
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effect of molecular structure on the photochemical stability of acceptor and donor polymers used in organic solar cells†
Suraj
Prasad
 a,
Zewdneh
Genene
b,
Cleber F. N.
Marchiori
a,
Shivam
Singh
a,
Leif K. E.
Ericsson
a,
Ergang
Wang
b,
C. Moyses
Araujo
a and
Ellen
Moons
*a
a,
Zewdneh
Genene
b,
Cleber F. N.
Marchiori
a,
Shivam
Singh
a,
Leif K. E.
Ericsson
a,
Ergang
Wang
b,
C. Moyses
Araujo
a and
Ellen
Moons
*a
aDepartment of Engineering and Physics, Karlstad University, SE-65188 Karlstad, Sweden. E-mail: ellen.moons@kau.se
bDepartment of Chemistry and Chemical Engineering, Chalmers University of Technology, SE-412 96 Göteborg, Sweden
First published on 2nd September 2024
Abstract
The limited operational lifetime of organic solar cells remains an obstacle to their commercial development and is largely due to the poor intrinsic photostability of the conjugated molecules that constitute the photoactive layer. Here, we selected a series of state-of-the-art donor and acceptor materials including PBDB-T, Y5, PF5-Y5, and PYT to study their photostability under AM1.5 simulated sunlight in ambient conditions. Their properties are monitored over time, using various spectroscopy techniques, including UV-Vis absorption, Fourier-transform infrared (FTIR), and X-ray and ultraviolet photoelectron spectroscopy (XPS and UPS). We found that the absorption spectra of Y5 and PYT films remain almost intact even after 30 hours of light exposure in air, while the PF5-Y5 and PBDB-T films undergo rapid photobleaching. The absorption losses observed in blend films of PBDB-T with Y5 and with PF5-Y5 can be understood as composed of contributions from the separate blend components that are similar to the absorption losses in neat films. The new peaks emerging in the FTIR spectra of PBDB-T, PF5-Y5, and their blend films witness the formation of new carbonyl groups, while these are absent in the spectra of the Y5 and PYT films. The XPS C 1s spectra of the PF5-Y5 and PBDB-T films confirm this carbonyl formation and the S 2p spectra reveal that sulphone groups are formed after 30 hours of exposure of these films. These results confirm that films of Y5 and the copolymer PYT are significantly more resistant to photooxidation, compared to the copolymer PF5-Y5. The comparison of these results suggests that the benzo[1,2-b:4,5-b′]dithiophene moiety with alkylated thiophenes as side chains (BDT-T) accelerates the photodegradation of PBDB-T and PF5-Y5. The replacement of the BDT-T unit by thiophene contributes to the enhanced stability of PYT, demonstrating that the nature of the co-monomer has a significant effect on the intrinsic photostability of Y5-based copolymers. These new insights are expected to stimulate the design of stable donors and acceptor polymers for the development of long-lived OPV devices.
1. Introduction
Organic photovoltaics (OPV) is a low-cost solar energy technology based on thin films of molecular semiconductor materials that has interesting advantages compared to more conventional PV technologies, related to their light weight, flexibility, and large-scale manufacturability.1–6 In recent years, OPV technologies have gained significant attention due to the remarkable improvements in power conversion efficiency (PCE), achieved with the novel small-molecule non-fullerene acceptors (NFAs). Chemical functionalization of the NFAs provides key advantages compared to the fullerene-based acceptors, such as tunability of their energy gap7–9 to obtain enhanced visible/near-IR absorption,10,11 and high open circuit voltages with minimal losses due to non-radiative recombination.12,13 In particular, using the Y-series of NFAs, that are based on A–DA′D–A configurations, where A stands for the electron acceptor moiety and D stands for the electron donor moiety, PCE values above 18% have been reported for binary14,15 and ternary16–18 bulk heterojunction solar cells. Despite this progress in performance, a remaining issue that OPV faces is the limited device lifetime.19–21 The lifetime of OPV devices is determined by many different types of degradation. One of them is the photostability of each of the active layer materials, which undergo photochemical reactions under ambient conditions. In such conditions, degradation has been associated with photooxidation of the active layer materials. We and others have shown that the main cause of photodegradation in polymer:fullerene active layers could be assigned to the photooxidation of fullerene derivatives.22–26 Through a joint experimental–computational study, based on IR, X-ray photoelectron and absorption spectroscopy, we have shown that several fullerene photooxidation products are formed, including dicarbonyls and anhydrides.22 Even organic solar cells that are encapsulated or operated in inert conditions degrade under operation, typically observed as a sharp initial decrease of electrical performance, so-called burn-in losses.27,28 For NFA-based solar cells under inert conditions, burn-in effects are found to result from the instability of interfaces and electrodes.27,29–33 Another common cause of performance degradation during device operation is related to changes in morphology during solar cell operation, in particular the aggregation of the NFA. Several strategies have been proposed to overcome the issues related to morphological instability of the active layer in donor/NFA heterojunction solar cells. One of them is the use of polymerized acceptors, which have led to all-polymer solar cells with promising thermal and mechanical stability.34–36 Another strategy is to mix multiple derivatives of the acceptor molecule to suppress crystallization.37,38Recent photostability studies have focused on the donor polymers. Recently, Wang et al. studied the photodegradation of donor polymers PM6, D18, and PTQ10 (see Fig. S12 for molecular structures, ESI†) blended with NFA Y6.39 They have found that PM6 and D18 are the dominant contributors to the degradation in these blends upon light-soaking in air, as evident from optical spectroscopy measurements. The authors assigned the major cause of this donor polymer degradation to the backbone twisting of the (BDT-T) thiophene motif in PM6 and D18, in contrast to the more photostable PTQ10 where this motif is absent. The relation between molecular structure and photo-oxidative stability has been studied by the Ratcliff group for push–pull polymers based on alkyl thienyl-substituted benzodithiophene (BDT-T)40 by a combination of optical and chemical analysis by X-ray photoelectron spectroscopy. The results show that alkyl chain oxygen addition and sulfur oxidation are prevalent degradation pathways and that the latter is generally initiated after saturation of the former. Moreover, this comprehensive study elucidates that the specific pull components of BDT-T push polymers influence the relative stabilities, which cannot simply be predicted by the redox properties of the components.
Recently, the photostability of small molecule NFA materials has gained interest. In acceptor–donor–acceptor (A–D–A) NFAs, the two exocyclic vinyl bridges that connect the end-group acceptor moieties to the central fused-ring donor unit are prone to chemical and photochemical reactions.41,42 The poor photostability of ITIC and ITIC-M (see Fig. S12 for molecular structures, ESI†) is reported to be due to an initial photo-induced conformational change that facilitates the vinyl linkage between the core and the end groups of the acceptors.28 In contrast, the IDTBR family of NFAs (Fig. S12, ESI†) was found to show good photostability, giving superior long-term device stability. Liu et al. investigated the photodegradation pathway of A–D–A NFAs and found that photoisomerization of the vinyl group is responsible for the irreversible photooxidation.43 Moreover, in order to expand the absorption range towards the infrared region, a benzothiadiazole (BT) unit was incorporated into the central core of ITIC to create an electron-deficient-core-based fused structure (D–A′–D).44 Considerable efforts have been invested in improving the PCE of NFA-based organic solar cells, but there has been a lack of thorough exploration of their intrinsic photochemical stability and the degradation mechanism of thin films.44–47
Here, we report on the results of a systematic investigation of the photodegradation of donor polymer PBDB-T, small molecule acceptor Y5, and the copolymer acceptors PF5-Y5 and PYT thin films. All samples were intentionally subjected to white light illumination (AM 1.5) from a solar simulator in ambient conditions. The effect of photodegradation on their properties was studied, using UV-vis absorption spectroscopy, Fourier-transform infrared spectroscopy (FTIR), X-ray photoelectron spectroscopy (XPS), ultraviolet photoelectron spectroscopy (UPS), and atomic force microscopy (AFM). The UV-vis results reveal that both PBDB-T and PF5-Y5 exhibit a higher photobleaching rate in comparison to Y5 and PYT. The appearance of new carbonyl peaks in the IR spectra of PBDB-T and PF5-Y5 films provides evidence of photo-oxidation reactions, as further confirmed by C 1s as well as S 2p core-level XPS spectra. In summary, our findings reveal the contribution of the BDT-T moiety to an accelerated photodegradation rate of both donor and Y-type acceptor copolymers. Indeed, the substitution of the BDT-T moiety with a thiophene in the Y-type acceptor results in the improved photostability of the copolymer PYT.
2. Experimental
2.1 Materials and sample preparation
The donor polymer PBDB-T (PCE12) and small molecule NFA Y5 were purchased from 1-Material and Sigma Aldrich, respectively. The polymer acceptors PF5-Y5 and PYT are synthesized with an alternating electron-deficient Y5 unit and donor moieties thienyl-benzodithiophene (BDT-T) and thiophene, respectively. The synthesis of PF5-Y5 and PYT is described in detail in previous publications.46,48 Chlorobenzene was purchased from Sigma Aldrich (anhydrous, 99.8%). All solutions and thin films were prepared inside a N2-filled glove box (O2 < 0.1 ppm, H2O < 0.1 ppm) under yellow light (MB200MOD, MBraun Inert gas-Systeme GmbH, Germany). Solutions were prepared with a concentration of 17.5 mg ml−1 in chlorobenzene, stirred overnight at 80 °C, and then cooled down to room temperature before spin coating. The choice of chlorobenzene as the solvent is motivated by its predominant use in device preparation for this blend system.46 Thin films were spin coated at 3000 rpm for 60 seconds on suitable substrates: glass for UV-vis spectroscopy, KBr for FTIR spectroscopy, SiOx/Si for AFM, and glass/ITO for XPS and UPS. The glass and glass/ITO substrates were cleaned with acetone and isopropanol for 20 minutes each in an ultrasonic bath and then treated with UV-ozone for 20 minutes. KBr substrates, diameter 13 mm (Thermo Fisher Scientific, USA) were used as received, and n-type SiOx/Si(001) substrates were cleaned using a standard RCA method without the final hydrofluoric acid etching step.49 Intentional photodegradation was done by exposing films under a solar simulator (AM 1.5) (Sol2A, model 94022A, Oriel Instruments) in air. For UV-vis spectroscopy the films were exposed for 10 min, 2 hours, 11 hours, 20 hours, and 30 hours, for FTIR, 11 hours and 30 hours, and for XPS and UPS for 2 hours and 30 hours, respectively.2.2 Experimental methods
UV-vis absorption spectra were collected in transmission mode in the range 300–900 nm with a step size of 1 nm, using a Cary 5000 UV-vis-NIR spectrophotometer (Agilent Technologies, USA), equipped with Cary WinUV 6.1 software. Fourier-transform infrared (FT-IR) spectra were recorded in transmission mode in the range 600–4000 cm−1 with a step size of 4 cm−1, using an INVENIO S spectrometer (Bruker, France) purged with dry air, working with OPUS 8.5 software. Morphology and thickness measurements were performed using a Nanoscope 8 Multimode AFM (Bruker, France), in tapping mode with a Si tip (Model: RTESPA-300) from Bruker. The thickness of the spin-coated films on SiOx/Si substrates was measured by scanning the AFM tip across a scratch in the film.In-house XPS and UPS measurements were done in a UHV system (ÓMICRON, Germany) using a non-monochromated Al Kα X-ray source (PSP Vacuum Technology-TX400/2, UK), operated at 150 W, and a He I UV-source (HIS 13-FOCUS, Germany), respectively. A Scienta SES 100 electron analyzer was used with pass energies of 50 eV and 2 eV for XPS and UPS, respectively. For secondary electron cutoff (SECO) measurements by UPS, a negative 4.55 V bias was applied to the sample holder. The pressure was 1 × 10−9 mbar during XPS and 2 × 10−7 mbar during UPS measurements. For in-house XPS measurements, the binding energy was referenced to the Fermi level (Ef) as measured from a sputtered Au sample. High-resolution core-level XPS was performed at the FlexPES50 beamline at the synchrotron facility MAX IV in Lund, Sweden, using de-focused light and a Scienta DA-30L (W) electron analyzer at normal emission with pass energy 50 eV. The photon energies are given in Fig. S9 (ESI†). The base pressure was 3 × 10−10 mbar. Spectra were fitted with a combination of 30% Lorentzian and 70% Gaussian-shaped peaks using CasaXPS.51 The S 2p3/2 and S 2p1/2 components were fitted with a fixed peak separation of 1.18 eV, maintaining a height ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, and equivalent full width at half maximum (FWHM).52 A Shirley background subtraction was used for quantifications.53
:1, and equivalent full width at half maximum (FWHM).52 A Shirley background subtraction was used for quantifications.53
Density functional theory calculations for Y5 and PF-Y5 model structures were performed for aiming to facilitate the peak assignment. For Y5, the model structure consists of a full Y5 conjugate backbone but with alky side chains reduced to methyl. The PF5-Y5 model structure consists of a monomer with long alkyls replaced by methyl groups. After a geometry optimization, the core level binding energies were obtained by approximating it to the core-orbitals energy absolute values obtained from the self-consistent field calculation. The calculations were performed at the M06/6-311G**54,55 theory level using a periodic continuum model SMD and chlorobenzene (ε = 5.70) as a solvent to mimic the dielectric environment of the materials. Due to the intrinsic limitations of DFT and the minimalistic model structures used for the calculations, a global (rigid) shift of calculated core orbital energies was needed to adjust it to the experimental data. All the calculations were performed using the Gaussian16 (Rev C.01)56 package at the Tetralith supercomputer funded by the National Academic Infrastructure for Supercomputing in Swedish (NAISS).
3. Results and discussion
Fig. 1(a)–(d) represents the molecular structures of PBDB-T, Y5, PF5-Y5, and PYT, respectively. As indicated in the figure, the polymer acceptors PF5-Y5 and PYT consist of alternating electron-deficient Y5 units and donor moieties, thienyl-benzodithiophene (BDT-T) and thiophene, respectively. The UV-Vis absorption spectra of spin-coated films of PBDB-T, Y5, PF5-Y5, and PYT on glass substrates are shown in Fig. 1(e)–(h), respectively. The spectra are collected after exposure to one-sun-equivalent illumination (AM 1.5) in ambient conditions for different lengths of time from 10 min to 30 hours. The spectrum of unexposed PBDB-T shows the typical double absorption maximum at 580 nm and 620 nm. The spectra of unexposed Y5, PF5-Y5, and PYT films exhibit absorption maxima at 801 nm, 780 nm, and 796 nm, respectively. Both absorption peaks at 580 nm and 620 nm, as well as the smaller peaks at 361 nm and 421 nm in the PBDB-T spectrum, decrease in intensity with increasing exposure time (Fig. 1e). The 620 nm peak decreases faster than the 580 nm peak and cannot be resolved after exposure times longer than 2 hours, resulting in a broader absorption band with its maximum around 586 nm.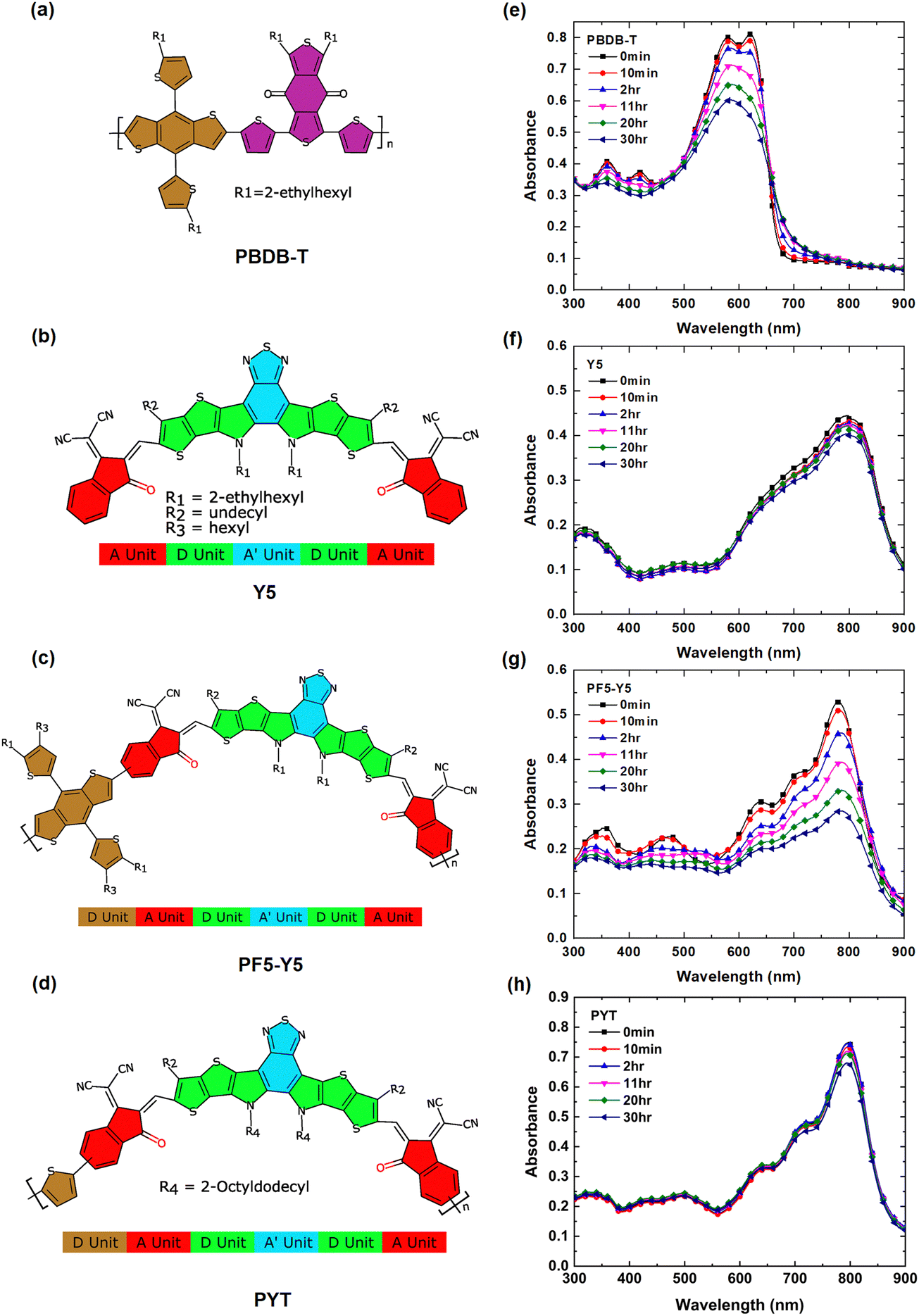 | ||
| Fig. 1 Molecular structures of (a) PBDB-T, (b) Y5, (c) PF5-Y5, and (d) PYT. UV-vis spectra of (e) PBDB-T, (f) Y5, (g) PF5-Y5, and (h) PYT films exposed in ambient air to AM 1.5 illumination for 0 min, 10 min, 2 hours, 11 hours, 20 hours, and 30 hours. | ||
The enhanced absorption in the tail states could be possibly due to the presence of sub-energy gap states and energetic disorder in PBDB-T upon photodegradation.39,40 In the PF5-Y5 spectrum, the decrease in absorbance is most prominent for the main absorption peak at 780 nm, but also the absorption bands at shorter wavelengths decrease with increasing exposure time, resulting in a spectrum with less distinct features after long-term exposure. In addition, the main absorption peak of PF5-Y5 does not undergo any significant shift upon degradation for 30 hours, while the absorption onset red-shifts by 21 nm (Fig. S1, ESI†). In contrast, the absorption intensity of the main peak Y5 at 801 nm and PYT at 796 nm films remained nearly unchanged after 30 hours of exposure (decrease less than 10% of the original absorbance). It is important to note that the one-sun-equivalent illumination (AM 1.5) that was used in this study contains high-energy ultraviolet photons. It is known that UV light is the main source of photodegradation and can cause breaking of the double bonds in the π-conjugated system.57,58 The observed photobleaching is interpreted as a loss of chromophores, probably as a consequence of the breaking of double bonds. The UV-vis absorption spectra demonstrate that the PBDB-T and PF5-Y5 films are photobleaching significantly faster than Y5 and PYT films during the first 30 hours of exposure in the presence of light and air. The rate of photobleaching is represented by the slope in the graphs shown in Fig. S2 (ESI†). The remaining absorbance after 30 hours of exposure for Y5, PYT, PF5-Y5, and PBDB-T is 91.5% (801 nm), 90.9% (796 nm), 46.4% (780 nm), and 31.8% (620 nm) of the original absorbance, respectively.
The PBDB-T:Y5 and PBDB-T:PF5-Y5 blend films show similar photobleaching behavior as the single component films, as seen in the UV-Vis absorption spectra shown in Fig. S3 (ESI†). The absorption peak at 626 nm in the PBDB-T:Y5 spectrum is mainly originating from the absorption of the PBDB-T component and it exhibits, as expected, a higher rate of photodegradation than the absorption peak at 782 nm, which originates from the Y5 component. The relative degradation rate of the blend and pure component films is shown in Fig. S2 (ESI†). The results indicate that even when dispersed in a blend film, the absorbance of Y5 remains at 90% (782 nm), whereas for PF5-Y5, the absorbance remains at 70% (779 nm) after 30 hours of exposure. A comparison of the photobleaching rates shows that PF5-Y5 degrades slower in PBDB-T:PF5-Y5 blend films compared to in the neat films.
To investigate the photodegradation products, IR spectra were measured of PBDB-T, Y5, PF5-Y5, and PYT films spin coated on KBr substrates and prepared under the same conditions as for UV-vis spectroscopy.
In Fig. 2, the FTIR spectra are shown in the range of 1000 cm−1 to 1900 cm−1 for PBDB-T, Y5, PF5-Y5, and PYT films and their evolution with exposure time. The wide range FTIR spectra from 1000 cm−1 to 3600 cm−1 are shown in Fig. S4 (ESI†). In the PBDB-T spectrum strong peaks can be identified in the 2850–2950 cm−1 range (CH2 and CH3 stretch vibrations from the side chains), a sharp peak at 1649 cm−1 (carbonyl stretch of the quinone group), a broad absorption band centered at 1455 cm−1, as well as four sharp peaks in the 1400–1100 cm−1 range for C–H rocking and scissoring modes.22,59–63 With increasing exposure time, the intensity of the quinone carbonyl stretch drops and a new distinct peak develops in the carbonyl region, centered at 1712 cm−1, indicative of the formation of a carboxylic acid or ketone, and a tail reaching until 1800 cm−1, as well as a weak shoulder at the low wavenumber side of the original carbonyl peak. The contribution to the tail towards the higher wavenumbers could indicate the additional formation of acyl anhydrides, (RCO2)2O, on two neighboring carbons in the conjugated backbone, as has been observed in photo-oxidized PC60BM.22 Such a contribution to a high wavenumber tail in the carbonyl region is even more pronounced for PF5-Y5 (Fig. 2c), as will be discussed below. Most other peaks in the 1400–1100 cm−1 region decrease in relative intensity and only a minor effect is observed on the CH2 and CH3 stretch vibrations after long exposure times (Fig. S4, ESI†). The result indicates that the quinone and most of the backbone are affected by the photooxidation of PBDB-T, breaking some bonds and forming several new types of carbonyl bonds.
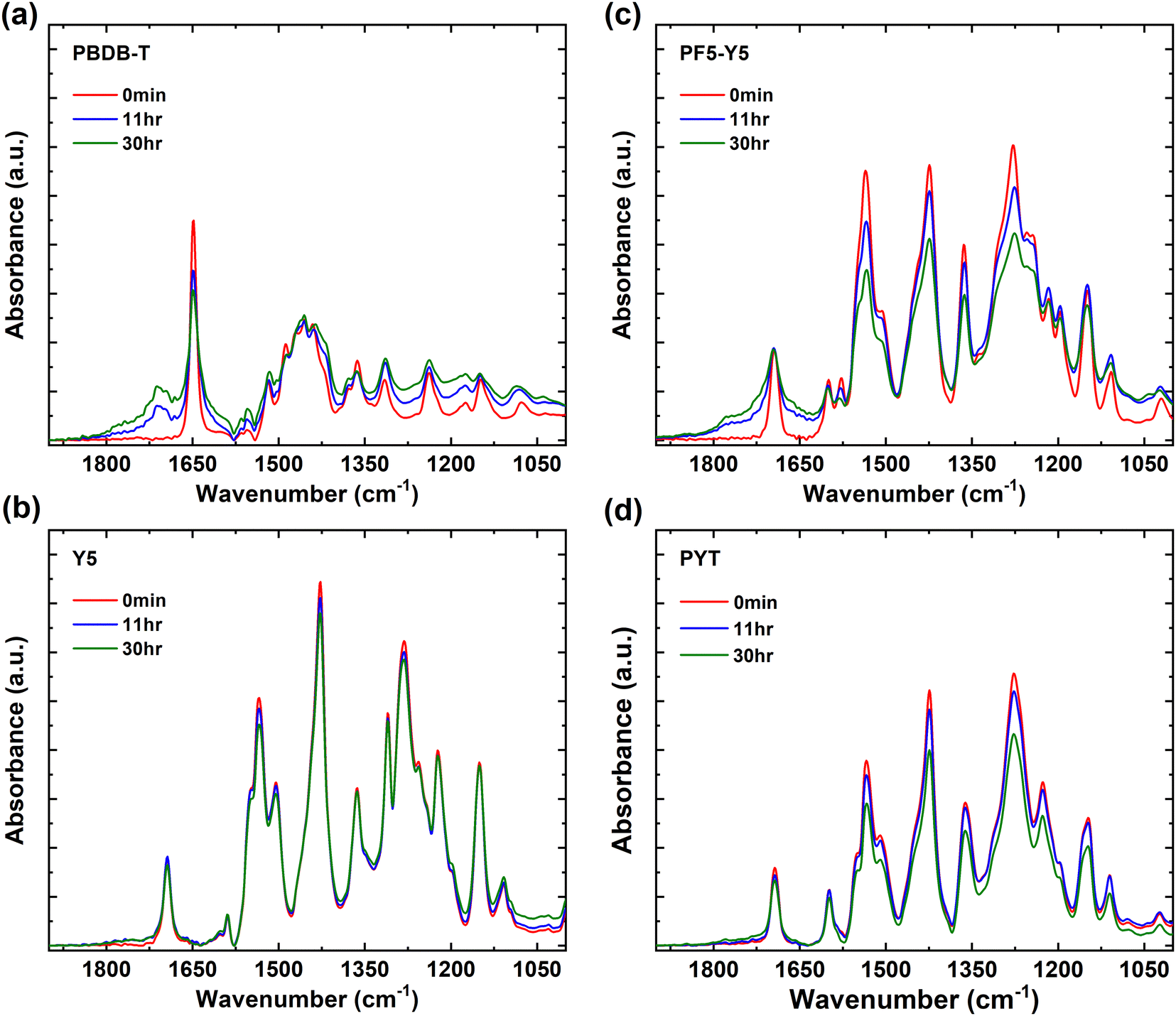 | ||
| Fig. 2 FTIR spectra of (a) PBDB-T, (b) Y5, (c) PF5-Y5, and (d) PYT films, unexposed (0 min) and after exposure to light in air for exposure times of 11 hours and 30 hours. | ||
In the Y5 and PYT spectra (Fig. 2b, d and Fig. S4, ESI†), we identify the CH2 and CH3 stretch vibrations in the 2850–2950 cm−1 region, the CN stretch of the cyano groups at 2216 cm−1, the carbonyl stretch at 1693 cm−1, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretch vibrations at 1535 cm−1 and 1428 cm−1, as well as five sharp peaks in the 1400–1100 cm−1 range.60–63 Interestingly, no new peaks appear in the IR spectra of Y5 and PYT upon exposure to light. For PYT, the peak intensities in the 1500–1100 cm−1 region decrease slightly at long exposure times, but for Y5 no significant changes are observed even after 30 hours of exposure.
C stretch vibrations at 1535 cm−1 and 1428 cm−1, as well as five sharp peaks in the 1400–1100 cm−1 range.60–63 Interestingly, no new peaks appear in the IR spectra of Y5 and PYT upon exposure to light. For PYT, the peak intensities in the 1500–1100 cm−1 region decrease slightly at long exposure times, but for Y5 no significant changes are observed even after 30 hours of exposure.
As expected, the main peaks in the PF5-Y5 spectrum overlap with the corresponding absorption peaks in the Y5 and PYT spectra, apart from a few small differences around 1278 cm−1, where several peaks observed in the Y5 spectrum merge into a broader absorption band in the PF5-Y5 spectrum. After exposure of PF5-Y5 the spectrum exhibits increased absorption between 1600 cm−1 and 1820 cm−1, growing as shoulders on both sides of the original carbonyl peak with increasing exposure time, while the intensity of the original carbonyl peak remains unchanged. Like in the case of PBDB-T, the high wavenumber contribution could be indicative of the formation of acyl anhydrides, (RCO2)2O, whose asymmetric and symmetric stretch CO vibrations are found at about 1750 ± 5 cm−1 and 1820 ± 5 cm−1, respectively. Additionally, the weak increment of the peak at 1043 cm−1 could be a contribution from the anhydride C–O stretch, expected between 1060 and 1035 cm−1, or alternatively the formation of oxidized sulfur compounds.64,65 Most other absorption peaks in the 1600–1100 cm−1 region decrease in intensity, as does also the cyano CN stretch peak at 2216 cm−1. The peak intensity ratios (30 hours/0 min) of PF5-Y5 and PYT (Fig. 2c and d) in the range 1100–1600 cm−1 are compared in Fig. S5 (ESI†) and are on average 0.80 for PYT (80% of the original intensity remains after 30 hours exposure) and about 0.75 for PF5-Y5, strengthening the faster degradation of PF5-Y5 as compared to PYT, also in this region. Additionally, with exposure of PF5-Y5, an increased IR absorbance over a broad range is observed in the high wavenumber region (3000 cm−1 to 3800 cm−1). While this could include contributions from hydroxyl groups (3218 cm−1), it can, however, not solely explain the increase of the background absorption.61,62 The FTIR spectral changes demonstrate that PF5-Y5 photo-oxidizes by forming new carbonyls and sulfur oxides. The photooxidation is accompanied by bond breaking in the whole molecule, affecting also the cyano groups and the side chains. This stands in strong contrast to the FTIR spectra of Y5 and PYT films, which show that Y5 and PYT are significantly more resistant to photooxidation than PBDB-T and PF5-Y5 films. This is in agreement with the weaker or nearly absent photobleaching observed in the UV-vis absorption spectra of Y5 and PYT films. However, when comparing the two copolymers PF5-Y5 and PYT, it is prominent that the BDT-T moiety in the copolymer PF5-Y5 increases the susceptibility to photooxidation of the Y5 moiety, while the thiophene co-monomer in PYT does not have such an effect. This may be related to the fact that the thiophene in PYT is directly bound to the Y5 moiety, which is a strong electron withdrawing group, while the thiophenes in the BDT-T BDT unit of PF5-Y5 are attached to the benzodithiophene (BDT) fused ring, which is an electron donating group. Therefore, the thiophene in PYT will be more electron deficient than the thiophenes in BDT-T, and can therefore be expected to be less vulnerable for oxidation.
While the BDT-T unit, which is present both in PBDB-T and in PF5-Y5, can be a site of photooxidation in these copolymers, we cannot exclude that the other co-monomer (Y5 in the case of PF5-Y5) is also taking part in the photooxidation reaction. Indeed, the long wavelength region of the UV-vis spectrum of PF5-Y5 is a signature of the Y5 monomer and it strongly photobleaches. The BDT-T unit can affect the distribution of charges in the polymer backbone, which in turn can affect the reactivity of specific sites for photooxidation.66 The FTIR spectra for the blend films of PBDB-T:Y5 and PBDB-T:PF5-Y5 are shown in Fig. S6 (ESI†), for different durations of exposure to light in air.
When comparing Y5 with the two copolymers, one could at first sight believe that the reason for its surprising photostability is related to the ability of the small molecule to pack in a film and form dense aggregates. Such dense packing could hinder the diffusion of the degradation agents, such as O2, into the film, and hence decrease the degree of photooxidation. To examine the morphology and surface roughness of the PBDB-T, Y5, PF5-Y5 and PYT films, and blends PBDB-T:Y5 and PBDB-T:PF5-Y5, AFM was used. AFM images of the films indeed show that Y5 films are significantly rougher, implying that the above-mentioned aggregation may indeed take place and hinder the formation of a smooth film (Fig. S7, ESI†).
As shown in Fig. S7 (ESI†), the morphology of the pure components showed a slight decrease in roughness after light exposure, while in the case of the PBDB-T:Y5 and PBDB-T:PF5-Y5 blend films a slight increase in roughness was observed. The pure PBDB-T film shows a smooth surface with elongated grain-like structures of the order of 10–20 nm in diameter and the roughness decreased from 1.5 nm to 1.0 nm after 30 hours of exposure. Comparatively, the Y5 film exhibited a significantly rougher surface, compared to the polymer films with larger irregularly shaped structures of 200–500 nm in size. After being exposed for 30 hours, the surface roughness of Y5 reduced from 7.5 nm to 5.5 nm. For PF5-Y5 the film surface was the smoothest and a network of small round structures of 10–20 nm could be observed. Its roughness decreased from 0.82 nm to 0.61 nm. For PYT the film is slightly rougher and features are larger (20–50 nm) and less distinct than in PF5-Y5 films. Its roughness decreased from 2.1 nm to 1.8 nm. The PBDB-T:Y5 blend film is much smoother than the pure Y5 film and shows a grain-like structure that looks similar to pure polymer films. The roughness values increase very slightly from 2.1 to 2.3 nm. The PBDB-T:PF5-Y5 blend film is only slightly rougher than the PF5-Y5 film and shows similar elongated surface structures than the PBDB-T film. Its roughness increases also here very slightly from 0.9 to 1.1 nm. In conclusion, film roughness is not significantly affected by 30 hours exposure to light in air.
To further understand the effect of photooxidation on the chemical composition of the films, wide survey spectra and C 1s, O 1s, S 2p, and N 1s core level XPS spectra were measured in-house for PBDB-T, Y5, and PF5-Y5 films on ITO substrates. The survey scans are shown in Fig. S8 (ESI†). The approximate atomic percentages of C, O, S, and N in these films, extracted from these survey scans, are listed in ESI,† Table S3, along with some remnant traces of In from the substrate ITO.
In order to accurately assign the components in the XPS spectra, high-resolution core level XPS spectra of the unexposed Y5 and PF5-Y5 films were acquired at the soft X-ray spectroscopy beamline FlexPES at the MAX IV synchrotron. These spectra are presented in Fig. S9 (ESI†), along with the calculated core-orbitals energies, aiming at confirming the assignments. The full description of the core-orbitals for each molecule can be found in Tables S1 and S2 (ESI†).
Fig. 3 shows the C 1s and S 2p spectra of unexposed and 30 hours exposed samples. Fig. 3a and b shows the C 1s spectra of PBDB-T for 0 hours and 30 hours. The peak at 285.1 eV of the C 1s spectrum corresponds to carbon from the side chain and aromatic ring. The peak at 286.6 eV has a contribution from C–S and CO moieties. Upon photodegradation for 30 hours, the main peak decreases in intensity (see Fig. S10, ESI†) and shifts to higher binding energy at 285.2 eV, and a new feature (Fig. 3b, green component) arises at 288.6 eV. This high binding energy component is likely due to the formation of even more strongly oxidized carbon, such as anhydride, confirming the changes in the high wavenumber tail of the carbonyl peak in the FT-IR spectra.67 The formation of this anhydride was also observed in photo-oxidized PC60BM.22 The peak at 286.6 eV also shifts to 0.1 eV towards the higher binding energy of C–S bond upon photodegradation. The O 1s spectra of PBDB-T are shown in Fig. S10c (ESI†) for 0 hours, 2 hours, and 30 hours. The increment in the O 1s main peak intensity at 532.4 eV confirms the addition of oxygen on the PBDB-T polymer surface upon photodegradation, in agreement with the formation of new carbonyl groups observed in FTIR and the new component in the C 1s spectra.68,69 It is worth noting that no change is observed in the C 1s spectra of Y5 (Fig. 3e and f). Fig. 3i and j show the C 1s spectra of PF5-Y5. The peak at 286.2 eV contains contributions from CO, CN, and C–S, as shown by the synchrotron measurements and confirmed by DFT calculations (see Fig. S9 and Table S2, ESI†). Regarding the photodegradation, the C 1s spectra exhibit similar behavior as those of PBDB-T. For PF5-Y5 the O 1s spectrum (Fig. S10j, ESI†) changes in a similar way to that of PBDB-T, namely a severe increase of the O 1s intensity and a broadening after 30 hours. Fig. 3c and d show the S 2p spectra for PBDB-T films exposed for 0 hours and 30 hours, respectively. For PBDB-T samples, the doublets S 2p1/2 and S 2p3/2 are observed at 165.31 eV and 164.13 eV and assigned to the sulfur in the thiophene (C–S).67 Upon photodegradation for 30 hours, the intensities of the S 2p1/2 and S 2p3/2 peaks decrease and the peaks shift by 0.29 eV towards the higher binding energy (S 2p1/2 and S 2p3/2 at 165.60 eV and 164.42 eV, respectively) while an additional doublet appears at 169.30 eV and 169.12 eV. The chemical shift of the S 2p spectra towards higher binding energy indicates that the S 2p core level electron gets bound stronger, which is in agreement with an electron-deficiency on the S upon oxidation of the sulfur. The appearance of the new peak at the higher binding energy indicates the formation of oxidized sulfur (SOx), such as sulphones, upon photodegradation. Again, no change was observed in the S 2p spectra even after 30 hours of light exposure of the Y5 film.
 | ||
| Fig. 3 In-house C 1s (rows 1 and 2) and S 2p XPS (rows 3 and 4) spectra of PBDB-T (a–d), Y5 (e–h), and PF5-Y5 (i–l) films illuminated for 0 hours (rows 1 and 3) and 30 hours (rows 2 and 4) under AM 1.5 solar simulator light in ambient conditions. | ||
For the PF5-Y5 film, once more the deconvolution of the spectrum acquired at the synchrotron facility, together with calculation, assisted the assignment of the two doublets (see Fig. S9 and Table S2, ESI†). The doublet of S 2p1/2 and S 2p3/2 at 164.92 eV and 163.74 eV originates from the thiophene rings (C–S) in the molecule (Fig. 3k and l).67 The decrease in intensity and chemical shift by 0.2 eV towards higher binding energy (165.12 eV and 163.94 eV) upon photodegradation indicate bond breakage in one of the thiophenes, possibly in the BDT-T moiety of the PF5-Y5. An additional feature appears at higher binding energy, at 168.92 eV and 167.74 eV, from the oxidized sulfur (SOx), for instance sulphone, as in the case of PBDB-T, which is also observed in the FTIR spectra at 1043 cm−1.64,65 The additional doublet at higher binding energy at 165.66 eV and 164.48 eV is from N–S in the benzothiadiazole group.33,70 Upon exposure, a minor change is observed in the peaks of N–S at 165.86 eV and 164.68 eV. These results conclude that upon photodegradation the benzothiadiazole core group is not affected, whereas the thiophenes are involved in the photooxidation reaction. This is particularly prominent in the case of PBDB-T and PF5-Y5, while Y5 remains intact, confirming that the degradation is mainly due to the presence of the BDT-T moiety. Based on the present evidence from XPS we cannot conclude if the reaction site(s) for photooxidation of PF5-Y5 consist uniquely of the thiophenes in the BDT-T moiety or also involve thiophenes in the Y5 moiety. In a systematic study by the Ratcliff group,40,64 the prevalent degradation mechanisms of alkylthienyl-benzodithiophene based push–pull polymers were investigated and found to consist of alkyl chain oxygen addition and sulfur oxidation, which is in agreement with our observations for PBDB-T photooxidation. As expected, and in agreement with the FTIR spectroscopy results, no major change was observed in the N 1s XPS spectra upon exposure to the Y5 and PF5-Y5 films (Fig. S10g and k, respectively, ESI†). This is another indication that the core acceptor benzothiadiazole group is unaffected upon photodegradation.
In the O 1s spectra of Y5 (Fig. S10f, ESI†), the intensity of the main peak does not change upon exposure. This is remarkably different from the strong increase in peak intensities observed in the O 1s spectra of PF5-Y5 and PBDB-T films (Fig. S10j and c, respectively, ESI†). The only change observed in the O 1s spectrum in Fig. S10f (ESI†) concerns a weak secondary peak at lower binding energy for the unexposed sample. This peak is most likely originating from the ITO substrate. Indeed, as also confirmed by an In 3d signal seen in the survey scan (Fig. S8, ESI†), the coverage of Y5 on the ITO substrate is probably incomplete.
To understand the effect of photodegradation on the frontier energy levels, we now determine the ionization potential, i.e., the position of the highest occupied molecular orbital (HOMO) with respect to vacuum, and the work function from the valence band (VB) spectra of PBDB-T, Y5, and PF5-Y5, measured by UPS. The work function values are determined from the secondary electron cut-off (SECO) of the UPS spectra, presented in Fig. S11 (ESI†), and are given in Table S5 (ESI†). Fig. 4a shows the valence band spectrum of PBDB-T upon 0 hours, 2 hours, and 30 hours exposure. The estimated positions of the valence band onset of PBDB-T, measured with respect to the Fermi level (EF), are 1.06 eV, 0.61 eV, and 0.57 eV for 0 hours, 2 hours, and 30 hours, respectively. This simultaneous shift of the VB onset towards the Fermi level by 0.45 eV and the 0.5 eV increase in work function, observed after 2 hours of exposure, can be understood as indicative of the development of a strong upward band bending at the film surface. We note here that this band bending causes a (global) shift of all core levels towards higher binding energies, which was corrected for in the presented XPS spectra. After 30 hours of exposure, the work function has further increased (Fig. S11, ESI†) and the density of states (DOS) of the VB has drastically decreased, so that the VB onset is hard to determine from the UPS spectrum in Fig. 4a. From the energy level diagrams of PBDB-T, drawn in Fig. 4d, we can conclude that after 2 hours of exposure, the ionization potential does not change significantly, while at longer exposure times (30 hours), the ionization potential of PBDB-T increases from 5.09 eV to approximately 5.47 eV, which is indicative of surface dipoles.
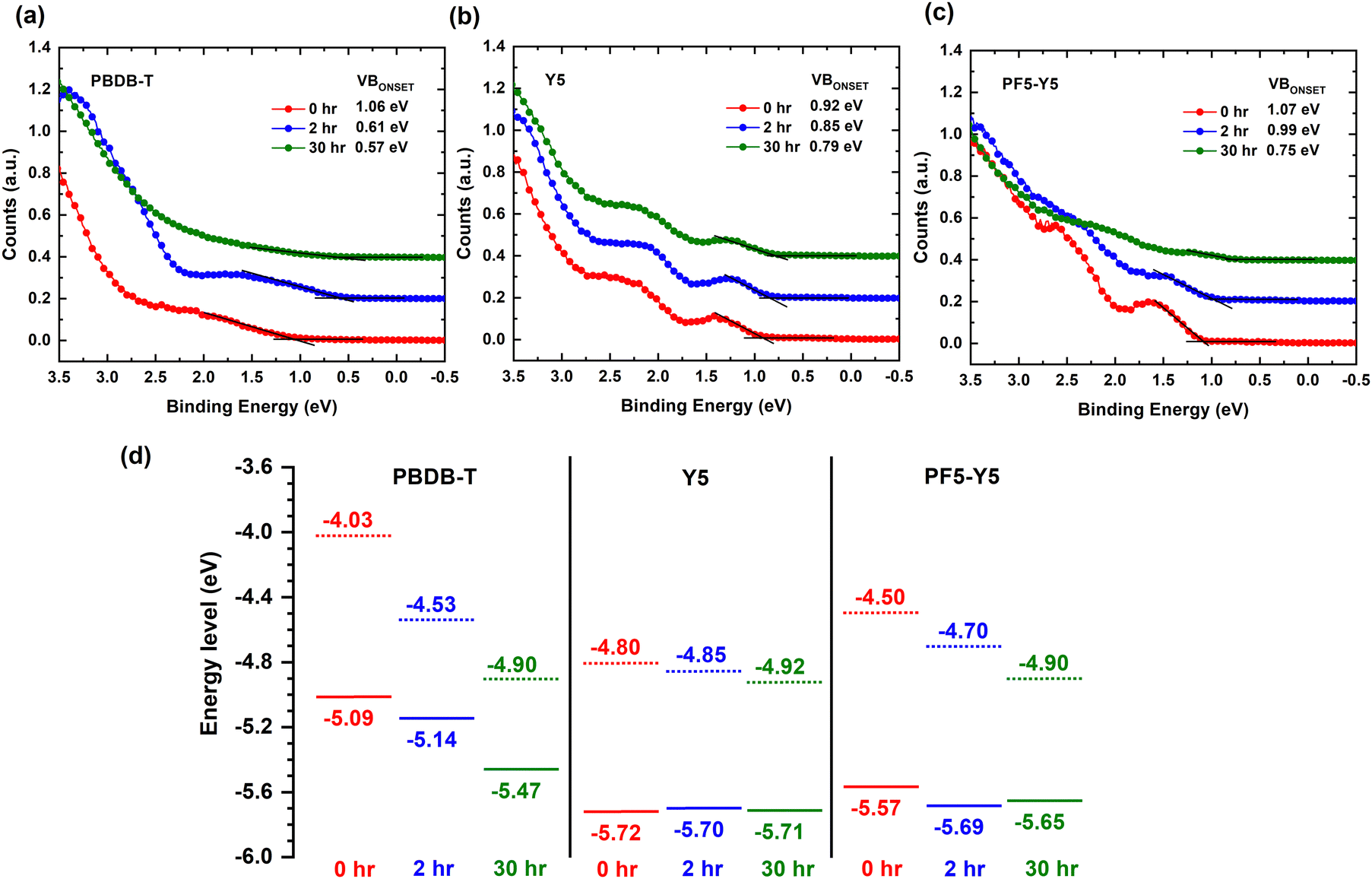 | ||
| Fig. 4 Evolution of the valence band spectra, measured by UPS, of (a) PBDB-T, (b) Y5, and (c) PF5-Y5 upon exposure for 0 hours, 2 hours, and 30 hours. (d) Energy level diagrams (drawn with respect to a constant vacuum level) of PBDB-T, Y5, and PF5-Y5, constructed using the work function and valence band onset values extracted from the UPS spectra (a)–(c). | ||
For Y5, the VB onsets after 0 hours, 2 hours, and 30 hours of exposure, measured with respect to EF, are 0.92 eV, 0.85 eV, and 0.79 eV, respectively (Fig. 4b). Together with the increase in the work function of Y5 by 0.12 eV (Fig. S11, ESI†), this 0.13 eV decrease of VB onset position with respect to EF yields an unchanged ionization potential for Y5 after 30 hours of exposure (see Fig. 4d). The energy level shifts for Y5 are much smaller than for PBDB-T, in agreement with the lack of significant chemical changes in composition found in the FTIR spectra and XPS core level spectra for Y5. In the case of PF5-Y5 (Fig. 4c), the valence band onsets, measured with respect to EF, are found at 1.07 eV, 0.99 eV, and 0.75 eV for the 0 hours, 2 hours, and 30 hours exposed films, respectively. PF5-Y5 films follow a similar trend as PBDB-T, but with a smaller shift of VB onset towards EF of 0.08 eV and 0.32 eV after 2 hours and 30 hours exposure, respectively, while exhibiting a corresponding 0.2 eV and 0.4 eV increase in work function. This is indicative of a combination of a weak upward band bending and the formation of surface dipoles. While the ionization energy for Y5 remains the same, it changes slightly from 5.57 eV to 5.65 eV for PF5-Y5 upon photodegradation from 0 hours to 30 hours. The shape of the VB spectrum of unexposed PF5-Y5 looks similar to that of Y5. However, the DOS of the VB of PF5-Y5 decreases drastically after 30 hours of exposure, in a similar way as in the case of PBDB-T, making the VB onset hard to determine accurately. This is in line with the results from UV-vis, FTIR, and XPS spectroscopy that show that PBDB-T and PF5-Y5 photobleach and photooxidize faster than Y5. While the DOS of the occupied states are strongly affected by photodegradation for PF5-Y5 and PBDB-T, the HOMO position is only significantly affected for the donor polymer. We note that after exposure, the energy level positions of PF5-Y5 and Y5 become quite similar, as do their UV-vis spectra. This could possibly be an indication of the breaking of conjugation in PF5-Y5 between the Y5 and the BDT-T donor unit, making the electronic structure of the degraded PF5-Y5 copolymer more similar to that of Y5, at the same time as the photooxidation of the BDT-T unit leads to a decreased DOS of VB, in a similar way to PBDB-T.
4. Conclusions
In this study, we have investigated the photodegradation of films of the donor polymer PBDB-T, the polymeric acceptors PF5-Y5 and PYT, and the small molecule acceptor Y5 in ambient conditions under AM 1.5 illumination. We found that Y5 and PYT films demonstrate excellent photostability, while fast photobleaching is observed for PBDB-T and PF5-Y5 films in the visible spectral range (300–900 nm). The IR spectra of Y5 and PYT films show no signatures of oxidation products after exposure. The PBDB-T and PF5-Y5 undergo photooxidation leading to the formation of new carbonyl bonds, as observed in the transmission IR spectra, which is in agreement with the increased oxygen and decreased carbon content in the film surface measured by XPS and the appearance of a new high BE component in the C 1s core level spectra. Moreover, the S 2p core level spectra of both polymers show that photooxidation also happens on the thiophenes (C–S), while for Y5 neither the C 1s nor the S 2p spectra show any changes, even after 30 hours exposure. This could suggest that the thiophene units, including those in the BDT-T unit that is present both in PBDB-T and in PF5-Y5 but absent in Y5, are affected by photooxidation. The N 1s core level spectra confirm that the benzothiadiazole in the Y5 core group (N–S) of PF5-Y5 is not affected upon photooxidation. The UPS results reveal that the PBDB-T develops a strong band bending after 2 hours exposure, while 30 hours exposure leads to an increased ionization potential, associated with surface dipoles. PF5-Y5 undergoes a similar but weaker growth of band bending and surface dipoles, compared to PBDB-T, while no such changes are observed for the Y5 film. For both PBDB-T and PF5-Y5 films, the DOS of the VB is strongly affected by the degradation. Only for the donor polymer the HOMO position is significantly shifted upon photodegradation, while the ionization potential of Y5 and PF5-Y5 is not significantly affected. The results suggest that the BDT-T moiety affects the stability of PBDB-T and PF5-Y5 negatively. Furthermore, we have shown that the replacement of the BDT-T moiety in PF5-Y5 with a thiophene moiety in PYT strongly improves the intrinsic material stability. Hence, we conclude that the presence of the BDT-T moiety accelerates the photodegradation process. By contributing to a better understanding of the role of specific moieties, like BDT-T, on the photostability of conjugated copolymers, our results may inspire the design of new donor and acceptor molecules for use in organic solar cells with increased operational lifetime.Author contributions
ZG and EW designed and synthesized the polymerized small molecule acceptors. SP prepared all samples, carried out the degradation, measured absorption and IR spectroscopy and wrote the first draft of the manuscript. SS measured the in-house XPS and UPS spectra. EM, LE and CFNM measured the high-resolution XPS spectra at MAX IV. CFNM prepared the calculated XPS spectra. CFNM, LE, EM, MA, and EW supervised the work and contributed to the discussions and interpretation of the results and to the writing of the manuscript. All authors contributed to the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors have no conflicts of interests.Acknowledgements
The authors would like to thank Dr Hanmin Zhang for contributions to the analysis of XPS and UPS results and Dr Leandro R. Franco for fruitful discussions. EM is grateful to Dr Andreas Opitz for sharing the beamtime on the FlexPES beamline at MAX IV Laboratory under Proposal 20220348 and the FlexPES beamline staff for their assistance. Research conducted at MAX IV, a Swedish national user facility, is supported by the Swedish Research Council under contract 2018-07152, the Swedish Governmental Agency for Innovation Systems under contract 2018-04969, and Formas under contract 2019-02496. MA and CM thank the National Academic Infrastructure for Supercomputing in Sweden (NAISS) and the National Supercomputer Centre (NSC) at Linköping University, partially funded by the Swedish Research Council through Grant Agreement No. 2022-06725 and 2018-05973, for providing the computational resources. EM acknowledges the Swedish Energy Agency (contract 48598-1) and the Swedish Research Council (contract 2021-04798) for financial support of the project. EW acknowledges the Swedish Research Council (contract 2019-04683), the Swedish Research Council Formas (contract 2020-01201), and the Swedish Energy Agency (contract P2021-90067) for financial support. The authors are grateful to the Knut and Alice Wallenberg Foundation (grant number 2016.0059) for the financial support of their collaborative work.References
- K. A. Mazzio and C. K. Luscombe, The future of organic photovoltaics, Chem. Soc. Rev., 2015, 44, 78–90 RSC.
- O. Inganäs, Organic Photovoltaics over Three Decades, Adv. Mater., 2018, 30, 1800388 CrossRef.
- G. Li, R. Zhu and Y. Yang, Polymer solar cells, Nat. Photonics, 2012, 6, 153–161 CrossRef CAS.
- Z. Hu, J. Wang, X. Ma, J. Gao, C. Xu, K. Yang, Z. Wang, J. Zhang and F. Zhang, A critical review on semitransparent organic solar cells, Nano Energy, 2020, 78, 105376 CrossRef CAS.
- Y. Li, G. Xu, C. Cui and Y. Li, Flexible and Semitransparent Organic Solar Cells, Adv. Energy Mater., 2018, 8, 1701791 CrossRef.
- R. Søndergaard, M. Hösel, D. Angmo, T. T. Larsen-Olsen and F. C. Krebs, Roll-to-roll fabrication of polymer solar cells, Mater. Today, 2012, 15, 36–49 CrossRef.
- X. Zhao, C. Yao, T. Liu, J. C. Hamill Jr, G. O. Ngongang Ndjawa, G. Cheng, N. Yao, H. Meng and Y. L. Loo, Extending the Photovoltaic Response of Perovskite Solar Cells into the Near-Infrared with a Narrow-Bandgap Organic Semiconductor, Adv. Mater., 2019, 31, 1904494 CrossRef CAS PubMed.
- J. Zhang, H. S. Tan, X. Guo, A. Facchetti and H. Yan, Material insights and challenges for non-fullerene organic solar cells based on small molecular acceptors, Nat. Energy, 2018, 3, 720–731 CrossRef CAS.
- S. Li, W. Liu, C. Z. Li, M. Shi and H. Chen, Efficient Organic Solar Cells with Non-Fullerene Acceptors, Small, 2017, 13, 1701120 CrossRef.
- H. Yao, Y. Cui, R. Yu, B. Gao, H. Zhang and J. Hou, Design, Synthesis, and Photovoltaic Characterization of a Small Molecular Acceptor with an Ultra-Narrow Band Gap, Angew. Chem., Int. Ed., 2017, 56, 3045–3049 CrossRef CAS.
- Y. Lin, J. Wang, Z.-G. Zhang, H. Bai, Y. Li, D. Zhu and X. Zhan, An Electron Acceptor Challenging Fullerenes for Efficient Polymer Solar Cells, Adv. Mater., 2015, 27, 1170–1174 CrossRef CAS.
- J. Hou, O. Inganäs, R. H. Friend and F. Gao, Organic solar cells based on non-fullerene acceptors, Nat. Mater., 2018, 17, 119–128 CrossRef CAS PubMed.
- C. Yan, S. Barlow, Z. Wang, H. Yan, A. K. Y. Jen, S. R. Marder and X. Zhan, Non-fullerene acceptors for organic solar cells, Nat. Rev. Mater., 2018, 3, 18003 CrossRef CAS.
- L. Zhu, M. Zhang, J. Xu, C. Li, J. Yan, G. Zhou, W. Zhong, T. Hao, J. Song and X. Xue, Single-junction organic solar cells with over 19% efficiency enabled by a refined double-fibril network morphology, Nat. Mater., 2022, 21, 656–663 CrossRef CAS PubMed.
- H. Meng, W. Jing, X. Xu, L. Yu and Q. Peng, Nickel(II) Nitrate Hole-Transporting Layers for Single-Junction Bulk Heterojunction Organic Solar Cells with a Record 19.02% Efficiency, Angew. Chem., 2023, 135, e202301958 CrossRef.
- K. Jiang, J. Zhang, C. Zhong, F. R. Lin, F. Qi, Q. Li, Z. Peng, W. Kaminsky, S. H. Jang and J. Yu, Suppressed recombination loss in organic photovoltaics adopting a planar–mixed heterojunction architecture, Nat. Energy, 2022, 7, 1076–1086 CrossRef.
- G. Wu, X. Xu, C. Liao, L. Yu, R. Li and Q. Peng, Improving Cooperative Interactions Between Halogenated Aromatic Additives and Aromatic Side Chain Acceptors for Realizing 19.22% Efficiency Polymer Solar Cells, Small, 2023, 2302127 CrossRef CAS.
- M. Deng, X. Xu, Y. Duan, L. Yu, R. Li and Q. Peng, Y-Type Non-Fullerene Acceptors with Outer Branched Side Chains and Inner Cyclohexane Side Chains for 19.36% Efficiency Polymer Solar Cells, Adv. Mater., 2023, 35, 2210760 CrossRef CAS.
- Y. Zhang, I. D. W. Samuel, T. Wang and D. G. Lidzey, Current Status of Outdoor Lifetime Testing of Organic Photovoltaics, Adv. Sci., 2018, 5, 1800434 CrossRef.
- L. Duan and A. Uddin, Progress in Stability of Organic Solar Cells, Adv. Sci., 2020, 7, 1903259 CrossRef CAS.
- M. Riede, D. Spoltore and K. Leo, Organic Solar Cells-The Path to Commercial Success, Adv. Energy Mater., 2021, 11, 2002653 CrossRef CAS.
- I. E. Brumboiu, L. K. E. Ericsson, V. Blazinic, R. Hansson, A. Opitz, B. Brena and E. Moons, Photooxidation of PC60BM: new insights from spectroscopy, Phys. Chem. Chem. Phys., 2022, 24, 25753–25766 RSC.
- V. Blazinic, L. K. E. Ericsson, I. Levine, R. Hansson, A. Opitz and E. Moons, Impact of intentional photo-oxidation of a donor polymer and PC70BM on solar cell performance, Phys. Chem. Chem. Phys., 2019, 21, 22259–22271 RSC.
- A. Distler, P. Kutka, T. Sauermann, H. J. Egelhaaf, D. M. Guldi, D. Di Nuzzo, S. C. J. Meskers and R. A. J. Janssen, Effect of PCBM on the Photodegradation Kinetics of Polymers for Organic Photovoltaics, Chem. Mater., 2012, 24, 4397–4405 CrossRef CAS.
- T. Heumueller, W. R. Mateker, A. Distler, U. F. Fritze, R. Cheacharoen, W. H. Nguyen, M. Biele, M. Salvador, M. von Delius, H. J. Egelhaaf, M. D. McGehee and C. J. Brabec, Morphological and electrical control of fullerene dimerization determines organic photovoltaic stability, Energy Environ. Sci., 2016, 9, 247–256 RSC.
- E. M. Speller, J. D. McGettrick, B. Rice, A. M. Telford, H. K. H. Lee, C.-H. Tan, C. S. De Castro, M. L. Davies, T. M. Watson, J. Nelson, J. R. Durrant, Z. Li and W. C. Tsoi, Impact of Aggregation on the Photochemistry of Fullerene Films: Correlating Stability to Triplet Exciton Kinetics, ACS Appl. Mater. Interfaces, 2017, 9, 22739–22747 CrossRef CAS.
- L. Duan, Y. Zhang, M. He, R. Deng, H. Yi, Q. Wei, Y. Zou and A. Uddin, Burn-In Degradation Mechanism Identified for Small Molecular Acceptor-Based High-Efficiency Nonfullerene Organic Solar Cells, ACS Appl. Mater. Interfaces, 2020, 12, 27433–27442 CrossRef CAS.
- A. J. Clarke, J. Luke, R. Meitzner, J. Wu, Y. Wang, H. K. Lee, E. M. Speller, H. Bristow, H. Cha and M. J. Newman, Non-fullerene acceptor photostability and its impact on organic solar cell lifetime, Cell Rep. Phys. Sci., 2021, 2, 100498 CrossRef CAS.
- P. Jiang, L. Hu, L. Sun, Z. A. Li, H. Han and Y. Zhou, On the interface reactions and stability of nonfullerene organic solar cells, Chem. Sci., 2022, 13, 4714–4739 RSC.
- Y. Jiang, L. Sun, F. Jiang, C. Xie, L. Hu, X. Dong, F. Qin, T. Liu, L. Hu, X. Jiang and Y. Zhou, Photocatalytic effect of ZnO on the stability of nonfullerene acceptors and its mitigation by SnO2 for nonfullerene organic solar cells, Mater. Horiz., 2019, 6, 1438–1443 RSC.
- F. Qin, W. Wang, L. Sun, X. Jiang, L. Hu, S. Xiong, T. Liu, X. Dong, J. Li, Y. Jiang, J. Hou, K. Fukuda, T. Someya and Y. Zhou, Robust metal ion-chelated polymer interfacial layer for ultraflexible non-fullerene organic solar cells, Nat. Commun., 2020, 11, 4508 CrossRef CAS PubMed.
- S. Park and H. J. Son, Intrinsic photo-degradation and mechanism of polymer solar cells: the crucial role of non-fullerene acceptors, J. Mater. Chem. A, 2019, 7, 25830–25837 RSC.
- S. Yoon, N. Schopp, D. G. Choi, H. Wakidi, K. Ding, H. Ade, H. Vezin, G. N. M. Reddy and T. Q. Nguyen, Influences of Metal Electrodes on Stability of Non-Fullerene Acceptor-Based Organic Photovoltaics, Adv. Funct. Mater., 2023, 2308618 CrossRef.
- W. Yang, Z. Luo, R. Sun, J. Guo, T. Wang, Y. Wu, W. Wang, J. Guo, Q. Wu, M. Shi, H. Li, C. Yang and J. Min, Simultaneous enhanced efficiency and thermal stability in organic solar cells from a polymer acceptor additive, Nat. Commun., 2020, 11, 1218 CrossRef CAS.
- Q. Fan, W. Su, S. Chen, T. Liu, W. Zhuang, R. Ma, X. Wen, Z. Yin, Z. Luo and X. Guo, A non-conjugated polymer acceptor for efficient and thermally stable all-polymer solar cells, Angew. Chem., Int. Ed., 2020, 59, 19835–19840 CrossRef CAS.
- Q. Fan, W. Su, S. Chen, W. Kim, X. Chen, B. Lee, T. Liu, U. A. Méndez-Romero, R. Ma, T. Yang, W. Zhuang, Y. Li, Y. Li, T. S. Kim, L. Hou, C. Yang, H. Yan, D. Yu and E. Wang, Mechanically Robust All-Polymer Solar Cells from Narrow Band Gap Acceptors with Hetero-Bridging Atoms, Joule, 2020, 4, 658–672 CrossRef CAS.
- S. Hultmark, S. H. K. Paleti, A. Harillo, S. Marina, F. A. A. Nugroho, Y. Liu, L. K. E. Ericsson, R. Li, J. Martín, J. Bergqvist, C. Langhammer, F. Zhang, L. Yu, M. Campoy-Quiles, E. Moons, D. Baran and C. Müller, Suppressing Co-Crystallization of Halogenated Non-Fullerene Acceptors for Thermally Stable Ternary Solar Cells, Adv. Funct. Mater., 2020, 30, 2005462 CrossRef CAS.
- S. H. K. Paleti, S. Hultmark, J. Han, Y. Wen, H. Xu, S. Chen, E. Järsvall, I. Jalan, D. R. Villalva, A. Sharma, J. I. Khan, E. Moons, R. Li, L. Yu, J. Gorenflot, F. Laquai, C. Müller and D. Baran, Hexanary blends: a strategy towards thermally stable organic photovoltaics, Nat. Commun., 2023, 14, 4608 CrossRef CAS.
- Y. Wang, J. Luke, A. Privitera, N. Rolland, C. Labanti, G. Londi, V. Lemaur, D. T. W. Toolan, A. J. Sneyd, S. Jeong, D. Qian, Y. Olivier, L. Sorace, J.-S. Kim, D. Beljonne, Z. Li and A. J. Gillett, The critical role of the donor polymer in the stability of high-performance non-fullerene acceptor organic solar cells, Joule, 2023, 7, 810–829 CrossRef CAS.
- M. A. Anderson, A. Hamstra, B. W. Larson and E. L. Ratcliff, Distinguishing photo-induced oxygen attack on alkyl chain versus conjugated backbone for alkylthienyl-benzodithiophene (BDTT)-based push–pull polymers, J. Mater. Chem. A, 2023, 11, 17858–17871 RSC.
- T. Liu, Q. C. Burlingame, M. R. Ivancevic, X. Liu, J. Hu, B. P. Rand and Y. L. Loo, Photochemical Decomposition of Y-Series Non-Fullerene Acceptors Is Responsible for Degradation of High-Efficiency Organic Solar Cells, Adv. Energy Mater., 2023, 13, 2300046 CrossRef CAS.
- Y. Che, M. R. Niazi, R. Izquierdo and D. F. Perepichka, Mechanism of the Photodegradation of A–D–A Acceptors for Organic Photovoltaics, Angew. Chem., Int. Ed., 2021, 60, 24833–24837 CrossRef CAS PubMed.
- Z. X. Liu, Z.-P. Yu, Z. Shen, C. He, T. K. Lau, Z. Chen, H. Zhu, X. Lu, Z. Xie and H. Chen, Molecular insights of exceptionally photostable electron acceptors for organic photovoltaics, Nat. Commun., 2021, 12, 3049 CrossRef CAS.
- J. Yuan, Y. Zhang, L. Zhou, C. Zhang, T. K. Lau, G. Zhang, X. Lu, H.-L. Yip, S. K. So, S. Beaupré, M. Mainville, P. A. Johnson, M. Leclerc, H. Chen, H. Peng, Y. Li and Y. Zou, Fused benzothiadiazole: a building block for n-type organic acceptor to achieve high-performance organic solar cells, Adv. Mater., 2019, 31, 1807577 CrossRef.
- G. Li, X. Zhang, L. O. Jones, J. M. Alzola, S. Mukherjee, L. W. Feng, W. Zhu, C. L. Stern, W. Huang and J. Yu, Systematic merging of nonfullerene acceptor π-extension and tetrafluorination strategies affords polymer solar cells with >16% efficiency, J. Am. Chem. Soc., 2021, 143, 6123–6139 CrossRef CAS.
- Q. Fan, Q. An, Y. Lin, Y. Xia, Q. Li, M. Zhang, W. Su, W. Peng, C. Zhang and F. Liu, Over 14% efficiency all-polymer solar cells enabled by a low bandgap polymer acceptor with low energy loss and efficient charge separation, Energy Environ. Sci., 2020, 13, 5017–5027 RSC.
- Y. Liu, Q. Fan, H. Liu, I. Jalan, Y. Jin, J. V. Stam, E. Moons, E. Wang, X. Lu and O. Inganas, In Situ Optical Spectroscopy Demonstrates the Effect of Solvent Additive in the Formation of All-Polymer Solar Cells, J. Phys. Chem. Lett., 2022, 13, 11696–11702 CrossRef CAS PubMed.
- Z. Genene, J. W. Lee, S. W. Lee, Q. Chen, Z. Tan, B. A. Abdulahi, D. Yu, T. S. Kim, B. J. Kim and E. Wang, Polymer Acceptors with Flexible Spacers Afford Efficient and Mechanically Robust All-Polymer Solar Cells, Adv. Mater., 2022, 34, 2107361 CrossRef CAS PubMed.
- W. Kern, Handbook of semiconductor wafer cleaning technology, Noyes Publication, New Jersey, 1993, pp. 111–196 Search PubMed.
- A. Preobrajenski, A. Generalov, G. Öhrwall, M. Tchaplyguine, H. Tarawneh, S. Appelfeller, E. Frampton and N. Walsh, FlexPES: a versatile soft X-ray beamline at MAX IV Laboratory, J. Synchrotron Radiat., 2023, 30, 831–840 CrossRef CAS PubMed.
- N. Fairley, V. Fernandez, M. Richard-Plouet, C. Guillot-Deudon, J. Walton, E. Smith, D. Flahaut, M. Greiner, M. Biesinger, S. Tougaard, D. Morgan and J. Baltrusaitis, Systematic and collaborative approach to problem solving using X-ray photoelectron spectroscopy, Appl. Surf. Sci. Adv., 2021, 5, 100112 CrossRef.
- J. H. Scofield, Hartree-Slater subshell photoionization cross-sections at 1254 and 1487 eV, J. Electron Spectrosc. Relat. Phenom., 1976, 8, 129–137 CrossRef CAS.
- A. Proctor and P. M. Sherwood, Data analysis techniques in X-ray photoelectron spectroscopy, Anal. Chem., 1982, 54, 13–19 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, Self-consistent molecular orbital methods. A basis set for correlated wave functions, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- A. Classen, T. Heumueller, I. Wabra, J. Gerner, Y. He, L. Einsiedler, N. Li, G. J. Matt, A. Osvet, X. Du, A. Hirsch and C. J. Brabec, Revealing Hidden UV Instabilities in Organic Solar Cells by Correlating Device and Material Stability, Adv. Energy Mater., 2019, 9, 1902124 CrossRef CAS.
- C. H. Peters, I. Sachs-Quintana, J. P. Kastrop, S. Beaupre, M. Leclerc and M. D. McGehee, High efficiency polymer solar cells with long operating lifetimes, Adv. Energy Mater., 2011, 1, 491–494 CrossRef CAS.
- U. Dettinger, H. J. Egelhaaf, C. J. Brabec, F. Latteyer, H. Peisert and T. Chassé, FTIR Study of the Impact of PC[60]BM on the Photodegradation of the Low Band Gap Polymer PCPDTBT under O2 Environment, Chem. Mater., 2015, 27, 2299–2308 CrossRef CAS.
- J. Guo, Y. Wu, R. Sun, W. Wang, J. Guo, Q. Wu, X. Tang, C. Sun, Z. Luo and K. Chang, Suppressing photo-oxidation of non-fullerene acceptors and their blends in organic solar cells by exploring material design and employing friendly stabilizers, J. Mater. Chem. A, 2019, 7, 25088–25101 RSC.
- R. M. Silverstein and G. C. Bassler, Spectrometric identification of organic compounds, J. Chem. Educ., 1962, 39, 546 CrossRef CAS.
- J. Coates, Interpretation of infrared spectra, a practical approach, Encyclopedia of analytical chemistry, 2000, vol. 12, pp. 10815–10837 Search PubMed.
- J. Zhang, J. Guan, Y. Zhang, S. Qin, Q. Zhu, X. Kong, Q. Ma, X. Li, L. Meng, Y. Yi, J. Zheng and Y. Li, Direct Observation of Increased Free Carrier Generation Owing to Reduced Exciton Binding Energies in Polymerized Small-Molecule Acceptors, J. Phys. Chem. Lett., 2022, 13, 8816–8824 CrossRef CAS.
- K. E. Watts, T. Nguyen, B. J. Tremolet de Villers, B. Neelamraju, M. A. Anderson, W. A. Braunecker, A. J. Ferguson, R. E. Larsen, B. W. Larson, Z. R. Owczarczyk, J. R. Pfeilsticker, J. E. Pemberton and E. L. Ratcliff, Stability of push–pull small molecule donors for organic photovoltaics: spectroscopic degradation of acceptor endcaps on benzo[1,2-b:4,5-b′]dithiophene cores, J. Mater. Chem. A, 2019, 7, 19984–19995 RSC.
- M. Manceau, A. Rivaton, J. L. Gardette, S. Guillerez and N. Lemaître, The mechanism of photo- and thermooxidation of poly(3-hexylthiophene) (P3HT) reconsidered, Polym. Degrad. Stab., 2009, 94, 898–907 CrossRef CAS.
- I. Jalan, C. F. N. Marchiori, Z. Genene, A. Johansson, C. M. Araujo, E. Wang, J. Van Stam and E. Moons, Donor–acceptor polymer complex formation in solution confirmed by spectroscopy and atomic-scale modelling, J. Mater. Chem. C, 2023, 11, 9316–9326 RSC.
- Q. Zhang, Y. Chen, X. Liu and M. Fahlman, In situ near-ambient pressure X-ray photoelectron spectroscopy reveals the effects of water, oxygen and light on the stability of PM6:Y6 photoactive layers, J. Mater. Chem. C, 2023, 11, 3112–3118 RSC.
- J. Kettle, Z. Ding, M. Horie and G. Smith, XPS analysis of the chemical degradation of PTB7 polymers for organic photovoltaics, Org. Electron., 2016, 39, 222–228 CrossRef CAS.
- S. Kim, M. A. M. Rashid, T. Ko, K. Ahn, Y. Shin, S. Nah, M. H. Kim, B. Kim, K. Kwak and M. Cho, New Insights into the Photodegradation Mechanism of the PTB7-Th Film: Photooxidation of π-Conjugated Backbone upon Sunlight Illumination, J. Phys. Chem. C, 2020, 124, 2762–2770 CrossRef CAS.
- M. Gora, W. Krzywiec, J. Mieczkowski, E. C. Rodrigues Maia, G. Louarn, M. Zagorska and A. Pron, Alternating copolymers of diketopyrrolopyrrole or benzothiadiazole and alkoxy-substituted oligothiophenes: spectroscopic, electrochemical and spectroelectrochemical investigations, Electrochim. Acta, 2014, 144, 211–220 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma00447g |
| This journal is © The Royal Society of Chemistry 2024 |