
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA road for macroporous silicon stabilization by ultrathin ALD TiO2 coating†
Bachar
Al Chimali‡
 a,
Irene
Carrasco‡
ab,
Thomas
Defforge
a,
Romain
Dailleau
a,
Lisa
Monnier
ac,
Kaushik
Baishya
d,
Jan M.
Macak
de,
Gael
Gautier
a and
Brice
Le Borgne
*a
a,
Irene
Carrasco‡
ab,
Thomas
Defforge
a,
Romain
Dailleau
a,
Lisa
Monnier
ac,
Kaushik
Baishya
d,
Jan M.
Macak
de,
Gael
Gautier
a and
Brice
Le Borgne
*a
aGREMAN UMR-CNRS 7347, INSA Centre Val de Loire, Université de Tours, 37071 Tours Cedex 2, France. E-mail: brice.leborgne@univ-tours.fr
bInstitut Supérieur des Matériaux et Mécaniques Avancées du Mans (ISMANS Groupe CESI), 44 avenue Frédéric Auguste Bartholdi, 72000 Le Mans, France
cInterfaces, Confinement, Matériaux et Nanostructures (ICMN), CNRS-Université d’Orléans, UMR 7374, 1b, Rue de la Férollerie, C.S. 40059, 45071 Orléans Cedex 2, France
dCentral European Institute of Technology, Brno University of Technology, Purkynova 123, 612 00 Brno, Czech Republic
eCenter of Materials and Nanotechnologies, Faculty of Chemical Technology, University of Pardubice, Nam. Cs. Legii 565, 53002 Pardubice, Czech Republic
First published on 22nd October 2024
Abstract
Macroporous silicon films have great potential for a plethora of applications in optoelectronics and microelectronics. However, such layers are too electrically and chemically unstable to be used in fuel cells, supercapacitors or any devices requiring the use of an electrolyte. This is due to their high surface-to-volume ratio, which makes them prone to chemical reactions, such as photo-oxidation, especially in aqueous media. In this work, we investigated how to exploit the capabilities of macroporous silicon while avoiding its oxidation. To do so, we explored the influence of ultrathin TiO2 films by atomic layer deposition (ALD) onto the walls of silicon macropores, created by electrochemical etching from n-type wafers. Using microscopy and optical analysis, we demonstrate the achievability of ALD coating on macroporous silicon, as well as the stability of these films against oxidation. In particular, we show that 5 ALD cycles that correspond to less than 1 nm thin coating are sufficient to passivate the silicon surface. The coated and uncoated layers were analyzed and compared before and after exposure to water and sunlight. The monitoring of the Si–O–Si band area evolution over 29 days gave no evidence of photo-corrosion. In addition, the wettability of the samples did not change after functionalization. Finally, to investigate the oxidation prevention for photocatalytic applications, we showed that methylene blue degradation rates were significantly increased (by 50% on average) for 10 nm TiO2 ALD-coated porous silicon samples when compared to natural degradation. Interestingly, layers thinner than 1 nm also showed enhanced catalytic kinetics for short times (t < 40 min).
Introduction
As stated a few years ago by Rasson and Francis,1 porous silicon's (PSi) semi-conducting and optical properties, as well as its large specific surface area, make it interesting for numerous applications. Among them, we can point out electronic components, which mainly benefit from its dielectric properties. The obtainable relative dielectric constant varies from 2 to 6 depending on PSi's porosity.2 Moreover, PSi's uses are industrially viable.3 Despite these advantages, it is still suffering from poor chemical stability in aqueous media. PSi is indeed prone to oxidation, which prevents drawing the quintessence from it. The oxidation of silicon (Si) is responsible for the formation of a thin silicon dioxide (SiO2) layer at the surface of the sample, i.e. inside the pores. This mechanism occurs spontaneously (on native oxide) or under photo-thermal oxidative conditions,4 as is often the case in plasmonic-based photocatalysis applications.5 Overall, this leads to instability of the material (enhanced by its high surface-to-volume ratio), which results in a change in the optical properties,6 a reduction in the specific surface area7 and a modification of the electrical properties, impeding the use of PSi for the design of photoelectrodes. Several studies have shown that PSi has potential in water-splitting to produce hydrogen8 and artificial photosynthesis (AP)9 for the production of fuels, i.e. the conversion of CO2 into added-value chemicals produced by combining a catalysis reaction and solar energy.The most conventional method to stabilize PSi is to oxidize it on purpose, by performing annealing at a high temperature (>800 °C) to form stable Si–O bonds.10 Here, the aim is to find alternative ultrathin materials to passivate PSi, as the presence of a stable SiO2 layer leads to reduced performances of the photo-electrodes because it tends to convert the PSi skeleton to silicon oxide. Moreover, high-temperature annealing can lead to PSi damage due to thermal stress. Consequently, when it is required to stabilize PSi without oxidizing it, thermal hydrosilylation has been often preferred. It allows the formation of Si–C bonds and can be performed using chemical treatments with 1-dodecene,11 microwave-assisted hydrosilylation,12 electrochemical grafting,13 or thermal carbonization.14 These methods are the most common ways to protect macroporous silicon from oxidation without drastically changing its properties. It is therefore necessary to add functional materials if other properties are required. To do so, the recent trend is to use ALD or CVD of metal oxides to protect the porous media.15
To date, different kinds of nanostructured silicon films have been passivated using ALD, such as the nanoparticle-based ones16 or randomly etched n-doped films,17 but they are present in “thick” layers (>10 nm). Thus, advanced studies focusing on the stabilization of PSi layers by ultrathin pores coatings are still in their infancy and mainly deal with specific applications.18,19
PSi oxidation mechanism has been studied since its discovery. The three main techniques are infrared spectroscopy,20,21 spectroscopic ellipsometry22,23 and optical refractive index measurements.24 The last two are generally preferred when the thickness of silicon dioxide grown at the surface of the substrate is monitored. It is still challenging to perform ellipsometry on macroporous silicon25 and optical measurements do not allow knowing the kind of bonds present at the silicon surface. Consequently, this study favored the use of FTIR spectroscopy to monitor the Si–O–Si bonds on the surface of the coated and uncoated samples. Moreover, FTIR allows the detection of Si–H bonds, which are useful for knowing about the surface chemistry of the samples.
The oxidation mechanisms between planar and PSi are very similar. Due to its outstanding surface-to-volume ratios and high surface energies, macroporous silicon is much more prone to degradation associated with spontaneous oxidation than planar silicon.26 Surface photo-oxidation causes various physicochemical changes in PSi films or planar silicon. These changes can be advantageous as they can lead to an increase in the emission quantum yield,27 but they can be also disadvantageous because of the strong deterioration of long-range charge transport.28
Consequently, despite recent advances in the field,29–31 there is still a gap in the literature regarding the passivation of high-specific surface materials by atomic layer deposition (ALD) for applications in aqueous media. One of the reasons is that it is still challenging to coat porous materials conformally. A brilliant review paper from Rongé et al.,32 suggested that the undesired oxidation could be prevented by coating the pores with a thin passivating layer using ALD, and many recent studies confirmed this assumption.
If the coating material used to passivate silicon also has photocatalytic properties, the resulting photoelectrode could potentially perform a carbon dioxide reduction reaction (CO2RR) in water. This reaction, if properly controlled, allows producing renewable fuels. Its mastery is nowadays one of the main investigation fields. Many catalysts have been studied to perform CO2RR and other molecule-splitting applications. However, some of them contain non-Earth abundant materials, such as Ru, Pt, Ir33–35 or need expensive synthesis processes,36 impeding the market introduction of renewable gas compared to lower-cost fossil-based fuels. A very common material that matches all the criteria for the development of PSi/ALD industrially compatible fabrication of PEC cells for solar fuel production is titanium dioxide (TiO2). Here, TiO2 is chosen because (i) it is well-known for its outstanding photocatalytic properties;37 (ii) the possibility of being deposited by ALD38 in a highly conformal way; (iii) the Earth-abundancy of titanium;39 (iv) its non-toxicity40 and (v) its extremely high chemical stability. Additionally, TiO2 is suitable for antireflection purposes41 and has been used previously in the photovoltaic industry.42
For these reasons, it remains interesting to investigate the stabilizing capabilities of ALD TiO2 films on PSi, in view of the design of future PEC cells. Previous achievements based on a few nm-thick TiO2,17 Al2O3,16 or 5 nm/5 nm Al2O3/TiO215 coatings showed that PSi stabilization is possible. In particular, it is worth investigating the stabilizing effect for very thin coatings, i.e. below 5 nm.
In this work, the stability of the coated PSi after exposure to aqueous medium at pH = 7 was probed using Fourier transform infrared (FTIR) spectroscopy and compared with bare PSi. The samples were directly exposed to sunlight by placing them by a window for 29 days (approx. 700 h). Moreover, the wetting properties of the coated samples show that macro-PSi/TiO2 composites are hydrophilic, which matches the requirements of photocatalysis cells, encouraging the development of such devices. Here, we employed ALD based on an emerging stop-flow strategy to allow successful ultrathin TiO2 coating of PSi.
Additionally, an evaluation of the photocatalytic capabilities of ultrathin TiO2 films was performed by monitoring the degradation of methylene blue (MB). It showed enhanced photocatalytic activities even for a few angstrom-thick layers.
Previous works showed that ultrathin coating (typically below 2 nm) can protect porous materials against photocorrosion.43 Ultrathin coating can also increase their chemical and temperature stability44,45 and improve the biological response of the substrate.46,47 On the other hand, other works showed that “thicker” (more than 20 nm) ALD layers also improve the photocurrent48 and photocatalytic49,50 response of different materials. In the latter case, however, the ALD coatings were prepared on already partially photoactive materials. Here, we report a way to both stabilize and enhance the photocatalytic activity of n-type PSi films via subnanometric TiO2 coatings.
Experimental
Fabrication of porous silicon samples
Silicon wafers (0.49–0.505 Ω cm resistivity, 〈100〉 orientation, double-side polished, boron-doped) were purchased from Siltronic (Germany). Aqueous hydrofluoric acid (HF) was 50 wt% concentrated.The PSi samples were prepared by electrochemical etching of a double-sided polished heavily doped n-type silicon using a cell purchased from AMMT Gmbh (Germany), which uses platinum electrodes, and a potentiostat as the current source, as previously described. The electrolyte used for the porosification was 46![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) deionized (DI) water and an HF solution. Cetyltrimethylammonium chloride (CTAC), a cationic surfactant, was used to obtain a better sample quality related to hydrogen release. Note that double-side polished wafers are not necessary to obtain the desired quality of PSi, but this choice ensures a sufficient level of reflection during the FTIR spectroscopy measurements. To perform the etching, a constant voltage of 10 V was applied between two electrodes for 60 min while placing the silicon sample between them. Lightening the backside of the sample ensured the generation of photocarriers. Samples were then rinsed with DI water and dried under nitrogen flow or at ambient air. Afterwards, parts of the samples were immediately characterized while others underwent ALD oxide coating.
:1 (v/v) deionized (DI) water and an HF solution. Cetyltrimethylammonium chloride (CTAC), a cationic surfactant, was used to obtain a better sample quality related to hydrogen release. Note that double-side polished wafers are not necessary to obtain the desired quality of PSi, but this choice ensures a sufficient level of reflection during the FTIR spectroscopy measurements. To perform the etching, a constant voltage of 10 V was applied between two electrodes for 60 min while placing the silicon sample between them. Lightening the backside of the sample ensured the generation of photocarriers. Samples were then rinsed with DI water and dried under nitrogen flow or at ambient air. Afterwards, parts of the samples were immediately characterized while others underwent ALD oxide coating.
Atomic layer deposition
ALD was performed using a BENEQ TFS 200 deposition tool. TiCl4 (STREM Chemicals, Inc., U.S.A) and DI H2O precursors (oxygen source) were fed into the reaction chamber from corresponding canisters at a temperature of 21 °C (cleanroom temperature). The samples were placed into the reactor, right after their etching. ALD processes were performed by using the prolonged exposure mode in a stop-flow reactor chamber. In this configuration, the chamber is isolated from the pump during precursor pulses, which allows precursors to penetrate deeply into the pores, ensuring a homogeneous coating throughout the whole depth of the porous matrix, which has a high aspect ratio. Each ALD full cycle is composed of a starting TiCl4 half-cycle and a subsequent water precursor half-cycle. Each TiCl4 half-cycle starts with the closing of the automatic pressure control (APC) valve followed by a pulse of the precursor with a duration of 0.25 seconds for TiCl4. After a 5-second delay to allow the precursor to react, the APC valve is fully opened for 10 seconds. This step was repeated twice before starting the next half-cycle to ensure the removal of both the unreacted precursor molecules and the reaction byproducts. Full cycles were repeated a given number of times depending on the growth rate and desired thickness. In our case, the full cycles were repeated 5, 21 or 210 times for samples groups B, C and D, respectively, with a deposition temperature of 300 °C. During the deposition, the gas pressure was kept lower than 5 mbar in the reaction chamber. A capillary tube fitted to the reactor was used to control the flow of precursors injected into the ALD chamber. The N2 injection rate was 300 standard cubic centimetres per minute (sccm) into the precursor lines and 150 sccm into the outer reaction chamber.Scanning electron microscopy
A JEOL JSM-7900F high-resolution field-emission scanning electron microscope (SEM) was used to observe and measure the thickness and pore diameter of the PSi layers.Contact angle measurements
Contact angle measurements were performed using a KRUSS GmBH DSA100 Drop Shape Analyzer using the sessile drop technique. For each sample, a 2 μL of DI water droplet was placed onto the sample's surface with a syringe before taking a picture. Then, the DSA3 software analysed the image to extract the contact angles. The measurement was performed on three water droplets for each sample type (naturally oxidized flat silicon, PSi and TiO2-coated PSi).Fourier transform infrared spectroscopy
FTIR spectroscopy was used under vacuum (<1 hPa) with a Vertex 70v spectrometer from Bruker Optics equipped with a glow bar radiation source and a DLaTGS MIR (4000–400 cm−1) detector. Spectra were recorded in the reflection mode with a resolution of 6 cm−1 and 20 scans. The reference sample consisted of a flat silicon sample (not etched) of the same type of silicon as the one used for the fabrication of PSi. The native oxide was not removed prior to FTIR measurements. Note that the incident radiation crosses the film and is reflected by the polished backside of the sample, which means that the data correspond to a measurement of the radiation intensity that crosses the sample two times. The area of the Si–O–Si bands was estimated by numerical integration of the spectrum in the 1000–1200 cm−1 region. No baseline was performed in the wavelength range of integration.First, FTIR measurement was performed to define the t0 of the experiment. For samples from group A, it was performed right after the etching, while for groups B to D right after ALD. After the first measurement, samples were immersed in water and placed by a window, directly exposed to sunlight. At regular temporal intervals, samples were inspected using the following protocol: (i) N2-flow drying for 30 s; (ii) hot plate drying at 50 °C for 1 min to ensure moisture removal; (iii) FTIR spectroscopy measurements. The protocol was kept the same for all sample groups.
X-ray photoelectron spectroscopy
The surface chemical composition of the films was evaluated by X-ray photoelectron spectroscopy (XPS, Kratos Axis Supra) analysis. A monochromatic Al Kα (1486.7 eV) X-ray source operated at 250 W was used. The binding energy scale was referenced to adventitious carbon (284.8 eV) and no charging neutralizer was used during the measurements.Methylene blue degradation
The photocatalytic properties of the different structures were studied by following the degradation of an aqueous solution of methylene blue under light exposure. Samples from groups B to D were placed in a beaker containing 9 mL of a MB solution (5.6 × 10−3 mol L−1) and were irradiated with a lamp (100 mW cm−2) for 110 min. The control solution did not contain any sample. The photocatalytic degradation was evaluated by monitoring the absorption of MB at 662 nm. The absorption spectra were acquired with a FLAME-S-XR1-ES UV-vis spectrometer (200–1034 nm) connected to a computer equipped with OceanView software. Measurements were performed every 10 min placing the MB solution in a cuvette of 4.5 cm3. 3 samples of each group and 3 fresh control solutions were studied under the same conditions in order to compare the obtained results.Results and discussion
Structural characterization
Silicon was etched to obtain a 30 μm thick macroporous layer (see Fig. 1a). Macropores are preferred in order to ensure water penetration as the films are meant to operate in the aqueous medium. The pores’ morphology exhibited vertical macropores and thinner secondary pores (branches). Indeed, as the backside of the sample was lightened during the etching step, the photo-generated charge carriers migrated towards the backside side of the sample, leading to the formation of macroporous structures.51 The branches are typical for etching at low current densities and/or for high HF concentrations. Such a structure ensures efficient gas evacuation during the etching step and such pore design is necessary for the photoelectrochemical cell operation.49 | ||
| Fig. 1 SEM images of a group D sample showing the overall geometry of the macroporous silicon layer (a) and details of the TiO2 layer at the top (b) and the bottom of the pores (c). | ||
20 samples were prepared and separated into 4 groups (A, B, C and D) of 5 samples each. Samples from group A were kept “blank”, whereas those from groups B, C and D were coated using ALD with 5, 21 and 210 cycles of TiO2, respectively. The number of cycles was adjusted to obtain thicknesses of below 1 nm, approximately 1 and 10 nm, respectively. The deposition temperature was set to 300 °C, which usually leads to anatase structure, generally considered to be the most efficient photocatalytic structure of TiO2.50
Subsequent characterization of a group D sample by SEM proved that the TiO2 film is conformal and the deposition successfully covered the bottom of the vertical pores, whilst penetrating also into the horizontally branched ones (Fig. 1b and c). The thickness of the film (approx. 10 nm) is homogeneous to the bottom of the pores.
Fig. 1c shows a partial delamination of the TiO2, which we ascribe to the sample dicing process. However, TiO2 is present down to the bottom of the pore. For the samples that underwent 5 and 21 ALD cycles, the given thicknesses (less than 1 nm and ∼1 nm, respectively) are an approximation from the data obtained by ellipsometry and transmission electron microscopy (TEM) on flat silicon substrates (see Fig. S2, ESI†).
The survey spectra of the surface of the sample after ALD are presented in Fig. 2a. The O 1s and Ti 2p spectra of samples from groups B, C and D are presented in Fig. 2b, c and d, respectively. The Ti 2p spectra are very similar for the three samples. The films exhibit Ti 2p3/2 peaks at 458.2, 457.2 and 457 eV, and Ti 2p1/2 peaks at 463.9, 462.9 and 462.8 eV. None of them shows asymmetries at their low energy side that could indicate the presence of Ti3+.
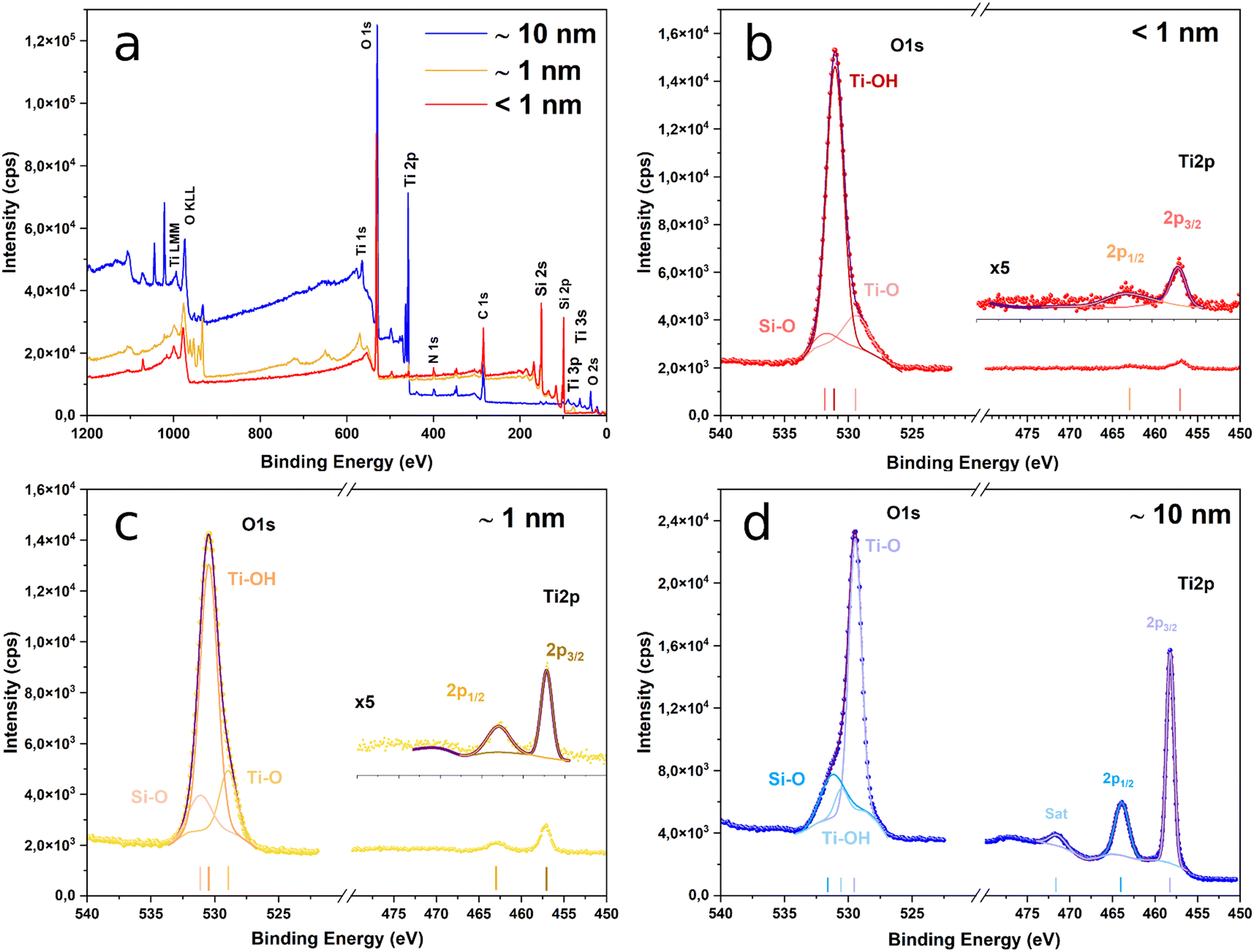 | ||
| Fig. 2 XPS spectra of various studied samples (a) and detailed high resolution XPS spectra of the O 1s and the Ti 2p peaks for the various PSi layers coated with sub-1 nm (b); about 1 nm (c); and about 10 nm (d) ALD TiO2 layers. In (b)–(d), solid dots correspond to the experimental data, whereas the solid purple line corresponds to the fit. | ||
The position of the Ti 2p photoelectron peaks and the spin–orbit splitting of the 2p1/2–2p3/2 doublet is about 5.7 in all the cases, which are both characteristics of TiO2.52 Furthermore, the spectrum of the group D sample exhibits a strong satellite peak at ∼13.3 eV from the 2p3/2 peak, which is also a distinctive feature of TiO2.53 The low intensity of the Ti 2p line for groups B and C samples prevents the observation of this peak in their corresponding spectra. On the contrary, the O 1s line presents significant differences among the various samples. For 210 ALD cycles, the spectrum shows a peak at ∼529.5 eV with a strong asymmetry at high binding energies.
The peak can be deconvoluted into three components at 531.2, 530.6 and 529.5 eV. The latest is the most significant and is ascribed to oxygen bound to tetravalent Ti ions, whereas the other two indicate the presence of Ti–OH54 and Si–O55 bonds at the surface of the films. For groups C and B samples, however, peaks located at ∼530.4 eV and ∼531 eV, respectively, and the shoulder at the lower energies side appear since in this case the main contribution comes from the Ti–OH bonds and not from the Ti–O ones. This is explained because of the low thickness of the TiO2 film for these two samples (∼1 nm and <1 nm), which reduces the weight in the XPS spectra of the Ti–O bonds coming from TiO2 while increasing the influence of other species that are present at the surface. In a similar fashion, the low thickness of the coatings explains the appearance of the Si-related lines in the photoelectron spectra of these two samples that, however, are hardly visible for 210 ALD cycles (Fig. S3, ESI†).
This is expected, because, in this case, the thickness is at the limit of the typical values for the information depth of the XPS technique (∼10 nm) and so the signal comes mainly from the TiO2 layer, whilst for the other two, the signal contains information from the PSi substrate. The presence of Si–O bonds in the spectra of the three samples indicates the formation of native SiO2 at the surface of the PSi substrate prior to TiO2 covering. Its presence is inherent to the process as the oxide appears during the few minutes between the PSi etching and the beginning of the ALD step. However, we can see that the native oxide does not seem to affect the protective role of TiO2 (see following sections).
ALD deposition of TiO2 on SiO2 by successive pulses of TiCl4 and H2O occurs through a complicated chain of reactions happening on the surface,56 which evolve as the number of cycles increases. Gu and Tripp show that during the first cycle, TiCl4 vapours react with 100% of the isolated SiOH (silanol) groups present on the surface of silica leading to a combination of Ti–O, Si–O–Ti and Si–O–Ti–Cl species. The addition of water vapor in the second cycle leads to the cleavage of some of the freshly formed Si–O–Ti bonds, resulting in the regeneration of silanol groups available to react with TiCl4. However, as the number of ALD cycles increases and the TiO2 structure propagates, the number of Si–O–Ti bonds cleaved by the water vapour decreases. After a few cycles, the reacting surface corresponds only to titania (the SiO2 layer is fully covered) and the TiCl4 adsorption occurs via direct reaction with TiO2 and polymerization with the adsorbed water layer. We can then consider that, at this stage, all the Si–O–Si bonds are saturated, i.e., not available for further reactions. As a result, the growth of the new SiO2 layers is inhibited, leading to a fully stabilized surface even though the deposition occurs in the presence of H2O.
Protective role of TiO2 films against oxidation process in water
Wetting properties were first studied for flat silicon, bare PSi and TiO2-coated PSi (∼10 nm, 210 cycles) by water contact angle measurements. Fig. S4 (ESI†) presents the obtained results. The porosification of these samples or the ALD coating of the porous structure showed relatively small effects on the wetting properties, with all contact angles in the range 20°–45°. This wide range is most likely due to the morphological inhomogeneity at its surface. It is probably not related to possible contamination by adsorption of hydrocarbons because the samples were only exposed to deionised water during the experiments. We can conclude that the TiO2 coating has no influence on the water penetration into the pores. Consequently, after both porosification and ALD coating, the substrates remained hydrophilic, which makes them suitable for applications in aqueous media.This allowed us to assess the impact of the TiO2 films on the oxidation process in water by submerging the samples into the water and measuring their FTIR spectra during the process, monitoring the evolution of the Si–O band (1000–1200 cm−1) over 700 hours (i.e. 29 days). A representative example of the spectra recorded prior to exposure to oxidation conditions, and after the 700 hours of water immersion is presented in Fig. 3a. The full FTIR spectra are available in Fig. S5 (ESI†). For the uncoated samples (group A) there is a remarkable increase of the Si–O–Si band when exposing samples to oxidative conditions, and this is true even for only 30 minutes of water immersion. In contrast, the FTIR spectra of samples coated with TiO2 thin films do not show significant differences, regardless of the duration of the water exposure.
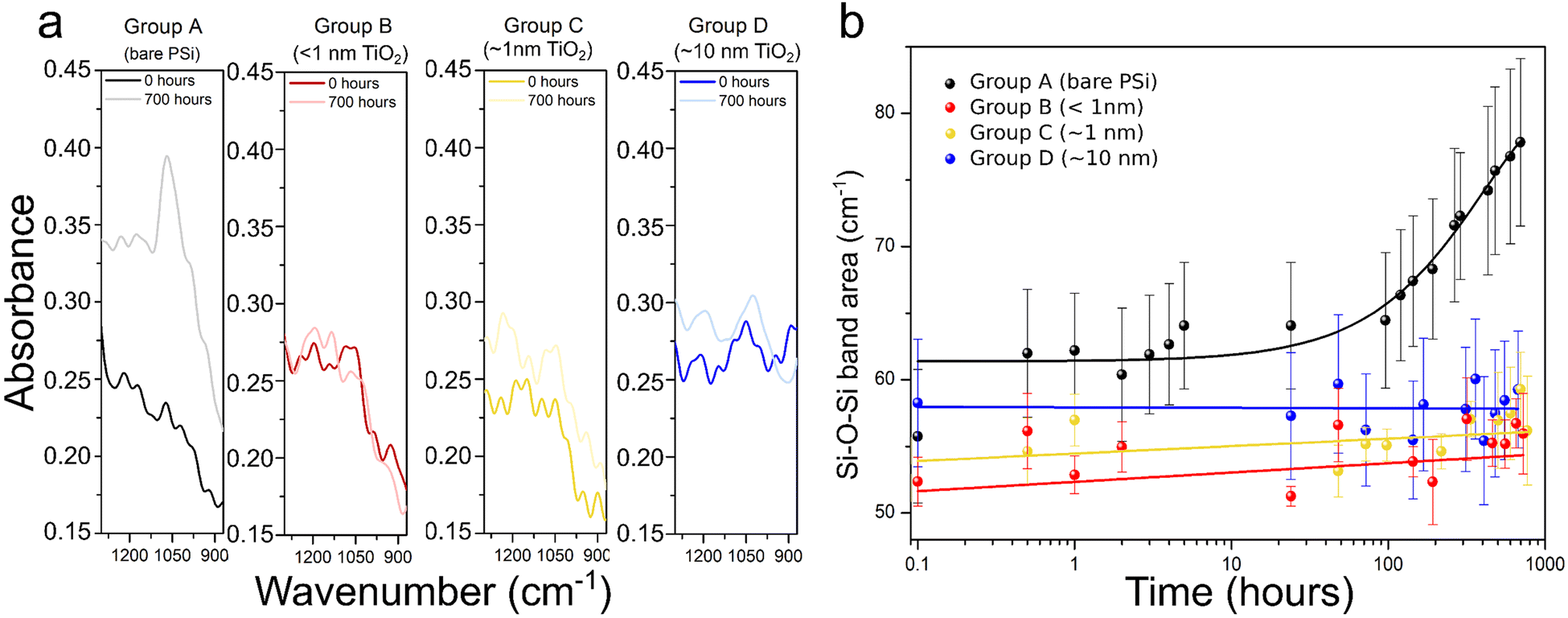 | ||
| Fig. 3 Details of the Si–O–Si band of the FTIR spectra recorded for oxidation times of 0 and 700 hours (a) and evolution of the Si–O–Si band area as a function of the oxidation time (b). Lines in b are only a guide to the eye. Experiments were conducted in water. | ||
The Si–O–Si band area as a function of the oxidation time is shown in Fig. 3b. Each value in the graph represents the mean of the calculated values of the band area for the 5 samples comprising each set of samples. For group A samples the Si–O band increases significantly right after exposure to the oxidation conditions.
After that, an induction time, in which the area increases only slightly is observed, followed by an exponential growth after ≈96 hours of immersion into water. For samples from groups B, C and D, this first oxidation upon immersion was not noticed. The signal of the Si–O–Si band was increasing only slightly over time. Although we cannot conclude what would happen after the 29 days of water exposure measurement, the chemical stability of TiO2 likely ensures a longer-term passivation of porous silicon within aqueous media. Indeed, the kinetics of Si–O–Si bond formation are not even measurable. These observations confirm that the PSi layer was protected from oxidation thanks to the TiO2 layer even for only 5 ALD cycles (i.e. less than 1 nm-thick layer).
It is to be noted that ALD of metal oxides is known to passivate surfaces such as flat silicon. The passivation materials are usually wide band gap semiconductors (e.g. ZrO2, Ta2O3, HfO2) or insulators (e.g. Al2O3) and can be used as dielectrics in microelectronic devices.57 However, to our knowledge they do not show photocatalytic activity. Ultrathin TiO2 was chosen in this work for its known photocatalytic effects and its interesting optical properties,58,59 also used for solar cell fabrication.
Influence of the coating on methylene blue degradation
The influence of the number of TiO2 ALD cycles on the photocatalytic activity of n-type PSi/TiO2 was explored. The degradation of MB was studied by monitoring the evolution of its absorption peak at 662 nm (see Fig. S6, ESI†) on group B, C and D samples and a control solution. Group A samples (uncoated) were not tested because PSi is known to have, if not protected, no photocatalytic activity due to corrosion.60 Since the catalytic activity depends on the contact area with the reactants, we did not consider a comparison with flat silicon samples.It must be considered that the lamp used for the experiment emits both UV and visible light (see Fig. S6, ESI†), which results in the heating of the solutions (in our case from 20 °C to 43 °C for 2 hours of measurements) and hence, potentially self-decomposition of MB occurs. Moreover, MB molecules can absorb light in the region of 500–700 nm leading to the formation of singlet and triplet species by electronic transition and intersystem crossing,61 causing luminescence quenching. In addition, MB can also undergo light-driven self-decomposition to a certain extent.62 The combination of these phenomena can explain why degradation appears also in the control solution that contains only MB.
Fig. 4a shows the percentage decomposition of MB as a function of time, whereas Fig. 4b presents the plot of 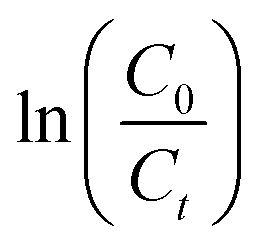 as a function of time. The percentage of decomposition was calculated using the relation,
as a function of time. The percentage of decomposition was calculated using the relation,
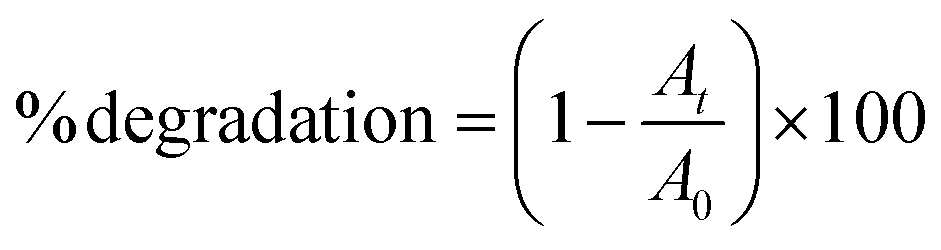 | (1) |
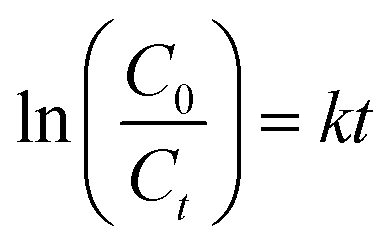 | (2) |
 | ||
| Fig. 4 Degradation rates of group B, C and D samples and control solution containing only MB (a). Plot of the MB decomposition in logarithmic scale as a function of time (b). | ||
Groups B and C samples follow very similar degradation kinetics, whilst the fastest degradation corresponds to the 10 nm-thick coating layers (group D), with an apparent rate constant that is 46% higher than that for the photofading process, (see Table S1 for the values of the resulting apparent rate constants, ESI†). Interestingly, all samples exhibit a faster degradation than the control solution, with very similar degradation kinetics that follows a linear behaviour, which means that TiO2 films that are as thin as a few angstroms still produce short-term photocatalytic effects. However, the trend for groups B and C samples clearly deviates from the pseudo-first kinetics after 20 and 40 min from the beginning of the light exposition, respectively. For both groups, at long times (t > 80 min) the degradation kinetics becomes comparable to that of the control solution. The deviation from linearity is particularly high for group B samples, reaching up to 88% at the end of the experiment. This can be explained because of the very low thickness of the TiO2 films: at short times, there are plenty of sites available at the surface for the adsorption of MB molecules and, as the dye is decomposed, new free ones rapidly replace them. This process, however, alters the TiO2, and since the films are thinner than 1 nm, after a certain time no more catalyst is left and the only mechanism of degradation of MB is photofading, leading to much slower decomposition rates.
Note that the electrical properties are not investigated here as they were previously studied in depth on planar silicon.63 However, performing this study on coated PSi would constitute an original work. Nevertheless, it is expected that TiO2 coatings of PSi will form nanostructures possessing unique optical, structural and electrical properties that can be used for the development of electrical/optical devices.
Conclusions
In this work, we used atomic layer deposition of TiO2 to chemically stabilize and protect PSi, which is otherwise prone to photo-oxidation. We demonstrated that sub-nanometric ALD coatings are able to fully – and conformally – coat the pores and, thanks to FTIR spectroscopy, we demonstrated that they can prevent oxidation even in aqueous media. Moreover, our experiments point out that this passivation function takes place in spite of the presence of a thin layer of native SiO2 between the surface of PSi and the TiO2. In addition, by monitoring the degradation of methylene blue in the presence of functionalized samples, we proved that these ultrathin TiO2 layers on macroporous Si show photocatalytic activity. Again, the native oxide layer occurring prior to ALD coating does not prevent the interaction between TiO2 and PSi.Our study provides a contribution towards the production of stable photoactive electrodes based on porous films using a very small amount of catalysts. The stability of PSi should no longer considered as a serious bottleneck for water-splitting of CO2 reduction applications.
Author contributions
Bachar Al Chimali: ALD, FTIR and MB degradation investigation, formal analysis, writing – original draft. Irene Carrasco: FTIR investigation, FTIR validation, FTIR and XPS formal analysis, writing – original draft, review and editing. Thomas Defforge: PSi etching methodology and validation. Romain Dailleau: ALD methodology and validation. Lisa Monnier: contact angle investigation and formal analysis. Kaushik Baishya: XPS investigation. Jan M. Macak: conceptualization, resources, supervision, writing – review and editing. Gaël Gautier: conceptualization, resources, supervision, writing – review and editing. Brice Le Borgne: conceptualization, supervision, formal analysis, writing – original draft, review and editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors want to acknowledge Dr Damien Valente and Dr Virginie Grimal for their technical support. The authors also thank Mr Thierry Halbout from STMicroelectronics for his technical support on the ALD tool. Dr Le Borgne thanks Pr. Joaquim P. Leitão and Dr Rui N. Pereira (i3n, Universidade de Aveiro) for their help in designing the experiment. We acknowledge CzechNanoLab Research Infrastructure (LM2023051), supported by the Ministry of Education, Youth and Sports of the Czech Republic, for the XPS measurements. We thank the ASCENT+ team at Tyndall Institute (Ireland) for its contribution to TEM observations, provided in the supplementary information. This project has received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement 871130. The French Agence Nationale de la Recherche supported this work through the TempoScopy project, grant ANR-19-CE42-0008.References
- J. Rasson and L. A. Francis, J. Phys. Chem. C, 2018, 122, 331–338 CrossRef.
- A. J. Simons, T. I. Cox, M. J. Uren and P. D. J. Calcott, Thin Solid Films, 1995, 255, 12–15 CrossRef.
- L. Canham, in Handbook of Porous Silicon, ed. L. Canham, Springer International Publishing, Cham, 2014, pp. 733–740 DOI:10.1007/978-3-319-05744-6_74.
- G. Mauckner, K. Thonke and R. Sauer, J. Phys.: Condens. Matter, 1993, 5, L9 CrossRef.
- X. Zhang, Y. Chen, R.-S. Liu and D. P. Tsai, Reports on progress in physics, Physical Society, Great Britain, 2013, vol. 76, p. 046401 Search PubMed.
- W. Theiß, Surf. Sci. Rep., 1997, 29, 91–192 CrossRef.
- M. Morita, T. Ohmi, E. Hasegawa, M. Kawakami and M. Ohwada, J. Appl. Phys., 1990, 68, 1272–1281 CrossRef.
- K. Oh, L. Joanny, F. Gouttefangeas, B. Fabre, V. Dorcet, B. Lassalle-Kaiser, A. Vacher, C. Mériadec, S. Ababou-Girard and G. Loget, ACS Appl. Energy Mater., 2019, 2, 1006–1010 CrossRef.
- J. M. Smieja, E. E. Benson, B. Kumar, K. A. Grice, C. S. Seu, A. J. Miller, J. M. Mayer and C. P. Kubiak, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15646–15650 CrossRef PubMed.
- C. Pacholski, M. Sartor, M. J. Sailor, F. Cunin and G. M. Miskelly, J. Am. Chem. Soc., 2005, 127, 11636–11645 CrossRef CAS PubMed.
- D. F. Shriver and M. A. Drezdzon, The Manipulation of Air-Sensitive Compounds, Wiley, 2nd edn, 1986 Search PubMed.
- R. Boukherroub, A. Petit, A. Loupy, J.-N. Chazalviel and F. Ozanam, J. Phys. Chem. B, 2003, 107(48), 13459–13462 CrossRef CAS.
- T. Dubois, F. Ozanam and J. N. Chazalviel, Proc. - Electrochem. Soc., 1997, 296–310 CAS.
- J. Salonen, A. M. Kaukonen, J. Hirvonen and V.-P. Lehto, J. Pharm. Sci., 2008, 97, 632–653 CrossRef CAS.
- P. Roy, C. Badie, J.-B. Claude, A. Barulin, A. Moreau, J. Lumeau, M. Abbarchi, L. Santinacci and J. Wenger, ACS Appl. Mater. Interfaces, 2021, 4(7), 7199–7205 CAS.
- A. Botas, J. Leitão, B. Falcão, M. Wiesinger, F. Rauh, J. P. Teixeira, H. Wiggers, M. Stutzmann, R. Ferreira and R. N. Pereira, ACS Appl. Nano Mater., 2020, 3(6), 5033–5044 CrossRef CAS.
- L. Santinacci, M. W. Diouf, M. K. Barr, B. Fabre, L. Joanny, F. Gouttefangeas and G. Loget, ACS Appl. Mater. Interfaces, 2016, 8, 24810–24818 CrossRef CAS.
- E. Chistè, G. Ischia, M. Gerosa, P. Marzola, M. Scarpa and N. Daldosso, Nanomaterials, 2020, 10(3), 463 CrossRef.
- S. Weis, R. Körmer, M. Jank, M. Lemberger, M. Otto, H. Ryssel, W. Peukert and L. Frey, Small, 2011, 7, 2853–2857 CrossRef CAS.
- B. Bardet, D. De Sousa Meneses, T. Defforge, J. Billoué and G. Gautier, Phys. Chem. Chem. Phys., 2016, 18, 18201–18208 RSC.
- J. Riikonen, M. Salomäki, J. van Wonderen, M. Kemell, W. Xu, O. Korhonen, M. Ritala, F. MacMillan, J. Salonen and V.-P. Lehto, Langmuir, 2012, 28, 10573–10583 CrossRef PubMed.
- B. Fodor, E. Agocs, B. Bardet, T. Defforge, F. Cayrel, D. Alquier, M. Fried, G. Gautier and P. Petrik, Microporous Mesoporous Mater., 2016, 227, 112–120 CrossRef.
- M. Fried, H. Wormeester, E. Zoethout, T. Lohner, O. Polgár and I. Bársony, Thin Solid Films, 1998, 313–314, 459–463 CrossRef.
- E. V. Astrova and V. A. Tolmachev, J. Mater. Sci. Eng. B, 2000, 69–70, 142–148 CrossRef.
- J. L. M. Villanueva, D. R. V. Huanca and A. F. Oliveira, Mater. Chem. Phys., 2023, 307, 128070 CrossRef.
- V. Lehmann, Electrochemistry of Silicon: Instrumentation, Science, Materials and Applications, 2002 Search PubMed.
- A. M. P. Botas, R. A. S. Ferreira, R. N. Pereira, R. J. Anthony, T. Moura, D. J. Rowe and U. Kortshagen, J. Phys. Chem. C, 2014, 118, 10375–10383 CrossRef.
- R. N. Pereira, S. Niesar, W. B. You, A. F. da Cunha, N. Erhard, A. R. Stegner, H. Wiggers, M. G. Willinger, M. Stutzmann and M. S. Brandt, J. Phys. Chem. C, 2011, 115, 20120–20127 CrossRef.
- S. Ng, J. Prášek, R. Zazpe, Z. Pytlíček, Z. Spotz, J. R. Pereira, J. Michalička, J. Přikryl, M. Krbal, H. Sopha, J. Hubálek and J. M. Macák, ACS Appl. Mater. Interfaces, 2020, 12, 33386–33396 CrossRef CAS.
- T. Imrich, R. Zazpe, H. Krýsová, Š. Paušová, F. Dvorak, J. Rodriguez-Pereira, J. Michalicka, O. Man, J. M. Macak, M. Neumann-Spallart and J. Krýsa, J. Photochem. Photobiol., A, 2021, 409, 113126 CrossRef CAS.
- B. D. Wiltshire, M. Alijani, H. Sopha, D. Pavliňák, L. Hromadko, R. Zazpe, S. M. Thalluri, E. Kolibalova, J. M. Macak and M. H. Zarifi, ACS Appl. Mater. Interfaces, 2023, 15, 18379–18390 CrossRef CAS PubMed.
- J. Rongé, T. Bosserez, D. Martel, C. Nervi, L. Boarino, F. Taulelle, G. Decher, S. Bordiga and J. A. Martens, Chem. Soc. Rev., 2014, 43, 7963–7981 RSC.
- R. Kuriki, K. Sekizawa, O. Ishitani and K. Maeda, Angew. Chem., Int. Ed., 2015, 54, 2406–2409 CrossRef CAS.
- H. Wang, L. Zhang, Y. Zhou, S. Qiao, X. Liu and W. Wang, Appl. Catal., B, 2020, 263, 118331 CrossRef CAS.
- S. Sato, T. Morikawa, T. Kajino and O. Ishitani, Angew. Chem., Int. Ed., 2013, 52, 988–992 CrossRef CAS PubMed.
- A. B. Getsoian, Z. Zhai and A. T. Bell, J. Am. Chem. Soc., 2014, 136, 13684–13697 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- I. Iatsunskyi, M. Jancelewicz, G. Nowaczyk, M. Kempiński, B. Peplińska, M. Jarek, K. Załęski, S. Jurga and V. Smyntyna, Thin Solid Films, 2015, 589, 303–308 CrossRef CAS.
- N. Holden, T. Coplen, J. Bohlke, L. Tarbox, J. Benefield, J. Laeter, P. Mahaffy, G. O’Connor, E. Roth, D. Tepper, T. Walczyk, M. Wieser and S. Yoneda, Pure Appl. Chem., 2018, 90, 1833–2092 CrossRef CAS.
- J. Bacova, P. Knotek, K. Kopecka, L. Hromadko, J. Capek, P. Nyvltova, L. Bruckova, L. Schröterova, B. Sestakova, J. Palarcik, M. Motola, D. Cizkova, A. Bezrouk, J. Handl, Z. Fiala, E. Rudolf, Z. Bilkova, J. M. Macak and T. Rousar, Int. J. Nanomed., 2022, 17, 4211–4225 CrossRef CAS.
- G. San Vicente, A. Morales and M. T. Gutierrez, Thin Solid Films, 2001, 391, 133–137 CrossRef CAS.
- US Pat., 3, 533, 850, 1970 Search PubMed.
- H. Sopha, A. T. Tesfaye, R. Zazpe, J. Michalicka, F. Dvorak, L. Hromadko, M. Krbal, J. Prikryl, T. Djenizian and J. M. Macak, FlatChem, 2019, 17, 100130 CrossRef CAS.
- R. Pessoa and M. Fraga, Front. Nanosci., 2019, 291–307, DOI:10.1016/B978-0-08-102572-7.00011-8.
- M. Motola, J. Capek, R. Zazpe, J. Bacova, L. Hromadko, L. Bruckova, S. Ng, J. Handl, Z. Spotz, P. Knotek, K. Baishya, P. Majtnerova, J. Prikryl, H. Sopha, T. Rousar and J. M. Macak, ACS Appl. Bio Mater., 2020, 3, 6447–6456 CrossRef CAS.
- S. Ng, M. Krbal, R. Zazpe, J. Prikryl, J. Charvot, F. Dvořák, L. Strizik, S. Slang, H. Sopha, Y. Kosto, V. Matolin, F. K. Yam, F. Bures and J. M. Macak, Adv. Mater. Interfaces, 2018, 5, 1701146 CrossRef.
- J. W. Shin, S. Oh, S. Lee, J. G. Yu, J. Park, D. Go, B. C. Yang, H. J. Kim and J. An, ACS Appl. Mater. Interfaces, 2019, 11, 46651–46657 CrossRef CAS.
- M. Juntunen, J. Heinonen, V. Vähänissi, P. Repo, D. Valluru and H. Savin, Nat. Photonics, 2016, 10, 777–781 CrossRef CAS.
- P. Kočí, M. Isoz, M. Plachá, A. Buzková Arvajová, M. Václavík, M. Svoboda, E. Price, V. Novák and D. Thompsett, Catal. Today, 2019, 320, 165–174 CrossRef.
- J. Zhang, P. Zhou, J. Liu and J. Yu, Phys. Chem. Chem. Phys., 2014, 16, 20382–20386 RSC.
- T. Osaka, K. Ogasawara and S. Nakahara, J. Electrochem. Soc., 1997, 144, 3226 CrossRef.
- M. Murata, K. Wakino and S. Ikeda, J. Electron Spectrosc. Relat. Phenom., 1975, 6, 459–464 CrossRef.
- P. Stefanov, M. Shipochka, P. Stefchev, Z. Raicheva, V. Lazarova and L. Spassov, J. Phys.: Conf. Ser., 2008, 100, 012039 CrossRef.
- J. Pouilleau, D. Devilliers, H. Groult and P. Marcus, J. Mater. Sci., 1997, 32, 5645–5651 CrossRef.
- R. Methaapanon and S. F. Bent, J. Phys. Chem. C, 2010, 114, 10498–10504 CrossRef.
- W. Gu and C. P. Tripp, Langmuir, 2005, 21, 211–216 CrossRef PubMed.
- J. Ren, G. Zhou, Y. Hu, H. Jiang and D. W. Zhang, Appl. Surf. Sci., 2008, 254, 7115–7121 CrossRef.
- Z. Lin, C. Jiang, C. Zhu and J. Zhang, ACS Appl. Mater. Interfaces, 2013, 5, 713–718 CrossRef CAS PubMed.
- K. M. Gad, D. Vössing, A. Richter, B. Rayner, L. M. Reindl, S. E. Mohney and M. Kasemann, IEEE J. Photovolt., 2016, 6, 649–653 Search PubMed.
- J. Su, H. Yu, X. Quan and Q. Zhao, Sep. Purif. Technol., 2012, 96, 154–160 CrossRef.
- E. Subramanian, S. Subbulakshmi and C. Murugan, Mater. Res. Bull., 2014, 51, 128–135 CrossRef.
- S. Shahabuddin, N. Muhamad Sarih, S. Mohamad and J. Joon Ching, Polymers, 2016, 8, 27 CrossRef PubMed.
- M. E. Dufond, M. W. Diouf, C. Badie, C. Laffon, P. Parent, D. Ferry, D. Grosso, J. C. S. Kools, S. D. Elliott and L. Santinacci, Chem. Mater., 2020, 32, 1393–1407 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: (1) ALD window. (2) TEM images of TiO2 films for samples from groups B and D. (3) Detailed high-resolution XPS spectrum of the Si 2s and 2p peaks. (4) Water contact angle measurements. (5) Complete FTIR spectra (400 to 4000 cm-1). (6) Evolution of the absorption band at 620 nm of MB in aqueous solution. (7) Luminescence spectra. (8) Apparent rate constants for MB degradation. (9) ESI References. See DOI: https://doi.org/10.1039/d4ma00654b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |