
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Study of self-assembly of mixed-ligand metal–organic cages by high-resolution mass spectrometry†
Kang
Tong
a,
Jia
Jia
 *a,
Rongfu
Huang
a and
Jin
Luo
b
*a,
Rongfu
Huang
a and
Jin
Luo
b
aCollege of Material and Chemistry & Chemical Engineering, Chengdu University of Technology, Chengdu 610059, China. E-mail: jiajia17@cdut.edu.cn
bAnalytical and Testing Centre of Sichuan Province, Chengdu, China
First published on 29th May 2024
Abstract
In this study, we scrutinized the self-assembly dynamics of mixed-ligand MOCs employing HR-MS. Our findings unveiled the remarkable coexistence of tetrahedron and cigar configurations under specific conditions, providing novel insights into multi-linker self-assembly processes.
As discrete and porous supramolecular entities, metal–organic cages (MOCs) manifest through the self-assembly of metal nodes (comprising metal clusters or naked ions) and electron-rich organic ligands.1,2 The inherent diversity of metal nodes and ligands engenders a profusion of MOCs, each with distinct topological structures and configurations. This diversity significantly broadens the spectrum of MOC species and their potential applications, thereby elevating MOCs to a realm of considerable allure within the materials landscape.3 Yuan et al.4 explored the realm of Zr-MOCs, constructing a series of tetrahedral Zr-MOCs such as V4E6 or V4F4 (where V denotes the vertex, E signifies the edge, and F denotes the face) through the hydrolysis of bis(cyclopentadienyl)zirconium dichloride (Cp2ZrCl2, with Cp representing μ5-C5H5) coordinated with oriented dicarboxylate linkers. This seminal work has inaugurated a new era in Zr-MOC research, characterized by the incorporation of multifunctional groups into the Zr-MOC framework. Such advancements underscore the remarkable potential of Zr-MOCs across a spectrum of applications including gas separation,5 catalysis,6 sensing,7–10 and beyond.
In stark contrast to metal–organic frameworks (MOFs),11,12 MOCs are constrained by the spatial orientation of their metal nodes or linkers, fostering the convergence of intermediates into mono-molecular structures rather than extensive polymers during self-assembly processes.13 A striking disparity in properties between the two lies in solubility: MOFs typically exhibit insolubility,14,15 while MOCs display a propensity for dissolution in a wide array of solvents. This solubility characteristic not only facilitates the fabrication of membranes, particularly for applications such as molecular separation,16 but also enables the elucidation of liquid interactions with MOCs, thus broadening avenues for characterization and exploration in liquid environments.
In the realm of Zr-MOCs, the prevailing notion of tetrahedral structures has been challenged by mounting evidence supporting their multi-configurational nature. Bloch17 not only delineated the existence of both cigar and tetrahedral configurations but also unveiled the intricate modulation of Zr-MOC configurations by linker parameters. Zhou's work18 further corroborated these findings, demonstrating the profound influence of temperature, solvent composition, counter ions, and coordination angles on the Zr-MOC configuration. Recent strides19 have introduced the strategy of linker exchange as a pioneering method to attain pure configurations of Zr-MOCs. Consequently, our understanding of the self-assembly principles governing homoleptic cages, synthesized via single linkers, has become more nuanced. The quest for comprehensive functionality has propelled investigations into more sophisticated structures. Through the integration of mixed-linkers, heteroleptic cages have shown remarkable potential in catalysis20 and host–guest chemistry,21 eliciting considerable excitement within scientific circles. The incorporation of multi-linkers into self-assembly systems for the development of advanced materials represents a captivating avenue of exploration, promising transformative breakthroughs in materials science and beyond.
Hollway's22 investigations delineate two distinct self-assembly paradigms for multi-linkers: social self-sorting and narcissistic self-sorting. Notably, heteroleptic cages emerge exclusively through the social self-sorting mechanism. Building upon the multi-linker strategy, Lee23 engineered a remarkable photocatalyst for CO2 reduction, comprising H2ReTC and H2BPDC. Similarly, Qi24 devised a CO2-reducing photocatalyst, Ir-MOC-NH2, following Lee's footsteps. Echoing these endeavours, Choe25 ventured further by amalgamating six different linkers for self-assembly, resulting in an exponential proliferation of heteroleptic cage species. The pursuit of novel strategies prompts a pertinent inquiry: do different linkers modulate the configuration of heteroleptic cages within the mixed-linker framework? This question underscores the evolving landscape of supramolecular chemistry, offering tantalizing prospects for unravelling the intricate interplay between linkers and their resultant structures.
According to Bloch's classification,17 three distinct types of Zr-MOCs have been identified: the C type (cigar), C/T type (cigar/tetrahedron), and T type (tetrahedron) (Scheme 1). Given the susceptibility of Zr-MOC configurations to alterations under synthetic conditions, the meticulous selection of linkers with exclusive self-assembly conformations in diverse environments becomes paramount.
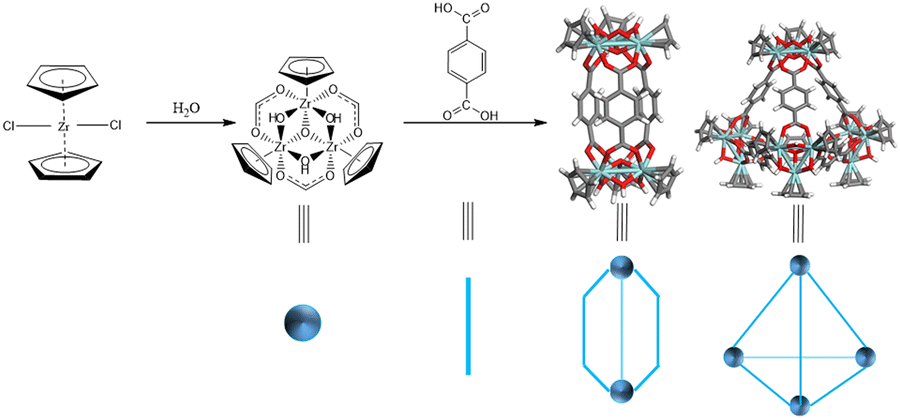 | ||
| Scheme 1 Zr-metal cluster and BDC form C and T type Zr-MOCs. | ||
It is noteworthy that variations in temperature26,27 (ranging from 25 °C to 65 °C) and solvent composition26 (DMA and DMF) have been shown to directly influence conformational distinctions. To address this, we devised an experimental protocol involving the cross-tuning of temperature and solvent for self-assembly. Specifically, we followed a previously reported method, wherein Cp2ZrCl2 and linkers were introduced into solvent at a ratio of 3/2 equivalents. Subsequently, the temperature was incrementally adjusted from 25 °C to 65 °C, and the solvent transitioned from DMF to DMA. HR-MS using ESI-Q-TOF was employed to identify the configurations of homoleptic cages. Although the [C]+1 and [T]+2 occupied the same spectral position, subtle nuances in isotopic distribution and the distances between isotopic peaks served as decisive evidence for determination. Specifically, the distance between isotopic peaks for the [C] type was 1.0 Da, while for the [T] type, it was 0.5 Da (refer to Fig. 1(a)). Notably, homoleptic cages formed with L1 (AA, adipic acid), L2 (H2BDC, 1,4-dicarboxybenzene), and L3 (H2BDC-NH2, 2-aminoterephthalic acid) exhibited conformational selectivity in crossover experiments (refer to Fig. 1(b–d)). Consequently, these linkers were chosen as representative candidates for multi-linker self-assembly. Of particular interest, L3, a subtetrahedral linker, demonstrated a mixed configuration exclusively under conditions of 65 °C/DMF.
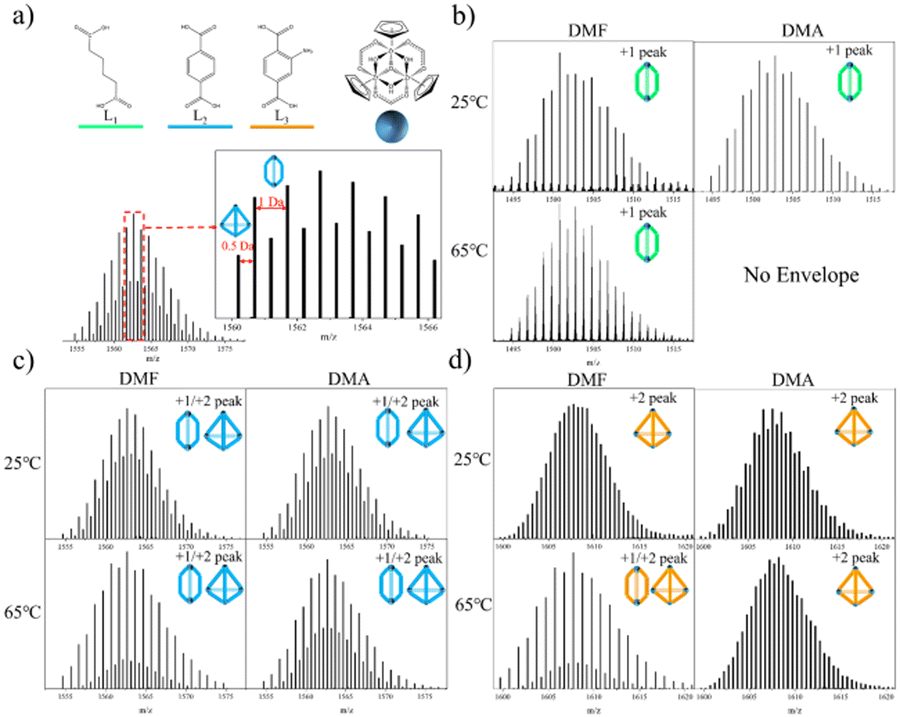 | ||
| Fig. 1 (a) Bridging linker, L1, L2, L3, zirconium cluster and determination of cigar and tetrahedron. (b) ESI-Q-TOF analysis of Zr-MOCs with L1. (c) ESI-Q-TOF analysis of Zr-MOCs with L2. (d) ESI-Q-TOF analysis of Zr-MOCs with L3. | ||
Upon mixing equimolar solutions of L1 and L3 in DMA, the reaction proceeded at 65 °C, yielding the powder sample. Subsequently, the sample was dissolved and diluted in MeOH/H2O (9/1), and its composition was analysed via HR-MS (refer to Fig. 2). Surprisingly, the equimolar mixture of L1 and L3 did not exhibit the anticipated normal multicomponent distribution observed for a previously reported28 homoleptic cage ZrT-1-NH2. Instead, it yielded three distinct clusters of +1 peak envelopes at 1537.7701 m/z, 1572.7505 m/z, and 1607.7288 m/z. Through meticulous simulation and comparison, we ascertained the molecular formulas of these peak envelopes to be C50H55O20N1Zr6, C52H52O20N2Zr6, and C54H49O20N3Zr6, respectively. Notably, the differences in m/z between each peak envelope were approximately 35 Da, correlating with the difference between the molecular compositions of L1 and L3. Consequently, we have designated these two novel compositions as ZrC-(L1)2(L3)1 and ZrC-(L1)1(L3)2, respectively.
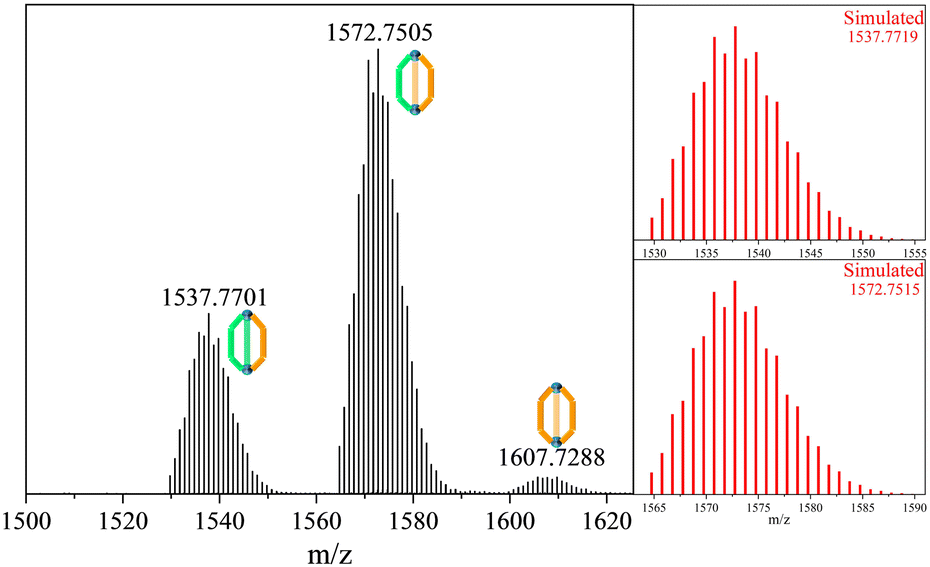 | ||
| Fig. 2 Experimental and simulated MS spectra of heteroleptic cages with equivalents of L1 and L3. | ||
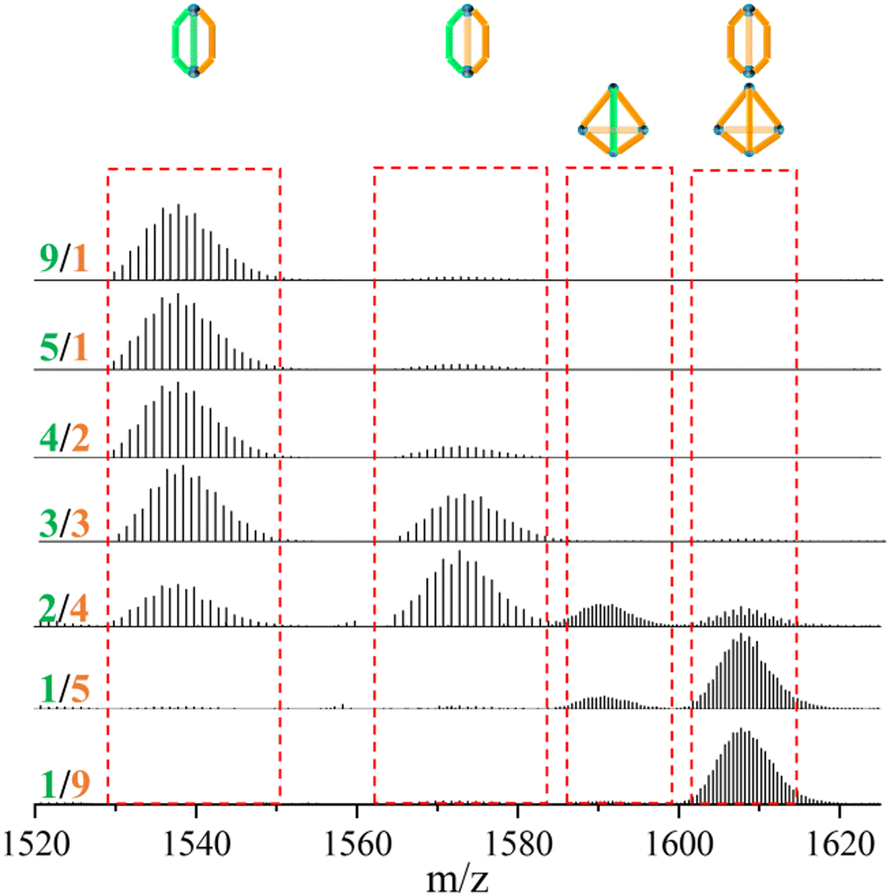
In contrast to the tetrahedron homoleptic cage ZrT-1-NH2, the peak envelope at m/z 1607.7288 exhibited a cigar conformation under the same conditions. To delve deeper into the intricacies of multi-linker self-assembly, we systematically adjusted the ratios of L1 and L3 (ranging from 1/9 to 9/1) for synthesis and analysis (refer to Fig. 3). As the ratio of L3 increased, the envelope of ZrC-(L1)2(L3)1 gradually diminished, while the peak envelope of ZrC-(L1)1(L3)2 initially intensified before subsequent weakening. Simultaneously, the peak envelope at m/z 1607.7288 exhibited a gradual increase, concomitant with the corresponding conversion of configuration. When the ratio of L1/L3 reached 1/5, the peak envelope at m/z 1607.7288 displayed features indicative of mixed configuration. With a continued increase in the ratio of L3, the characteristic envelope at m/z 1607.7288 transitioned to the tetrahedral ZrT-1-NH2 configuration.
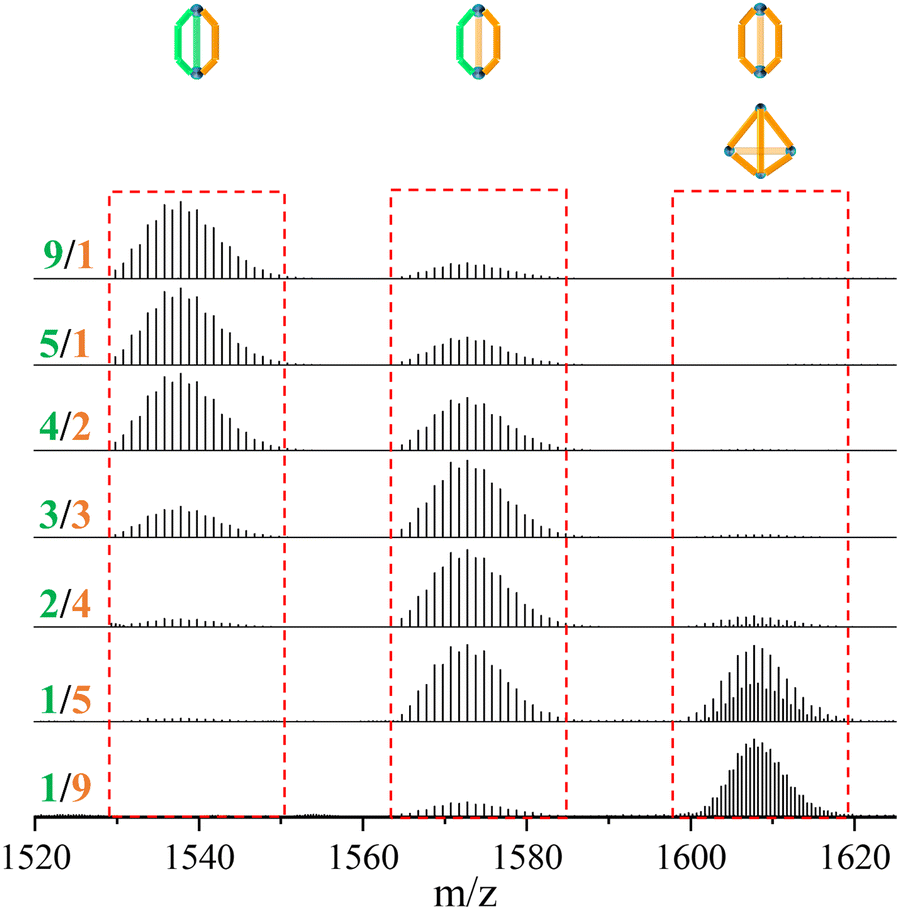 | ||
| Fig. 3 Proportional experiment of multi-linkers with L1 and L3 at 65 °C in DMA. | ||
DFT calculations were employed to elucidate the stability of ZrC-(L1)2(L3)1 and ZrC-(L1)1(L3)2. The MM2 (minimized energy) values of ZrC-(L1)2(L3)1 and ZrC-(L1)1(L3)2 were determined to be −909.25 eV and −913.99 eV, respectively (refer to Fig. S1 and S2, ESI†). This analysis suggested that ZrC-(L1)1(L3)2 exhibits greater stability compared to ZrC-(L1)2(L3)1. The transformation of the envelope characteristic at m/z 1607.7288 likely indicates that the configuration of the homoleptic cage is also influenced by the presence of multi-linkers, underscoring the intricate interplay between different linker compositions and resultant cage configurations.
As per our prior observations, all heteroleptic cages with L1 exhibited a cigar conformation. However, we pondered whether it is possible to prepare a heteroleptic tetrahedron with L1. Drawing inspiration from Zhou's work,18 which suggests that lower temperatures facilitate the conversion of cigar to tetrahedron, we replicated experiments at 25 °C (refer to Fig. 4). Maintaining an equal content of L1/L3, ZrC-(L1)1(L3)2 and ZrC-(L1)2(L3)1 remained the primary components. However, notable differences emerged: ZrC-(L1)2(L3)1 predominated as the major product at 65 °C, whereas ZrC-(L1)1(L3)2 exhibited higher relative content at 25 °C. Upon increasing the ratio of L1 and L3 to 2/4 or 1/5, we detected a +2 peak envelope at 1590.2425 m/z. Through simulation, we successfully synthesized the heteroleptic tetrahedron ZrT-(L1)1(L3)5 (with the formula: C106H103O40N5Zr12). We speculate that the influence of temperature on the configuration of Zr-MOCs may originate from the balance of kinetic and thermodynamic factors. At low temperatures, kinetic control dominates, favoring the formation of the thermodynamically metastable tetrahedron configuration; while at high temperatures, thermodynamic control dominates, favoring the formation of the stable cigar configuration. Moreover, the increased mobility of ligands at high temperatures may reduce the spatial hindrance required for the formation of the highly symmetric tetrahedron configuration.
 | ||
| Fig. 4 Proportional experiment of multi-linkers with L1 and L3 at 25 °C in DMA. | ||
As we explored the multi-linker self-assembly process in DMF, regardless of the varied conditions, we didn’t observe any signal of ZrT-(L1)1(L3)5 (refer to Fig. S3 and S4, ESI†). This absence might be attributed to the size disparity between DMA and DMF molecules.26 We propose that the solvent effect on the configuration of Zr-MOCs can be attributed to the differences in the solvation of ligands and intermediates. DMA, with its larger size and higher polarity, may provide a more favorable solvation environment for the ligands and intermediates, thus promoting the formation of the tetrahedron configuration. To further investigate, we introduced L2 into the experiment. Given the binary configuration selectivity of L2, the self-assembly products became more intricate upon its addition. Overall, under conditions of 25 °C/DMA, a variety of multicomponent products coexisted with tetrahedron configurations. When the ratio of L2 surpassed its maximum threshold (exceeding 5/1), both tetrahedron and cigar configurations coexisted within the system, as evidenced by Fig. S5–S11 in the ESI.† Conversely, under conditions of 65 °C/DMA, all products displayed mixed configurations, except under conditions where the ratio of L2 fell below its minimum threshold (less than 1/5), as illustrated in Fig. S12–S18 in the ESI.†
Under conditions of 25 °C/DMF, the configuration and components were similar to those observed under conditions of 25 °C/DMA (refer to Fig. S19–S25, ESI†). However, under conditions of 65 °C/DMF, a mixed configuration of tetrahedron and cigar emerged, particularly with extreme L3 (L2/L3 = 1/9) content. Additionally, even a small increment in L2 led to the complete conversion of the L3 homoleptic cage into a tetrahedron, as depicted in Fig. S26–S32 (ESI†), and XRD further confirmed its tetrahedral structure, as shown in Fig. S33 in the ESI.† These findings underscore the complex interplay between temperature, solvent composition, and the presence of different linkers in multi-linker self-assembly processes, shedding light on the nuanced mechanisms underlying heteroleptic cage formation.
Conclusions
This study presents a thorough investigation into the intricate self-assembly dynamics of heteroleptic Zr-MOCs. Through the introduction of mixed-linker systems, the research uncovers binary configuration selectivity, enhancing the complexity of self-assembly products. Notably, our findings reveal the coexistence of tetrahedron and cigar configurations under specific conditions, offering fresh insights into multi-linker self-assembly processes. These discoveries not only deepen our understanding of supramolecular chemistry but also pave the way for the development and synthesis of advanced materials with promising applications in catalysis, sensing, and beyond.Conflicts of interest
There are no conflicts to declare.Notes and references
- H. Vardhan, M. Yusubov and F. Verpoort, Coord. Chem. Rev., 2016, 306, 171–194 CrossRef CAS.
- B. Lee, B. Go, B. Jung and J. Park, Small Struct., 2023, 2308393 Search PubMed.
- S. Lee, H. Jeong, D. Nam, M. S. Lah and W. Choe, Chem. Soc. Rev., 2021, 50, 528–555 RSC.
- G. Liu, Z. Ju, D. Yuan and M. Hong, Inorg. Chem., 2013, 52, 13815–13817 CrossRef CAS PubMed.
- W. Fan, S. B. Peh, Z. Zhang, H. Yuan, Z. Yang, Y. Wang, K. Chai, D. Sun and D. Zhao, Angew. Chem., Int. Ed., 2021, 60, 17338–17343 CrossRef CAS PubMed.
- M. Sun, Q. Q. Wang, C. Qin, C. Y. Sun, X. L. Wang and Z. M. Su, Chem. – Eur. J., 2019, 25, 2824–2830 CrossRef CAS PubMed.
- S. Chen, S. Cheng, L. Zhao, C. Sun, C. Qin and Z. Su, New J. Chem., 2020, 44, 21255–21260 RSC.
- Z. Gao, J. Jia, W. Fan, T. Liao and X. Zhang, Chin. Chem. Lett., 2022, 33, 4415–4420 CrossRef CAS.
- K. Jin, D. Moon, M. Kim and J. Park, Sens. Actuators, B, 2023, 393, 134205 CrossRef CAS.
- J. Yuan, X. Fan, J. Yang and X. Zhang, Chin. Chem. Lett., 2023, 34, 108155 CrossRef CAS.
- Q. Zhai, Y. Ren, H. Wang, C. Liu, Z. Li and H. Jiang, Dalton Trans., 2024, 53, 5836–5843 RSC.
- J. Chen, N. Yao, Y. Tang, L. Xie, X. Zhuo and Z. Jiang, Dalton Trans., 2024, 53, 5900–5910 RSC.
- B. S. Pilgrim and N. R. Champness, ChemPlusChem, 2020, 85, 1842–1856 CrossRef CAS PubMed.
- A. Yu, X. Liang, C. Hao, X. Hu, J. Li, X. Bo, D. Du and Z. Su, Dalton Trans., 2024, 53, 6275–6281 RSC.
- Y. Wang, Z. Song, Y. Liu, Y. Chen, J. Li, L. Li and J. Yao, Dalton Trans., 2024, 53, 6802–6808 RSC.
- X. Guo, S. Xu, Y. Sun, Z. Qiao, H. Huang and C. Zhong, J. Membr. Sci., 2021, 632, 119354 CrossRef CAS.
- A. J. Gosselin, G. E. Decker, B. W. McNichols, J. E. Baumann, G. P. A. Yap, A. Sellinger and E. D. Bloch, Chem. Mat., 2020, 32, 5872–5878 CrossRef CAS.
- Z. Xiao, H. F. Drake, Y. H. Rezenom, P. Cai and H.-C. Zhou, Small Struct., 2021, 3, 2100133 CrossRef.
- M. G. Sullivan, H. K. Welgama, M. R. Crawley, A. E. Friedman and T. R. Cook, Chem. Mat., 2023, 36, 567–574 CrossRef.
- J. Jiao, C. Tan, Z. Li, Y. Liu, X. Han and Y. Cui, J. Am. Chem. Soc., 2018, 140, 2251–2259 CrossRef CAS PubMed.
- S. C. Li, L. X. Cai, M. Hong, Q. Chen and Q. F. Sun, Angew. Chem., Int. Ed., 2022, 61, e202204732 CrossRef CAS PubMed.
- L. R. Holloway, P. M. Bogie and R. J. Hooley, Dalton Trans., 2017, 46, 14719–14723 RSC.
- H. S. Lee, S. Jee, R. Kim, H.-T. Bui, B. Kim, J.-K. Kim, K. S. Park, W. Choi, W. Kim and K. M. Choi, Energy Environ. Sci., 2020, 13, 519–526 RSC.
- X. Qi, R. Zhong, M. Chen, C. Sun, S. You, J. Gu, G. Shan, D. Cui, X. Wang and Z. Su, ACS Catal., 2021, 11, 7241–7248 CrossRef CAS.
- D. Nam, J. Kim, E. Hwang, J. Nam, H. Jeong, T.-H. Kwon and W. Choe, Matter, 2021, 4, 2460–2473 CrossRef CAS.
- X. Chen, S.-B. Li, Z.-Y. Liu and Y.-T. Zhang, J. Solid State Chem., 2021, 296, 121998 CrossRef CAS.
- Z. Niu, L. Wang, S. Fang, P. C. Lan, B. Aguila, J. Perman, J. G. Ma, P. Cheng, X. Li and S. Ma, Chem. Sci., 2019, 10, 6661–6665 RSC.
- G. Liu, Y. Di Yuan, J. Wang, Y. Cheng, S. B. Peh, Y. Wang, Y. Qian, J. Dong, D. Yuan and D. Zhao, J. Am. Chem. Soc., 2018, 140, 6231–6234 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma00406j |
| This journal is © The Royal Society of Chemistry 2024 |