
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCabazitaxel-loaded redox-responsive nanocarrier based on D-alpha-tocopheryl-chitosan and hyaluronic acid for improved anti-tumor efficacy in DMBA-induced breast cancer model†
Abhishek
Jha
 a,
Manish
Kumar
a,
Pooja
Goswami
b,
Kanchan
Bharti
a,
Manjit
Manjit
a,
Ashutosh
Gupta
c,
Sudheer
Moorkoth
c,
Biplob
Koch
*b and
Brahmeshwar
Mishra
*a
a,
Manish
Kumar
a,
Pooja
Goswami
b,
Kanchan
Bharti
a,
Manjit
Manjit
a,
Ashutosh
Gupta
c,
Sudheer
Moorkoth
c,
Biplob
Koch
*b and
Brahmeshwar
Mishra
*a
aDepartment of Pharmaceutical Engineering & Technology, Indian Institute of Technology (BHU), Varanasi - 221005, Uttar Pradesh, India. E-mail: bmishra.phe@iitbhu.ac.in; abhishekjha.phe17@itbhu.ac.in; manishkumar.rs.phe19@itbhu.ac.in; kanchan.bharti.rs.phe18@itbhu.ac.in; manjit.rs.phe20@itbhu.ac.in
bGenotoxicology and Cancer Biology Laboratory, Department of Zoology Institute of Science, Banaras Hindu University, Varanasi - 221005, Uttar Pradesh, India. E-mail: biplob@bhu.ac.in; pooja1428@bhu.ac.in
cDepartment of Pharmaceutical Quality Assurance, Manipal College of Pharmaceutical Sciences, Manipal Academy of Higher Education, Manipal - 576104, Karnataka, India. E-mail: ashutoshgupta086@gmail.com; moorkoth.s@manipal.edu
First published on 13th September 2024
Abstract
The study involved the formulation of cabazitaxel loaded D-alpha-tocopheryl succinate/chitosan conjugate (CSVE) and hyaluronic acid (HA) based redox-responsive nanoparticles crosslinked using 3,3′-dithiodipropionic acid (DTPA). The nanoparticle surface was functionalized with cetuximab (Cmab) to give CSVE/HA/DTPA/Cmab NP for EGFR targeted delivery of the payload. The formulations were subjected to particle analysis, morphological assessment, solid-state characterization, and in vitro drug release studies. The results showed cationic, sub-200 nm sized spherical particles with the glutathione-responsive release of cabazitaxel. In vitro studies revealed a marked decrease in the IC50 value, improved cellular uptake, and a superior apoptotic effect. To determine the in vivo efficacy of the formulation, pharmacokinetic assessment, tumor regression analysis, and survival analysis were performed. The nanoparticles showed improved pharmacokinetic and anti-tumor efficacy compared to free cabazitaxel. The prepared nanoparticles demonstrated immense potential in targeted delivery of the payload for enhanced breast cancer therapy.
1. Introduction
Breast cancer is the most prevalent type of cancer accounting for over 25 percent of total cancer cases worldwide. According to GLOBOCAN 2020, there were an estimated 2.3 million new cases of breast cancer globally in 2020 with approximately 685![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths in the same year. The age-standardized mortality rate for breast cancer was reported to be 13.0 per 100000 women and an estimated 5-year prevalence of breast cancer (both sexes, all ages) of around 7.8 million cases.1 This makes breast cancer a significant health concern and underscores the importance of ongoing efforts in the research to understand its prevalence and the development of effective treatment strategies. Radiation therapy and chemotherapy are the traditional therapeutic strategies in case the surgical removal is not feasible. However, poor clinical results, toxic effects, and drug resistance are the major limitations of traditional treatment strategies.
000 deaths in the same year. The age-standardized mortality rate for breast cancer was reported to be 13.0 per 100000 women and an estimated 5-year prevalence of breast cancer (both sexes, all ages) of around 7.8 million cases.1 This makes breast cancer a significant health concern and underscores the importance of ongoing efforts in the research to understand its prevalence and the development of effective treatment strategies. Radiation therapy and chemotherapy are the traditional therapeutic strategies in case the surgical removal is not feasible. However, poor clinical results, toxic effects, and drug resistance are the major limitations of traditional treatment strategies.
Nanoparticles have shown great promise in ameliorating the limitations associated with traditional treatment strategies for breast cancer.2,3 They can utilize the EPR effect to enter the tumor cells.4 Their submicron size improves cellular uptake and facilitates intracellular delivery of therapeutic agents.5,6 To improve their selectivity, the surface of nanoparticles can be functionalized with specific ligands,7,8 to ensure precise intracellular delivery of therapeutics via receptor mediated endocytosis.9,10 To further improve the precise delivery of therapeutics at the target site, nanoparticles responsive to physiological stimuli can be designed.11–14 Recently, redox responsive nanosystems have gained enormous attention. They release their payload selectively in the presence of elevated levels of reducing agents like glutathione (GSH) in cancer cells which triggers the disassembly of nanoparticles.11,15,16 This selectivity prevents the premature drug release in blood circulation, minimizes off-target toxicity and ensures a higher concentration of the drug at the target site. Redox-responsive nanoparticles can also address multidrug resistance by facilitating intracellular accumulation of the drug payload, thereby enhancing the efficacy of cancer treatments.
Natural polysaccharides as biomaterials have shown immense potential in the development of nanosized drug delivery platforms17 owing to their biocompatibility, biodegradability, and non-toxicity.18,19 Polysaccharides such as chitosan and their derivatives have been extensively explored for the development of nanomedicine responsive to tumor microenvironments.20–22 Chitosan, a cationic biopolymer derived from chitin, when used in the fabrication of nanoparticles, interacts with negatively charged cell membranes and enhances their cellular internalization.23,24 Chitosan can also be modified to achieve specific properties for drug delivery applications.12,25 Conjugating a hydrophobic moiety like D-alpha-tocopherol (vitamin E) on the hydrophilic chitosan backbone may make the conjugate amphiphilic for improving the loading of poorly aqueous soluble drugs.26–28 Vitamin E also modulates the signaling pathways such as the PI3K/AKT/mTOR pathway involved in cancer progression and drug resistance.29 Besides, the ability of vitamin E to neutralize reactive oxygen species (ROS) that promotes drug resistance in cancer cells also contributes to overcoming drug resistance by modulating the tumor microenvironment.30–33 Another amphiphilic derivative of vitamin E is D-alpha-tocopheryl polyethylene glycol 1000 succinate (TPGS) which provides steric stabilization to nanoparticles, prevents aggregation, and enhances cellular uptake.34,35 It forms a hydration layer around the nanoparticles, reducing particle–particle interactions, and provides stealth against immune recognition, thereby prolonging their circulation in bloodstream.36 TPGS also provides a reactive terminal functional group for ligand conjugation on the surface of nanomedicine.35 Moreover, CSVE and TPGS together can form a mixed micellar structure due to a common hydrophobic core (vitamin E) that provides a better hydrophobic microenvironment for encapsulation and stabilization of hydrophobic drugs in their core.26,34 CSVE/TPGS-based mixed micelles can also be combined with other polysaccharides like hyaluronic acid to create a hybrid nanostructure with improved performance.37 Hyaluronic acid (HA), an anionic polysaccharide, can enhance the stability of CSVE/TPGS-based micelles as a carrier.38,39 The positive and negative charges of chitosan and hyaluronic acid interact to form a complex that enhances self-assembly into nanoparticles through ionic gelation.40 Hyaluronic acid (HA) also has the ability to interact with CD44 receptors, which are overexpressed on cancer cell surfaces.13,38,39 Thus, incorporation of HA into nanocarriers can provide targeted drug delivery to the tumor.13,41 HA also enhances the penetration of nanoparticles into tumor tissue, ensuring better distribution of therapeutic payload in the complex tumor microenvironment. Furthermore, crosslinking of these hybrid structures can be achieved using a crosslinker like 3,3′-dithiodipropionic acid (DTPA, a redox-responsive linker)42,43 that utilizes the free amine group of the CSVE structure to form a stable and compact nanostructure with redox-responsive characteristics.
In the present study, cabazitaxel, an FDA-approved antineoplastic agent, was used as the model hydrophobic drug.44 It disrupts microtubule dynamics in cancer cells, inhibiting mitosis and causing apoptosis.45 Currently approved for metastatic castration-resistant prostate cancer, it is used in patients who have previously received docetaxel-based chemotherapy.46,47 However, studies are ongoing to explore its use in other cancer types and its potential in combination with other therapeutic agents.48 Also, cabazitaxel belonging to BCS Class IV drugs exhibits limited aqueous solubility and poor permeability.49 In addition, associated toxicity to major organs due to non-selective distribution other than the tumor site is another major challenge. Thus, strategies to improve the properties of cabazitaxel for smoothening the delivery is required which can be attained by formulating their nanoparticles. The use of nanoparticles may address the above issues and ensure higher therapeutic response by minimizing off-target distribution, prolonging systemic circulation, controlling drug release and facilitating accumulation at the tumor site.49,50 The nanoparticles can be modified with a stimuli responsive and targeting moiety to further specify their delivery to the tumor microenvironment and cancer cells, thereby reducing off-target distribution and drug-associated adverse effects.51–53 The current study involves the synthesis of vitamin-E conjugated chitosan (CSVE). The synthesized CSVE and HA were used for the development of TPGS-COOH stabilized sub-200 nm sized self-assembled hybrid nanoparticles. The prepared particles were further crosslinked with DTPA to endow redox-responsive characteristics. The particles were subsequently functionalized by conjugating cetuximab on their surface to further improve the selectivity of the nanoparticles toward cancer cells (details in Fig. S1, ESI†).
2. Materials and methods
2.1. Materials
D-Alpha-tocopheryl polyethylene glycol 1000 succinate (TPGS) was obtained as a gift sample from Antares Health Products, St. Charles, USA. Cabazitaxel was a gift sample from Hetero Labs, India. Sodium hyaluronate (HA) with an average molecular weight of ∼9.4 kDa was provided as a gift sample by Shandong Topscience Biotech Co. Ltd, Shandong, PRC. All the other chemicals and solvents used in the experiments were of high purity.The MDA-MB-231, T47D, and HCT116 cell lines were procured from NCCS Pune, India. DMEM (Dulbecco's Modified Eagle Medium) and 12-well and 6-well cell culture plates were procured from Genetix (Genetics Biotech Asia Pvt. Ltd). 96-well plates and T-25 flasks were obtained from Eppendorf. Penicillin–streptomycin, Trypsin-EDTA and FBS (fetal bovine serum) were purchased from Gibco.
2.2. Formulation of CSVE-based nanoparticles
The hybrid CSVE/HA nanoparticles (CSVE/HA-NP) were prepared using the solvent emulsification–evaporation method. Briefly, the CSVE conjugate (detailed in the ESI†) and TPGS were dissolved in 10 ml of distilled water and kept under stirring. CBT (5 mg ml−1) in chloroform was added to the CSVE/TPGS solution and probe-sonicated using a 6 mm probe at 70% amplitude for 7 minutes in an ice-bath.28 The solution was kept under stirring at 200 rpm for 3 h for the organic solvent to evaporate. The anionic TPGS and cationic CSVE form a stable micellar assembly due to their amphiphilic nature and common hydrophobic group. Subsequently, 5 ml of HA solution in distilled water (2 mg ml−1) was added dropwise and stirred for 2 h.54To prepare redox-responsive nanoparticles (CSVE/HA/DTPA NP), only 2.5 ml of HA (2 mg ml−1) was used, while DTPA was used as the crosslinker according to a previously reported method with minor modifications.14,55 For this, 72 mg of sulfo-NHS and 64 mg of EDC were dissolved in 2.5 ml of water then 12 mg of DTPA was added and stirred for 30 min. This solution was added dropwise into the CSVE/HA nanoparticle suspension and stirred for 3 h. For the preparation of targeted nanoparticles (CSVE/HA/DTPA/Cmab), CSVE/HA/DTPA was incubated with NHS and EDC for 30 minutes followed by the addition of cetuximab (2.5 mg) and stirred for 3 h.56 The nanoparticles were washed thrice with distilled water using Amicon® Ultra centrifugal filters (MWCO 30 kDa). The washed nanoparticles were then subjected to lyophilization and stored at 4–8 °C until further use. In preparing coumarin-6 (CM6) loaded nanoparticles, 0.3 mg of CM6 was added instead of cabazitaxel to 1 ml of chloroform mentioned above. Also, for preparing chitosan-based hybrid nanoparticles, CSVE was replaced with water soluble CSO.
2.3. Characterization of CSVE-based nanoparticles
2.4. Drug release studies
Drug release studies were performed using the dialysis method.57 Herein, the nanoparticle suspensions equivalent to 5 mg of cabazitaxel were loaded in a sealed pouch of dialysis membrane (Spectra/Por® 7, 1000 Da MWCO) and immersed in 50 ml of the medium. 1 ml of the medium was collected at predetermined time intervals. The samples were then filtered with 0.1 µm pore size membrane filters and analyzed using the HPLC-based analytical method. The drug release was investigated in phosphate buffered saline (pH 7.4), acetate buffer (pH 5.5), and acetate buffer (pH 5.5)/GSH (10 mM).2.5. Stability of lyophilized nanoparticles
To assess the stability of the prepared formulations, washed and purified CSVE/HA/DTPA NP and CSVE/HA/DTPA/Cmab NP were lyophilized and packed in airtight borosilicate glass vials and kept in a refrigerator at 4 °C for predetermined intervals. After day 7, 15, 30, 90, and 180, the lyophilized formulation was resuspended in deionized water and assessed for hydrodynamic particle size, zeta potential, PDI, and entrapment efficiency.2.6. In vitro cell line studies
To evaluate the mitochondrial distribution pattern, a similar protocol was followed up to the treatment stage. Following treatment, the cells were stained with MitoTracker™ Red at a 1 µM concentration and the nuclei were counterstained using Hoechst33342 at a 10 µg ml−1 concentration for 30 minutes. After staining, the cells were again washed with PBS and the images were taken using a fluorescence microscope in phase contrast, blue and red channels.61
For quantitative assessment of apoptosis, Alexa Fluor 488 Annexin V/PI double stained MDA-MB-231 cells were subjected to flow cytometry after being exposed to IC50 concentrations of the formulations. For this, 2 × 105 cells were seeded in 6-well cell culture plates and incubated for 24 h. Thereafter, the cells were treated with IC50 concentrations of the formulations. After 24 h, the cells were harvested in 1 mM EDTA in PBS (pH 7.4). Further, the treated and control cells were processed according to the manufacturer protocol for Alexa Fluor 488 Annexin V/PI staining (Thermo Fisher Scientific, Invitrogen Bioservices India Pvt. Ltd). After staining, the samples were analyzed by flow cytometry to evaluate apoptosis.63
2.7. In vivo studies
All in vivo experiments were carried out in accordance with the guidelines of CPCSEA (a Committee for the Purpose of Control and Supervision of Experiments on Animals, Regd. No. 2123/GO/Re/S/21/CPCSEA). The protocols for in vivo studies were approved by the Institutional Animal Ethics Committee (IAEC) (Approval number: IIT(BHU)/IAEC/2022/011) prior to the conduction of experiments. The animals were procured from the animal facility, Institute of Medical Sciences (BHU), Varanasi-221005 (542/GO/ReBi/S/02/CPCSEA).where r indicates the average radius of the tumor tissue.
 :1 (v/v) mixture of acetonitrile and methanol (200 µl) was added into the homogenate and mixed. The homogenate was separated by centrifugation and the supernatant was analyzed for drug concentration using the HPLC method.
:1 (v/v) mixture of acetonitrile and methanol (200 µl) was added into the homogenate and mixed. The homogenate was separated by centrifugation and the supernatant was analyzed for drug concentration using the HPLC method.
2.8. Statistical analysis
The results were reported as mean ± standard deviation (SD). The statistical significance of data was determined using the t test (non-parametric), one-way ANOVA and two-way ANOVA at p values of *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. All statistical analyses were performed using GraphPad Prism 9.0.0 for Windows (GraphPad Software, San Diego, CA, USA).3. Results and discussion
3.1. Formulation of CSVE-based nanoparticles
The prepared conjugate of CSO and VES (confirmed by NMR and FTIR; see details in the ESI,† Fig. S2 and S3) was used to prepare CSVE based nanoparticles. The presence of the same hydrophobic group (VES) and the net cationic and anionic charges of CSVE and TPGS-COOH, respectively, allows the formation of a stable micellar structure under aqueous conditions. The drug (CBT) was loaded in the stable hydrophobic core of the micellar structure which was further stabilized by ionic gelation through the addition of anionic hyaluronic acid. The stable hybrid nanoparticles formed were then crosslinked using DTPA to endow redox-responsive release behavior via carbodiimide chemistry. To impart cancer selective characteristics, the particles were surface-functionalized by conjugating Cmab to the carboxyl group of TPGS-COOH present on the surface of the nanoparticles.The formulation composition and characterized parameters are given in Table 1. The hydrodynamic sizes determined by DLS of CSO/HA NP, CSVE/HA NP, CSVE/HA/DTPA NP, and CSVE/HA/DTPA/Cmab NP were 190 ± 23 nm, 179 ± 32 nm, 164 ± 29 nm, and 203 ± 43 nm, respectively. The amphiphilic nature of the CSVE conjugate combined with the identical hydrophobic segment (VES) of TPGS-COOH allows for the formation of a compactly packed micellar structure that leads to a lower size of CSVE/HA NP. The size of CSVE/HA/DTPA NP and CSVE/HA NP did not show major differences. However, the size of CSVE/HA/DTPA/Cmab NP exhibited a marked increase which may be attributed to the conjugation of Cmab on the surface of the nanoparticles.
| Formulation | Composition (mg) | Particle size (nm) | PDI | Zeta potential (ζ) | Entrapment efficiency (%) | Drug loading (%) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CSO | CSVE | HA | TPGS-COOH | CBT | DTPA | Cmab | ||||||
| *Results are shown as mean ± SD (n = 6). | ||||||||||||
| CSO/HA NP | 50 | — | 10 | 10 | 5 | — | — | 190 ± 23 | 0.265 ± 0.084 | 19.2 ± 2.35 | 53.24 ± 3.57 | 4.71 ± 0.19 |
| CSVE/HA NP | — | 50 | 10 | 10 | 5 | — | — | 179 ± 32 | 0.238 ± 0.071 | 16.4 ± 3.72 | 62.35 ± 2.91 | 5.06 ± 0.13 |
| CSVE/HA/DTPA NP | — | 50 | 5 | 10 | 5 | 12 | — | 164 ± 29 | 0.268 ± 0.045 | 12.1 ± 2.08 | 64.49 ± 3.53 | 4.98 ± 0.07 |
| CSVE/HA/DTPA/Cmab NP | — | 50 | 5 | 10 | 5 | 12 | 2.5 | 203 ± 43 | 0.167 ± 0.063 | 11.9 ± 2.57 | 63.77 ± 3.96 | 4.85 ± 0.14 |
However, the zeta potential (ζ) showed a different trend, with CSVE/HA/DTPA NP exhibiting the highest zeta potential, followed by CSVE/HA/DTPA/Cmab NP, CSO/HA NP, and the least in CSVE/HA NP (Table 1). This observation may be due to the higher amount of HA (an anionic polysaccharide) being used in CS/HA NP and CSVE/HA NP. The lower ζ in CSVE/HA NP than in CSO/HA NP may be due to the crosslinking via amide bond formation between chitosan and VES. Similar results have been previously reported where ζ has exhibited a reduction due to the conjugation of moieties with the amine groups of CSO.27,72 The polydispersity index (PDI) of all formulations was less than 0.3, indicating monodisperse systems.
Additionally, TEM, SEM, and AFM images confirmed the formulation of well-structured spherical nanoparticles (Fig. 1). The particle sizes of CSO/HA NP, CSVE/HA NP, CSVE/HA/DTPA NP, and CSVE/HA/DTPA/Cmab NP observed by TEM were 170 ± 12 nm, 135 ± 19 nm, 126 ± 14 nm, and 175 ± 9 nm, respectively. The size observed in TEM was smaller than that observed in DLS which could be due to the difference in measurement conditions, as TEM analysis was employed for dried samples, while DLS was used to measure the hydrodynamic size of the particles and it takes into account the solvated water molecules on the surface.73 The SAED pattern for all nanoparticles exhibited diffused rings, reflecting the amorphous state of nanoparticles. The particles of spherical anisotropy were confirmed by SEM (i.e., CSO/HA NP: 145 ± 24, CSVE/HA NP: 136 ± 22 nm, CSVE/HA/DTPA NP: 117 ± 21 and CSVE/HA/DTPA/Cmab NP: 152 ± 29) and were of size similar to TEM results. TEM and SEM results followed the trend of DLS results, as desired. AFM images further confirmed the smooth surface spherical morphology of formulated nanoparticles. The results of SEM, TEM and AFM were in good correlation. The entrapment efficiency of formulated nanoparticles ranged between 53 and 65%. The nanoparticles formulated using the synthesized CSVE/HA had higher entrapment efficiency than CSO/HA nanoparticles. The higher entrapment efficiency of CSVE based formulations than that of CSO/HA NP may be attributed to the amphiphilic nature of the synthesized CSVE which may improve the drug loading capacity for hydrophobic drugs in the core of the micellar structure.72 The degree of Cmab conjugation to the carboxyl group of TPGS-COOH was confirmed by the Bradford assay (details in the ESI†) which showed 71.635 ± 4.23% of added Cmab was conjugated onto the surface, indicating efficient surface functionalization.
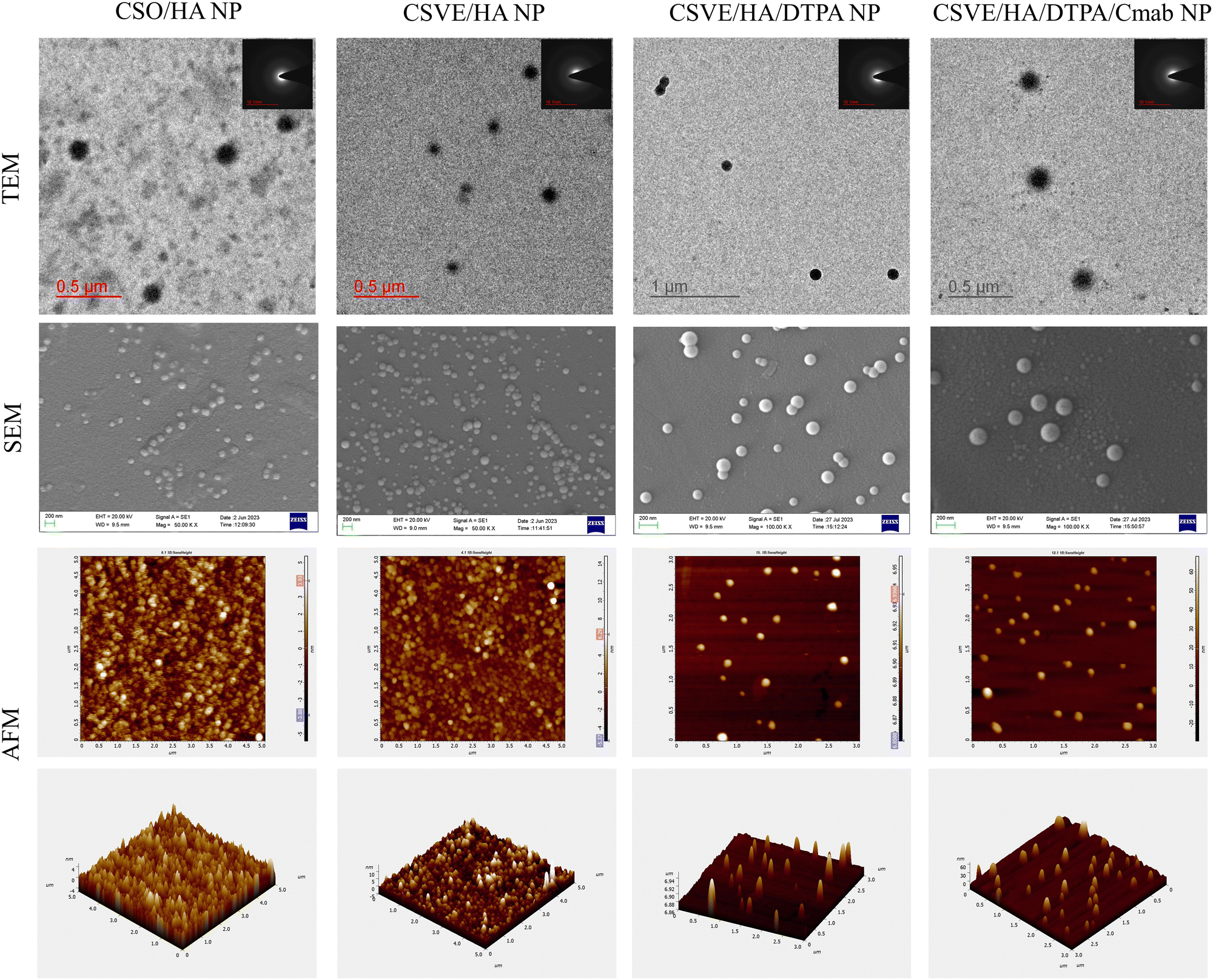 | ||
| Fig. 1 Morphological assessment (TEM, SEM, and AFM) of CSO/HA NP, CSVE/HA NP, CSVE/HA/DTPA NP, and CSVE/HA/DTPA/Cmab NP. | ||
3.2. Solid-state characterization (FTIR/XRD/DSC/XPS)
The FTIR spectra of CBT, CSVE, TPGS, HA, CSVE/HA NP, CSVE/HA/DTPA NP and CSVE/HA/DTPA/Cmab NP are shown in Fig. 2A (details in the ESI†). The characteristic peaks observed for CBT were similar to those previously reported for CBT.74–76 The FTIR spectrum of CSVE/HA nanoparticles exhibited merged peaks of CSVE and HA and a similar trend was shown by CSVE/HA/DTPA NP and CSVE/HA/DTPA/Cmab NP. The characteristic peaks of crystalline CBT were masked by CSVE and HA, indicating complete encapsulation of the drug in the prepared nanoparticles.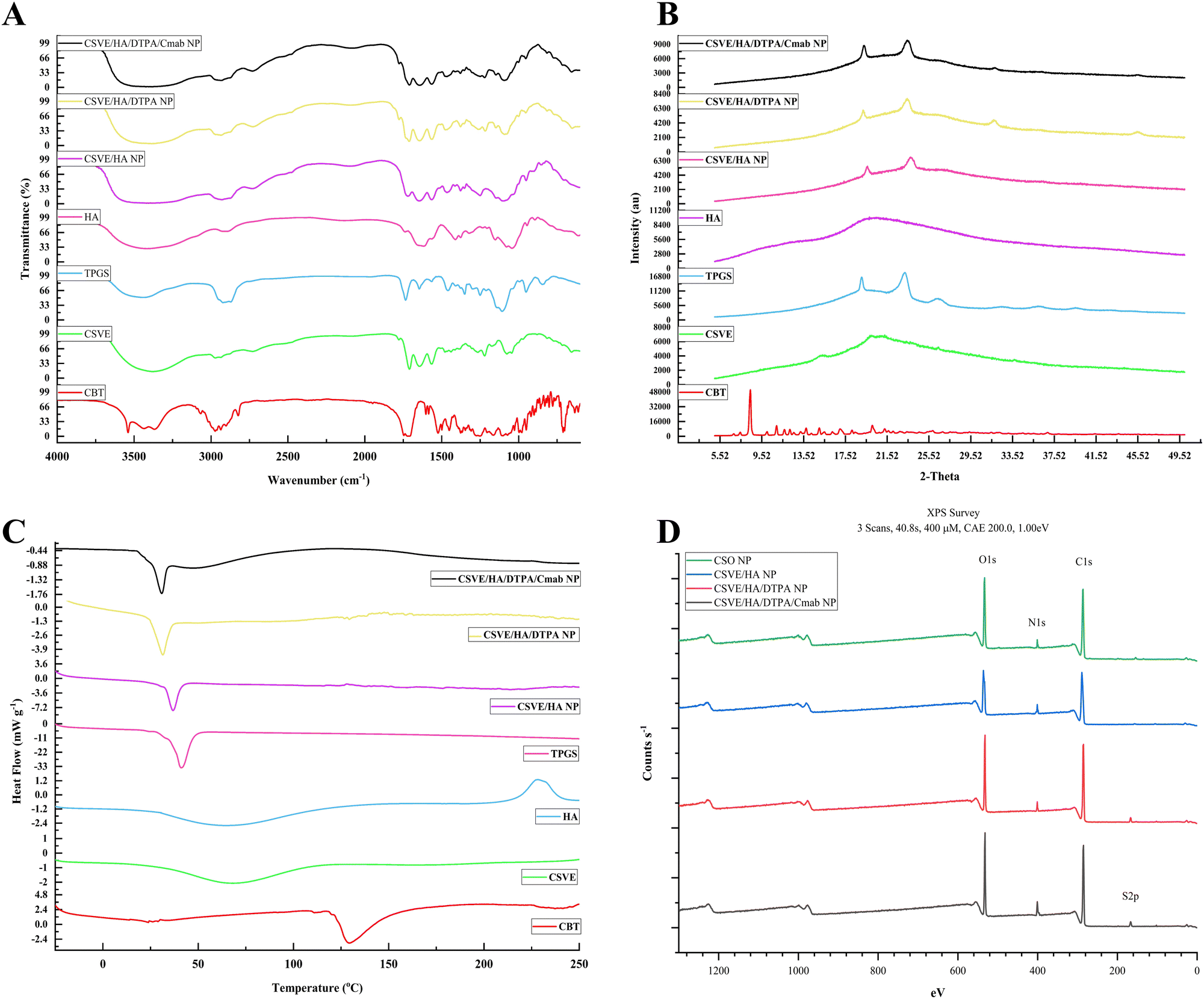 | ||
| Fig. 2 Solid-state characterization; (A) FTIR, (B) XRD, and (C) DSC of cabazitaxel (CBT), CSVE, HA, TPGS, CSVE/HA NP, CSVE/HA/DTPA NP, and CSVE/HA/DTPA/Cmab NP. (D) XPS spectra of CSO NP, CSVE/HA NP, CSVE/HA/DTPA NP, and CSVE/HA/DTPA/Cmab NP. | ||
The X-ray diffraction pattern (Fig. 2B) of CBT revealed its crystalline nature and its peaks were similar to previously reported peaks.76 TPGS revealed two distinct peaks of a high intensity, while CSVE had an amorphous halo diffraction pattern with very small intensity peaks. CSVE/HA NP had peaks corresponding to TPGS and CSVE/HA/DTPA NP exhibited four peaks due to TPGS and the crosslinker. Meanwhile, the reduction in the intensity of these peaks in the case of CSVE/HA/DTPA/Cmab NP may be due to the surface conjugation of Cmab. The absence of CBT peaks in nanoparticles was due to the peak dominating effects of CSVE and HA (details in the ESI†).
The thermogram (Fig. 2C) of CBT showed a sharp endothermic peak which was absent in nanoparticles (details in the ESI†). The characteristic peak of TPGS however was present due to its presence on the nanoparticle surface with a minor shift attributed to nanosized particles.
The XPS spectra of CSO/HA NP and CSVE/HA NP showed signature C1s, N1s, and O1s peaks as shown in Fig. 2D. The presence of S2p and a marked increase in N1s signals indicate the successful DTPA mediated crosslinking and Cmab conjugation on the CSVE/HA/DTPA/Cmab NP surface (details in the ESI†).
3.3. Drug release study
The release study was conducted under conditions corresponding to the systemic circulation (pH 7.4), acidic tumor microenvironment (pH 5.5), and intracellular elevated ROS and reduced pH conditions (pH 5.5 + GSH). The redox responsive CSVE/HA/DTPA NP and CSVE/HA/DTPA/Cmab NP showed a marked increase in drug release in the presence of low pH and glutathione (GSH) (details in the ESI,† Fig. S5). Similar results were observed for a redox responsive nanoparticle prepared using DTPA due to cleavage by glutathione (GSH).53 The release rate was highest in pH 5.5 + GSH (10 mM) followed by pH 5.5 and pH 7.4. Also, T50 values for CSVE/HA/DTPA/Cmab NP at pH 7.4, pH 5.5 and pH 5.5 + GSH were 15 h, 8.5 h, and 2.5 h, respectively, exhibiting a similar trend to CSVE/HA/DTPA NP (Fig. 3A). The pH-responsive trend in the release behavior of all the particles can be attributed to protonation of the amino groups on CS at lower pH values responsible for enhanced electrostatic repulsion and thus easy diffusion of the loaded drug from NPs.72,77 However, the increased release of the drug in the media comprising low pH and GSH is due to the cleavage of the disulfide bonds of the crosslinker DTPA in response to GSH.15,16,42 The GSH-responsive cleavage of the disulfide linkage was further confirmed using Ellman's reagent which showed a rapid release of the free thiol group in CSVE/HA/DTPA NP in the first 90 min when exposed to acetate buffer (pH 5.5) + GSH (details in the ESI,† Fig. S6). When the CSVE/HA/DTPA NP were exposed to acetate buffer (pH 5.5) + GSH, the disulfide linkage of DTPA cleaves to release the free thiol group, resulting in disassembly of the crosslinked polysaccharide nanoparticles consequently increasing the rate of cabazitaxel release.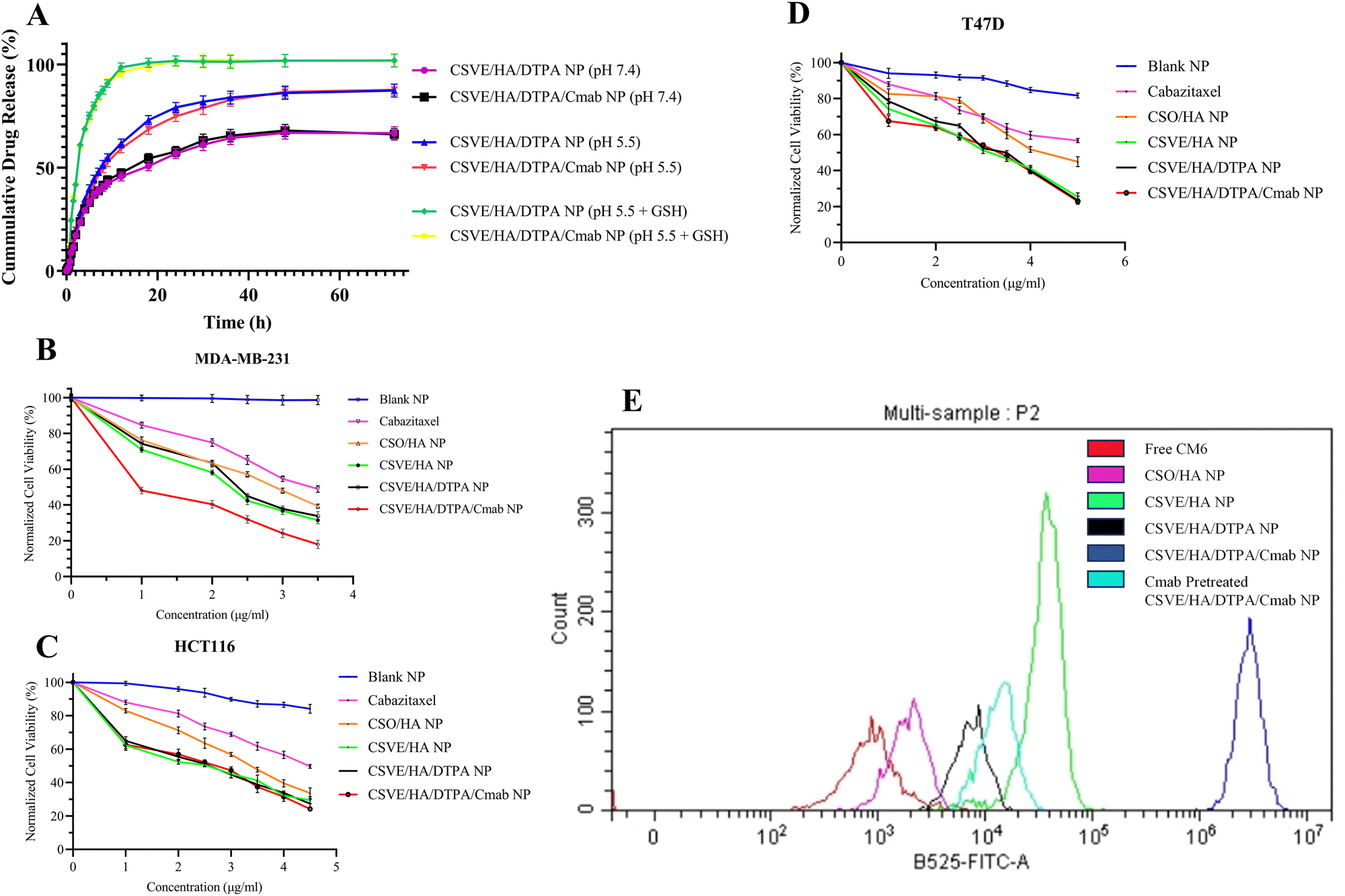 | ||
| Fig. 3 (A) Drug release profiles of CSVE/HA/DTPA NP and CSVE/HA/DTPA/Cmab NP in pH 7.4 phosphate buffer saline, pH 5.5 acetate buffer, and pH 5.5 acetate buffer and glutathione (10 mM). (B) In vitro cell viability of MDA-MB-231 cells after treatment with cabazitaxel-loaded formulations for 24 h (n = 3). (C) In vitro cell viability of HCT116 cells after treatment with cabazitaxel-loaded formulations for 24 h (n = 3). (D) In vitro cell viability of T47D cells after treatment with cabazitaxel-loaded formulations for 24 h (n = 3). (E) Quantitative assessment of the intracellular uptake of green fluorescent CM6 in MDA-MB-231 cells treated with free CM6, CSO/HA NP, CSVE/HA NP, CSVE/HA/DTPA NP, CSVE/HA/DTPA/Cmab NP, and Cmab pre-treated CSVE/HA/DTPA/Cmab NP at a 0.2 µg ml−1 concentration of CM6 for 6 h using flow cytometry. The blue peak shows the highest cellular uptake of CM6 in CM6-loaded CSVE/HA/DTPA/Cmab NP treated cells compared to free CM6, CSO/HA NP, CSVE/HA NP, CSVE/HA/DTPA NP, and CSVE/HA/DTPA/Cmab NP treated cells. | ||
The drug release kinetic modelling was also performed for CSVE/HA/DTPA/Cmab NP which followed Weibull/Gompertz at pH 7.4, logistic/probit at pH 5.5 and Weibull/logistic under pH 5.5 + GSH (details in the ESI,† Table S1). This suggested controlled drug release via a slow drug diffusion mechanism from spherical particles at pH 7.4.78,79 At pH 5.5, an initial faster release followed by controlled drug release via a diffusion mechanism was observed.80,81 This could be due to the pH responsiveness of the prepared nanoparticles that resulted in an initial faster release of the drug from the nanoparticle surface directly exposed to media followed by controlled release via drug diffusion from the core to the outer surface and then to the media. Meanwhile, in the case of pH 5.5 + GSH, the release was faster via an erosion and diffusion mechanism in response to GSH responsible for breakage of the linkage, loosening of the nanoparticle assembly and subsequent dissolution of the matrix and drug.80,82
The prepared nanoparticles were also investigated for storage stability (see details in the ESI,† Fig. S7). Slight changes in the particle size (hydrodynamic diameter), polydispersity index, zeta potential, and entrapment efficiency were observed. However, the differences were non-significant and thus the prepared nanoparticles were stable during their storage, suggesting their suitability for long-term use.
3.4. In vitro study
 | ||
| Fig. 4 Fluorescence microscopy images of MDA-MB-231 cells after 6 h incubation with the fluorescent CM6-loaded formulations. The blue fluorescence from DAPI channels shows Hoechst33342 stained nuclei, while the third column shows an overlay image of Hoechst and GFP. The fourth column represents an overlay image of phase contrast, GFP, and Hoechst33342. Each scale bar represents 100 µm. The GFP channel showed a higher level of green fluorescence in CM6 loaded CSVE/HA/DTPA/Cmab NP treated cells compared to free CM6 and CM6-loaded CSVE/HA/DTPA NP treated cells due to receptor-mediated cellular uptake of CM6 in the cytoplasm. The CM6-loaded CSVE/HA NP also exhibited superior intracellular localization of CM6 compared to CM6-loaded CSO/HA NP which may be attributed to the amphiphilic nature of the CSVE conjugate. The Cmab pretreated cells exhibited reduced uptake of CM6 which may be due to the competitive binding of Cmab to the EGFR. | ||
 | ||
| Fig. 5 (A) Assessment of mitochondrial membrane potential using JC-1 dye in control and treated MDA-MB-231 cells after treatment with cabazitaxel, CSVE/HA/DTPA NP, CSVE/HA/DTPA/Cmab NP, and Cmab pretreated CSVE/HA/DTPA/Cmab NP. The CSVE/HA/DTPA/Cmab NP treated group showed maximum depolarization (green fluorescence of JC-1 monomer) in cells. In contrast, the control group exhibited an accumulation of JC-1 monomers in negatively charged and energized normal mitochondria to spontaneously form J aggregates (red fluorescence). (B) and (C) Quantitative detection of mitochondrial membrane potential in MDA-MB-231 after treatment with the formulations through flow cytometry. The upper right and lower right quadrants represent cells with normal mitochondrial membrane potential and depolarized mitochondria, respectively. The CSVE/HA/DTPA/Cmab NP-treated cells showed maximum mitochondrial depolarization. (D) Pattern of mitochondrial distribution in MDA-MB-231 cells using MitoTracker™ Red after treatment with cabazitaxel, CSVE/HA/DTPA NP, CSVE/HA/DTPA/Cmab NP, and Cmab pretreated CSVE/HA/DTPA/Cmab NP. The uniform distribution of mitochondria was seen in control cells represented by the yellow arrow, while aggregated mitochondria were observed in the treatment group represented by the white arrows. The CSVE/HA/DTPA/Cmab treated cells showed the highest mitochondrial aggregation. Statistical analysis with significance: ****p < 0.0001, ***p < 0.001, and **p < 0.01. | ||
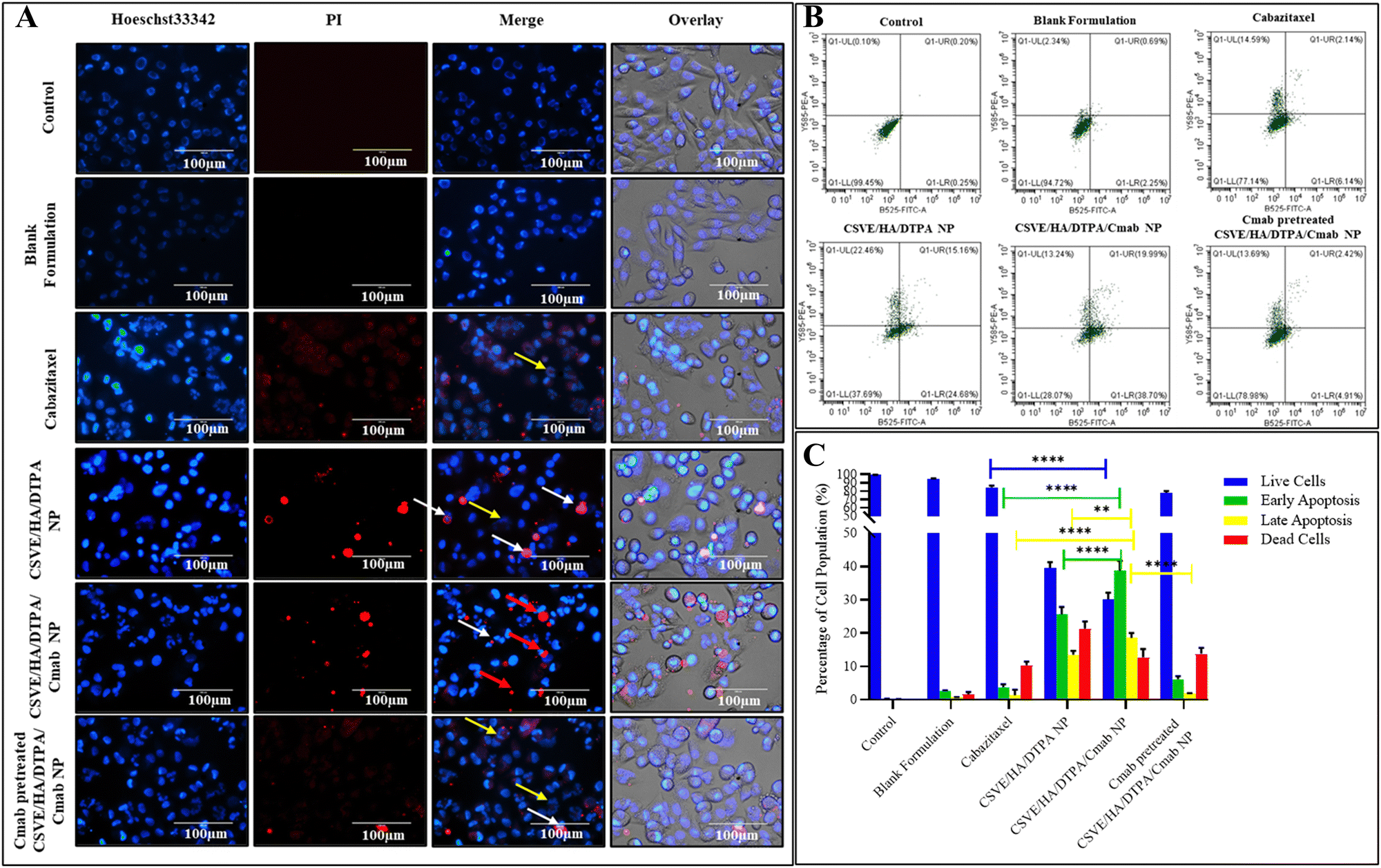 | ||
| Fig. 6 (A) Combined images of Hoechst33342/PI-stained MDA-MB-231 cells after treatment with the formulations. The yellow, white, and red arrows represent early apoptotic, late apoptotic, and necrotic cells. Cells emitting blue fluorescence are Hoechst positive, while those with red fluorescence are PI positive. (B) and (C) Quantitative assessment of apoptosis by Annexin V/PI dual staining through flow cytometry in treated MDA-MB-231 cells. Annexin V/PI dual staining discriminates the percentage of live (lower left quadrant), early apoptotic (lower right quadrant), late apoptotic (upper right quadrant), and necrotic or dead (upper left quadrant) cells. Statistical analysis with significance: ****p < 0.0001 and **p < 0.01. | ||
3.5. In vivo study
669 ng ml−1) followed by CSVE/HA/DTPA/Cmab NP (18369 ng ml−1) and CSVE/HA/DTPA NP (18119 ng ml−1). However, the area under the curve for CSVE/HA/DTPA NP and CSVE/HA/DTPA/Cmab NP was three- to four-fold that of CBT. This could be due to the higher clearance of CBT from systemic circulation as compared to CSVE/HA/DTPA NP and CSVE/HA/DTPA/Cmab NP. A t1/2 of only 11.34 h was observed for CBT, while t1/2 values for CSVE/HA/DTPA NP and CSVE/HA/DTPA/Cmab NP were 30.74 h and 38.36 h, respectively. The mean retention time (MRT) of the drug was also shorter for CBT (13.56 h) than those for CSVE/HA/DTPA NP (40.40 h) and CSVE/HA/DTPA/Cmab NP (50.84 h). The results indicate that the prepared nanoparticles provided longer systemic circulation than CBT, allowing the sustained drug concentration to reach the target site and leverage its action, which could contribute to a better therapeutic response.
 | ||
| Fig. 7 Concentration of cabazitaxel in the plasma, tumor, liver, lungs, spleen and kidneys after intravenous administration of cabazitaxel, CSVE/HA/DTPA NP, and CSVE/HA/DTPA/Cmab NP. | ||
| Parameters | Unit | Cabazitaxel | CSVE/HA/DTPA NP | CSVE/HA/DTPA/Cmab NP |
|---|---|---|---|---|
| t 1/2 | h | 11.3409 | 30.7382 | 38.3640 |
| T max | h | 0.25 | 0.25 | 0.25 |
| C max | ng ml−1 | 18669.4540 |
18119.0039 |
18369.1091 |
| C 0 | ng ml−1 | 23473.1886 |
21774.7884 |
22217.6985 |
| AUC 0-t | ng ml−1 h−1 | 96771.0839 |
247526.4187 |
226707.1421 |
| AUC 0-inf_obs | ng ml−1 h−1 | 103915.5047 |
358035.5039 |
369966.0585 |
| AUMC 0-inf_obs | ng ml−1 h−2 | 1408825.881 |
14463854.26 |
18809769.52 |
| MRT 0-inf_obs | H | 13.5574 | 40.3978 | 50.8419 |
| Cl_obs | (mg kg−1)/(ng ml−1) h−1 | 9.6232 | 2.7930 | 2.7029 |
| F rel | — | 3.44 | 3.56 |
 | ||
| Fig. 8 (A) Body weight and (B) tumor volume of normal SD rats, tumor-bearing SD rats, and formulation treated tumor-bearing SD rats over the 28-day treatment period; (C) survival rates of tumor-bearing SD rats after treatment with formulations (day 0 represents the day of administration of the first dose after the tumor volume reached ∼500 mm3); (D) morphology of tumors harvested at the end of the study. | ||
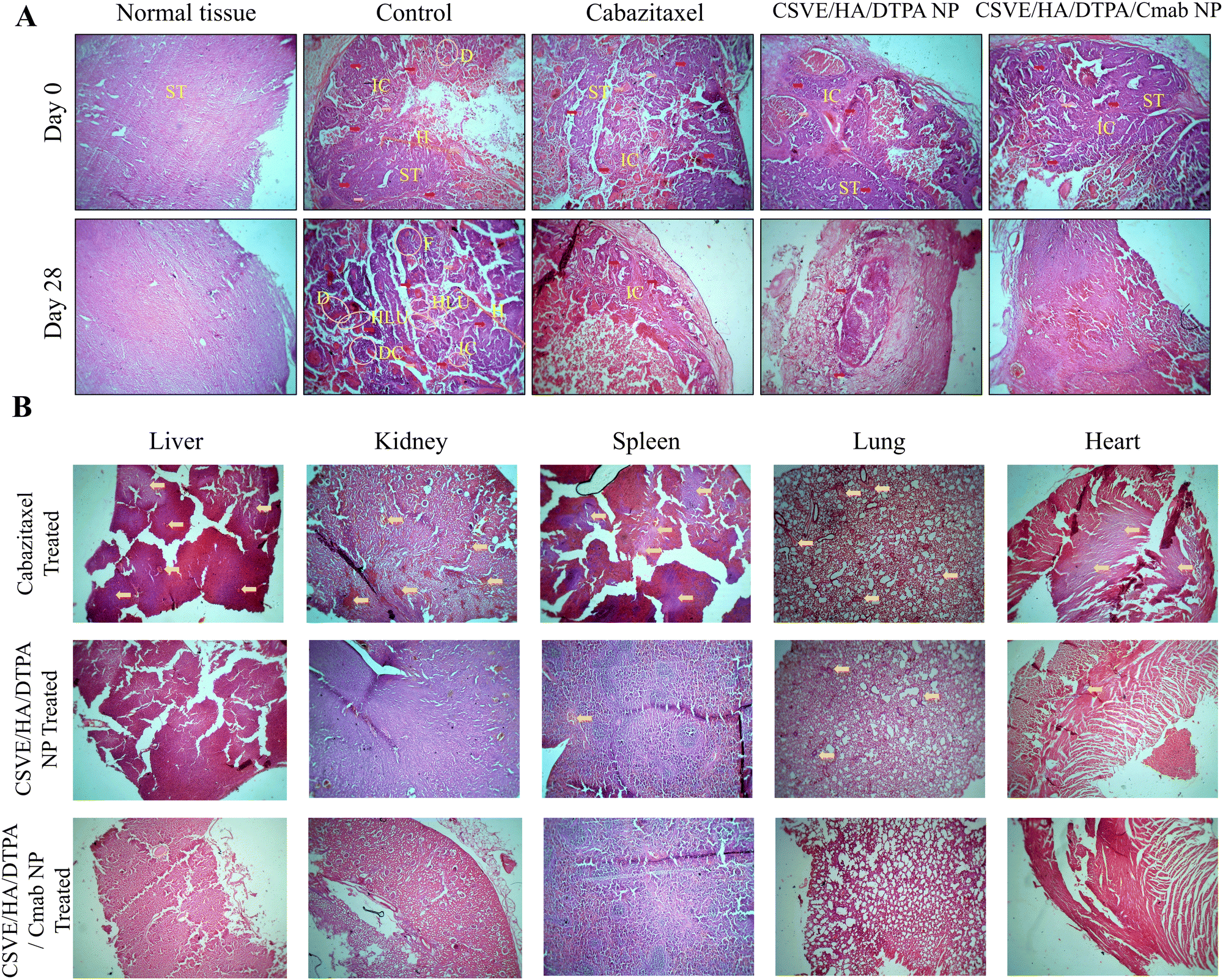 | ||
| Fig. 9 (A) Histological assessment of tumors isolated from rats after the tumor regression study using H&E stain (magnification, 4×; scale bar, 100 µm). (B) Histological images of organ tissues after 28 days of treatment. Invasive carcinoma (IC), stromal tissue (ST), proliferated and expanded terminal lobular units (HLU), dilated ducts with inspissated secretions (D), ductal carcinoma (DC), mucin (yellow arrow), fibroadenoma (F) and breast hyperplasia (H). | ||
The histological investigation of all major organs including the liver, kidneys, spleen, lungs and heart was also conducted to determine the safety of the prepared formulation as compared to free cabazitaxel. Free cabazitaxel showed major signs of toxicity (yellow arrow, Fig. 9B) in all major organs. In contrast, CSVE/HA/DTPA NP and CSVE/HA/DTPA/Cmab NP showed few or no signs of toxicity, suggesting the lower distribution of the free drug from nanoparticles to all major organs. Therefore, the prepared nanoformulations were safe and reduced the organ toxicity probably by minimizing the drug distribution to tissue and organs other than cancer.
4. Conclusion
Novel GSH-responsive cetuximab anchored chitosan/vitamin-E succinate conjugate-based hybrid nanoparticles were developed by an ionic gelation method. The particles exhibited spherical morphology with a sub-200 nm size, cationic surface charges, and redox responsive drug release. In vitro evaluations showed significantly superior cytotoxicity against MDA-MB-231 cells, enhanced cellular uptake, and reduced mitochondrial membrane potential in comparison to free CBT. The particles induced higher apoptosis than the free drug. The in vivo studies showed better performance of the nanoparticles than free CBT, with improved half-life, AUC, and MRT. Tumor regression analysis, survival and histological analysis indicated that CSVE/HA/DTPA/Cmab NP treated groups elicited better treatment response and lesser signs of toxicity in tissues than the free drug. Overall, the prepared hybrid receptor-targeted nanoparticles responsive to intracellular ROS species like GSH have immense potential as a targeted anticancer drug delivery platform. However, these preliminary findings should be extrapolated to extensive and advanced investigation to determine the clinical viability of CSVE/HA/DTPA/Cmab NP as an effective treatment modality for breast cancer. The major hurdle towards the translational potential of the tumor microenvironment responsive system is the variable response of the system over the wide range of stimuli from various microenvironments seen in different cancers. The ability of the nanocarrier to release therapeutics in response to a broader range of redox stimuli in in vitro and in vivo settings may be studied in the future.Data availability
All data are included in the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the Ministry of Education, Govt. of India and IIT (BHU), Varanasi, India for providing teaching assistantship. The authors are thankful to the Central Instrumentation Facility (CIF), IIT (BHU), Varanasi and SATHI-BHU, Varanasi for providing the instrumental support for the characterization of the formulations. The author BK acknowledges Banaras Hindu University for providing funding under the IoE scheme (File No. R/Dev/D/IoE/Incentive/2021-22/32449). The graphical abstract was made using elements from Servier Medical Art by Servier licensed under a Creative Commons Attribution 3.0 Unported License (https://smart.servier.com).References
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries, Ca-Cancer J. Clin., 2021, 71, 209–249, DOI:10.3322/caac.21660.
- M. T. Manzari, Y. Shamay, H. Kiguchi, N. Rosen, M. Scaltriti and D. A. Heller, Targeted drug delivery strategies for precision medicines, Nat. Rev. Mater., 2021, 6, 351–370, DOI:10.1038/s41578-020-00269-6.
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Engineering precision nanoparticles for drug delivery, Nat. Rev. Drug Discovery, 2021, 20, 101–124, DOI:10.1038/s41573-020-0090-8.
- D. Fan, Y. Cao, M. Cao, Y. Wang, Y. Cao and T. Gong, Nanomedicine in cancer therapy, Signal Transduction Targeted Ther., 2023, 8, 1–34, DOI:10.1038/s41392-023-01536-y.
- P. Foroozandeh and A. A. Aziz, Insight into Cellular Uptake and Intracellular Trafficking of Nanoparticles, Nanoscale Res. Lett., 2018, 13, 339, DOI:10.1186/s11671-018-2728-6.
- M. E. Davis, Z. (Georgia) Chen and D. M. Shin, Nanoparticle therapeutics: an emerging treatment modality for cancer, Nat. Rev. Drug Discovery, 2008, 7, 771–782, DOI:10.1038/nrd2614.
- D. A. Richards, A. Maruani and V. Chudasama, Antibody fragments as nanoparticle targeting ligands: a step in the right direction, Chem. Sci., 2016, 8, 63–77, 10.1039/C6SC02403C.
- E. da S. Santos, K. A. B. Nogueira, L. C. C. Fernandes, J. R. P. Martins, A. V. F. Reis, J. de, B. V. Neto, I. J. da, S. Júnior, C. Pessoa, R. Petrilli and J. O. Eloy, EGFR targeting for cancer therapy: Pharmacology and immunoconjugates with drugs and nanoparticles, Int. J. Pharm., 2021, 592, 120082, DOI:10.1016/j.ijpharm.2020.120082.
- D. Geethakumari, A. Bhaskaran Sathyabhama, K. Raji Sathyan, D. Mohandas, J. V. Somasekharan and S. T. Puthiyedathu, Folate functionalized chitosan nanoparticles as targeted delivery systems for improved anticancer efficiency of cytarabine in MCF-7 human breast cancer cell lines, Int. J. Biol. Macromol., 2022, 199, 150–161, DOI:10.1016/j.ijbiomac.2021.12.070.
- S. Idoudi, Y. Hijji, T. Bedhiafi, H. M. Korashy, S. Uddin, M. Merhi, S. Dermime and N. Billa, A novel approach of encapsulating curcumin and succinylated derivative in mannosylated-chitosan nanoparticles, Carbohydr. Polym., 2022, 297, 120034, DOI:10.1016/j.carbpol.2022.120034.
- S. Chen, X. Ji, M. Zhao, J. Jin, H. Zhang and L. Zhao, Construction of chitooligosaccharide-based nanoparticles of pH/redox cascade responsive for co-loading cyclosporin A and AZD9291, Carbohydr. Polym., 2022, 291, 119619, DOI:10.1016/j.carbpol.2022.119619.
- L. Jia, Z. Li, D. Zheng, Z. Li and Z. Zhao, A targeted and redox/pH-responsive chitosan oligosaccharide derivatives based nanohybrids for overcoming multidrug resistance of breast cancer cells, Carbohydr. Polym., 2021, 251, 117008, DOI:10.1016/j.carbpol.2020.117008.
- P. Song, Z. Lu, T. Jiang, W. Han, X. Chen and X. Zhao, Chitosan coated pH/redox-responsive hyaluronic acid micelles for enhanced tumor targeted co-delivery of doxorubicin and siPD-L1, Int. J. Biol. Macromol., 2022, 222, 1078–1091, DOI:10.1016/j.ijbiomac.2022.09.245.
- M. Zhao, P. Li, H. Zhou, L. Hao, H. Chen and X. Zhou, pH/redox dual responsive from natural polymer-based nanoparticles for on-demand delivery of pesticides, Chem. Eng. J., 2022, 435, 134861, DOI:10.1016/j.cej.2022.134861.
- A. Sood, A. Gupta, R. Bharadwaj, P. Ranganath, N. Silverman and G. Agrawal, Biodegradable disulfide crosslinked chitosan/stearic acid nanoparticles for dual drug delivery for colorectal cancer, Carbohydr. Polym., 2022, 294, 119833, DOI:10.1016/j.carbpol.2022.119833.
- F. Wang, J. Li, C. Chen, H. Qi, K. Huang and S. Hu, Preparation and synergistic chemo-photothermal therapy of redox-responsive carboxymethyl cellulose/chitosan complex nanoparticles, Carbohydr. Polym., 2022, 275, 118714, DOI:10.1016/j.carbpol.2021.118714.
- Q. Li, X. Liu, C. Yan, B. Zhao, Y. Zhao, L. Yang, M. Shi, H. Yu, X. Li and K. Luo, Polysaccharide-Based Stimulus-Responsive Nanomedicines for Combination Cancer Immunotherapy, Small, 2023, 19, 2206211, DOI:10.1002/smll.202206211.
- M. Li, Y. Zhao, W. Zhang, S. Zhang and S. Zhang, Multiple-therapy strategies via polysaccharides-based nano-systems in fighting cancer, Carbohydr. Polym., 2021, 269, 118323, DOI:10.1016/j.carbpol.2021.118323.
- S. Mizrahy and D. Peer, Polysaccharides as building blocks for nanotherapeutics, Chem. Soc. Rev., 2012, 41, 2623–2640, 10.1039/C1CS15239D.
- F. A. Taher, S. A. Ibrahim, A. A. El-Aziz, M. F. Abou El-Nour, M. A. El-Sheikh, N. El-Husseiny and M. M. Mohamed, Anti-proliferative effect of chitosan nanoparticles (extracted from crayfish Procambarus clarkii, Crustacea: Cambaridae) against MDA-MB-231 and SK-BR-3 human breast cancer cell lines, Int. J. Biol. Macromol., 2019, 126, 478–487, DOI:10.1016/j.ijbiomac.2018.12.151.
- H. Amin, M. A. Amin, S. K. Osman, A. M. Mohammed and G. Zayed, Chitosan nanoparticles as a smart nanocarrier for gefitinib for tackling lung cancer: Design of experiment and in vitro cytotoxicity study, Int. J. Biol. Macromol., 2023, 246, 125638, DOI:10.1016/j.ijbiomac.2023.125638.
- F. V. Ataabadi, F. Oveissi, M. Etebari and A. Taheri, Preparation of chitosan nanoparticles for simultaneous drug delivery of dacarbazine and enoxaparin in melanoma, Carbohydr. Polym., 2023, 316, 121041, DOI:10.1016/j.carbpol.2023.121041.
- P. Manivasagan, S. Bharathiraja, N. Q. Bui, I. G. Lim and J. Oh, Paclitaxel-loaded chitosan oligosaccharide-stabilized gold nanoparticles as novel agents for drug delivery and photoacoustic imaging of cancer cells, Int. J. Pharm., 2016, 511, 367–379, DOI:10.1016/j.ijpharm.2016.07.025.
- F.-Q. Hu, X. Wu, Y.-Z. Du, J. You and H. Yuan, Cellular uptake and cytotoxicity of shell crosslinked stearic acid-grafted chitosan oligosaccharide micelles encapsulating doxorubicin, Eur. J. Pharm. Biopharm., 2008, 69, 117–125, DOI:10.1016/j.ejpb.2007.09.018.
- Y.-Z. Du, L. Wang, H. Yuan and F.-Q. Hu, Linoleic acid-grafted chitosan oligosaccharide micelles for intracellular drug delivery and reverse drug resistance of tumor cells, Int. J. Biol. Macromol., 2011, 48, 215–222, DOI:10.1016/j.ijbiomac.2010.11.005.
- J. Dou, H. Zhang, X. Liu, M. Zhang and G. Zhai, Preparation and evaluation in vitro and in vivo of docetaxel loaded mixed micelles for oral administration, Colloids Surf., B, 2014, 114, 20–27, DOI:10.1016/j.colsurfb.2013.09.010.
- Y. Chen, S. Feng, W. Liu, Z. Yuan, P. Yin and F. Gao, Vitamin E Succinate-Grafted-Chitosan Oligosaccharide/RGD-Conjugated TPGS Mixed Micelles Loaded with Paclitaxel for U87MG Tumor Therapy, Mol. Pharmaceutics, 2017, 14, 1190–1203, DOI:10.1021/acs.molpharmaceut.6b01068.
- A. M. Itoo, M. Paul, B. Ghosh and S. Biswas, Oxaliplatin delivery via chitosan/vitamin E conjugate micelles for improved efficacy and MDR-reversal in breast cancer, Carbohydr. Polym., 2022, 282, 119108, DOI:10.1016/j.carbpol.2022.119108.
- Y. Yu, L. Hou, H. Song, P. Xu, Y. Sun and K. Wu, Akt/AMPK/mTOR pathway was involved in the autophagy induced by vitamin E succinate in human gastric cancer SGC-7901 cells, Mol. Cell. Biochem., 2017, 424, 173–183, DOI:10.1007/s11010-016-2853-4.
- L. Liang and L. Qiu, Vitamin E succinate with multiple functions: A versatile agent in nanomedicine-based cancer therapy and its delivery strategies, Int. J. Pharm., 2021, 600, 120457, DOI:10.1016/j.ijpharm.2021.120457.
- T. Weber, H. Dalen, L. Andera, A. Nègre-Salvayre, N. Augé, M. Sticha, A. Lloret, A. Terman, P. K. Witting, M. Higuchi, M. Plasilova, J. Zivny, N. Gellert, C. Weber and J. Neuzil, Mitochondria Play a Central Role in Apoptosis Induced by α-Tocopheryl Succinate, an Agent with Antineoplastic Activity: Comparison with Receptor-Mediated Pro-Apoptotic Signaling, Biochemistry, 2003, 42, 4277–4291, DOI:10.1021/bi020527j.
- R. G. Tuguntaev, S. Chen, A. S. Eltahan, A. Mozhi, S. Jin, J. Zhang, C. Li and X.-J. Liang, P-gp Inhibition and Mitochondrial Impairment by Dual-Functional Nanostructure Based on Vitamin E Derivatives To Overcome Multidrug Resistance, ACS Appl. Mater. Interfaces, 2017, 9, 16900–16912, DOI:10.1021/acsami.7b03877.
- F. A. Boratto, M. S. Franco, A. L. B. Barros, G. D. Cassali, A. Malachias, L. A. M. Ferreira and E. A. Leite, Alpha-tocopheryl succinate improves encapsulation, pH-sensitivity, antitumor activity and reduces toxicity of doxorubicin-loaded liposomes, Eur. J. Pharm. Sci., 2020, 144, 105205, DOI:10.1016/j.ejps.2019.105205.
- W. Jiang, Q. Fan, J. Wang, B. Zhang, T. Hao, Q. Chen, L. Li, L. Chen, H. Cui and Z. Li, PEGylated phospholipid micelles containing D-α-tocopheryl succinate as multifunctional nanocarriers for enhancing the antitumor efficacy of doxorubicin, Int. J. Pharm., 2021, 607, 120979, DOI:10.1016/j.ijpharm.2021.120979.
- L. Manthalkar, S. Bhattacharya, K. Hatware, P. Sreelaya, D. Shah, A. Jain and N. Phatak, Fabrication of D-α-tocopheryl polyethylene glycol 1000 succinates and human serum albumin conjugated chitosan nanoparticles of bosutinib for colon targeting application; in vitro–in vivo investigation, Int. J. Biol. Macromol., 2023, 253, 127531, DOI:10.1016/j.ijbiomac.2023.127531.
- S. Behzadi, V. Serpooshan, W. Tao, M. A. Hamaly, M. Y. Alkawareek, E. C. Dreaden, D. Brown, A. M. Alkilany, O. C. Farokhzad and M. Mahmoudi, Cellular uptake of nanoparticles: journey inside the cell, Chem. Soc. Rev., 2017, 46, 4218–4244 RSC https://rsc.66557.net/en/content/articlehtml/2017/cs/c6cs00636a (accessed November 27, 2023).
- M. A. Anbardan, S. Alipour, G. R. Mahdavinia and P. F. Rezaei, Synthesis of magnetic chitosan/hyaluronic acid/κ-carrageenan nanocarriers for drug delivery, Int. J. Biol. Macromol., 2023, 253, 126805, DOI:10.1016/j.ijbiomac.2023.126805.
- Z. Lu, L. Ma, L. Mei, K. Ren, M. Li, L. Zhang, X. Liu and Q. He, Micellar nanoparticles inhibit the postoperative inflammation, recurrence and pulmonary metastasis of 4T1 breast cancer by blocking NF-κB pathway and promoting MDSCs depletion, Int. J. Pharm., 2022, 628, 122303, DOI:10.1016/j.ijpharm.2022.122303.
- H. Wang, P. Agarwal, S. Zhao, R. X. Xu, J. Yu, X. Lu and X. He, Hyaluronic acid-decorated dual responsive nanoparticles of Pluronic F127, PLGA, and chitosan for targeted co-delivery of doxorubicin and irinotecan to eliminate cancer stem-like cells, Biomaterials, 2015, 72, 74–89, DOI:10.1016/j.biomaterials.2015.08.048.
- T. Nalini, S. K. Basha, A. M. Sadiq and V. S. Kumari, In vitro cytocompatibility assessment and antibacterial effects of quercetin encapsulated alginate/chitosan nanoparticle, Int. J. Biol. Macromol., 2022, 219, 304–311, DOI:10.1016/j.ijbiomac.2022.08.007.
- Y. Zhang, R. Ma, C. You, X. Leng, D. Wang, S. Deng, B. He, Z. Guo, Z. Guan, H. Lei, J. Yu, Q. Zhou, J. Xing and Y. Dong, Hyaluronic acid modified oral drug delivery system with mucoadhesiveness and macrophage-targeting for colitis treatment, Carbohydr. Polym., 2023, 313, 120884, DOI:10.1016/j.carbpol.2023.120884.
- H. Vakilzadeh, J. Varshosaz, M. Dinari, M. Mirian, V. Hajhashemi, N. Shamaeizadeh and H. M. Sadeghi, Smart redox-sensitive micelles based on chitosan for dasatinib delivery in suppressing inflammatory diseases, Int. J. Biol. Macromol., 2023, 229, 696–712, DOI:10.1016/j.ijbiomac.2022.12.111.
- J. Wang, J. Liu, D.-Q. Lu, L. Chen, R. Yang, D. Liu and B. Zhang, Diselenide-crosslinked carboxymethyl chitosan nanoparticles for doxorubicin delivery: Preparation and in vivo evaluation, Carbohydr. Polym., 2022, 292, 119699, DOI:10.1016/j.carbpol.2022.119699.
- A. K. Pandey, N. Piplani, T. Mondal, A. Katranidis and J. Bhattacharya, Efficient delivery of hydrophobic drug, Cabazitaxel, using Nanodisc: A nano sized free standing planar lipid bilayer, J. Mol. Liq., 2021, 339, 116690, DOI:10.1016/j.molliq.2021.116690.
- C. Villanueva, F. Bazan, S. Kim, M. Demarchi, L. Chaigneau, A. Thiery-Vuillemin, T. Nguyen, L. Cals, E. Dobi and X. Pivot, Cabazitaxel: a novel microtubule inhibitor, Drugs, 2011, 71, 1251–1258, DOI:10.2165/11591390-000000000-00000.
- C. J. Paller and E. S. Antonarakis, Cabazitaxel: a novel second-line treatment for metastatic castration-resistant prostate cancer, DDDT, 2011, 5, 117–124, DOI:10.2147/DDDT.S13029.
- G. Nightingale and J. Ryu, Cabazitaxel (jevtana): a novel agent for metastatic castration-resistant prostate cancer, P T, 2012, 37, 440–448 Search PubMed.
- A. Koutras, F. Zagouri, G.-A. Koliou, E. Psoma, I. Chryssogonidis, G. Lazaridis, D. Tryfonopoulos, A. Kotsakis, N. K. Kentepozidis and E. Razis, Phase 2 study of Cabazitaxel as second-line treatment in patients with HER2-negative metastatic breast cancer previously treated with taxanes—a Hellenic Cooperative Oncology Group (HeCOG) Trial, Br. J. Cancer, 2020, 123, 355–361 CrossRef CAS PubMed https://www.nature.com/articles/s41416-020-0909-4 (accessed November 27, 2023).
- N. Kommineni, S. Mahira, A. J. Domb and W. Khan, Cabazitaxel-loaded nanocarriers for cancer therapy with reduced side effects, Pharmaceutics, 2019, 11, 141 CrossRef CAS PubMed https://www.mdpi.com/1999-4923/11/3/141 (accessed August 19, 2024.
- Y. Sun, R. J. Lee, F. Meng, G. Wang, X. Zheng, S. Dong and L. Teng, Microfluidic self-assembly of high cabazitaxel loading albumin nanoparticles, Nanoscale, 2020, 12, 16928–16933, 10.1039/C9NR10941B.
- Vikas, A. K. Mehata, M. K. Viswanadh, A. K. Malik, A. Setia, P. Kumari, S. K. Mahto and M. S. Muthu, EGFR Targeted Redox Sensitive Chitosan Nanoparticles of Cabazitaxel: Dual-Targeted Cancer Therapy, Lung Distribution, and Targeting Studies by Photoacoustic and Optical Imaging, Biomacromolecules, 2023, 24, 4989–5003, DOI:10.1021/acs.biomac.3c00658.
- S. E. Park, N. S. El-Sayed, K. Shamloo, S. Lohan, S. Kumar, M. I. Sajid and R. K. Tiwari, Targeted Delivery of Cabazitaxel Using Cyclic Cell-Penetrating Peptide and Biomarkers of Extracellular Matrix for Prostate and Breast Cancer Therapy, Bioconjugate Chem., 2021, 32, 1898–1914, DOI:10.1021/acs.bioconjchem.1c00319.
- M. Wei, T. Lu, Z. Nong, G. Li, X. Pan, Y. Wei, Y. Yang, N. Wu, J. Huang, M. Pan, X. Li and F. Meng, Reductive response and RGD targeting nano-graphene oxide drug delivery system, J. Drug Delivery Sci. Technol., 2019, 53, 101202, DOI:10.1016/j.jddst.2019.101202.
- E. Lallana, J. M. Rios De La Rosa, A. Tirella, M. Pelliccia, A. Gennari, I. J. Stratford, S. Puri, M. Ashford and N. Tirelli, Chitosan/Hyaluronic Acid Nanoparticles: Rational Design Revisited for RNA Delivery, Mol. Pharmaceutics, 2017, 14, 2422–2436, DOI:10.1021/acs.molpharmaceut.7b00320.
- S. Fengli, Synthesis method of 3,3′-dithiodipropionic acid bis(N-hydroxyl succinimide ester), CN105315191A, 2016. https://patents.google.com/patent/CN105315191A/en (accessed November 26, 2023).
- S. Maya, L. G. Kumar, B. Sarmento, N. Sanoj Rejinold, D. Menon, S. V. Nair and R. Jayakumar, Cetuximab conjugated O-carboxymethyl chitosan nanoparticles for targeting EGFR overexpressing cancer cells, Carbohydr. Polym., 2013, 93, 661–669, DOI:10.1016/j.carbpol.2012.12.032.
- S. Zhai, X. Hu, Y. Hu, B. Wu and D. Xing, Visible light-induced crosslinking and physiological stabilization of diselenide-rich nanoparticles for redox-responsive drug release and combination chemotherapy, Biomaterials, 2017, 121, 41–54, DOI:10.1016/j.biomaterials.2017.01.002.
- F. Chen, J. Zhang, L. Wang, Y. Wang and M. Chen, Tumor pHe-triggered charge-reversal and redox-responsive nanoparticles for docetaxel delivery in hepatocellular carcinoma treatment, Nanoscale, 2015, 7, 15763–15779, 10.1039/C5NR04612B.
- S. Yin, J. Huai, X. Chen, Y. Yang, X. Zhang, Y. Gan, G. Wang, X. Gu and J. Li, Intracellular delivery and antitumor effects of a redox-responsive polymeric paclitaxel conjugate based on hyaluronic acid, Acta Biomater., 2015, 26, 274–285, DOI:10.1016/j.actbio.2015.08.029.
- X. Zhang, Y. Wang, G. Wei, J. Zhao, G. Yang and S. Zhou, Stepwise dual targeting and dual responsive polymer micelles for mitochondrion therapy, J. Controlled Release, 2020, 322, 157–169 CrossRef CAS PubMed https://www.sciencedirect.com/science/article/pii/S0168365920301632 (accessed November 28, 2023).
- H. Wang, W. Shi, D. Zeng, Q. Huang, J. Xie, H. Wen, J. Li, X. Yu, L. Qin and Y. Zhou, pH-activated, mitochondria-targeted, and redox-responsive delivery of paclitaxel nanomicelles to overcome drug resistance and suppress metastasis in lung cancer, J. Nanobiotechnol., 2021, 19, 152, DOI:10.1186/s12951-021-00895-4.
- H. Sun, S. Li, W. Qi, R. Xing, Q. Zou and X. Yan, Stimuli-responsive nanoparticles based on co-assembly of naturally-occurring biomacromolecules for in vitro photodynamic therapy, Colloids Surf., A, 2018, 538, 795–801, DOI:10.1016/j.colsurfa.2017.11.072.
- K. Vaghasiya, E. Ray, A. Sharma, O. P. Katare and R. K. Verma, Matrix Metalloproteinase-Responsive Mesoporous Silica Nanoparticles Cloaked with Cleavable Protein for “Self-Actuating” On-Demand Controlled Drug Delivery for Cancer Therapy, ACS Appl. Bio Mater., 2020, 3, 4987–4999, DOI:10.1021/acsabm.0c00497.
- P. Patel and J. Shah, Protective effects of hesperidin through attenuation of Ki67 expression against DMBA-induced breast cancer in female rats, Life Sci., 2021, 285, 119957, DOI:10.1016/j.lfs.2021.119957.
- K. Kolanjiappan and S. Manoharan, Chemopreventive efficacy and anti-lipid peroxidative potential of Jasminum grandiflorum Linn. on 7,12-dimethylbenz(a)anthracene-induced rat mammary carcinogenesis, Fundam. Clin. Pharmacol., 2005, 19, 687–693, DOI:10.1111/j.1472-8206.2005.00376.x.
- S. Li, P. E. Saw, C. Lin, Y. Nie, W. Tao, O. C. Farokhzad, L. Zhang and X. Xu, Redox-responsive polyprodrug nanoparticles for targeted siRNA delivery and synergistic liver cancer therapy, Biomaterials, 2020, 234, 119760, DOI:10.1016/j.biomaterials.2020.119760.
- Z. Wang, X. Xue, Y. He, Z. Lu, B. Jia, H. Wu, Y. Yuan, Y. Huang, H. Wang, H. Lu, K. S. Lam, T.-Y. Lin and Y. Li, Novel Redox-Responsive Polymeric Magnetosomes with Tunable Magnetic Resonance Property for In Vivo Drug Release Visualization and Dual-Modal Cancer Therapy, Adv. Funct. Mater., 2018, 28, 1802159, DOI:10.1002/adfm.201802159.
- K. Xiao, Q. Liu, N. Suby, W. Xiao, R. Agrawal, M. Vu, H. Zhang, Y. Luo, Y. Li and K. S. Lam, LHRH-Targeted Redox-Responsive Crosslinked Micelles Impart Selective Drug Delivery and Effective Chemotherapy in Triple-Negative Breast Cancer, Adv. Healthcare Mater., 2021, 10, 2001196, DOI:10.1002/adhm.202001196.
- R. Wang, H. Yang, A. R. Khan, X. Yang, J. Xu, J. Ji and G. Zhai, Redox-responsive hyaluronic acid-based nanoparticles for targeted photodynamic therapy/chemotherapy against breast cancer, J. Colloid Interface Sci., 2021, 598, 213–228, DOI:10.1016/j.jcis.2021.04.056.
- T. G. Buu, T. T. P. Nhung and N. T. Trang, Characterization of Crilin and Nanocurcumin's Synergistic Effect on Treatment for 7.12-Dimethylbenz[a]anthracene (DMBA)-Induced Breast Cancer Mice, VNU J. Sci.: Med. Pharm. Sci., 2018, 34 DOI:10.25073/2588-1132/vnumps.4099.
- V. Akhouri, M. Kumari and A. Kumar, Therapeutic effect of Aegle marmelos fruit extract against DMBA induced breast cancer in rats, Sci. Rep., 2020, 10, 18016, DOI:10.1038/s41598-020-72935-2.
- Y. Guo, M. Chu, S. Tan, S. Zhao, H. Liu, B. O. Otieno, X. Yang, C. Xu and Z. Zhang, Chitosan-g-TPGS Nanoparticles for Anticancer Drug Delivery and Overcoming Multidrug Resistance, Mol. Pharmaceutics, 2014, 11, 59–70, DOI:10.1021/mp400514t.
- B. Wei, M. He, X. Cai, X. Hou, Y. Wang, J. Chen, M. Lan, Y. Chen, K. Lou and F. Gao, Vitamin E succinate-grafted-chitosan/chitosan oligosaccharide mixed micelles loaded with C-DMSA for Hg2+ detection and detoxification in rat liver, Int. J. Nanomed., 2019, 14, 6917, DOI:10.2147/IJN.S213084.
- P. Chand, H. Kumar, N. Badduri, N. V. Gupta, V. G. Bettada, S. V. Madhunapantula, S. S. Kesharwani, S. Dey and V. Jain, Design and evaluation of cabazitaxel loaded NLCs against breast cancer cell lines, Colloids Surf., B, 2021, 199, 111535, DOI:10.1016/j.colsurfb.2020.111535.
- W. Cabri, D. Ciceri, L. Domenighini, A. Gambini and F. Peterlongo, A crystalline anhydrous form of cabazitaxel, process for the preparation and pharmaceutical compositions thereof, EP3060556A1, 2016. https://patents.google.com/patent/EP3060556A1/en (accessed November 26, 2023).
- A. K. Jangid, D. Pooja, P. Jain, S. V. K. Rompicharla, S. Ramesan and H. Kulhari, A nanoscale, biocompatible and amphiphilic prodrug of cabazitaxel with improved anticancer efficacy against 3D spheroids of prostate cancer cells, Mater. Adv., 2020, 1, 738–748, 10.1039/D0MA00189A.
- Q. Chen, C. Jia, Y. Xu, Z. Jiang, T. Hu, C. Li and X. Cheng, Dual-pH responsive chitosan nanoparticles for improving in vivo drugs delivery and chemoresistance in breast cancer, Carbohydr. Polym., 2022, 290, 119518, DOI:10.1016/j.carbpol.2022.119518.
- U. de J. Martín-Camacho, N. Rodríguez-Barajas, J. A. Sánchez-Burgos and A. Pérez-Larios, Weibull β value for the discernment of drug release mechanism of PLGA particles, Int. J. Pharm., 2023, 640, 123017, DOI:10.1016/j.ijpharm.2023.123017.
- S. Zhang, X. Fan, G. Zhang, W. Wang and L. Yan, Preparation, characterization, and in vitro release kinetics of doxorubicin-loaded magnetosomes, J. Biomater. Appl., 2022, 36, 1469–1483, DOI:10.1177/08853282211060544.
- A.-M. Craciun, M. L. Barhalescu, M. Agop and L. Ochiuz, Theoretical Modeling of Long-Time Drug Release from Nitrosalicyl-Imine-Chitosan Hydrogels through Multifractal Logistic Type Laws, Comput. Math. Methods Med., 2019, 2019, 4091464, DOI:10.1155/2019/4091464.
- K. Ghosal, A. Chandra, R. Rajabalaya, S. Chakraborty and A. Nanda, Mathematical modeling of drug release profiles for modified hydrophobic HPMC based gels, Die Pharmazie-An Int. J. Pharm. Sci., 2012, 67, 147–155 CAS https://www.ingentaconnect.com/content/govi/pharmaz/2012/00000067/00000002/art00009 (accessed February 9, 2024).
- C. Corsaro, G. Neri, A. M. Mezzasalma and E. Fazio, Weibull Modeling of Controlled Drug Release from Ag-PMA Nanosystems, Polymers, 2021, 13, 2897, DOI:10.3390/polym13172897.
- J. B. Minari, G. O. Ogar and A. J. Bello, Antiproliferative potential of aqueous leaf extract of Mucuna pruriens on DMBA-induced breast cancer in female albino rats, Egypt. J. Med. Hum. Genet., 2016, 17, 331–343, DOI:10.1016/j.ejmhg.2015.12.008.
- B. Singh, N. K. Bhat and H. K. Bhat, Partial Inhibition of Estrogen-Induced Mammary Carcinogenesis in Rats by Tamoxifen: Balance between Oxidant Stress and Estrogen Responsiveness, PLoS One, 2011, 6, e25125, DOI:10.1371/journal.pone.0025125.
- W. Wang, D. Luo, J. Chen, J. Chen, Y. Xia, W. Chen and Y. Wang, Amelioration of cyclophosphamide-induced myelosuppression during treatment to rats with breast cancer through low-intensity pulsed ultrasound, Biosci. Rep., 2020, 40, BSR20201350, DOI:10.1042/BSR20201350.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma00556b |
| This journal is © The Royal Society of Chemistry 2024 |