DOI:
10.1039/D4MA00610K
(Paper)
Mater. Adv., 2024,
5, 8787-8797
Fire-resistant propargyl ether networks derived from bio-based hydroxycinnamic acids†
Received
11th June 2024
, Accepted 3rd September 2024
First published on 12th September 2024
Abstract
Three bio-based propargyl ether thermosetting resins with trans-stilbene cores were synthesized from p-coumaric (CD), ferulic (FD), and sinapic (SD) acid, respectively. Differential scanning calorimetry (DSC) analysis of these materials indicated modest processability due to high melting points, short processing windows and large exotherms. To address this issue, a fourth resin with a more flexible bridging group (TD) was synthesized from p-coumaric acid and used as a blending agent. In parallel, CD was photochemically isomerized to the cis-isomer (PD) and blends of CD:PD were prepared. Cross-linked networks derived from the resins exhibited glass transition temperatures (Tgs) ranging from 285–330 °C (storage modulus) and char yields from 27–59% at 1000 °C under N2. The processable resin blends exhibited exceptional thermal stability due to a higher degree of cross-linking enabled by the structural diversity of the blends. The fire resistance of the networks was evaluated through microscale combustion calorimetry. The networks exhibited heat release capacity (HRC) values ranging from 43–103 J g−1 K−1, which classified them as either non-ignitable or self-extinguishing materials. The results demonstrate that abundant, bio-based hydroxycinnamic acids can serve as platform chemicals for the preparation of thermally stable, fire-resistant networks for aerospace applications.
Introduction
Thermosetting polymers are a class of materials that undergo cross-linking reactions to form robust, three-dimensional network structures. Typical thermoset networks are permanently cross-linked and cannot be melted or reformed.1 This characteristic makes them ideal for applications that require mechanical integrity across a broad temperature range and resistance to heat and chemicals. These networks are broadly used in composite materials, coatings, and adhesives.2–4 The majority of high-performance thermosetting resins including epoxies, cyanate esters, benzoxazines, and phthalonitriles, are derived from petroleum-based feedstocks.5–8 In contrast, a wide variety of monomers with unique functionality can be derived from plant extracts, produced via fermentation, or generated from crude biomass sources through valorization and chemical synthesis. Leveraging the diversity of bio-based molecules enables the discovery of new materials with novel properties from sustainable carbon sources.9 Much of the research on this topic is focused on the exploitation of bio-based polyphenolic compounds.9–16 For example, our group has previously studied thermosets derived from resveratrol and eugenol.11,12,17
In addition to well-defined phenolic precursors, crude bio-feedstocks like lignin are of interest as abundant and low-cost materials that can be leveraged for material applications.18,19 Lignin is a complex polymer found in the cell walls of plants and is the second most abundant biopolymer on Earth.20,21 Lignin-based thermosetting polymers have been studied and exploit in a number of applications.22 For example, lignin has been used extensively as a filler or reinforcing agent in epoxy composites, providing enhanced mechanical strength, improved thermal stability, and reduced cost.23–25 Recently, Song and co-workers described a lignin-based epoxy resin with a high glass transition temperature (193 °C) and outstanding mechanical properties (tensile strength = 98.3 MPa and flexural strength = 158.9 MPa).26 Lignin has also been chemically modified to produce polyols that were then cross-linked with isocyanates to form polyurethane foams. These materials exhibited improved thermal insulation properties, reduced flammability, and enhanced sustainability compared to traditional polyurethane foams.27–29
Valorization of lignin to generate well-defined phenolic compounds followed by chemical synthesis is an important route for the preparation of polyphenols. Examples include the synthesis of bisphenols from vanillin, creosol, guaiacol, and propylguaiacol.30–33 More recently, we developed a highly efficient approach to the synthesis of bisphenols from hydroxycinnamic acids including, p-coumaric, ferulic, and sinapic acids.34 These acids can be readily obtained through lignin depolymerization processes such as treatment with alkali or enzymatic hydrolysis followed by solvent extraction. Alternatively, molecules like p-coumaric acid can be efficiently produced from sugars through a fermentation process.35,36 The acids can then be converted to stilbene-based bisphenols through a process consisting of thermal decarboxylation followed by [Ru]-catalyzed metathesis.34
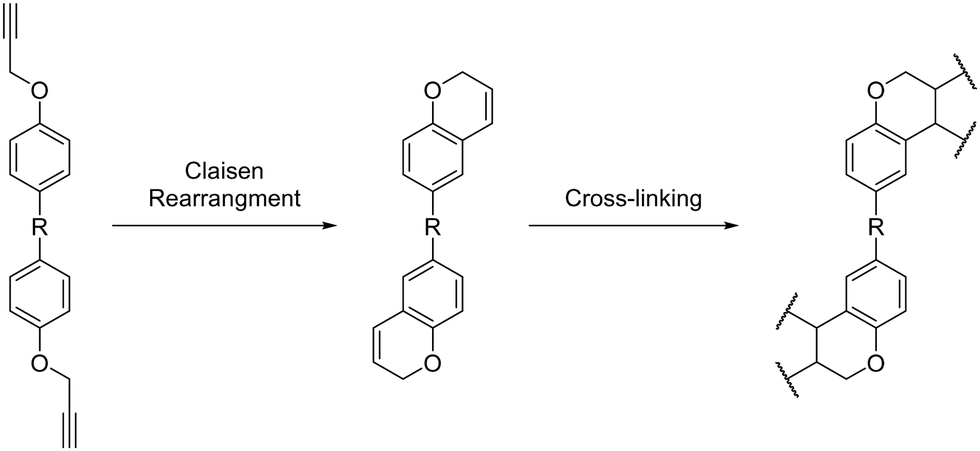
Propargyl ether monomers have garnered interest in recent years due to their ability to generate polymer networks with glass transition temperatures well above 300 °C while maintaining processability comparable to that of ubiquitous epoxy resins.37,38 For example, propargyl ether networks derived from biosynthetic resveratrol exhibited low melting points (39–81 °C, Tgs ranging from 358 °C to >400 °C and char yields at 1000 °C ranging from 54–66%.17 Propargyl ether monomers typically cure through a Claisen rearrangement, which forms a chromene ring that subsequently cross-links to form the thermoset network (Scheme 1).
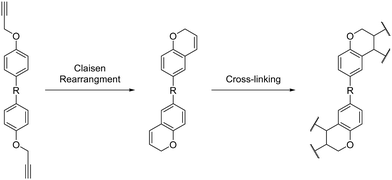 |
| | Scheme 1 General curing mechanism for bis(propargyl ether) thermosets. | |
To explore the properties of propargyl ethers synthesized from sustainable bisphenols the current work is focused on the synthesis, characterization, and cure chemistry of stilbene-based propargyl ethers derived from p-coumaric, ferulic, and sinapic acid. The bisphenols were converted to bis(propargyl ethers) through a Williamson-ether synthesis and then thermally cross-linked to form networks with high thermal stabilities and flame retardancy. To enhance the processability of the resin systems, blends with lower melting monomers derived from p-coumaric acid were prepared. The thermal properties of the networks were evaluated by differential scanning calorimetry (DSC), thermogravimetric analysis (TGA), thermomechanical analysis (TMA), and microscale combustion calorimetry (MCC). The properties of these materials were then compared to conventional networks prepared from petrochemical sources.
Experimental methods
Materials and methods
Unless otherwise noted, all reagents were purchased and used as received from the manufacturer without further purification. (E)-4,4′-(ethene-1,2-diyl)diphenol, (E)-4,4′-(ethane-1,2-diyl)bis(2-methoxyphenol), (E)-4,4′-(ethane-1,2-diyl)bis(2,6-dimethoxyphenol), and (E)-4,4′-(but-1-ene-1,3-diyl)diphenol were all synthesized according to procedures previously reported by our group.341H and 13C NMR spectra were recorded on a JEOL 400 MHz spectrometer. All NMR chemical shifts are reported in ppm downfield from tetramethylsilane and are referenced relative to the NMR solvent according to the literature values: 1H NMR (CDCl3 = 7.26 ppm, DMSO-d6 = 2.50 ppm, (CD3)2CO = 2.05 ppm); 13C NMR (CDCl3 = 77.16 ppm, DMSO-d6 = 39.52 ppm). Fourier transform infrared (FTIR) spectroscopy was performed using a Thermo Scientific Nicolet 6700 FTIR spectrometer equipped with a liquid nitrogen cooled MCTA detector. Experiments were performed using an attenuated total reflectance (ATR) smart accessory with a germanium window. Each FTIR spectrum is an average of 16 scans. Differential scanning calorimetry (DSC) analysis was conducted on a DSC Q200 (TA Instruments) under N2 at 50 mL min−1. Approximately 2–3 mg of sample were tested under a heating scan from −75 to 400 °C with a heating rate of 10 °C min−1. Thermogravimetric analysis (TGA) was performed on cured samples with a TGA Q5000 (TA Instruments) under N2 flow from 24 to 1000 °C with a heating rate of 10 °C min−1. Thermomechanical analysis (TMA) was performed with monolithic pucks, ∼300 mg, on a Discovery TMA 450 (TA Instruments) under N2 flow from 25 to 400 °C at 5 °C min−1. Microscale combustion calorimetry (MCC) studies were performed on an FTT FAA microcalorimeter with ultra-high purity N2 and 99.5% pure O2 gas. Cured samples were broken into small specimens (2–10 mg) with random shapes, which were then analyzed in triplicate by the MCC instrument at a constant heating rate of 1 °C s−1 according to ASTM D 7309 Method A. MCC tests were conducted with a nitrogen flow rate of 80 mL min−1 and oxygen flow rate of 20 mL min−1. The maximum heat release rate (Qmax), total heat release (THR), and heat release capacity (ηc) of the samples were obtained according to the method.
Synthesis of (E)-1,2-bis(4-prop-2-yn-1-yloxy)phenyl)ethane (CD)
To a solution of (E)-4,4′-(ethene-1,2-diyl)diphenol (0.804 g, 3.8 mmol) in 5 mL of acetone was added propargyl bromide (0.901 g, 574 μL, 7.6 mmol) and potassium carbonate (2 g, 14 mmol). The resulting red/orange solution was then heated to reflux and stirred for two days under N2. The resulting solids were filtered, and the filtrate placed in a −30 °C freezer overnight. The solids were taken up in 50 mL of CH2Cl2 and 50 mL of DI water. The organic layer was then washed with 3 × 100 mL DI water and dried over anhydrous MgSO4. Concentrating under reduced pressure yielded 0.285 g of the product as an off-white solid. The original filtrate was removed from the freezer and the crystals in the flask were collected to yield another 0.140 g of product (m.p. 175–179 °C). The total yield of CD was 0.425 g (39%). Single crystals suitable for X-ray crystallography were grown by slow evaporation from DCM (CCDC deposition number 2347707). 1H NMR (400 MHz, DMSO-d6) δ 7.52 (d, J = 8.7 Hz, 4H, Ar–H), 7.06 (s, 2H, C(sp2)–H), 6.98 (d, J = 8.9 Hz, 4H, Ar–H), 4.81 (s, 4H, CH2), 3.57 (s, 2H, C(sp)–H), 13C NMR (100 MHz, DMSO-d6) δ 157.1, 131.2, 127.9, 126.6, 115.6, 79.8, 78.8, 55.9.
Synthesis of (E)-1,2-bis(3-methoxy-4-(prop-2-yn-1-yloxy)phenyl)ethane (FD)
To a solution of (E)-4,4′-(ethane-1,2-diyl)bis(2-methoxyphenol) (1 g, 3.7 mmol) in 5 mL of acetone was added propargyl bromide (0.874 g, 557 μL, 7.3 mmol) and potassium carbonate (2 g, 14 mmol). The reaction and workup was conducted in a similar manner to that described above for CD to yield 0.533 g (42% yield) of FD as a light brown solid (m.p. 155–162 °C). 1H NMR (400 MHz, DMSO-d6) δ 7.23 (s, 2H, Ar–H), 7.09 (d, J = 8.5 Hz, 4H, Ar–H), 7.04 (d, J = 8.5 Hz 2H, C(sp2)–H), 4.78 (s, 4H, CH2), 3.82 (s, 6H, CH3), 3.56 (s, 2H, C(sp)–H), 13C NMR (100 MHz, CDCl3) δ 149.9, 146.5, 132.0, 127.1, 119.4, 114.5, 109.3, 78.6, 75.9, 56.9, 56.0.
Synthesis of (E)-1,2-bis(3,5-dimethoxy-4-(prop-2-yn-1-yloxy)phenyl)ethane (SD)
To a solution of (E)-4,4′-(ethane-1,2-diyl)bis(2,6-dimethoxyphenol) (1 g, 3.0 mmol) in 5 mL of acetone was added propargyl bromide (0.716 g, 456 μL, 7.3 mmol) and potassium carbonate (1.66 g, 12 mmol). The resulting red/orange solution was then heated to reflux and stirred for two days under N2. The resulting solids were taken up in 50 mL of CH2Cl2 and 50 mL of DI water. The organic layer was then washed with 3 × 100 mL DI water and dried over anhydrous MgSO4. Removal of the solvent under reduced pressure provided 0.85 g (69% yield) of SD as a red/orange solid (m.p. 165–170 °C). 1H NMR (400 MHz, DMSO-d6) δ 7.18 (s, 2H, C(sp2)–H), 6.92 (s, 4H, Ar–H), 4.60 (s, 4H, CH2), 3.82 (s, 12H, CH3), 3.44 (s, 2H, C(sp)–H), 13C NMR (100 MHz, DMSO-d6) δ 153.8, 135.1, 133.9, 128.6, 104.1, 80.4, 78.2, 59.7, 56.4.
Synthesis of (E)-4,4′-(but-1-ene-1,3-diyl)bis((prop-2-yn-1-yloxy)benzene) (TD)
To a 250 mL round bottom flask under a N2 atmosphere and equipped with a condenser, (E)-4,4′-(but-1-ene-1,3-diyl)diphenol (4.15 g, 17 mmol), propargyl bromide (4.93 g, 41.4 mol), potassium carbonate (5.96 g, 43.1 mmol), and acetone (85 mL) were added. The mixture was vigorously stirred with a magnetic stirring bar and heated to reflux for three days. The solution was filtered on a fritted funnel and the solids were washed with acetone. The solvent was removed from the filtrate under reduced pressure to yield the product as a thick orange oil (5.09 g, 93% yield). 1H NMR ((CD3)2CO, 400 MHz) δ: 7.34 (d, J = 8.9 Hz, 2H, Ar–H), 7.22 (d, J = 8.7 Hz, 2H, Ar–H), 6.91 (dd, J1 = 8.6 Hz J2 = 6.1 Hz 4H, Ar–H), 6.35 (m, 2H, C = H), 4.77 (dd, J1 = 7.0 Hz J2 = 2.4 Hz, 4H, CH2), 3.56 (m, 3H, C(sp)–H, CH), 1.36 (d, J = 6.9 Hz, 3H, CH3) 13C NMR (CDCl3, 100 MHz) δ: 156.9, 156.0, 139.0, 134.1, 131.0, 128.5, 127.6, 116.3, 115.4, 115.3, 79.9, 79.7, 78.7, 78.6, 56.3, 55.92, 55.90, 41.7, 22.1.
Synthesis of (Z)-1,2-bis(4-prop-2-yn-1-yloxy)phenyl)ethane (PD)
500 mg of CD were added to a 100 mL quartz flat-bottom flask. Dry MeCN (50 mL) was then added and the mixture sparged with N2 for one h. The flask was then placed in a Luzchem® LZC-4V photoreactor fitted with fourteen 355 nm, 8 W fluorescent lamps and irradiated with stirring for 18 h. Evaporation of the MeCN resulted in a viscous, gelatinous solid (m.p. 31–36 °C) that was confirmed to be a 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of cis
:1 mixture of cis![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) :trans isomers (Fig. S38, ESI†). 1H NMR (400 MHz, DMSO-d6) δ 7.23 (d, J = 8.7 Hz. 4H, Ar–H), 6.90 (d, J = 8.7 Hz, 4H, Ar–H), 6.49 (s, 2H, C(sp2)–H), 4.70 (s, 4H, CH2), 3.15 (s, 2H, C(sp)–H).
:trans isomers (Fig. S38, ESI†). 1H NMR (400 MHz, DMSO-d6) δ 7.23 (d, J = 8.7 Hz. 4H, Ar–H), 6.90 (d, J = 8.7 Hz, 4H, Ar–H), 6.49 (s, 2H, C(sp2)–H), 4.70 (s, 4H, CH2), 3.15 (s, 2H, C(sp)–H).
Synthesis of 1,3-dimethoxy-2-(prop-2-yn-1-yloxy)benzene (MPE)
2,6-Dimethoxyphenol (10 g, 0.052 mol), propargyl bromide (10 mL, 0.13 mol), potassium carbonate (36 g, 0.26 mol), and 400 mL acetone were added to a 1 L round bottom flask. The reaction mixture was heated to reflux and stirred for 30 h. The reaction solution was then cooled to room temperature and solids collected on a fritted funnel. The filtrate was then concentrated under reduced pressure and the resulting residue purified by silica flash chromatography using a 9:1 hexane/EtOAc mobile phase to yield the product as a yellow oil (9.0 g, 72% yield). 1H NMR (400 MHz, DMSO-d6) δ 6.96 (t, J = 8.3 Hz, 1H, Ar–H, 6.53 (d, J = 8.5 Hz, 2H, Ar–H), 4.66 (d, J = 2.5 Hz, 2H, CH2), 3.79 (s, 6H, CH3), 2.40 (t, J = 2.4 Hz, 1H, C(sp)–H).
Fabrication of monolithic samples
Cross-linked propargyl ether networks were formed by first pressing the monomers into pellets using a hydraulic press (∼10 tons, 10 min). The pellets were then placed into Cole-Parmer 0.35 mL aluminum weigh dishes. Preparation of 3:7, 1:1, and 7:3 cis:trans mixtures was accomplished by adding the necessary amount of pure trans isomer to the 9:1 mixture in DCM and then concentrating the solution to dryness. NMR spectroscopy was used to confirm the new cis:trans composition. Cure protocols for the preparation of the monolithic samples are summarized in Table 1.
Table 1 Cure protocols for bis(propargyl ether) thermoset networks
| Compound |
Cure protocol |
|
CD
|
200 °C (2 h), 230 °C (2 h), 260 °C (3 h), 280 °C (2 h) |
|
TD and 7:3 TD:CD |
200 °C (2 h), 220 °C (4 h), 240 °C (4 h), 280 °C (2 h) |
| 7:3 PD:CD |
80 °C (2 h), 160 °C (1 h), 200 °C (2 h), 260 °C (2 h), 280 °C (2 h) |
Results and discussion
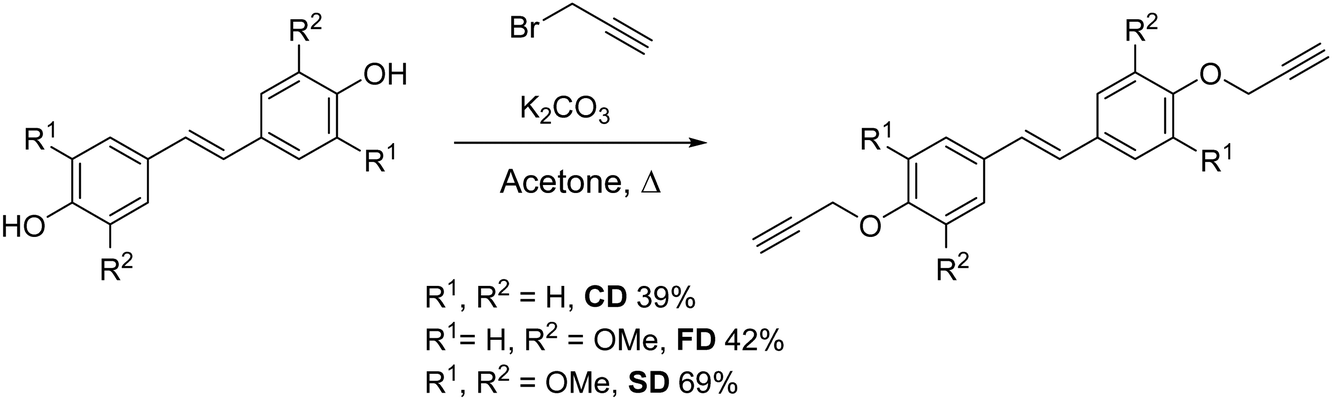
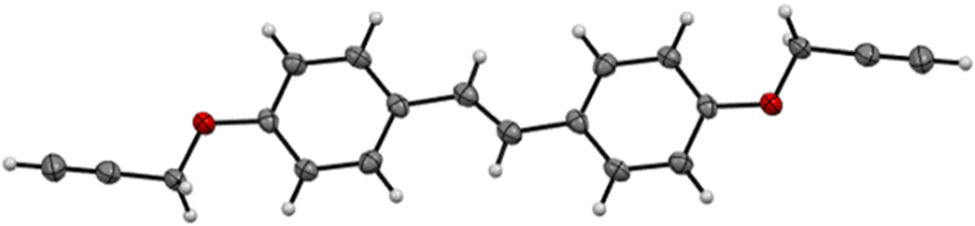
Three bis(propargyl ethers) were synthesized from hydroxycinnamic acid-derived bisphenols reported in our previous work.34 The bisphenols were allowed to react in the presence of K2CO3 to generate the propargyl ether monomers in 39–69% isolated yield (Scheme 2). The NMR spectra for CD, FD, and SD were consistent with the expected trans-stilbene structures (Fig. S30–S35, ESI†). In addition, a single crystal X-ray structural analysis of CD confirmed the NMR results and revealed staggered π-stacking between the aryl rings (Fig. 1 and Fig. S27, ESI†).
 |
| | Scheme 2 Synthesis of bis(propargyl ethers) from bio-derived, stilbene-based bisphenols. | |
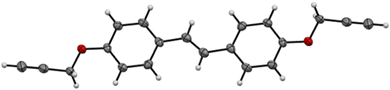 |
| | Fig. 1 X-ray structure of CD with thermal ellipsoids at 50% probability. | |
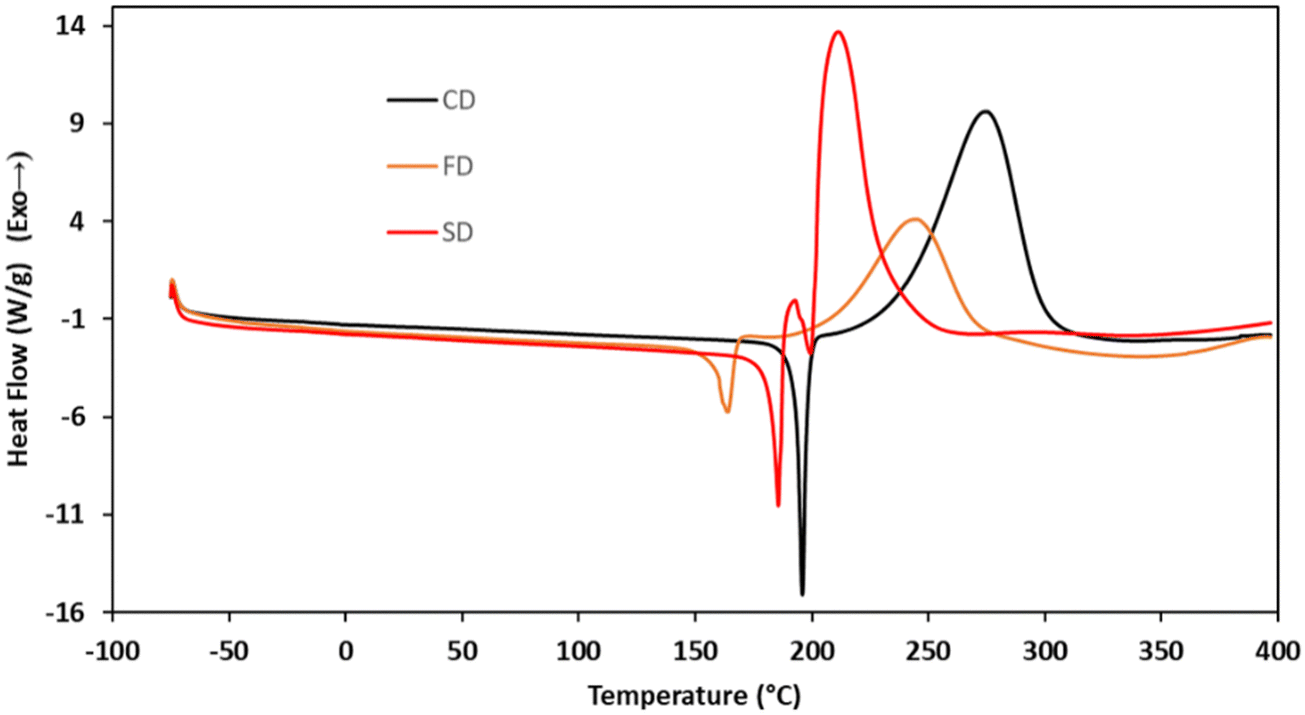
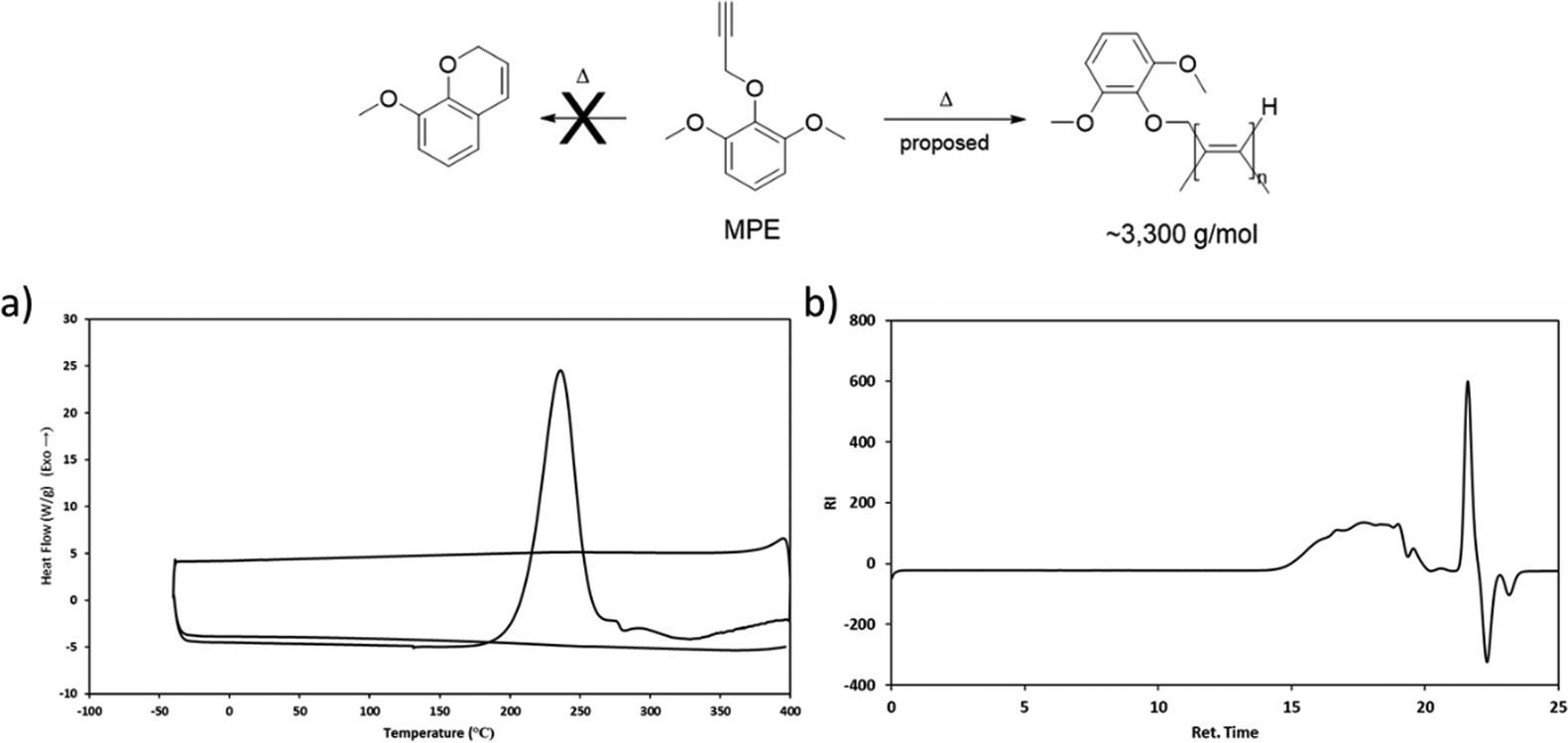
To elucidate the cure chemistry of CD, FD, and SD, DSC experiments were performed. CD melted with an onset temperature of 184 °C (peak at 195 °C), which was immediately followed by an exothermic cure reaction with a peak at 275 °C (Fig. 2, black). FD demonstrated a lower melting point with an onset temperature at 130 °C (peak at 164 °C), followed by an exotherm with an onset of 186 °C (peak at 245 °C) (Fig. 2, orange). SD had a melting point intermediate between CD and FD with an onset at 173 °C (peak at 186 °C) (Fig. 2, red). The decreased melting points of FD and SD compared to CD are attributed to the presence of the methoxy groups on the aryl rings. In the case of FD, the asymmetry induced by the ortho-methoxy group results in the lowest melting point of the propargyl ether monomers.39 A small exothermic event at 193 °C of unknown origin, followed by an exothermic cure reaction (peak at 211 °C) was observed for SD. Due to the presence of the ortho-methoxy groups, SD is unable to cross-link through a chromene ring like the other monomers. This would suggest that the exothermic event with sharp onset at ∼200 °C is a polymerization reaction that is not common for propargyl ethers. To probe this phenomenon, a monofunctional propargyl ether with two methoxy-groups in the positions ortho to the propargyl ether group (MPE) was synthesized (Fig. 3). DSC analysis of MPE indicated a large exotherm peaking at 240 °C (Fig. 3a), similar to that observed for SD. Further, the exothermic events for both SD and MPE had much steeper onsets compared to CD or FD, providing additional confirmation of a different cure mechanism for SD and MPE.
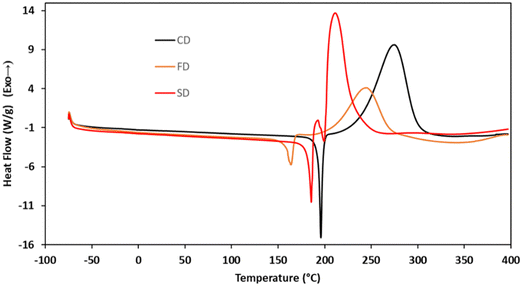 |
| | Fig. 2 DSC traces for monomers CD, FD, and SD. | |
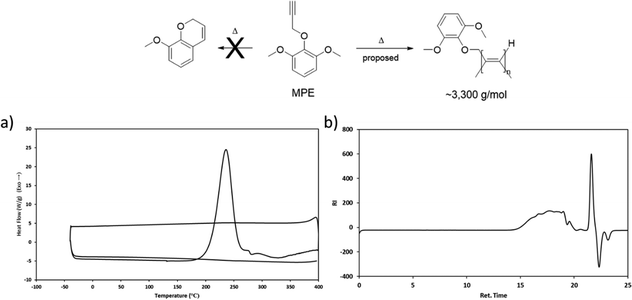 |
| | Fig. 3 (a) DSC profile of MPE and (b) GPC trace of cured MPE. | |
Polymerized samples of MPE were prepared utilizing conditions obtained from the DSC results. GPC analysis of these cured samples found an Mw of 3300 g mol−1, Mn of 1605 g mol−1 and PDI of 2.08, thus confirming the oligomerization of the propargyl ether through a mechanism other than chromene formation (Fig. 3b). Based on literature precedent, we hypothesize that both MPE and SD undergo a linear insertion polymerization at the terminal alkynes (Fig. 3).40 Given the close proximity of the exothermic cure reaction in SD and MPE to conventional cross-linking reactions in CD and FD, it seems likely that the general cross-linking mechanism of propargyl ethers is more complicated than typically reported. The evidence suggests that this class of thermosetting resins cross-links through a combination of linear insertion of alkynes, polymerization via chromene formation, and further cross-linking through residual alkenes.
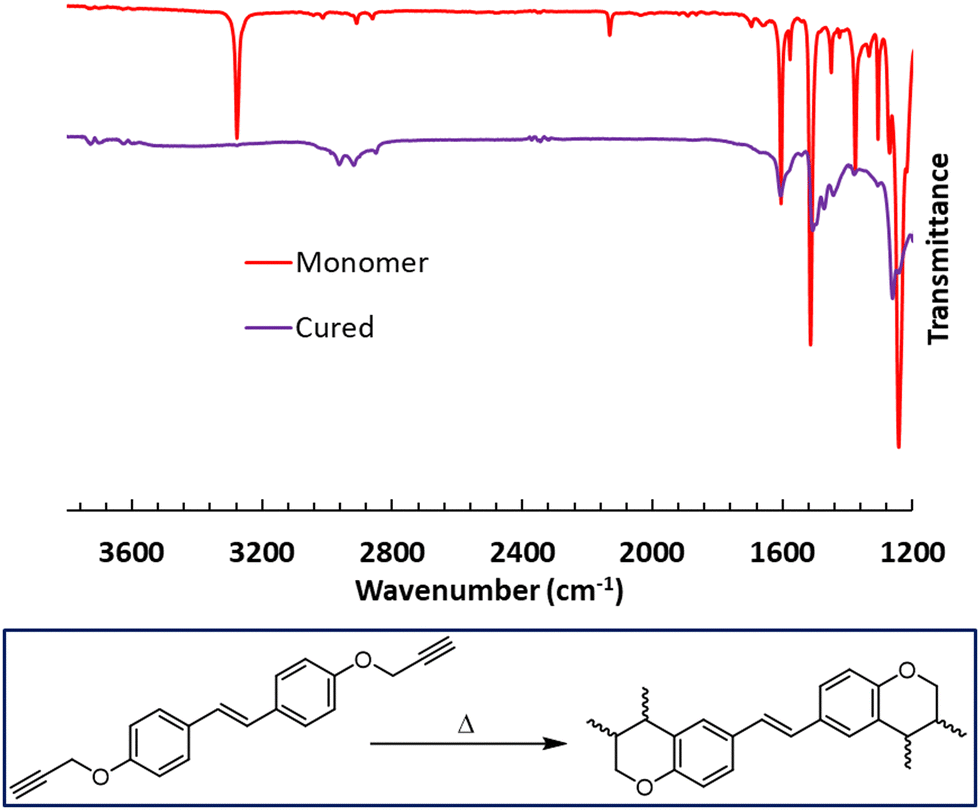
The cross-linking of the bis(propargyl ether) monomers was confirmed through FTIR spectroscopy by observing the loss of the –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–H stretch at ∼3300 cm−1 and the –CC– stretch at ∼2100 cm−1 (Fig. S28 and S29, ESI†). A comparative example for CD can be found in Fig. 4. The IR spectrum of cross-linked SD (Fig. S29, ESI†) confirms the consumption of the alkyne group during the alternative cross-linking reaction.
C–H stretch at ∼3300 cm−1 and the –CC– stretch at ∼2100 cm−1 (Fig. S28 and S29, ESI†). A comparative example for CD can be found in Fig. 4. The IR spectrum of cross-linked SD (Fig. S29, ESI†) confirms the consumption of the alkyne group during the alternative cross-linking reaction.
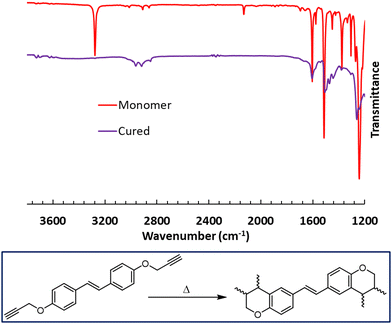 |
| | Fig. 4 FTIR spectra comparing CD and cross-linked CD. | |
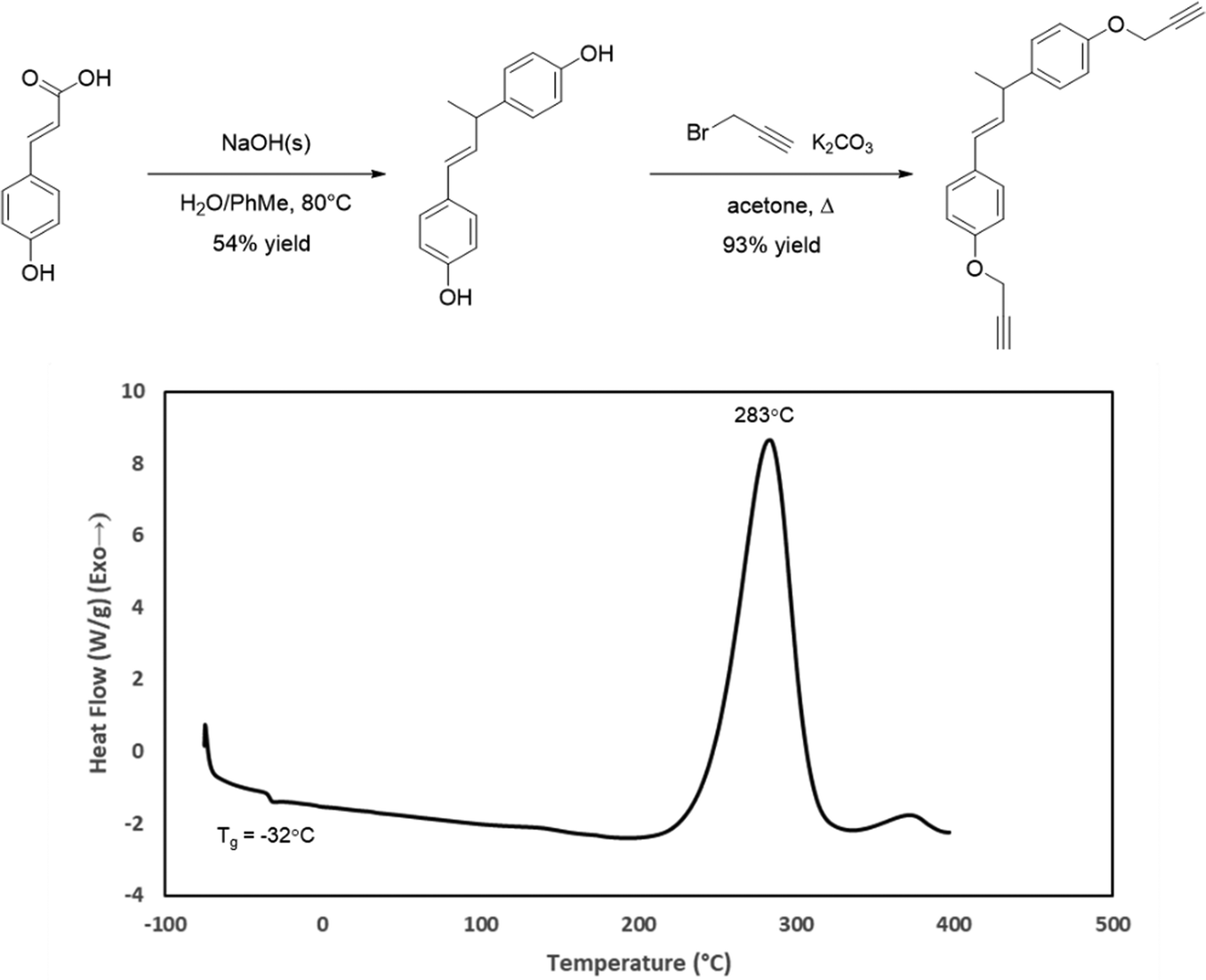
The DSC profiles for CD, FD, and SD indicated that they would be difficult to cure due to exceptionally short processing windows (∼0–10 °C) and large cure exotherms. In an effort to improve the utility of the resins for composite fabrication, we attempted to decrease the melting points and increase the processing window by incorporating a more flexible propargyl ether as a blending agent. In our previous work, we synthesized a bisphenol from p-coumaric acid by decarboxylation followed by thermal dimerization.34 To explore this bisphenol as a network scaffold, a propargyl ether was synthesized from the thermal dimer and designated as TD. DSC analysis of TD showed a Tg at −32 °C and a cure exotherm initiating around 200 °C (Fig. 5). This exceptional processing window suggested that TD would be useful as a blending agent. Blends of TD with CD ranging from 90–70% TD were all homogenous viscous liquids upon heating. However, blends with higher proportions of TD underwent phase separation. The resulting mixtures became unworkable clay-like pastes, even at elevated temperatures. Based on this result, the 7:3 TD:CD blend was selected for further study.
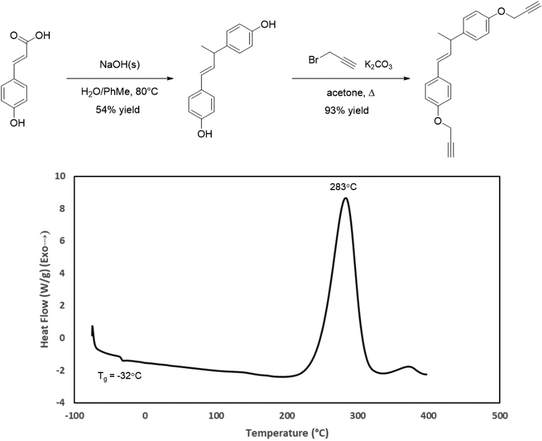 |
| | Fig. 5 Synthesis of TD from p-coumaric acid and its DSC profile. | |
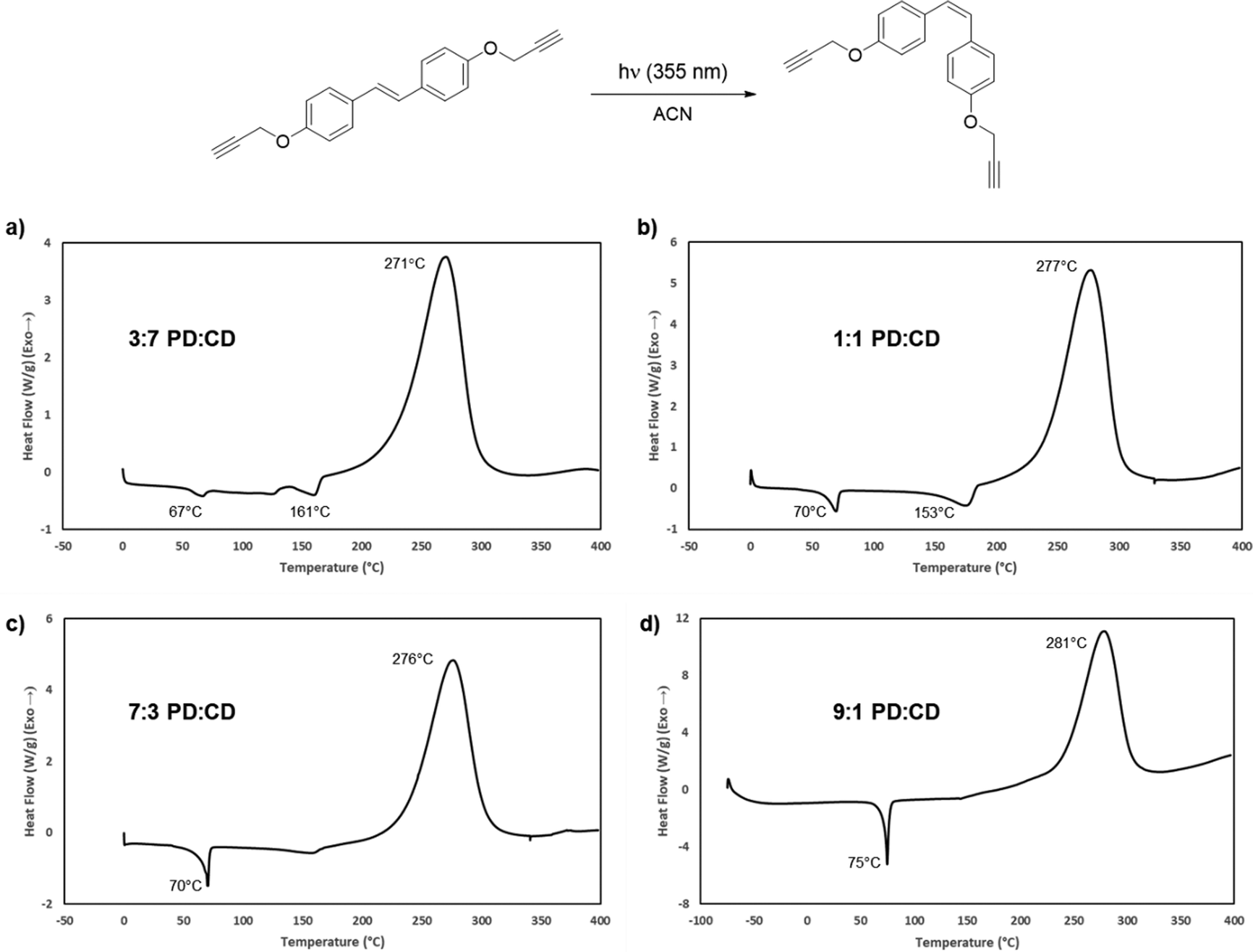
In addition to the use of TD as a blending agent, it was also of interest to explore the photoisomerization of CD as a mechanism to reduce the melting point of the resin system. A similar approach was successful for both cyanate esters and propargyl ethers derived from resveratrol.17,41 We previously studied the photochemical isomerization of the parent bisphenol to the cis-isomer, utilizing 355 nm light in dry MeCN. Unlike that work, irradiation of CD resulted in a much cleaner isomerization to the cis-isomer (PD). The only work-up required to obtain suitably pure resin samples was removal of the solvent under reduced pressure. This method resulted in a 9:1 PD:CD mixture. However, to determine the minimum amount of isomerization/energy input required, we prepared and evaluated several blends with variable amounts of PD. DSC analysis began with a 30% PD blend, which exhibited two discrete melting points at 67 °C and 161 °C (Fig. 6a). Increasing the concentration of the cis-isomer to obtain a 1:1 mixture also resulted in two distinct melting points (Fig. 6b). The first Tm occurred at a temperature (70 °C) higher than that observed for the 30% PD blend mixture while the second Tm decreased to 153 °C. Increasing the PD concentration to 70% resulted in a single Tm at 71 °C, 124 °C lower than pure CD, which afforded a large processing window (Fig. 6c). The 90% PD mixture exhibited a subtle increase in Tm to 75 °C (Fig. 6d). Attempts at obtaining pure PD (>95%) through either fractional crystallization or column chromatography were unsuccessful, and based on the DSC results, unnecessary. The 70% PD blend was utilized for further cross-linking and thermal studies.
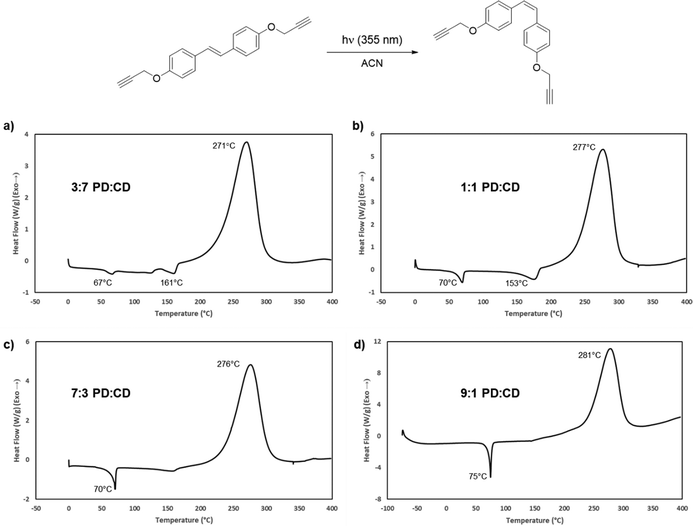 |
| | Fig. 6 Photoisomerization of bis(propargyl ether) CD to generate PD and the effect of isomer ratios on the DSC profile. (a) DSC trace of a 3:7 mixture of PD:CD. (b) DSC trace of a 1:1 mixture of PD:CD. (c) DSC trace of a 7:3 PD:CD mixture. (d) DSC trace of a 9:1 mixture of PD:CD. | |
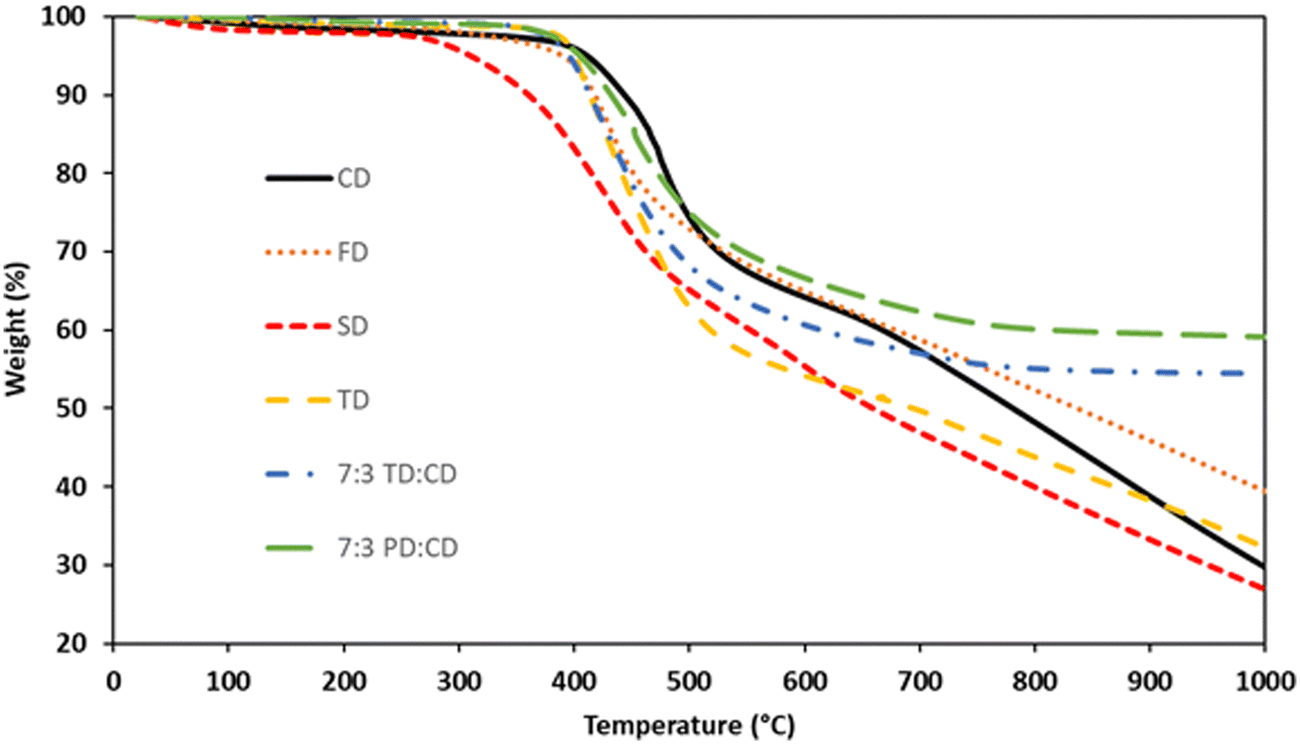
Samples for thermal analysis were prepared by cross-linking the bis(propargyl ethers) based on the information obtained from the DSC. The cure protocols are listed in Table 1. TGA data for the networks can be found in Fig. 7 and are summarized in Table 2. CD exhibited 5% weight loss (Td5) at 411 °C and a char yield of 30% at 1000 °C (N2). FD exhibited a Td5 at 390 °C and a char yield of 39% at 1000 °C while SD exhibited a Td5 at 310 °C and a char yield of 27% at 1000 °C (N2). As methoxy groups are added to the system, the degradation temperature decreases. A similar trend has previously been observed for cyanate ester resins with methoxy-groups ortho to the cross-linking moiety.41 However, the increase in char yield for FD compared to CD was surprising. If the methoxy groups are responsible for lower temperature decomposition mechanisms, one would expect a consistent trend in which the char yield decreases based on the number of methoxy-groups present. It doesn’t seem likely that methoxy-groups are more efficient at producing graphitic char compared to the conjugated stilbene core structure. Therefore, the difference in overall char yield must be due to the formation of a more robust network for FD compared to CD. Based on the DSC results, FD has a larger processing window and a broader cure exotherm, which extends the amount of cross-linking time prior to vitrification. In contrast, as described above, SD undergoes a different cure mechanism due to the lack of an open ortho-position next to the propargyl ether group. The lack of any significant processing window for SD coupled with a rapid exotherm likely results in a lower cross-link density network that is more susceptible to thermal degradation.
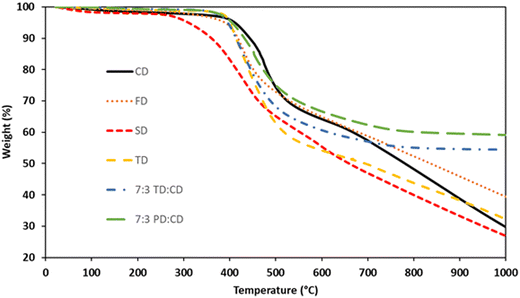 |
| | Fig. 7 TGA (N2) traces for bis(propargyl ether) networks. | |
Table 2 TGA (N2) data for bis(propargyl ether) networks
|
|
CD
|
FD
|
SD
|
TD
|
7:3 TD:CD |
7:3 PD:CD |
|
T
d5 (°C) |
411 |
390 |
310 |
402 |
395 |
405 |
| Char yield (1000 °C, %) |
30 |
39 |
27 |
32 |
54 |
59 |
TD exhibited a Td5 at 402 °C and a char yield of 32% at 1000 °C, values similar to CD. Based on the higher proportion of sp2 hybridized carbons in the CD network one might expect the char yield of TD to be lower than CD. However, TD enables a higher cross-link density compared to CD due to the large processing window for the former. Blending of TD and CD in a 7:3 mixture resulted in a significant improvement in overall thermal stability. The TD:CD network exhibited a Td5 at 395 °C, slightly lower than for either of the components, and a remarkable char yield of 54% at 1000 °C. Similarly, the 7:3 blend of PD:CD exhibited a Td5 at 405 °C and a char yield of 59% at 1000 °C. This result puts the thermal stability of the cis:trans mixture at comparable levels with highly cross-linked resveratrol-derived propargyl ethers (54–65%)17 and is a significant improvement compared to bisphenol A (BPA) derived propargyl ether networks (43% at 700 °C).42 The high char yields could be due to the blend of isomers allowing more complete network formation as a result of the greater degrees of freedom compared to the pure monomers. The difference between the PD:CD and TD:CD blends can be attributed to the higher degree of sp2 hybridized carbons in the former. Another interesting observation from the TGA data is the difference in slopes between the pure dimers and the blends. CD, FD, SD, and TD all exhibit significant weight loss (a downward slope in the TGA trace) as these materials approach 1000 °C. In contrast, the PD:CD and TD:CD blends exhibit near-zero slopes at 1000 °C, demonstrating the formation of thermally stable char formation.
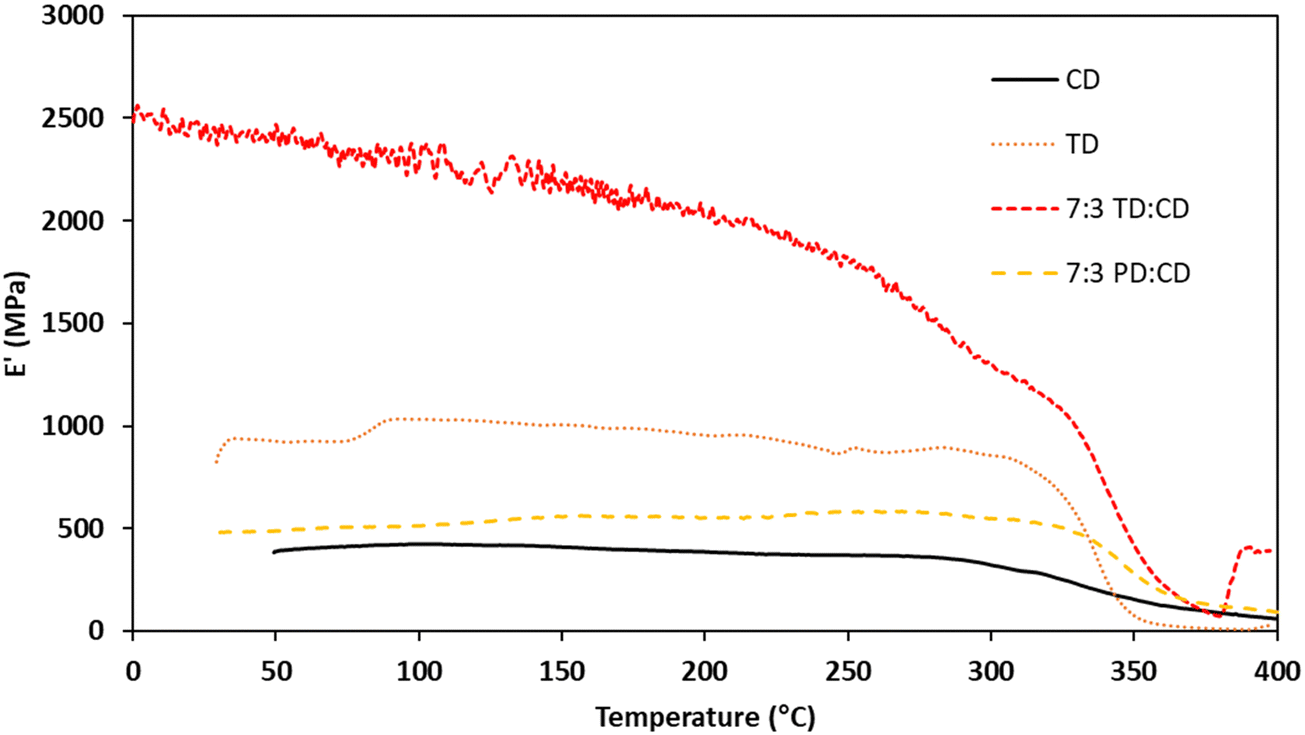
Thermomechanical analysis (TMA) was performed on cross-linked samples to evaluate mechanical properties across a broad temperature range. The TMA results for networks derived from CD, TD, and the 7:3 blends are summarized in Table 3 and graphically depicted in Fig. 8. Unfortunately, we were unable to obtain high quality, void-free samples for FD and SD. The Tgs as measured by the storage moduli (E′) ranged from 285–330 °C. Despite the rigid structure of the CD monomer, the derivative thermoset had the lowest E′ Tg (285 °C), likely due to a lower degree of cure. TD had an E′ Tg of 310 °C, which is 25 °C higher than CD despite the less rigid structure of the TD monomer. This result is attributed to the extended processing window for TD, which allowed for a higher degree of cure. The two blends had virtually identical E′ Tgs of ∼330 °C. This improvement is attributed to a higher cross-link density afforded by the structural diversity of the components. The Tgs as measured by the loss peak (E′′) ranged from 331–337 °C, which are lower than the highly cross-linked resveratrol propargyl ethers (384–389 °C),17 but higher than the analogous BPA propargyl ether (325 °C).43 The loss modulus results show that the bulk polymer chains/networks have similar rigidity regardless of the monomer and provides evidence that the significant differences in the E′ Tgs are a result of residual small molecules or oligomers trapped during vitrification. The TMA results considered alongside the TGA results highlight the power of targeting blends to enhance the properties of rigid high-temperature thermoset materials.
Table 3 TMA results for bis(propargyl ether) networks
|
|
CD
|
TD
|
7:3 TD:CD |
7:3 PD:CD |
|
T
g (E′, °C) |
285 |
310 |
328 |
330 |
|
T
g (E′′, °C) |
332 |
332 |
331 |
337 |
|
T
g (tanδ, °C) |
365 |
348 |
348 |
354 |
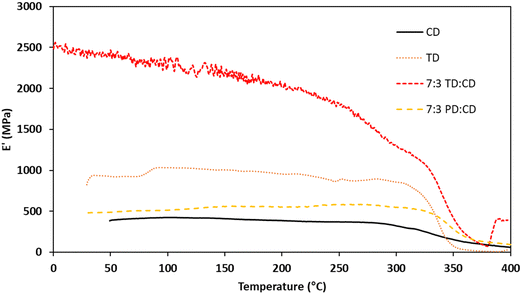 |
| | Fig. 8 TMA traces for bis(propargyl ether) networks. | |
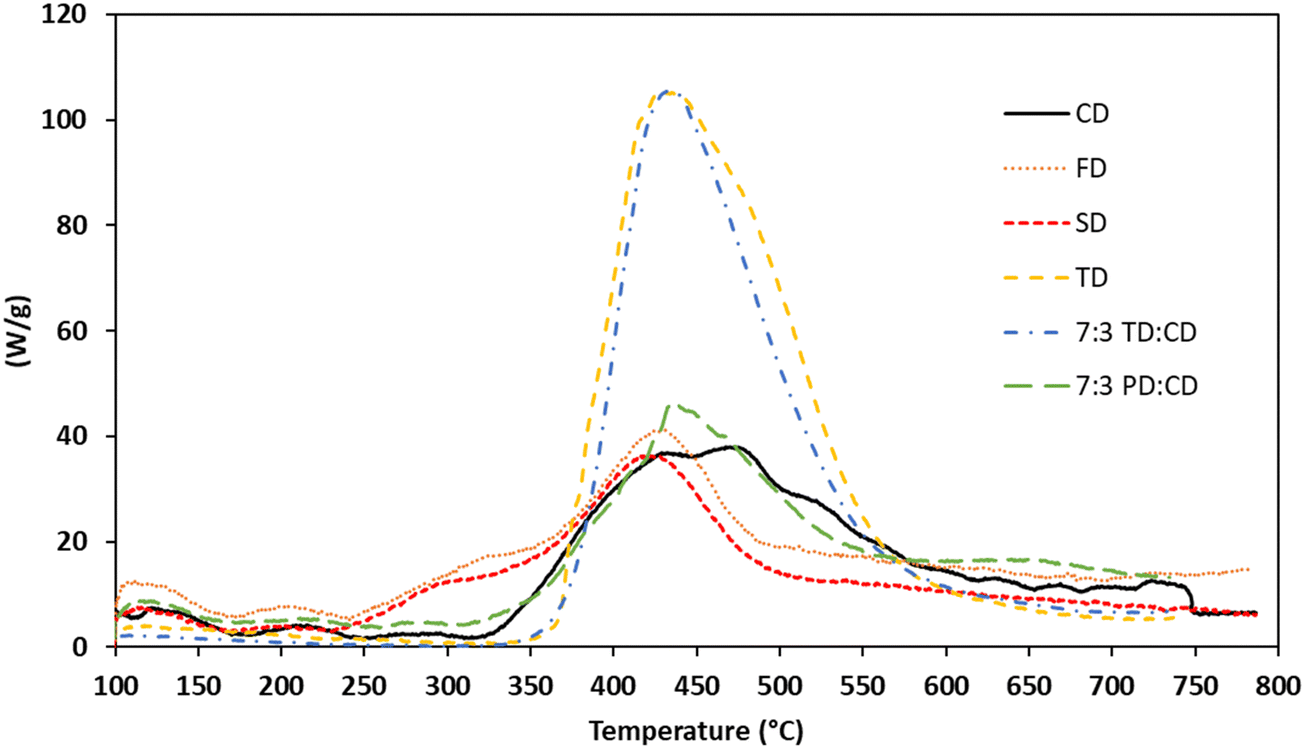
Based on the high Tgs and exceptional char yields of the networks, particularly the blends, it was of interest to explore the fire-resistance of these materials. Maximum heat release rate (Qmax), total heat release (THR), temperature at maximum heat release rate (Tmax), heat release capacity (HRC, ηc), and limiting oxygen index (LOI) were measured using microscale combustion calorimetry (MCC). The results of these experiments are listed in Table 4 while Qmax values are graphically depicted in Fig. 9. Similar to our previous report on propargyl ether thermosets derived from resveratrol,17 the Qmax values for all of the trans-stilbene networks (derived from CD, FD, and SD) were exceptionally low at around 40 W g−1. The Tmax values were consistent with the TGA data. CD exhibited the highest Tmax, while FD and SD exhibited substantially lower values due to the presence of methoxy groups. Heat release capacity (ηc) values for the thermosets were calculated to be 53, 48, and 43 kJ g−1 for CD, FD, and SD, respectively. These values, along with LOI values approaching 50, classify the trans-stilbene networks as non-ignitable, V-0 according to the UL-94 standard.44 In contrast, TD exhibited a much higher peak heat release at ∼102 W g−1 and a lower LOI at 39%. This is attributed to the less thermally stable structure of the bridging group between aromatic rings. Blending TD with CD did not improve the flammability. Of course, it should be noted that Qmax values of ∼100 W g−1 still place the networks in the self-extinguishing V-0/5 V category. The 7:3 blend of PD:CD exhibited MCC results in line with pure CD and the other stilbenic networks. Taking into account processability, Tg, thermal stability as measured by TGA, and fire-resistance, the 7:3 PD:CD blend is the most promising resin system for further development.
Table 4 MCC data for bis(propargyl ether) networks
|
|
CD
|
FD
|
SD
|
TD
|
7:3 TD:CD |
7:3 PD:CD |
|
Q
max (W g−1) |
37.9 ± 0.9 |
40.4 ± 1.1 |
39.4 ± 2.9 |
102.2 ± 3.3 |
102.6 ± 0 |
46.5 ± 1.2 |
|
T
Q
max
(°C) |
467.4 ± 1.2 |
428.2 ± 1.5 |
420.7 ± 4.2 |
441.3 ± 0 |
430.6 ± 3.0 |
436.2 ± 2.0 |
| THR (kJ g−1) |
6.4 ± 0.4 |
5.7 ± 0.1 |
4.5 ± 0.9 |
13.3 ± 0 |
11.5 ± 0 |
5.9 ± 0.5 |
| HRC (J g−1 K−1) |
53 ± 1 |
48 ± 1 |
43 ± 2 |
101 ± 4 |
103 ± 0 |
48 ± 2 |
| LOI |
46 |
47 |
49 |
39 |
39 |
47 |
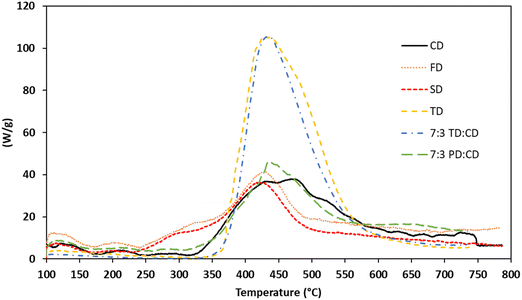 |
| | Fig. 9 MCC Traces for bis(propargyl ether) thermosetting resins and blends. | |
Conclusion
Three thermosetting propargyl ether resins with trans-stilbenic cores were prepared from bio-based hydroxycinnamic acids. Networks prepared from these resins exhibited high Tgs and excellent thermal stability, but suffered from poor processability due to the high melting points of the resins. To overcome this challenge, a more flexible bis(propargyl ether) monomer was prepared from p-coumaric acid by thermal dimerization followed by reaction with propargyl bromide. The new dimer, which contained an unsaturated, branched C4 bridging group (TD) was a liquid at ambient temperature and exhibited thermal properties comparable to the stilbenic coumaric acid-derived resin (CD). Taking advantage of the lower melting points of cis-alkenes compared to trans-alkenes, a cis-isomer of the p-coumaric acid resin (PD) was synthesized by photochemical isomerization. 7:3 blends of TD:CD or PD:CD resulted in processable resin mixtures with broad processing windows. Networks prepared from the blends had extraordinary thermal stability, exhibiting char yields of 54 and 59% at 1000 °C for the TD:CD and PD:CD networks, respectively. Analysis of the stilbenic networks by microscale combustion calorimetry revealed that these networks are non-ignitable, while pure TD or the TD:CD blend were self-extinguishing. Overall, the PD:CD blend exhibited the ideal combination of processability and performance. Future work on this topic should focus on studying the mechanical properties of these bio-based networks as well as the fabrication and evaluation of polymer matrix composites for aerospace and fire-retardant applications. Another interesting avenue of research is the impact of environmental UV irradiation on the structure, stability and mechanical properties of the networks containing cis-alkenes. This subject is currently being studied in our laboratory.
Data availability
The data supporting this article are included in the ESI.† Crystallographic data for compound CD have been deposited in the Cambridge Structural Database (CCDC deposition number 2347707†).
Conflicts of interest
A patent application, which includes some of the results of this study, has been submitted to the US Patent and Trademark Office.
Acknowledgements
The authors would like to thank the Office of Naval Research (ONR) for financial support of this work.
References
-
H. Dodiuk, Handbook of Thermoset Plastics, 3rd ed., William Andrew, 2013 Search PubMed.
-
J.-P. Pascault and R. J. J. Williams, In Thermosetting Polymers. Handbook of Polymer Synthesis, Characterization, and Processing, 2013, pp. 519–533 Search PubMed.
- K. Dušek and M. Dušková-Smrčková, Network structure formation during crosslinking of organic coating systems, Prog. Polym. Sci., 2000, 25(9), 1215–1260 CrossRef.
-
S. G. Advani and K. T. Hsiao, Manufacturing Techniques for Polymer Matrix Composites (PMCs), Elsevier Science, 2012 Search PubMed.
- B. Bilyeu, W. Brostow and K. P. Menard, Epoxy thermosets and their applications I: chemical structures and applications, J. Mater. Educ., 1999, 21(5/6), 281–286 CAS.
- A. Vashchuk, A. Fainleib, O. Starostenko and D. Grande, Application of ionic liquids in thermosetting polymers: Epoxy and cyanate ester resins, eXPRESS Polym. Lett., 2018, 12(10), 898–917 CrossRef CAS.
- N. Ghosh, B. Kiskan and Y. Yagci, Polybenzoxazines—New high performance thermosetting resins: Synthesis and properties, Prog. Polym. Sci., 2007, 32(11), 1344–1391 CrossRef CAS.
-
D. Augustine; M. S. Chandran; D. Mathew and C. R. Nair, In Polyphthalonitrile resins and their high-end applications, Thermosets, Elsevier, 2018, pp. 577–619 Search PubMed.
- S. Ma, T. Li, X. Liu and J. Zhu, Research progress on bio-based thermosetting resins, Polym. Int., 2016, 65(2), 164–173 CrossRef CAS.
- R. Auvergne, S. Caillol, G. David, B. Boutevin and J.-P. Pascault, Biobased thermosetting epoxy: present and future, Chem. rev., 2014, 114(2), 1082–1115 CrossRef CAS PubMed.
- M. Laskoski, J. S. Clarke, A. Neal, B. G. Harvey, H. L. Ricks-Laskoski, W. J. Hervey, M. N. Daftary, A. R. Shepherd and T. M. Keller, Sustainable high-temperature phthalonitrile resins derived from resveratrol and dihydroresveratrol, ChemistrySelect, 2016, 1(13), 3423–3427 CrossRef CAS.
- B. G. Harvey, C. M. Sahagun, A. J. Guenthner, T. J. Groshens, L. R. Cambrea, J. T. Reams, J. M. Mabry and A. High-Performance, Renewable Thermosetting Resin Derived from Eugenol, ChemSusChem, 2014, 7(7), 1964–1969 CrossRef CAS PubMed.
-
H. Fulcrand, L. Rouméas, G. Billerach, C. Aouf and E. Dubreucq, In Advances in Bio-based Thermosetting Polymers, Recent Advances in Polyphenol Research, 2019, pp. 285–334 Search PubMed.
- G.-G. Choi, S.-J. Oh, S.-J. Lee and J.-S. Kim, Production of bio-based phenolic resin and activated carbon from bio-oil and biochar derived from fast pyrolysis of palm kernel shells, Bioresour. Technol., 2015, 178, 99–107 CrossRef CAS.
- J. C. Domínguez, M. Oliet, M. V. Alonso, E. Rojo and F. Rodríguez, Structural, thermal and rheological behavior of a bio-based phenolic resin in relation to a commercial resol resin, Ind. Crops Prod., 2013, 42, 308–314 CrossRef.
- M. Fache, B. Boutevin and S. Caillol, Vanillin, a key-intermediate of biobased polymers, Eur. Polym. J., 2015, 68, 488–502 CrossRef CAS.
- M. D. Garrison, D. V. Lupton, J. E. Baca and B. G. Harvey, Heat-Resistant Polymer Networks Prepared from Processable Resveratrol-Based Propargyl Ether Monomers, ACS Appl. Polym. Mater., 2023, 5(3), 1737–1746 CrossRef CAS.
- O. Yu and K. H. Kim, Lignin to materials: A focused review on recent novel lignin applications, Appl. Sci., 2020, 10(13), 4626 CrossRef CAS.
- D. Stewart, Lignin as a base material for materials applications: Chemistry, application and economics, Ind. Crops Prod., 2008, 27(2), 202–207 CrossRef CAS.
- G. Neutelings, Lignin variability in plant cell walls: Contribution of new
models, Plant Sci., 2011, 181(4), 379–386 CrossRef CAS PubMed.
- D. Watkins, M. Nuruddin, M. Hosur, A. Tcherbi-Narteh and S. Jeelani, Extraction and characterization of lignin from different biomass resources, J. Mater. Res. Technol., 2015, 4(1), 26–32 CrossRef CAS.
- E. Feghali, K. M. Torr, D. J. van de Pas, P. Ortiz, K. Vanbroekhoven, W. Eevers and R. Vendamme, Thermosetting polymers from lignin model compounds and depolymerized lignins, Lignin Chem., 2020, 69–93 Search PubMed.
- H. A. Khalil, M. Marliana and T. Alshammari, Material properties of epoxy-reinforced biocomposites with lignin from empty fruit bunch as curing agent, BioResources, 2011, 6(4), 5206–5223 Search PubMed.
- D. Setua, M. Shukla, V. Nigam, H. Singh and G. Mathur, Lignin reinforced rubber composites, Polym. Compos., 2000, 21(6), 988–995 CrossRef CAS.
-
S. Piyanirund; W. Seangyen; P. Srinoppakhun and P. Dittanet In Reinforcement of epoxy resin by lignin, Materials Science Forum, Trans Tech Publ, 2021, pp. 151–155 Search PubMed.
- X. Song, Z.-P. Deng, C.-B. Li, F. Song, X.-L. Wang, L. Chen, D.-M. Guo and Y.-Z. Wang, A bio-based epoxy resin derived from p-hydroxycinnamic acid with high mechanical properties and flame retardancy, Chin. Chem. Lett., 2022, 33(11), 4912–4917 CrossRef CAS.
- X. Ma, J. Chen, J. Zhu and N. Yan, Lignin-based polyurethane: recent advances and future perspectives, Macromol. Rapid Commun., 2021, 42(3), 2000492 CrossRef CAS.
- N. Mahmood, Z. Yuan, J. Schmidt and C. C. Xu, Depolymerization of lignins and their applications for the preparation of polyols and rigid polyurethane foams: A review, Renewable Sustainable Energy Rev., 2016, 60, 317–329 CrossRef CAS.
- J. R. Gouveia, C. L. da Costa, L. B. Tavares and D. J. dos Santos, Synthesis of lignin-based polyurethanes: a mini-review, Mini-Rev. Org. Chem., 2019, 16(4), 345–352 CrossRef CAS.
- B. G. Harvey, A. J. Guenthner, H. A. Meylemans, S. R. Haines, K. R. Lamison, T. J. Groshens, L. R. Cambrea, M. C. Davis and W. W. Lai, Renewable thermosetting resins and thermoplastics from vanillin, Green Chem., 2015, 17(2), 1249–1258 RSC.
- H. Jiang, L. Sun, Y. Zhang, Q. Liu, C. Ru, W. Zhang and C. Zhao, Novel biobased epoxy resin thermosets derived from eugenol and vanillin, Polym. Degrad. Stab., 2019, 160, 45–52 CrossRef CAS.
- H. A. Meylemans, T. J. Groshens and B. G. Harvey, Synthesis of renewable bisphenols from creosol, ChemSusChem, 2012, 5(1), 206–210 CrossRef CAS PubMed.
- N. Van de Velde, S. Javornik, T. Sever, D. Štular, M. Šobak, Ž. Štirn, B. Likozar and I. Jerman, Bio-based epoxy adhesives with lignin-based aromatic monophenols replacing bisphenol A, Polymers, 2021, 13(22), 3879 CrossRef CAS.
- C. E. Zavala, N. A. Vest, J. E. Baca, D. D. Zhang, K. R. McClain and B. G. Harvey, Highly efficient synthesis of sustainable bisphenols from hydroxycinnamic acids, RSC Sustainability, 2023, 1(7), 1765–1772 RSC.
- J. Combes, N. Imatoukene, J. Couvreur, B. Godon, F. Brunissen, C. Fojcik, F. Allais and M. Lopez, Intensification of p-coumaric acid heterologous production using extractive biphasic fermentation, Bioresour. Technol., 2021, 337, 125436 CrossRef CAS.
- G. M. Borja, A. Rodriguez, K. Campbell, I. Borodina, Y. Chen and J. Nielsen, Metabolic engineering and transcriptomic analysis of Saccharomyces cerevisiae producing p-coumaric acid from xylose, Microb. Cell Fact., 2019, 18(1), 191 CrossRef.
- T. I. Kolesnikov, A. M. Orlova, F. V. Drozdov, A. I. Buzin, G. V. Cherkaev, A. S. Kechekyan, P. V. Dmitryakov, S. I. Belousov and A. A. Kuznetsov, New imide-based thermosets with propargyl ether groups for high temperature composite application, Polymer, 2022, 254, 125038 CrossRef CAS.
- C. H. Lin, C. M. Huang, T. I. Wong, H. C. Chang, T. Y. Juang and W. C. Su, High-Tg and low-dielectric epoxy thermosets based on a propargyl ether-containing phosphinated benzoxazine, J. Polym. Sci., Part A: Polym. Chem., 2014, 52(9), 1359–1367 CrossRef CAS.
- S. Zhao, X. Huang, A. J. Whelton and M. M. Abu-Omar, Renewable epoxy thermosets from fully lignin-derived triphenols, ACS Sustainable Chem. Eng., 2018, 6(6), 7600–7608 CrossRef CAS.
- J. Liu, J. W. Y. Lam and B. Z. Tang, Acetylenic Polymers: Syntheses, Structures, and Functions, Chem. Rev., 2009, 109(11), 5799–5867 CrossRef CAS.
- B. G. Harvey, A. J. Guenthner, W. W. Lai, H. A. Meylemans, M. C. Davis, L. R. Cambrea, J. T. Reams and K. R. Lamison, Effects of o-methoxy groups on the properties and thermal stability of renewable high-temperature cyanate ester resins, Macromolecules, 2015, 48(10), 3173–3179 CrossRef CAS.
- C. P. R. Nair, R. L. Bindu, K. Krishnan and K. N. Ninan, Bis propargyl ether resins: synthesis and structure–thermal property correlations, Eur. Polym. J., 1999, 35(2), 235–246 CrossRef CAS.
- S. K. Dirlikov, Propargyl-terminated Resins—A Hydrophobic Substitute for Epoxy Resins, High Perform. Polym., 1990, 2(1), 67–77 CrossRef CAS.
- Q. Xu, C. Jin, A. Majlingova and A. Restas, Discuss the heat release capacity of polymer derived from microscale combustion calorimeter, J. Therm. Anal. Calorim., 2018, 133(1), 649–657 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Joshua E.
Baca
,
Joshua E.
Baca