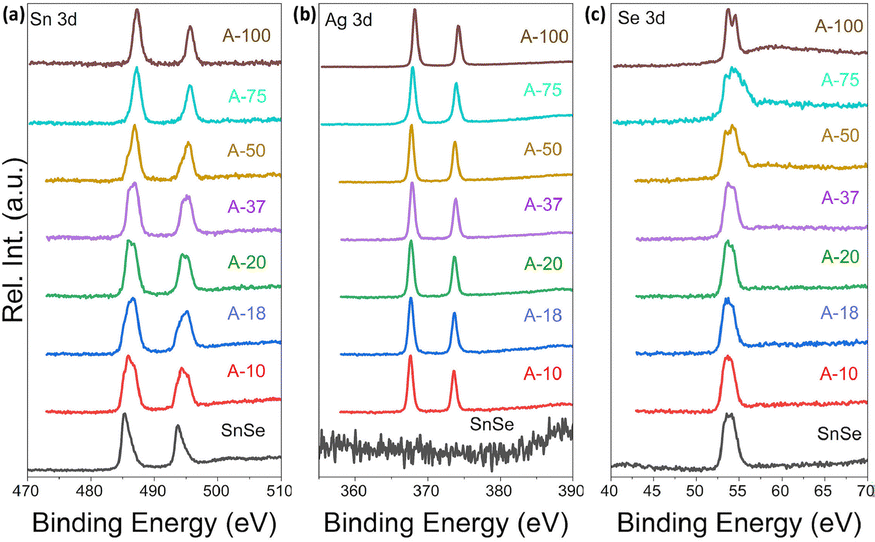 | ||
| Fig. 7 XPS spectra of (a) Sn_3d, (b) Ag_3d, and (c) Se_3d shell electrons in SnSe and selected Ag2Se/SnSe nanocomposites. | ||
Interestingly, the intensity of the peaks at 485.4 eV and 493.8 eV decreases with increasing Ag2Se content whereas that of the shoulder peaks at 487.2 eV and 495.6 eV increases. For composition with 75% Ag2Se (sample A-75), the peaks at 485.4 eV and 493.8 eV completely vanished with only the higher energy peaks remaining. A similar Sn-3d XPS spectrum is observed for the sample A-100, which nominally corresponds to the sample with complete cation exchange (i.e. 100% Ag2Se). The observed data further imply that during the cation exchange process, a fraction of Sn2+ is oxidized to Sn4+ presumably from surface oxidation of the SnSe nanorods, whereas the remaining Sn2+ ions are gradually replaced by Ag+ demonstrating the completeness of the cation exchange process. Indeed, the Ag-3d XPS spectra (Fig. 7(b)) of various composites (A-10 to A-100) revealed the presence of two isolated peaks at 373.6 eV and 367.6 eV, which correspond, respectively, to the Ag-3d3/2 and Ag-3d5/2 with a spin–orbit splitting of 6.0 eV, suggesting that the valence of Ag was +1.46 As the Ag2Se concentration increases, the peak positions of Ag-3d3/2 and Ag-3d5/2 states remain relatively unchanged. For the sample A-100, both isolated peaks appear at 374.2 eV (Ag-3d3/2) and 368.2 eV (Ag-3d5/2), which is still within the range reported for monovalent (+1) Ag in silver chalcogenides.47Fig. 7(c) depicts the Se-3d XPS spectra of the SnSe precursor and all SnSe/Ag2Se nanocomposites. One can notice the relative stability in both the SnSe precursors and the nanocomposites of the peak at 53.6 eV that is associated with the Se 3d5/2 state implying the stability of the Se2− anion during the cation exchange process. As the fraction of Ag2Se increases to 100%, two strong peaks are observed at 54.4 eV and 53.6 eV, which can be attributed to Se_3d3/2 and Se_3d5/2 states, indicating the presence of Se2− anions in the fully transformed Ag2Se sample.48
Optical properties
To assess the effect of Ag+ to Sn2+ cation exchange on the optical band gap, we have conducted diffuse reflectance measurements at 300 K (Fig. S2, ESI‡) on the SnSe precursor as well as on various SnSe/Ag2Se nanocomposites using a UV-vis-NIR spectrometer. Direct bandgap values extracted from the Tauc plots of the diffuse reflectance spectra (DRS) are displayed in Fig. 8 and the change in the band gap with increasing Ag2Se content is shown in Fig. S3 (ESI‡). The SnSe precursor showed a direct bandgap value of 1.02 eV, which is in agreement with previously reported values.49 The partial exchange of Sn2+ by Ag+ ions results in an initial marginal increase of the optical band gap of the resulting nanocomposites. For instance, the band gap value for the composite with 10% Ag2Se (sample A-10) is 1.09 eV, which corresponds to a 7% increase compared to that of the SnSe nanorod precursor. A further increase in the Ag2Se content led to a marginal drop in the band gap to 1.07 eV for the sample A-20 and to 1.06 eV for sample A-37, which are still larger than the band gaps of the SnSe precursor (Fig. 8(a)). This trend is quite unexpected since the band gap of pristine Ag2Se (∼0.1 eV) is considerably lower than that of SnSe, which is in the range 1.0–1.4 eV.50–55 One plausible explanation for this trend is that the initial substitutions of Sn2+ by two Ag+ ions lead to the formation of Ag+ interstitial solid-solution, Sn1−xAg2xSe, with an SnSe-structure type according to the defect eqn (2). | (2) |
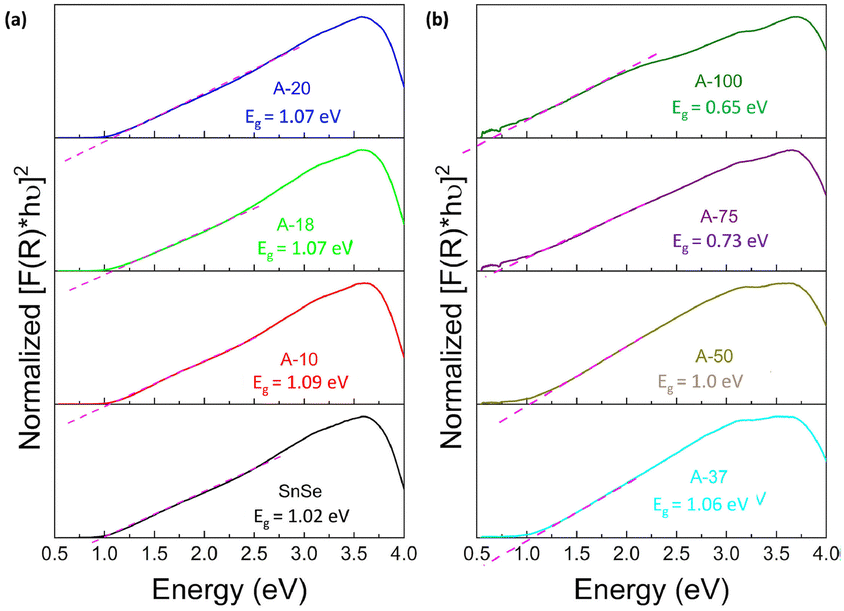 | ||
| Fig. 8 Tauc plots of the UV-vis-NIR diffuse reflectance spectra showing the optical band gap of Ag2Se/SnSe nanocomposites with (a) low Ag2Se content; and (b) high Ag2Se content. | ||
This conjecture is supported by the presence of diffraction peaks from only the SnSe phase on the XRD patterns of composites with low Ag2Se content (A-10 to A-20). Furthermore, a careful analysis of the lattice parameters (Table 3 and Table S2, ESI‡) of SnSe in selected samples with low Ag content shows an initial small expansion of the unit cell volume, which is consistent with the formation of interstitial Ag+ ions in the SnSe crystal lattice. Within this defected SnSe structure picture, the incorporation of Ag+ into the SnSe lattice could result in the formation of increasingly ionic bonding, M–Se (M = Sn/Ag) owing to the slightly higher electronegativity value of Ag (1.9) compared to Sn (1.8).56,57 This could explain the observed increase in the energy band gap of composites with 10% and 20% Ag2Se. The Sn1−xAg2xSe solid solution likely collapses with the substitution of a large fraction of Sn2+ ions (A-37 to A-75) resulting in a mixture of Sn-rich (SnSe) and Ag-rich (Ag2Se) phases (Fig. 2(g), (h), and (i)). This analysis is consistent with the observed rapid drop in the band gap value to 1.00 eV for sample A-50 and to 0.73 eV for sample A-75 (Fig. 8(b) and Fig. S3, ESI‡). Interestingly, the band gap value of 0.65 eV observed for sample A-100 (100% Ag2Se) is significantly larger than the reported value around 0.1 eV.50,58 We attribute such a large increase in the band gap to the nanometer scale particle size of Ag2Se obtained through the cation exchange reaction (Fig. 2e).
| Lattice parameters | SnSe | A-10 | A-18 | A-20 |
|---|---|---|---|---|
| a (Å) | 11.518 | 11.522 | 11.522 | 11.524 |
| b (Å) | 4.163 | 4.148 | 4.146 | 4.142 |
| c (Å) | 4.409 | 4.440 | 4.453 | 4.454 |
| V (Å3) | 211.415 | 212.216 | 212.751 | 212.632 |
Conclusions
In summary, we have developed a novel cation exchange method for a facile synthesis of Ag2Se/SnSe nanocomposites at room temperature. Using methanol as a solvent, we demonstrate the formation at 300 K of a highly crystalline Ag2Se phase through chemical transformation of SnSe nanorods with cation exchange of one Sn2+ ion by two Ag+ ions. The fraction of Ag2Se in the composites can be tuned by adjusting the Ag/Sn ratio in the starting mixture. XRD patterns and STEM images of selected composites revealed four stages in the transformation of SnSe nanorods to Ag2Se nanoparticles. For samples with low Ag content (A-10 to A-20), the ion exchange process results in the incorporation of Ag+ ions into the SnSe structure leading to the formation of a partial solid-solution with SnSe-structure type. A further increase in the Ag content results in the formation of a SnSe/Ag2Se core/shell structure for the sample A-37 followed by a random distribution of Sn-rich and Ag-rich nanodomains in composite with 50% Ag2Se (A-50) and finally to the phase segregation between Ag2Se and SnSe domains (A-75). The ability to achieve complete transformation of SnSe nanorods into highly crystalline Ag2Se nanoparticles at 300 K was rationalized using a combination of factors such as the relatively superior structural stability in polar solvent of Ag2Se compared to SnSe, and the strong binding affinity of the methanol molecule to the Sn2+ ion, which promotes its removal from octahedral voids in the SnSe crystal lattice paving the way for the incorporation of Ag+ ions.UV/vis diffuse reflectance measurement revealed an initial increase in the optical band gap for samples with low Ag content (A-10 to A-20), which is attributed to the formation of Ag+ interstitial solid-solution, Sn1−xAg2xSe, as well as the slightly enhanced (Sn/Ag)–Se ionic bonding. A further increase in the Ag2Se content results in a rapid drop in the band gap, which is consistent with the mixture between the wide band gap SnSe and the narrower band gap Ag2Se phase. This work demonstrates the potential of the gradual cation exchange process as a simple approach to engineer the optical properties of semiconductors, which can be leveraged for future material property engineering, especially in the fields of solar cells and optoelectronics.
Experimental section
Chemicals
Elemental selenium (Se) was purchased from Sigma-Aldrich. Sodium borohydride (NaBH4), tin(II) chloride dihydrate (SnCl2·2H2O), and silver nitrate (AgNO3) were purchased from Alfa Aesar. Sodium hydroxide (NaOH), methanol, and isopropyl alcohol (IPA) were purchased from Fisher Scientific. All reagents were used as received without additional purification.Synthesis of SnSe nanorods
The SnSe nanorods were synthesized using a modified surfactant-free route.32 First, Se powder, NaBH4, and deionized (DI) water were added in sequence in an empty beaker. The mixture experienced a fierce reaction which is manifested by the release of bubbles. This reaction can be described using the following chemical eqn (3):| 2Se + 4NaBH4 + 7H2O → 2NaHSe + Na2B4O7 + 14H2 | (3) |
In another empty beaker, a certain amount of NaOH was dissolved in DI water followed by the addition of SnCl2.2H2O into the NaOH solution. This mixture initially yielded some white precipitates, which gradually redissolved. The reaction can be described using the following chemical eqn (4):
| SnCl2 + 4NaOH → Na2SnO2 + 2H2O + 2NaCl | (4) |
Next, the two solutions were mixed, which resulted in the precipitation of black SnSe nanoparticles. The reaction can be described using the following chemical eqn (5):
| Na2SnO2 + NaHSe + H2O → SnSe + 3NaOH | (5) |
The as-synthesized product was washed in sequence and with centrifugation using diluted NaOH solution, DI water, and methanol. The purified SnSe product was dried in a vacuum at room temperature and stored for future use.
In a typical reaction towards the synthesis of 2 grams of SnSe precursor, 0.7989 g of Se was mixed with 0.7655 g NaBH4 in a beaker. Then, 60 ml of DI water was added, and magnetic stirring was performed. The mixture immediately turned into black and finally became a transparent and colorless solution (NaHSe). In a separate beaker, 4.856 g of NaOH was dissolved in 50 ml DI water under magnetic stirring. Then 2.2831 g of SnCl2·2H2O powder was added into the NaOH solution to make Na2SnO2. When the solid fully dissolved and became transparent, the as-synthesized NaHSe solution was injected to the Na2SnO2 solution. The mixture was kept under magnetic stirring for 2 hours at room temperature and then the synthesized SnSe precursor was collected by centrifugation. The product was washed several times using NaOH solution, DI water, and methanol and dried under vacuum.
Synthesis of SnSe–Ag2Se composite nanocrystals
For cation exchange reactions, a certain amount of SnSe (∼0.1 g) nanorods was added into a glass vial and dispersed in 4 ml of methanol. In a separate vial, the desired amount of AgNO3 powder was dissolved in methanol. The AgNO3 solution was subsequently added into the vial containing SnSe suspension and sealed. The mixture was placed under ultrasonic processing for 4 hours and underwent magnetic stirring for 2 days at room temperature. The final Ag2Se/SnSe nanocomposite was collected by centrifugation and washed with methanol several times. The final products were dried and kept under vacuum.Characterization
X-ray diffracted data were collected on a Rigaku Smart Lab, using monochromatic CuKα radiation (anode voltage 40 kV, emission current 44 mA) and a (D/teX 250) detector. XRD powder patterns were analyzed using PDXL software. Scanning electron microscopy (SEM) and energy dispersive X-Ray analyses (SEM-EDX) were carried out on a JEOL IT500 SEM. Further characterization on the nano-sized particles was performed using transmission electron microscopy (TEM), scanning transmission electron microscopy (STEM), STEM-EDX, and high-resolution TEM (HRTEM) with a ThermoFisher Scientific Talos F200X G2 S/TEM, operating with an accelerating voltage of 200 kV. For the TEM and HRTEM modes, the images were collected using a OneView camera, while for STEM and STEM-EDX, the images and elemental mappings were collected using 4 detectors: high-angle annular dark-field (HAADF), dark field-2 (DF2), dark field-4 (DF4), and bright field (BF). Samples for TEM/STEM experiments were prepared by dispersing powder samples of the Ag2Se/SnSe nanocomposite in isopropyl alcohol (IPA) using ultrasonic processing followed by the deposition of droplets on copper grids with carbon films. The loaded grids were dried in a vacuum with mild heating to remove the remaining IPA. HRTEM images were analyzed using Digital Micrograph software and STEM-EDX results were processed using Velox software. X-ray photoelectron spectroscopy (XPS) data were collected on a Kratos Axis Ultra XPS using a mono Al source (Al Kα, 1486.6 eV). The collected data were analyzed using Casa XPS software. Finally, the optical data (reflection spectrum) were collected on a JASCO V-770 UV-vis-NIR spectrophotometer.Author contributions
The manuscript was written through contributions from all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We gratefully acknowledge the financial support from the Department of Energy, Office of Basic Energy Science under Award # DE-SC-0018941. This work made use of the TEM and XPS from the Michigan Materials Characterization Center, (MC)2, purchased with funds from the University of Michigan College of Engineering and the National Science Foundation Awards DMR-0420785.References
- N. A. Moroz, A. Olvera, G. M. Willis and P. F. P. Poudeu, Nanoscale, 2015, 7, 9452–9456 RSC.
- A. A. Olvera, N. A. Moroz, P. Sahoo, P. Ren, T. P. Bailey, A. A. Page, C. Uher and P. F. P. Poudeu, Energy Environ. Sci., 2017, 10, 1668–1676 RSC.
- L. Maddi, K. Vinukonda, T. R. Gurugubelli and R. Koutavarapu, Electronics, 2023, 12, 1245 CrossRef CAS.
- S. K. Pandey, P. K. Mishra and D. Tiwary, J. Environ. Chem. Eng., 2022, 10, 107459 CrossRef CAS.
- M. Revathi, A. Pricilla Jeyakumari, R. Sridarane, M. Shkir, E. El Sayed Massoud and V. Sreedevi Gedi, J. Cluster Sci., 2022, 1–12 Search PubMed.
- X. Song, X. Li, Z. Chen and Z. Wang, Mater. Lett., 2020, 275, 128109 CrossRef CAS.
- M. Wang, P. F. Fang, Y. Chen, X. Y. Leng, Y. Yan, S. B. Yang, P. Xu and C. Yan, Adv. Funct. Mater., 2023, 2213902 CrossRef CAS.
- L. Zeng, H. Zhao, Y. Zhu, S. Chen, Y. Zhang, D. Wei, J. Sun and H. Fan, J. Mater. Chem. B, 2020, 8, 4093–4105 RSC.
- A. Olvera, T. P. Bailey, C. Uher and P. F. P. Poudeu, J. Mater. Chem. A, 2018, 6, 6997–7004 RSC.
- C. Zhou, Y. K. Lee, Y. Yu, S. Byun, Z.-Z. Luo, H. Lee, B. Ge, Y.-L. Lee, X. Chen and J. Y. Lee, Nat. Mater., 2021, 20, 1378–1384 CrossRef CAS PubMed.
- S. Chandra, U. Bhat, P. Dutta, A. Bhardwaj, R. Datta and K. Biswas, Adv. Mater., 2022, 34, 2203725 CrossRef CAS PubMed.
- S. Bano, S. I. Raj, A. Khalilullah, A. Jaiswal and I. Uddin, J. Photochem. Photobiol., A, 2021, 405, 112925 CrossRef CAS.
- L. De Trizio and L. Manna, Chem. Rev., 2016, 116, 10852–10887 CrossRef CAS PubMed.
- T. Ling, M. Jaroniec and S. Z. Qiao, Adv. Mater., 2020, 32, 2001866 CrossRef CAS PubMed.
- B. J. Beberwyck, Y. Surendranath and A. P. Alivisatos, J. Phys. Chem. C, 2013, 117, 19759–19770 CrossRef CAS.
- A. E. Powell, J. M. Hodges and R. E. Schaak, J. Am. Chem. Soc., 2016, 138, 471–474 CrossRef CAS PubMed.
- D. H. Ha, A. H. Caldwell, M. J. Ward, S. Honrao, K. Mathew, R. Hovden, M. K. A. Koker, D. A. Muller, R. G. Hennig and R. D. Robinson, Nano Lett., 2014, 14, 7090–7099 CrossRef CAS PubMed.
- E. A. Hernandez-Pagan, A. O'Hara, S. L. Arrowood, J. R. McBride, J. M. Rhodes, S. T. Pantelides and J. E. Macdonald, Chem. Mater., 2018, 30, 8843–8851 CrossRef CAS.
- D. H. Son, S. M. Hughes, Y. D. Yin and A. P. Alivisatos, Science, 2004, 306, 1009–1012 CrossRef CAS PubMed.
- Z. Z. Li, M. Saruyama, T. Asaka, Y. Tatetsu and T. Teranishi, Science, 2021, 373, 332 CrossRef CAS PubMed.
- J. M. Pietryga, D. J. Werder, D. J. Williams, J. L. Casson, R. D. Schaller, V. I. Klimov and J. A. Hollingsworth, J. Am. Chem. Soc., 2008, 130, 4879–4885 CrossRef CAS PubMed.
- M. Casavola, M. A. van Huis, S. Bals, K. Lambert, Z. Hens and D. Vanmaekelbergh, Chem. Mater., 2012, 24, 294–302 CrossRef CAS.
- K. Lambert, B. De Geyter, I. Moreels and Z. Hens, Chem. Mater., 2009, 21, 778 CrossRef CAS.
- Y. Justo, P. Geiregat, K. Van Hoecke, F. Vanhaecke, C. D. Donega and Z. Hens, J. Phys. Chem. C, 2013, 117, 20171–20177 CrossRef CAS.
- J. B. Zhang, J. B. Gao, C. P. Church, E. M. Miller, J. M. Luther, V. I. Klimov and M. C. Beard, Nano Lett., 2014, 14, 6010–6015 CrossRef CAS PubMed.
- J. B. Zhang, B. D. Chernomordik, R. W. Crisp, D. M. Kroupa, J. M. Luther, E. M. Miller, J. B. Gao and M. C. Beard, ACS Nano, 2015, 9, 7151–7163 CrossRef CAS PubMed.
- A. G. Butterfield, L. T. Alameda and R. E. Schaak, J. Am. Chem. Soc., 2021, 143, 1779–1783 CrossRef CAS PubMed.
- F. Huang, J. Ning, Z. Duan, A. A. Sergeev, A. Portniagin, S. V. Kershaw, J. Tian and A. L. Rogach, Chem. Mater., 2021, 33, 2398–2407 CrossRef CAS.
- G. Gariano, V. Lesnyak, R. Brescia, G. Bertoni, Z. Dang, R. Gaspari, L. De Trizio and L. Manna, J. Am. Chem. Soc., 2017, 139, 9583–9590 CrossRef CAS PubMed.
- L. De Trizio, H. Li, A. Casu, A. Genovese, A. Sathya, G. C. Messina and L. Manna, J. Am. Chem. Soc., 2014, 136, 16277–16284 CrossRef CAS PubMed.
- H. Li, R. Brescia, R. Krahne, G. Bertoni, M. J. Alcocer, C. D’Andrea, F. Scotognella, F. Tassone, M. Zanella and M. De Giorgi, ACS Nano, 2012, 6, 1637–1647 CrossRef CAS PubMed.
- G. Han, S. R. Popuri, H. F. Greer, J. W. G. Bos, W. Zhou, A. R. Knox, A. Montecucco, J. Siviter, E. A. Man and M. Macauley, Angew. Chem., Int. Ed., 2016, 55, 6433–6437 CrossRef CAS PubMed.
- V. Sidey, J. Phys. Chem. Solids, 2022, 171, 110992 CrossRef CAS.
- L. H. Ahrens, Geochim. Cosmochim. Acta, 1952, 2, 155–169 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- S. S. Ralsanov, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2020, 76, 38–40 CrossRef PubMed.
- M. S. Shirazi, A. Foroumadi, I. Saberikia and M. M. Farimani, Ultrason. Sonochem., 2022, 87, 106037 CrossRef PubMed.
- B. Pejjai, V. R. M. Reddy, K. Seku, M. R. Pallavolu and C. Park, New J. Chem., 2018, 42, 4843–4853 RSC.
- R. G. Parr and R. G. Pearson, J. Am. Chem. Soc., 1983, 105, 7512–7516 CrossRef CAS.
- H. Lee, B. Yoo, D. Kim, J. Cha, Y. K. Kang, S.-P. Cho, T. Hyeon, M.-G. Kim, M. G. Kanatzidis and I. Chung, J. Am. Chem. Soc., 2023, 145, 15951–15962 CrossRef CAS PubMed.
- S. H. Heo, S. Jo, H. S. Kim, G. Choi, J. Y. Song, J.-Y. Kang, N.-J. Park, H. W. Ban, F. Kim and H. Jeong, Nat. Commun., 2019, 10, 864 CrossRef PubMed.
- L. Huang, J. Lu, D. Ma, C. Ma, B. Zhang, H. Wang, G. Wang, D. H. Gregory, X. Zhou and G. Han, J. Mater. Chem. A, 2020, 8, 1394–1402 RSC.
- G. Zhang, G. Li, J. Wang, H. Tong, J. Wang, Y. Du, S. Sun and F. Dang, Adv. Energy Mater., 2022, 12, 2103910 CrossRef CAS.
- Y. K. Lee, Z. Luo, S. P. Cho, M. G. Kanatzidis and I. Chung, Joule, 2019, 3, 719–731 CrossRef CAS.
- L.-D. Zhao, C. Chang, G. Tan and M. G. Kanatzidis, Energy Environ. Sci., 2016, 9, 3044–3060 RSC.
- D. Lee, W. Park, Y. A. Kang, H. T. Lim, S. Park, Y. Mun, J. Kim and K.-S. Jang, ACS Appl. Mater. Interfaces, 2023, 15, 3047–3053 CrossRef CAS PubMed.
- V. Krylova, S. Žalenkienė, N. Dukstienė and J. Baltrusaitis, Appl. Surf. Sci., 2015, 351, 203–208 CrossRef CAS.
- C. Wang, H. Yang, Q. Wang, L. Qiao, X. Peng, J. Li, J. Han, Q. Wang, X. Li and Z. Wang, CrystEngComm, 2020, 22, 5296–5301 RSC.
- L. Qiu, X. Lai and J. Jian, Mater. Charact., 2021, 172, 110864 CrossRef CAS.
- P. Naumov, O. Barkalov, H. Mirhosseini, C. Felser and S. Medvedev, J. Phys.: Condens. Matter, 2016, 28, 385801 CrossRef CAS PubMed.
- C.-N. Zhu, P. Jiang, Z.-L. Zhang, D.-L. Zhu, Z.-Q. Tian and D.-W. Pang, ACS Appl. Mater. Interfaces, 2013, 5, 1186–1189 CrossRef CAS PubMed.
- Z. Li, L. Peng, Y. Fang, Z. Chen, D. Pan and M. Wu, Radiat. Phys. Chem., 2011, 80, 1333–1336 CrossRef CAS.
- V. Drozd, I. Nikiforova, V. Bogevolnov, A. Yafyasov, E. Filatova and D. Papazoglou, J. Phys. D: Appl. Phys., 2009, 42, 125306 CrossRef.
- W. Shi, M. Gao, J. Wei, J. Gao, C. Fan, E. Ashalley, H. Li and Z. Wang, Adv. Sci., 2018, 5, 1700602 CrossRef PubMed.
- R. Wang, M. Wei, G. Jiang, W. Liu and C. Zhu, Chem. Lett., 2014, 43, 693–695 CrossRef CAS.
- M. Pachecka, J. M. Sturm, R. W. E. van de Kruijs, C. J. Lee and F. Bijkerk, AIP Adv., 2016, 6, 075222 CrossRef.
- S. S. Batsanov, Russ. Chem. Rev., 1982, 51, 684 CrossRef.
- M.-A. Langevin, D. Lachance-Quirion, A. M. Ritcey and C. N. Allen, J. Phys. Chem. C, 2013, 117, 5424–5428 CrossRef CAS.
Footnotes |
| † Dedicated to Professor Michael Ruck on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma00394b |
| This journal is © The Royal Society of Chemistry 2024 |