
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Green fluorescent protein chromophore-based covalent organic polymers (GFPC-COPs): sensing of nitroaromatic organic pollutants and explosives†
Gulshan
Anjum
a,
Ashish
Kumar
a,
Gurunath
Ramanathan
 *a and
Jarugu Narasimha
Moorthy
*ab
*a and
Jarugu Narasimha
Moorthy
*ab
aDepartment of Chemistry, Indian Institute of Technology Kanpur, 208016, India. E-mail: moorthy@iitk.ac.in; gurunath@iitk.ac.in
bSchool of Chemistry, Indian Institute of Science Education and Research Thiruvananthapuram, Trivandrum 695551, India
First published on 22nd October 2024
Abstract
Green fluorescent protein (GFP) is extremely fluorescent in nature, and the chromophore that is responsible for the remarkable fluorescence has been identified as the benzylidene imidazolinone moiety. Emulation of high fluorescence, such as that observed in GFP in synthetic models, is thwarted by rapid bond rotation about C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–C bonds in the singlet-excited state. We have rationally designed and developed four covalent organic polymers (COPs) based on two di- and trihydroxy-substituted GFP chromophores, that is, (Z)-4-(4-hydroxybenzylidene)-1-(4-hydroxyphenyl)-2-phenyl-5-imidazolinone (DHI) and (Z)-4-(4-hydroxybenzylidene)-1,2-bis(4-hydroxyphenyl)-5-imidazolinone (THI), to emulate the fluorescence properties of GFP. Of the four polymers, two polymers based on DHI and THI were found to exhibit the highest solid-state fluorescence quantum yields of 83 and 73%, respectively. They are porous and their BET surface areas based on CO2 sorption studies at 195 K were found to be 107.9 and 164.8 m2 g−1. AFM analyses of the two polymeric materials revealed that the polymer based on DHI is comprised of nanosheets, while that based on THI exhibits a nanosphere morphology. It is shown that both polymers can be employed as sensory materials for the selective sensing of nitroaromatic compounds (NACs) containing acidic hydrogens, that is, nitrophenols, nitrothiophenols, and nitroanilines. The quenching efficiencies are found to be more than 80% for these NACs. For example, 4-nitroaniline (4-NA) and picric acid (TNP) are sensed at sub-ppm levels by the suspensions of the GFPC-COPs. Furthermore, the COP based on THI is demonstrated to serve as a mop for rapid adsorption of TNP; the latter is shown to be adsorbed by the COP by more than 50% from its solution in methanol within 10 min. The results exemplify the potential of innovative biomimetic design principles as applied to materials development and sensing technology.
C and C–C bonds in the singlet-excited state. We have rationally designed and developed four covalent organic polymers (COPs) based on two di- and trihydroxy-substituted GFP chromophores, that is, (Z)-4-(4-hydroxybenzylidene)-1-(4-hydroxyphenyl)-2-phenyl-5-imidazolinone (DHI) and (Z)-4-(4-hydroxybenzylidene)-1,2-bis(4-hydroxyphenyl)-5-imidazolinone (THI), to emulate the fluorescence properties of GFP. Of the four polymers, two polymers based on DHI and THI were found to exhibit the highest solid-state fluorescence quantum yields of 83 and 73%, respectively. They are porous and their BET surface areas based on CO2 sorption studies at 195 K were found to be 107.9 and 164.8 m2 g−1. AFM analyses of the two polymeric materials revealed that the polymer based on DHI is comprised of nanosheets, while that based on THI exhibits a nanosphere morphology. It is shown that both polymers can be employed as sensory materials for the selective sensing of nitroaromatic compounds (NACs) containing acidic hydrogens, that is, nitrophenols, nitrothiophenols, and nitroanilines. The quenching efficiencies are found to be more than 80% for these NACs. For example, 4-nitroaniline (4-NA) and picric acid (TNP) are sensed at sub-ppm levels by the suspensions of the GFPC-COPs. Furthermore, the COP based on THI is demonstrated to serve as a mop for rapid adsorption of TNP; the latter is shown to be adsorbed by the COP by more than 50% from its solution in methanol within 10 min. The results exemplify the potential of innovative biomimetic design principles as applied to materials development and sensing technology.
Introduction
Green fluorescent protein (GFP) from the Aequorea victoria, a jellyfish, is a protein which emits green fluorescent light upon exposure to UV light.1,2p-Hydroxybenzylideneimidazolinone (p-HBDI) has been identified as the chromophore unit that is responsible for the fluorescence of GFP.2 Because of its remarkable fluorescence (Φflca. 79%),3 GFP has been widely employed as a biological marker for gene expression2 and cell imaging applications.4 The isolated chromophore of GFP, however, does not fluoresce in its free state. It is fluorescent only when buried in the β-barrel structure of the protein, attesting to the fact that the protein enclosure plays a decisive role in the observed fluorescence of GFP;5,6 rotation around the exocyclic CC as well as C–C bonds of the chromophore in the free state has been implicated for efficient nonradiative decay of the singlet-excited state such that the fluorescence is suppressed.7,8 Evidently, the protein cage hinders the free rotation around the double bond of the GFP chromophore, which is held rigidly by H-bonds and other hydrophobic forces such that the radiative pathway is promoted to account for exceptionally high fluorescence quantum yield.6,9,10 Buoyed by the remarkable quantum yield of GFP, several approaches have been explored to restrict the free bond rotation in the synthetic mimics/analogs of GFP chromophores and enhance the fluorescence efficiency. These include the application of an electric field,11 creation of the β-barrel structure of the protein,12,13 synthesis of self-restricted rigid GFP chromophore analogues,14–16 encapsulation in bioconjugates17 and supramolecular hosts,12 entrapment in amphiphilic copolymers,18–20 inclusion in porous materials,21–24etc. GFP chromophores with enhanced fluorescence efficiency have been explored for sensing of metal ions such as Zn,25,26 and Hg,27 anions such as F−,28 and CN−,29 H2S in water as well as in living cells,30etc.
In our ongoing research focused on the development of functional porous organic materials in a bottom-up approach by rational design of molecular building blocks,31–34 we were enticed by the reports of the exploitation of the GFP chromophore in the construction of porous materials, that is, metal–organic frameworks (MOFs) and covalent porous organic polymers (POPs), to mimic the β-barrel environment of GFP to hinder bond rotations and exemplify photophysical properties akin to those of native GFP. Shustova and coworkers first constructed a MOF based on a structurally modified GFP chromophore and demonstrated replication of the β-barrel environment of the chromophore in a MOF by restricting the bond rotations; the MOF thus created was shown to exhibit emission λmax and lifetime akin to those of the native GFP. They also demonstrated approaches involving immobilization as well as the inclusion of the GFP chromophore in the MOF cavities and exemplified access to materials with tunable luminescence properties.21,22 Maji et al. reported the construction of a POP by employing a triiodo-arylidene imidazolinone and setting it up for a 3-fold Sonogashira coupling with 3-connecting 1,3,5-triethynylbenzene.23 The conjugated microporous polymer was thoroughly characterized and its photophysical properties were comprehensively established. The POP was shown to exhibit emission and lifetime similar to those of GFP. They also showed that the GFP chromophore can be immobilized on a nanoscale MOF-808 by postsynthetic modification.24 These studies sufficiently illustrate the fact that the utility of the GFP chromophore as a building block in the construction of porous materials permits access to luminescent porous materials. An incisive analysis of the GFP chromophore, that is, arylidene imidazolinone, reveals that the resonance delocalization should render the carbonyl oxygen electron-rich and hence an excellent hydrogen-bond acceptor, as also the N3 nitrogen with its sp2 hybridized lone pair. These attributes should make the chromophore extraordinarily sensitive to the environment. Indeed, solvatochromic studies on GFP chromophores have thoroughly established this feature.10 In this backdrop, we recognized that the studies of porous materials are largely limited to mimicking the photophysical properties of GFP, but any study concerned with the exploitation of the GFP chromophore-based porous materials for sensing and capturing guest species, by capitalizing on their interaction with the imidazolinone oxygen and nitrogen atoms, is singularly lacking.
In recent years, COPs have emerged as a class of porous materials with exceptional physicochemical stabilities.35–37 Their construction in diverse ways by a judicious choice of organic building blocks from an infinitely large pool limited only by one's imagination and a careful selection of organic reactions for covalent polymerization from a vast repertoire of transformations offers the opportunity to develop tailor-made materials for targeted applications. In continuation of our efforts on the development of porous functional materials by de novo rational design,31–34 we have been developing POPs as heterogeneous catalytic materials for diverse organic transformations in a recyclable fashion.38–41 Herein, we show that four polymers, namely, DOIDB, DOITP, TOIDB, and TOITP, constructed readily by Williamson etherification of the GFP chromophores, that is, di- and tri-hydroxyarylidene imidazolinones, with p-xylylene dibromide and 1,3,5-tris(p-bromomethylphenyl)benzene (Scheme 1), can be used for turn-off fluorescence sensing of nitrophenols (NPs), nitrothiophenols (NTPs) and nitroanilines (NAs) selectively. The latter are shown to be sensed in sub-ppm levels. Furthermore, the capture of 2,4,6-trinitrophenol (TNP) effectively by one of the polymers is also demonstrated. It is noteworthy that nitroaromatic compounds (NACs) such as nitroanilines, nitrophenols, and nitrothiophenols are important raw materials employed in agriculture, industry and defence applications; they are used in the synthesis of dyes, pharmaceuticals, pesticides, rubber, explosives, and paints. They indeed constitute highly toxic pollutants with several health hazards;42–48 2,4-dinitrophenol (2,4-DNP) and 2,4,6-trinitrophenol (TNP, picric acid) are explosives.48,49 The latter is a nonbiodegradable micropollutant with enhanced explosive power compared to 2,4,6-trinitrotoluene (TNT).50
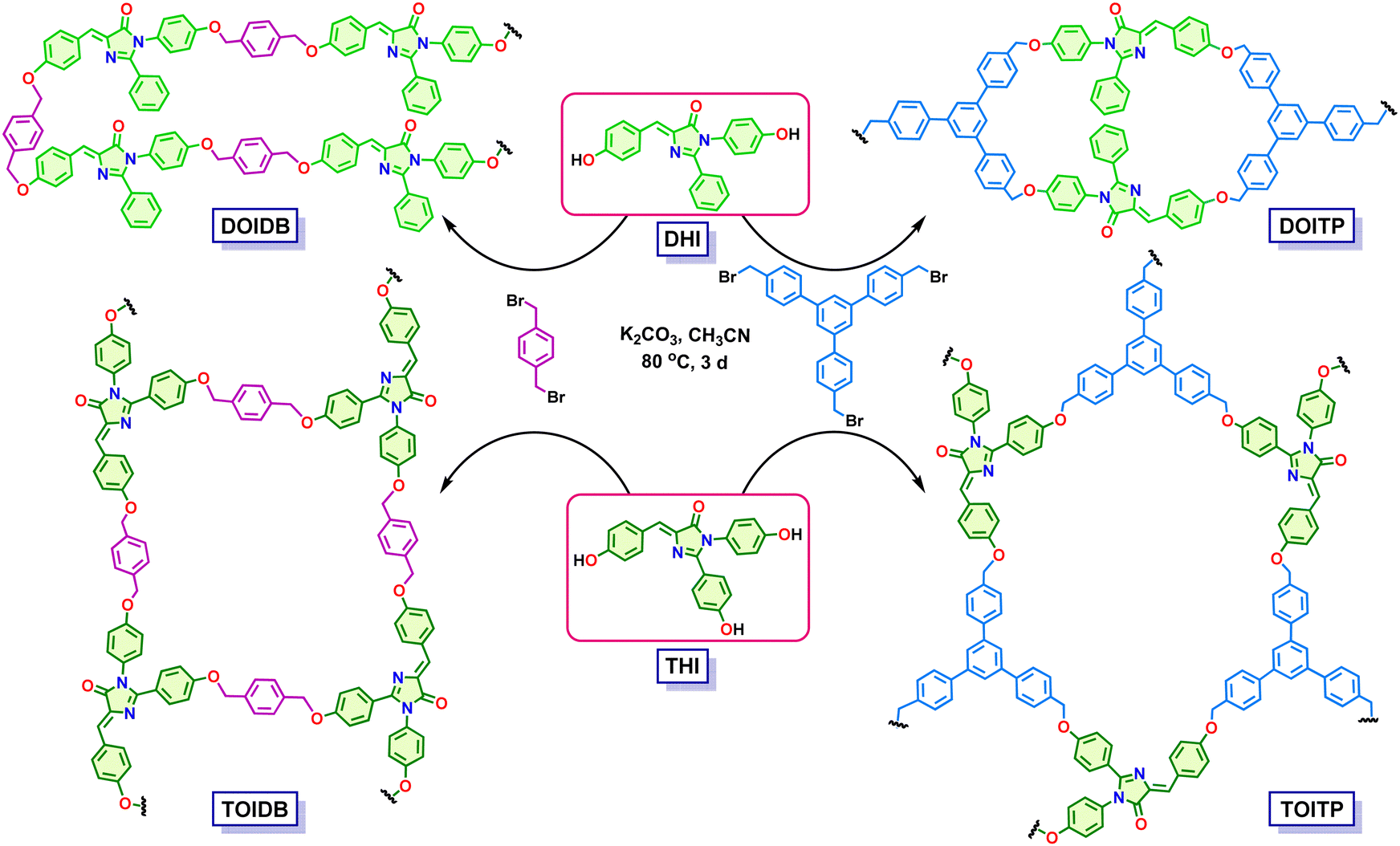 | ||
| Scheme 1 Synthesis of DOIDB, DOITP, TOIDB and TOITP polymers. | ||
Results and discussion
Synthesis of dihydroxy- and trihydroxy-imidazolinone monomers (DHI and THI)
The required GFP monomers, that is, (Z)-4-(4-hydroxybenzylidene)-1-(4-hydroxyphenyl)-2-phenyl-5-imidazolinone (DHI) and (Z)-4-(4-hydroxybenzylidene)-1,2-bis(4-hydroxyphenyl)-5-imidazolinone (THI), were synthesized by following a reported procedure involving Erlenmeyer azalactone51,52 reaction to obtain appropriately-substituted oxazolone starting from p-methoxybenzaldehyde and N-benzoylglycine derivatives, followed by reaction of the latter with p-anisidine and subsequent demethylation using BBr3; both phenols were isolated in excellent yields (Schemes S1 and S2, ESI†). UV-vis absorption and fluorescence emission spectra of DHI and THI in MeOH are shown in Fig. S1 (ESI†).Synthesis of covalent organic polymers
Williamson etherification of DHI and THI with two different 2- and 3-connecting bromomethylarenes, that is, 1,4-bis(bromomethyl)benzene and 1,3,5-tris(4-bromomethylphenyl)benzene in the presence of K2CO3 and CH3CN led to four different polymers. All the polymers based on the GFP chromophore, that is, arylidene imidazolinone, were obtained as insoluble materials after the reaction under reflux conditions over 3 d. The formation of polymers with structures like those shown in Scheme 1 was established by Fourier-transform infrared (FT-IR) spectroscopy and 13C solid-state NMR analyses. Their physical properties were investigated by thermogravimetric analyses (TGA), scanning electron microscopy-energy dispersive X-ray analysis (SEM-EDX), transmission electron microscopy (TEM) and X-ray photoelectron spectroscopy (XPS). The BET surface areas and porosity of the polymers were examined by N2 and CO2 gas sorption studies.The FT-IR spectra of all the product polymers reveal complete conversion of the starting materials to polymers as evidenced by the absence of absorptions due to C–Br stretches at 607 and 603 cm−1 in 1,4-bis(bromomethyl)benzene (BBMB) and 4,4′′-bis(bromomethyl)-5′-(4-(bromomethyl)phenyl)-1,1′:3′,1′′-terphenyl (TBMTP), respectively. At the same time, the IR spectra of the product polymers also reveal the absence of absorption bands due to O–H stretches in DHI and THI at 3361 and 3387 cm−1, respectively. The appearance of an absorption band due to C–O–C stretch in the range of 1238–1160 cm−1 in all the polymers attests to the successful formation of covalent organic polymers (Fig. 1a–c). Solid-state 13C NMR spectra were recorded for all the polymers to further establish their formation unambiguously. The spectra reveal the presence of different carbon atoms in the polymers. A prominent signal at 160 ppm is assigned to the imidazolinone carbonyl group. The peaks at 135.8, 127.7 and 115 ppm, assigned to different CC carbons, and the signal at 69.4 ppm, ascribed to the benzylic carbon bonded to the oxygen, confirm the successful formation of the polymers (Fig. 1d). The signals marked with asterisks in the region of 45–55 ppm appear to be those of the trapped solvent molecules.
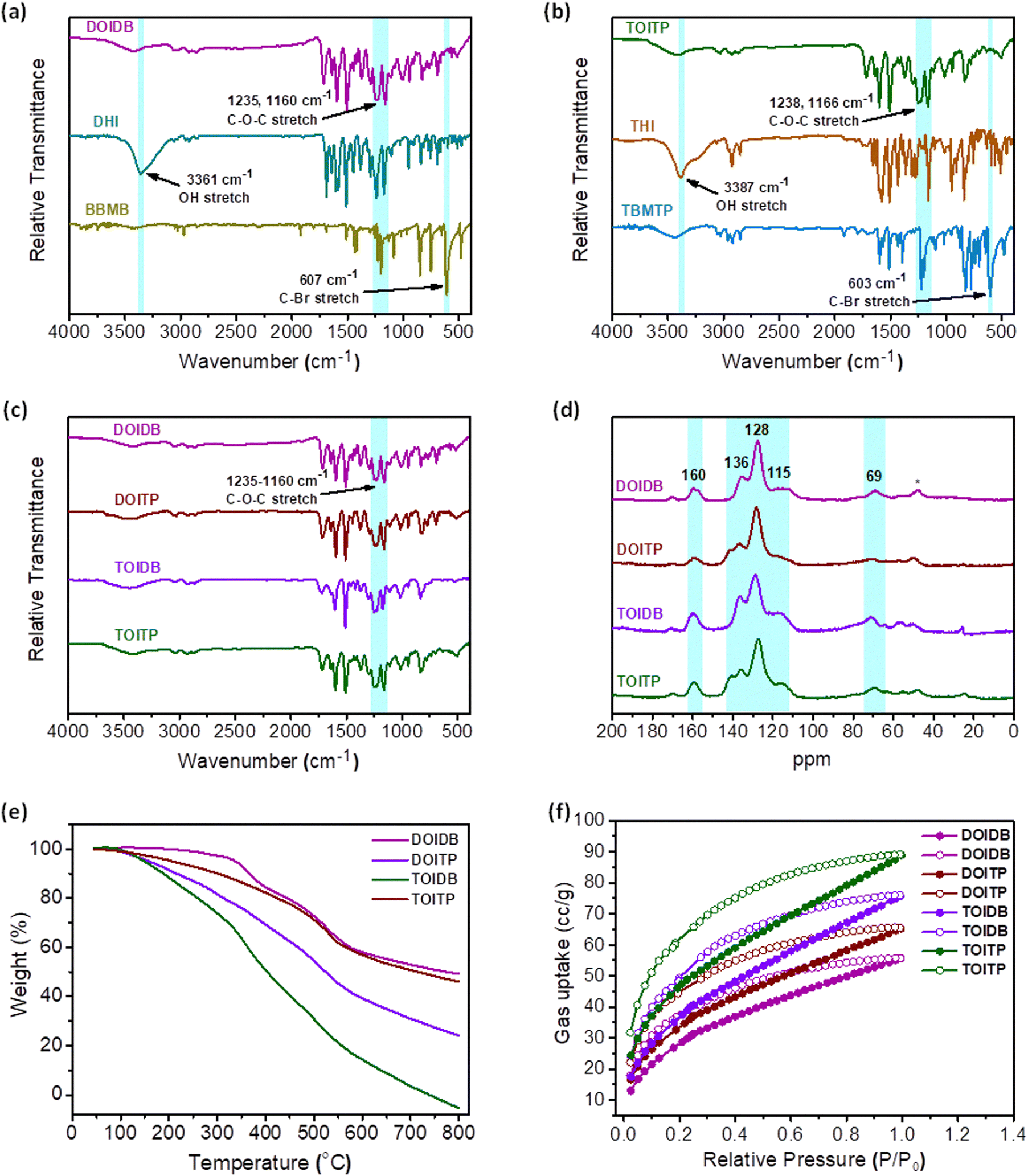 | ||
| Fig. 1 (a) and (b) FT-IR spectra of DOIDB and TOITP with their monomers and organic linkers. (c) FT-IR spectra, (d) solid-state 13C NMR, (e) TGA and (f) CO2 adsorption–desorption isotherms of DOIDB, DOITP, TOIDB, and TOITP. | ||
The TGA analyses under a N2 atmosphere show that DOIDB loses less than 5% up to 350 °C, while TOITP undergoes weight loss by up to 13% until 350 °C. It turns out that DOITP and TOIDB undergo more weight loss and show decomposition much faster. Otherwise, all polymers exhibit stability up to 350 °C (Fig. 1e). The chemical composition of the polymers was examined by XPS and SEM-EDX analyses. The full XPS survey spectra reveal peaks for C 1s, N 1s, and O 1s. The binding energy for C 1s in the XPS spectrum could be fitted to five distinct peaks at 284.2, 285.1, 286, 287, and 288.3 eV for DOIDB, and 284.4, 285.4, 286.3, 287.3 and 288.6 eV for TOITP, corresponding to the carbon atoms of the aromatic rings (C–C/CC/C–H), C–O/CN, CC–N/C–N, N–CO and CO bonds of imidazolinone of both the polymers, respectively. The band corresponding to the binding energy of N 1s could also be fitted likewise to three peaks at 398.4, 399.5, and 401 eV corresponding to CN–C, C–N/N–(CO) and CN of DOIDB and at 399.2, 400.4 and 401.8 for TOITP. The XPS spectra for O 1s can be deconvoluted into two peaks at 531.1 and 532.3 eV for DOIDB and 531.7 and 533 eV for TOITP corresponding to CO and C–O–C; these results indicate the existence of imidazolinone and ether bonds within the polymers, confirming the presence of the GFP chromophore within the polymeric matrix (Fig. S2, ESI†). From the elemental mapping and SEM-EDX analyses of both polymers, percentage ratios of C, N, and O could be evaluated (Fig. S3, ESI†).
The surface areas of all polymers DOIDB, DOITP, TOIDB, and TOITP based on the Brunauer–Emmett–Teller (BET) calculation model were determined for nitrogen at 77 K to be 8.2, 23.79, 9.82 and 9.2 m2 g−1, respectively (Fig. S4, ESI†). However, much better surface areas were determined for CO2 sorption at 195 K. The BET surface areas thus determined based on CO2 adsorption for all the polymers are 107.9, 121.9, 136.2 and 164.8 m2 g−1, respectively (Fig. 1f and Fig. S4, ESI†). It is noteworthy that the porosity observed by nitrogen sorption is unremarkable, while that by CO2 adsorption is moderate for polymers that contain hetero atoms. In view of the fact that the BET surface areas are uniformly lower and also because DOIDB is a 1-dimensional polymer, we have opted to term all the polymers as covalent organic polymers (COPs) instead of porous organic polymers (POPs) with subdued emphasis on porosity. Otherwise, it should be noted that COPs with low surface areas of the order of 7–40 m2 g−1 have indeed been termed POPs in the literature.53–55 The pore size distribution of all polymers calculated by nonlocal density functional theory (NLDFT) from N2 adsorption data, revealed that the pore sizes range from 0.77 nm to 1.32 nm (Fig. S5, ESI†). The surface morphologies were examined for two selected polymers by SEM after ultrasonication of the polymers for 1 h in MeOH. The SEM images show that DOIDB is comprised of sheets, while TOITP COP exhibits spherical morphology (Fig. 2a and e). As can be seen, the HRTEM image reveals the presence of thin layers and spheres similar to those observed in SEM (Fig. 2b and f). These observations are quite intriguing because DOIDB can only propagate as a 1-dimensional polymer, while TOITP should be expected to be a 2-dimensional layered material. The layered morphology revealed by microscopic analysis suggests that the 1D layers of DOIDB can undergo supramolecular self-assembly into nanosheets, while the nanosheets in the case of TOITP fold into nanospheres. These sheets and spheres were further investigated by atomic force microscopy (AFM). For this purpose, the methanolic colloidal suspensions of the polymers obtained by sonication for 120 min were drop-cast on a silicon wafer, and the morphology of the surface was examined. The height-profile diagrams reveal that the thickness of the sheets varies in the range of ca. 6–7 nm, while those of the spheres centre around 5–6 nm, signifying further the fact that DOIDB is made up of nanosheets (Fig. 2c and d) and that TOITP is comprised of nanospheres (Fig. 2g and h).
 | ||
| Fig. 2 (a) and (b) SEM and TEM images of DOIDB, respectively. (c) and (d) AFM image and height-profile diagram of the nanosheet of DOIDB. (e) and (f) SEM and TEM images of TOITP, respectively. (g) and (h) AFM image and height-profile diagram of the nanosphere of TOITP. | ||
Photophysical properties of the polymers
Solid-state fluorescence excitation spectra of the four COPs show that all polymers exhibit absorption maxima between 365–371 nm. Similarly, solid-state emission spectra reveal that all polymers exhibit emission maxima between 510–542 nm (Fig. 3a–d). The polymers, as expected, were found to be highly fluorescent under UV light (360 nm) (Fig. 3e). Fluorescence efficiencies were evaluated by determining their absolute solid-state emission quantum yields using an integrating sphere attached to the spectrofluorimeter. To begin with, the literature reported solid-state quantum yield for pyrene as the standard (Φflca. 68%)56 was established; the quantum yield using our integrating sphere attachment was determined to be 67.44% for excitation at 320 nm. Subsequently, the absolute quantum yields of emission of all polymers were determined; these were found to be 83, 44, 28, and 73% for DOIDB, DOITP, TOIDB, and TOITP, respectively, for excitation at λex 365 nm for DOIDB and DOITP and at λex 370 nm for TOIDB and TOITP. Two polymers, namely DOIDB and TOITP, exhibited the highest quantum yields of emission. These are also the two polymers that exhibited the lowest (DOIDB) and highest (TOITP) BET surface areas based on CO2 adsorption, which implies that close-packed polymers exhibit better fluorescence emission. Based on these properties in conjunction with the results of TGA analyses, our further studies, vide infra, were restricted to these polymers, i.e., DOIDB and TOITP.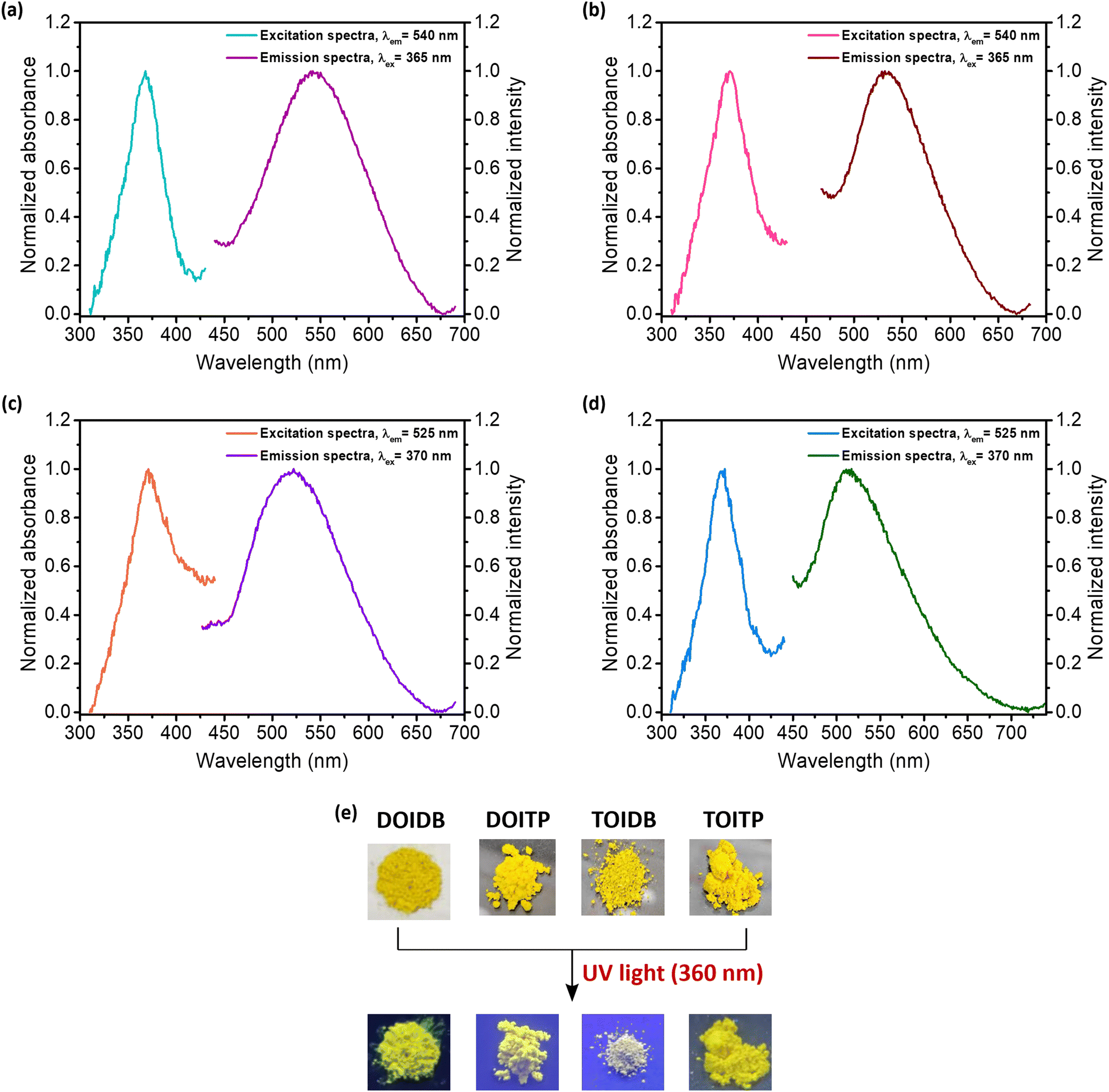 | ||
| Fig. 3 Solid-state fluorescence excitation and emission spectra of (a) DOIDB, (b) DOITP, (c) TOIDB and (d) TOITP, respectively. (e) Images of the polymers in room light and under exposure to 360 nm radiation. | ||
Solvatochromic properties of DOIDB and TOITP polymers
As mentioned earlier, arylidene imidazolinones are fluorescent in nature. The influence of solvents on their emission properties in the solid state was explored by suspending the finely ground polymers, that is, DOIDB and TOITP, in different solvents. Thus, 1.0 mg of each of the polymers was dispersed in 10 mL of a representative solvent, and subjected to ultrasonication for 60 min to obtain a homogeneous suspension; this corresponds to the concentration of 100 ppm based on mass/volume of the solvent, that is, 0.1 mg mL−1. The solvatochromic properties of the polymers were investigated using such suspensions. The fact that ultrasonication for 60 min does not affect the structures of the polymers was confirmed by removing the solvent and analysing the polymeric materials thus isolated back by IR and solid-state 13C NMR analyses (Fig. S6a and b, ESI†). While both polymers were found to be completely non-fluorescent in nonpolar solvents such as n-hexane, cyclohexane, and toluene, they were found to exhibit highly fluorescent behaviour in polar solvents such as DCM, THF, dioxane, DMSO, MeCN, iso-propanol, EtOH, MeOH, and water. The emission spectra of both polymers in different solvents are shown in Fig. S6 (ESI†). As can be seen, the emission λmax of DOIDB was found to shift bathochromically from 476 nm to 496 nm in going from DCM to H2O. In the same manner, the emission λmax of TOITP was also found to vary from 490 nm to 496 nm for DCM to H2O (Fig. S6c and d, ESI†).The emission properties of both polymer suspensions were compared with those of their monomers in a solvent such as methanol. The fluorescence excitation and emission spectra recorded for both polymers and their monomers in MeOH are shown in Fig. 4. What is noteworthy is that the excitation and emission spectra of both polymers DOIDB and TOITP compare very closely with their absorption maxima differing only by 2 nm (365 and 367 nm) and the emission maxima were likewise found to differ by 2 nm, appearing at 493 and 495 nm (Fig. 4a and b). When fluorescence emission properties were contrasted with those of their monomeric precursors, the polymers DOIDB and TOITP displayed bathochromically shifted emission maxima. The emission is red-shifted by 26–28 nm for both polymers relative to that of their precursor monomers (Fig. 4c and d). This attests to the fact that the chromophores are more stretched out in their conformations such that there exists more resonance delocalization. Furthermore, the emission λmax observed for the GFP chromophore in its protein barrel is 504 nm.3 This points to the fact that the chromophore in both polymers is somewhat twisted with a marginal resonance inhibition.
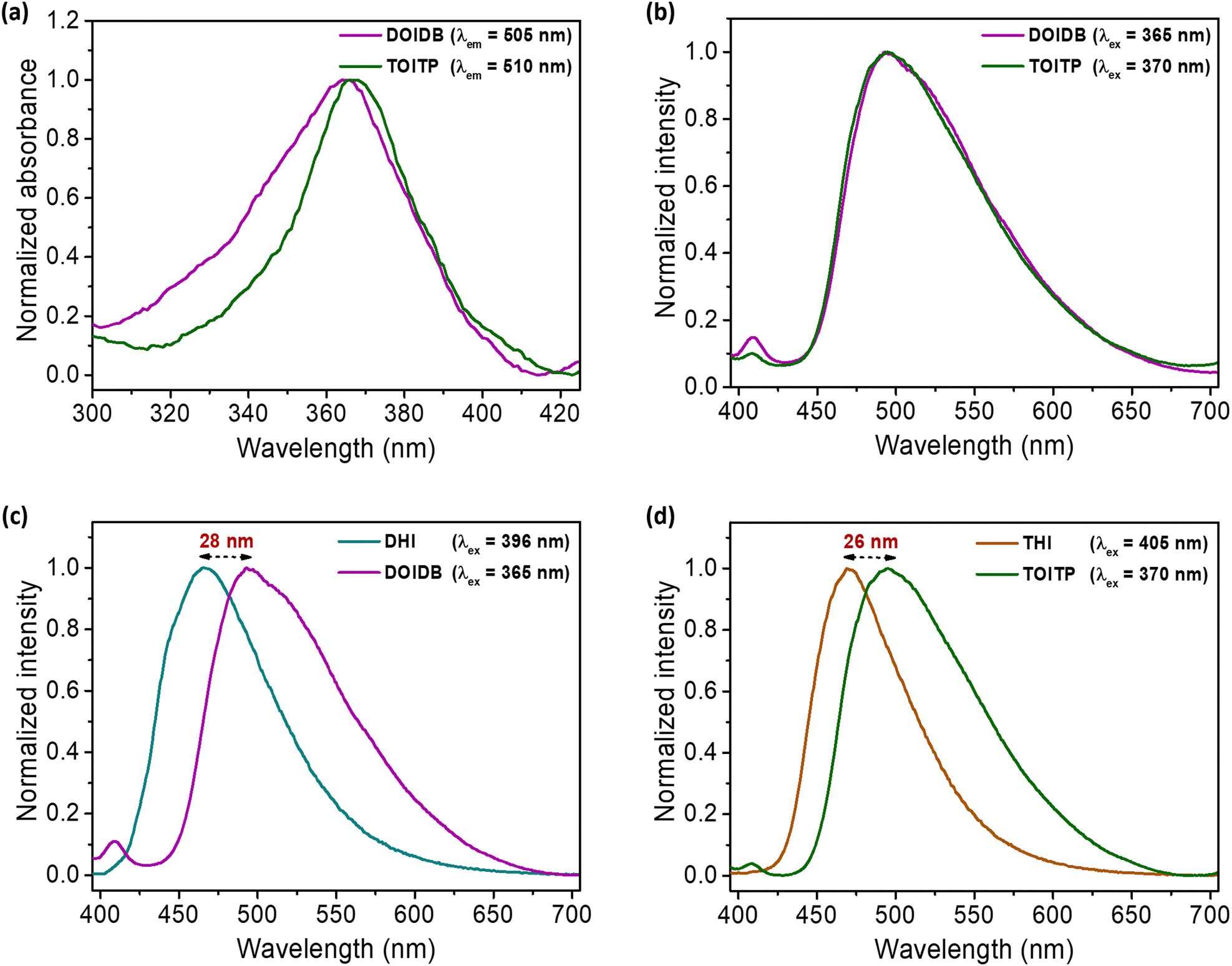 | ||
| Fig. 4 (a) Fluorescence excitation spectra of DOIDB and TOITP suspensions in MeOH. (b) Fluorescence emission spectra of DOIDB and TOITP suspensions in MeOH. (c) Comparison of the emission spectra of DHI and DOIDB in MeOH. (d) Comparison of the emission spectra of THI and TOITP in MeOH. | ||
‘Turn-off’ fluorescence sensing of 2,4,6-trinitrophenol (TNP) and other nitroaromatic organic pollutants by DOIDB and TOITP GFPC-COPs
The presence of pores in the COPs with exposed chromophores that exhibit high quantum yields of fluorescence were the prime reasons to examine the interaction of guest species with the imidazolinone moieties of the chromophores as reflected by fluorescence changes and develop sensing systems. We considered hazardous and explosive nitroaromatic compounds (NACs) for exploring their influence on the fluorescence properties of the polymer suspensions in MeOH. Amongst nitrobenzenes (NBs), 2-/3-/4-nitrophenols (NPs), 2,4-dinitrophenol (2,4-DNP) and 2,4,6-trinitrophenol (TNP) as analytes, the fluorescence was found to be influenced dramatically by all except the parent nitrophenol, implying the necessity of acidic phenol for turn-off fluorescence sensing. Indeed, 2,4,6-trinitrotoluene (TNT) was found not to affect the fluorescence (Fig. S7, ESI†). Clearly, the electron-deficient attribute of the analyte alone is not responsible for the quenching of the fluorescence of the polymers. Prompted by this observation, thiophenol and nitrothiophenols (NTPs), and aniline and nitroanilines (NAs) were examined for their quenching of the fluorescence of the polymers. It turns out that aniline, phenol, thiophenol and nitrobenzene alone were found to be poor quenchers of the fluorescence of the polymers, attesting to the fact that the analytes with acidic hydrogens only quench the fluorescence of the polymers (Fig. S7 and S8, ESI†). Thus, nitroaromatic compounds containing acidic hydrogens were found to selectively influence the fluorescence of GFPC-COPs remarkably. We also checked the sensitivity of DOIDB and TOITP polymers to different pH values (acidic, neutral and basic) by preparing the suspensions of the polymers in Milli-Q water and found that they are insensitive and that their fluorescence is unaffected (Fig. S9, ESI†).More elaborate fluorescence quenching experiments were carried out with incremental addition of the NACs to the suspensions of DOIDB and TOITP polymers in methanol separately. The suspension were prepared by dispersing 1 mg of each of the polymers in 10 mL of methanol, amounting to 100 ppm. With increasing conc. of the NACs, a progressive decrease in the fluorescence intensities of both the polymers was observed. The intensities fluorescence intensity of the suspensions of DOIDB and TOITP polymers were found to be quenched by ca. 88% and 86%, respectively, in the presence of TNP of ca. 200 μM concentration, and by ca. 88% and 84% in the presence of 4-nitroaniline (4-NA) of 200 μM concentration (Fig. 5). The fluorescence quenching data of the polymers were subjected to linear regression analyses using the Stern–Volmer equation to afford Stern–Volmer quenching rate constants (KSVs). The KSVs thus derived for all the NACs for excitation of the polymer suspensions at 365 nm (DOIDB) and 370 nm (TOITP) are given in Table 1. The KSV values for TNP and 4-nitroaniline are significantly higher than for all others. In these cases, the quenchers interact with the GFP fluorophores in both the ground and excited states; the competition experiment involving trinitrotoluene (TNT) and trinitrophenol (TNP), as monitored by UV-vis absorption spectroscopy, shows that TOITP binds TNP selectively, vide infra. Thus, static quenching due to binding in the ground state as well as competitive absorption by these nitroaromatic compounds with the GFP fluorophores of the polymers contribute to the efficient quenching of TNP.57,58
 | ||
| Fig. 5 (a) and (b) Fluorescence quenching of the suspensions of DOIDB and TOITP in MeOH, respectively, with increasing concentrations of 4-NA; the Stern–Volmer plots are shown in the insets. (c) and (d) Fluorescence quenching of the suspensions of DOIDB and TOITP in MeOH, respectively, with increasing concentrations of TNP; the corresponding Stern–Volmer plots are shown in the insets. | ||
| NACs | K SV (M−1) | η% | ||
|---|---|---|---|---|
| DOIDB | TOITP | DOIDB | TOITP | |
| a The efficiencies were determined for different NACs based on fluorescence intensity changes for the addition of 0.2 mM concentration of each of the analytes. | ||||
| 2-Nitroaniline (2-NA) | 4.6 × 103 | 4.7 × 103 | 51 | 51 |
| 3-Nitroaniline (3-NA) | 2.4 × 103 | 2.5 × 103 | 33 | 34 |
| 4-Nitroaniline (4-NA) | 4.9 × 104 | 2.9 × 104 | 88 | 84 |
| 2-Nitrophenol (2-NP) | 4.6 × 103 | 4.2 × 103 | 50 | 48 |
| 3-Nitrophenol (3-NP) | 8.9 × 102 | 1.2 × 103 | 16 | 20 |
| 4-Nitrophenol (4-NP) | 3 × 103 | 2.9 × 103 | 39 | 37 |
| 2,4-Dinitrophenol (2,4-DNP) | 1.8 × 104 | 1.5 × 104 | 57 | 51 |
| 2,4,6-Trinitrophenol (TNP) | 4.1 × 104 | 3.5 × 104 | 88 | 86 |
| 2-Nitrothiophenol (2-NTP) | 7.2 × 103 | 5.8 × 103 | 62 | 57 |
| 3-Nitrothiophenol (3-NTP) | 7.7 × 102 | 4.1 × 102 | 10 | 7 |
| 4-Nitrothiophenol (4-NTP) | 8.4 × 103 | 8.1 × 103 | 60 | 61 |
As can be seen, both polymers respond similarly. The KSV values for 4-NA and TNP are almost similar for both polymers (Fig. 5). A similar exercise was carried out for 2-/3-/4-nitroanilines (NAs), nitrophenols (NPs), and nitrothiophenols (NTPs) (Fig. S10–S15, ESI†). The quenching efficiencies, ηs, were also determined for all the analytes examined. These values are collected in Table 1 and Fig. 6. The maximum turn-off efficiency was observed for 4-NA among anilines, TNP among phenols, and 2/4-nitrothiophenol (2/4-NTP) among thiophenols. Glaringly, the analytes with the nitro group at the meta position perform poorly. Otherwise, the presence of a nitro group is found to be indispensable for efficient quenching.
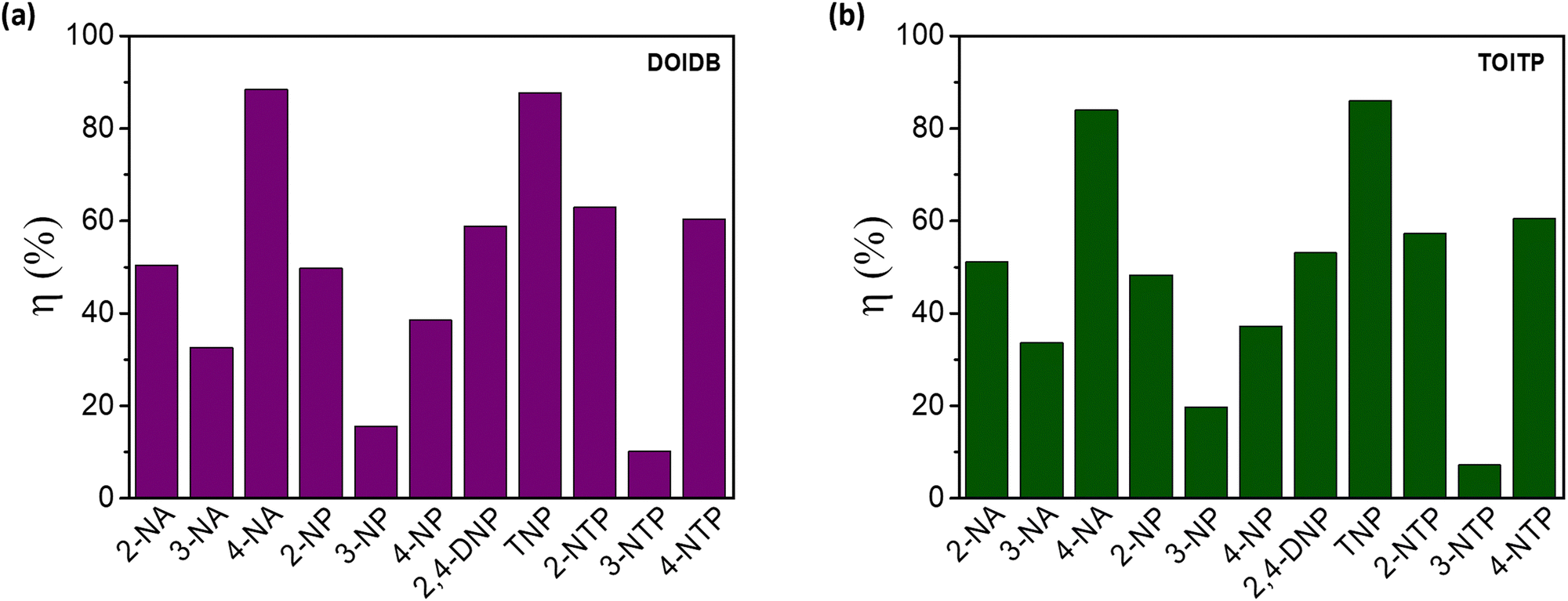 | ||
| Fig. 6 Bar diagrams for the quenching efficiencies of all NACs at 0.2 mM concentration of (a) DOIDB and (b) TOITP. | ||
Comparison of the quenching efficiencies (η%) of the monomers and polymers
To emphasize the importance of polymeric structures of the COPs, the quenching of the fluorescence of monomeric chromophores by TNP was investigated. For this purpose, the methoxy derivatives of the monomers of DOIDB and TOITP polymers were explored to avoid the effect of H-bonding of the OH groups on the sensing of TNP. The solutions of dimethoxy and trimethoxy monomers, i.e., DMI (0.26 mM) and TMI (0.24 mM), in MeOH equivalent to 100 ppm concentration were employed for quenching experiments. The efficiencies for the quenching of the fluorescence (η%) of the monomers in methanol were found to be substantially lower than those of the polymers. The η% values for the monomers were found to be 12% (DMI) and 37% (TMI) in comparison to 88% (DOIDB) and 86% (TOITP) for the respective polymers constructed therefrom. The quenching plots are shown in Fig. S16 (ESI†).Limits of detection (LOD) of nitrophenols and nitroanilines with DOIDB and TOITP
To evaluate the sensitivity limits of the polymers for the detection of representative nitroaniline, namely, 4-nitroaniline (4-NA) and nitrophenol, namely, 2,4,6-trinitrophenol (TNP), the quenching experiments were performed with very low concentrations of the analyte. Plots of quenching efficiencies (η%) versus the quencher concentrations reveal the limits of detection (LODs) for 4-NA to be 0.48 and 0.45 μM for DOIDB and TOITP, respectively. Similarly, the limits of detection for TNP were found to be 2.7 and 2.3 μM for DOIDB and TOITP, respectively. These values translate into 0.066 ppm and 0.062 ppm for 4-NA using DOIDB and TOITP, respectively. Similarly, the limits of detection for TNP turn out to be 0.62 and 0.52 ppm using DOIDB and TOITP polymers, respectively (Fig. 7). These limits of detection are quite comparable to some of the best sensory materials reported in the literature (Tables S1 and S2, ESI†).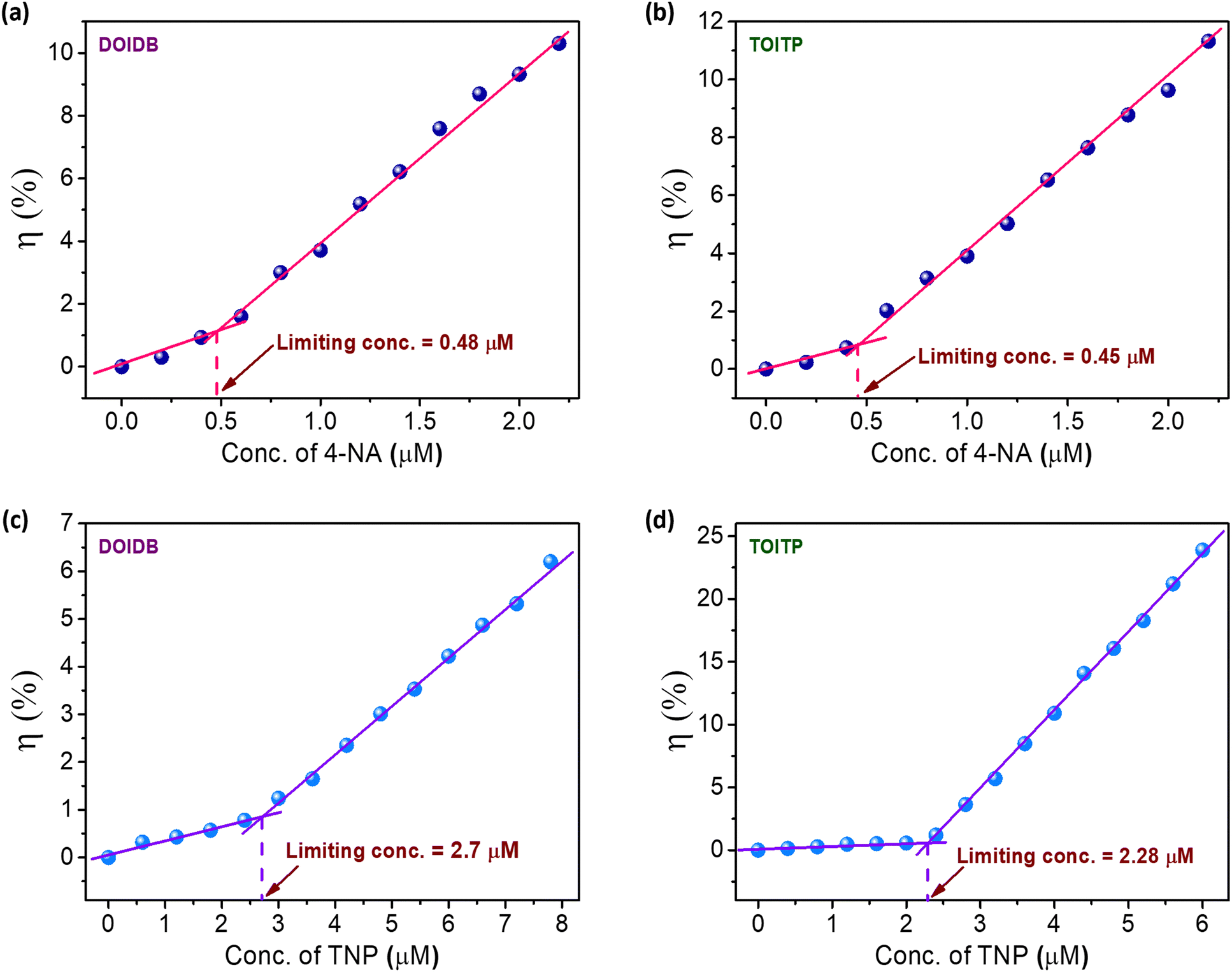 | ||
| Fig. 7 (a) and (b) Limits of detection of 4-NA using DOIDB and TOITP, respectively. (c) and (d) Limits of detection of TNP using DOIDB and TOITP, respectively. | ||
Adsorption of picric acid by the polymers
Buoyed by the remarkable efficiency of the fluorescence of both polymers by TNP, the capabilities of the polymers to adsorb TNP as a mop were examined. For this purpose, 10 mg of the polymer was immersed in 4 mL of 0.5 mM TNP solution in methanol, and the absorption of TNP was monitored periodically until the concentration of TNP remained constant. In the case of DOIDB, the conc. of TNP fluctuated with the equilibrium being achieved after 6 h of the addition of TNP solution to the polymer. However, in the case of TOITP, a rapid reduction in the conc. of TNP was observed within a few seconds with the equilibrium attained after 10 h. The adsorption efficiencies of TNP (η%) were found to be ca. 11% and 83.7% for DOIDB and TOITP, respectively (Fig. 8). Selective binding of TNP over TNT by TOITP was examined in methanol by suspending the polymer in the solutions of equimolar concentrations of TNP and TNT. As monitored by UV-vis absorption spectroscopy at different intervals of time, the concentration of TNP was found to decrease gradually while that of the TNT remained intact (Fig. S17, ESI†).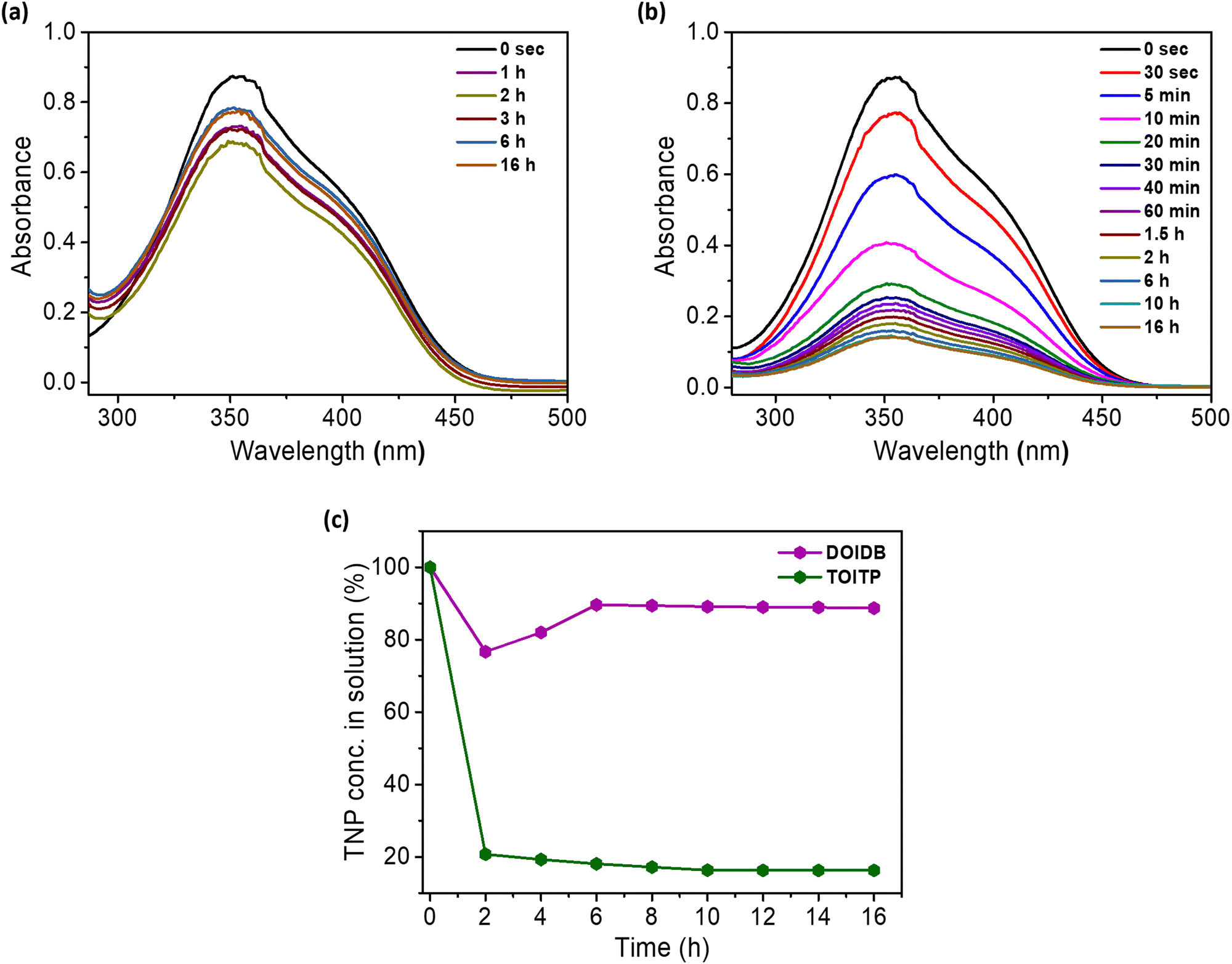 | ||
| Fig. 8 (a) and (b) UV-vis absorption spectra of TNP solutions with DOIDB and TOITP in MeOH, showing changes in the concentrations of TNP at different intervals of time. (c) Percentage of TNP left in the solution after the equilibrium is reached for each of the polymers. | ||
After the adsorption of TNP, the quantum yields of the polymers were determined to evaluate their binding in the void volumes of the polymers. Indeed, in the case of TOITP, the quantum yield was found to drop to 27% from 73%, while the change was found to be not significant in the case of DOIDB. This is completely in agreement with the expectation that DOIDB is not to show adsorption going by the fact that it is a 1-dimensional polymer unlike TOITP, which otherwise should be a 2D polymer with specific pockets for guest inclusion. A palpable fluorescence observed even after the binding of TNP should be reconciled with the fact that the imidazolinone moieties are not effectively hydrogen bonded to phenolic hydrogens.
Conclusions
Inspired by the high quantum yield of GFP, we rationally designed, synthesized and comprehensively characterized four covalent organic polymers (COPs), that is, DOIDB, DOITP, TOIDB and TOITP, based on two different GFP chromophores. BET surface areas of the polymers show that two of them exhibit respectable porosities of 107.9 and 164.8 m2 g−1, based on CO2 sorption studies at 195 K. A thorough investigation of their photophysical properties shows that all polymers are emissive in nature, but two of them, that is, DOIDB and TOITP, exhibit very high fluorescence emission with quantum yields of 83 and 73%, respectively, akin to the natural GFP (79%). The polymeric matrices of the COPs create an environment similar to that of the protein environment extant to the chromophore in natural GFP, restricting the rotations of C–C and CC bonds to offset the nonradiative deactivation channel of the excited states. It is shown that the fluorescent metal-free organic polymers can be exploited as sensory materials to permit the sensing of specific analytes selectively, namely, nitroaromatic compounds containing acidic hydrogens, by efficient turn-off fluorescence quenching. The electron-rich imidazolinone moiety of the GFP chromophore in DOIDB and TOITP polymers facilitates detection of 4-nitroaniline (4-NA) and at sub-ppm levels, i.e., 0.066 and 0.062 ppm, respectively, and of 2,4,6-trinitrophenol (TNP) in 0.62 and 0.52 ppm, respectively. It is noteworthy that trinitrotoluene (TNT), which lacks acidic hydrogen, does not affect the fluorescence of DOIDB and TOITP polymers. It is further shown that TOITP serves as a mop for rapid adsorption of TNP; the latter is shown to be adsorbed by TOITP by more than 50% from its solution in methanol within 10 min. To the best of our knowledge, the reported results constitute the first of their kind for the demonstration of application of polymers constructed from GFP chromophores for selective turn-off sensing of nitroaromatic compounds and organic pollutants.
Data availability
Synthesis of DHI and THI, UV-vis absorption and fluorescence emission spectra of DHI and THI in MeOH, XPS, elemental mapping, SEM-EDX analyses, and adsorption–desorption isotherms of N2 and CO2 of polymers. Pore size distribution of all polymers. IR and solid-state 13C NMR spectra of the polymers before and after sonication in MeOH. Emission spectra of DOIDB and TOITP in different solvents. Fluorescence emission spectra of DOIDB and TOITP suspensions with increasing concentrations of NACs. Sensitivity of the polymers DOIDB and TOITP to different pHs, Fluorescence quenching spectra of the monomers (DMI and TMI), and selective absorption of TNP over TNT by TOITP. Comparison of the performances of DOIDB and TOITP polymers in the sensing of 4-nitroaniline and TNP with those of other materials reported previously in the literature. 1H and 13C NMR spectra of the compounds are available in the ESI.†Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
G. A. thankfully acknowledges the University Grants Commission (UGC), New Delhi for a research fellowship. A. K. acknowledges the Council of Scientific and Industrial Research (CSIR), New Delhi for the CSIR research associate fellowship (Project no. 09/092(1053)/2020-EMR-I). J. N. M. is thankful to the Science and Engineering Research Board (SERB), India, for generous financial support through a J. C. Bose Fellowship (SB/S2/JCB-52/2014).References
- M. Chalfie, Y. Tu, G. Euskirchen, W. W. Ward and D. C. Prasher, Science, 1994, 263, 802–805 CrossRef PubMed.
- M. Zimmer, Chem. Rev., 2002, 102, 759–781 CrossRef PubMed.
- R. Y. Tsien, Annu. Rev. Biochem., 1998, 67, 509–544 CrossRef PubMed.
- R. M. Hoffman, J. Biomed. Opt., 2005, 10, 41202 CrossRef.
- J. Dong, K. M. Solntsev and L. M. Tolbert, J. Am. Chem. Soc., 2006, 128, 12038–12039 CrossRef PubMed.
- S. T. Hsu, G. Blaser and S. E. Jackson, Chem. Soc. Rev., 2009, 38, 2951–2965 RSC.
- S. Rafiq, B. K. Rajbongshi, N. N. Nair, P. Sen and G. Ramanathan, J. Phys. Chem. A, 2011, 115, 13733–13742 CrossRef.
- J. Chang, M. G. Romei and S. G. Boxer, J. Am. Chem. Soc., 2019, 141, 15504–15508 CrossRef.
- S. L. Maddalo and M. Zimmer, Photochem. Photobiol., 2006, 82, 367–372 CrossRef.
- L. Yang, S. Nian, G. Zhang, E. Sharman, H. Miao, X. Zhang, X. Chen, Y. Luo and J. Jiang, Sci. Rep., 2019, 9, 11640 CrossRef PubMed.
- J. W. Park and Y. M. Rhee, J. Am. Chem. Soc., 2016, 138, 13619–13629 CrossRef CAS PubMed.
- A. Baldridge, S. R. Samanta, N. Jayaraj, V. Ramamurthy and L. M. Tolbert, J. Am. Chem. Soc., 2011, 133, 712–715 CrossRef CAS PubMed.
- J. Zhang, H. Li, B. Lin, X. Luo, P. Yin, T. Yi, B. Xue, X. L. Zhang, H. Zhu and Z. Nie, J. Am. Chem. Soc., 2021, 143, 19317–19329 CrossRef CAS.
- H. Deng, C. Yu, L. Gong and X. Zhu, J. Phys. Chem. Lett., 2016, 7, 2935–2944 CrossRef CAS PubMed.
- M. Jiang, Z. He, Y. Zhang, H. H. Y. Sung, J. W. Y. Lam, Q. Peng, Y. Yan, K. S. Wong, D. Williams and Y. Zhao, J. Mater. Chem. C, 2017, 5, 7191–7199 RSC.
- J. R. M. Ferreira, C. I. C. Esteves, M. M. B. Marques and S. Guieu, Molecules, 2023, 28, 234 CrossRef CAS.
- C. K. Wong, A. J. Laos, A. H. Soeriyadi, J. Wiedenmann, P. M. G. Curmi, J. J. Gooding, C. P. Marquis, M. H. Stenzel and P. Thordarson, Angew. Chem., Int. Ed., 2015, 127, 5407–5412 CrossRef.
- Y. J. Zheng, G. L. Li, H. Deng, Y. Su, J. H. Liu and X. Zhu, Polym. Chem., 2014, 5, 2521–2529 RSC.
- H. Deng, Y. Su, M. Hu, X. Jin, L. He, Y. Pang, R. Dong and X. Zhu, Macromolecules, 2015, 48, 5969–5979 CrossRef CAS.
- W. Fan, H. Deng, L. Zhu, C. Tu, Y. Su, L. Shi, J. Yang, L. Zhou, L. Xu and X. Zhu, Biomater. Sci., 2019, 7, 2421–2429 RSC.
- D. E. Williams, E. A. Dolgopolova, P. J. Pellechia, A. Palukoshka, T. J. Wilson, R. Tan, J. M. Maier, A. B. Greytak, M. D. Smith, J. A. Krause and N. B. Shustova, J. Am. Chem. Soc., 2015, 137, 2223–2226 CrossRef CAS.
- E. A. Dolgopolova, T. M. Moore, W. B. Fellows, M. D. Smith and N. B. Shustova, Dalton Trans., 2016, 45, 9884–9891 RSC.
- A. Singh, D. Samanta, M. Boro and T. K. Maji, Chem. Commun., 2019, 55, 2837–2840 RSC.
- A. Singh, S. Karmakar, I. M. Abraham, D. Rambabu, D. Dave, R. Manjithaya and T. K. Maji, Inorg. Chem., 2020, 59, 8251–8258 CrossRef CAS PubMed.
- Y. Li, L. Shi, L. X. Qin, L. L. Qu, C. Jing, M. Lan, T. D. James and Y. T. Long, Chem. Commun., 2011, 47, 4361–4363 RSC.
- A. Csomos, E. Kovács, M. Madarász, Z. F. Fülöp, A. Fülöp, G. Katona, B. Rózsa and Z. Mucsi, Sens. Actuators, B, 2024, 398, 134753 CrossRef CAS.
- L. Shi, Y. Li, Z. Liu, T. D. James and Y. T. Long, Talanta, 2012, 100, 401–404 CrossRef.
- X. Y. Liu, L. Shi, Z. Ding and Y. T. Long, RSC Adv., 2014, 4, 53557–53560 RSC.
- A. L. Mirajkar, L. L. Mittapelli, G. N. Nawale and K. R. Gore, Sens. Actuators, B, 2018, 265, 257–263 CrossRef.
- L. L. Mittapelli, G. N. Nawale, S. P. Gholap, O. P. Varghese and K. R. Gore, Sens. Actuators, B, 2019, 298, 126875 CrossRef.
- P. Chandrasekhar, A. Mukhopadhyay, G. Savitha and J. N. Moorthy, Chem. Sci., 2016, 7, 3085–3091 RSC.
- P. Chandrasekhar, A. Mukhopadhyay, G. Savitha and J. N. Moorthy, J. Mater. Chem. A, 2017, 5, 5402–5412 RSC.
- A. Mukhopadhyay, V. K. Maka, G. Savitha and J. N. Moorthy, Chem, 2018, 4, 1059–1079 Search PubMed.
- V. K. Maka, P. Tamuly, S. Jindal and J. N. Moorthy, Appl. Mater. Today, 2020, 19, 100613 CrossRef.
- M. G. Mohamed, A. F. M. EL-Mahdy, M. G. Kotp and S.-W. Kuo, Mater. Adv., 2022, 3, 707–733 RSC.
- S. T. Wang, H. T. Li, H. N. Huang, X. H. Cao, X. D. Chen and D. P. Cao, Chem. Soc. Rev., 2022, 51, 2031–2080 RSC.
- N. Naz, M. H. Manzoor, S. M. G. Naqvi, U. Ehsan, M. Aslam and F. Verpoort, Appl. Mater. Today, 2024, 38, 102198 CrossRef.
- C. Yadav, V. K. Maka, S. Payra and J. N. Moorthy, J. Catal., 2020, 384, 61–71 CrossRef CAS.
- C. Yadav, V. K. Maka, S. Payra and J. N. Moorthy, ACS Appl. Polym. Mater., 2020, 2, 3084–3093 CrossRef CAS.
- A. Karn, C. Yadav, A. K. Sahoo and J. N. Moorthy, ChemCatChem, 2023, 15, e202300727 CrossRef CAS.
- P. Tamuly and J. N. Moorthy, ACS Appl. Mater. Interfaces, 2024, 16, 3348–3358 CrossRef CAS.
- K. Palpandi and N. Raman, New J. Chem., 2020, 44, 8454–8462 RSC.
- F. A. Alharthi, H. K. Aldubeikl, H. S. Alanazi, W. S. Al-Nafaei and I. Hasan, Nanomaterials, 2023, 13, 362 CrossRef CAS.
- M. T. Uddin, M. M. Alam, A. M. Asiri, M. M. Rahman, T. Toupance and M. A. Islam, RSC Adv., 2019, 10, 122–132 RSC.
- D. Thangavelu, Y. Chen, P. Annamalai, M. Ramadoss and V. Narayanan, ACS Appl. Mater. Interfaces, 2022, 14, 6518–6527 CrossRef.
- M. G. Choi, M. J. Cho, H. Ryu, J. Hong and S.-K. Chang, Dyes Pigm., 2017, 143, 123–128 CrossRef.
- Y. Lu, J. Mao, Z. Wang, Y. Qin and J. Zhou, Catalysts, 2020, 10, 746 CrossRef.
- Y. Nailwal, M. Devi and S. K. Pal, ACS Appl. Polym. Mater., 2022, 4, 2648–2655 CrossRef.
- S. Sau, F. Banerjee and S. K. Samanta, ACS Appl. Nano Mater., 2023, 6, 11679–11688 CrossRef.
- W. Che, G. Li, X. Liu, K. Shao, D. Zhu, Z. Su and M. R. Bryce, Chem. Commun., 2018, 54, 1730–1733 RSC.
- K. Y. Chen, Y. M. Cheng, C. H. Lai, C. C. Hsu, M. L. Ho, G. H. Lee and P. T. Chou, J. Am. Chem. Soc., 2007, 129, 4534–4535 CrossRef CAS PubMed.
- L. Wu and K. Burgess, J. Am. Chem. Soc., 2008, 130, 4089–4096 CrossRef CAS.
- W. Du, Y. Qin, C. Ni, W. Dai and J. Zou, ACS Appl. Polym. Mater., 2020, 2, 5121–5128 CrossRef CAS.
- O. Yildirim, A. Tsaturyan, A. Damin, S. Nejrotti, V. Crocellà, A. Gallo, M. R. Chierotti, M. Bonomo and C. Barolo, ACS Appl. Mater. Interfaces, 2023, 15, 15819–15831 CrossRef CAS.
- S. Bera, K. Garg and S. K. Samanta, ACS Appl. Nano Mater., 2024, 7, 1797–1803 CrossRef CAS.
- T. Iwasaki, S. Murakami, Y. Takeda, G. Fukuhara, N. Tohnai, Y. Yakiyama, H. Sakurai and N. Kambe, Chem. – Eur. J., 2019, 25, 14817–14825 CrossRef CAS.
- A. S. Tanwar, R. Parui, R. Garai, M. A. Chanu and P. K. Iyer, ACS Meas. Sci. Au, 2022, 2, 23–30 CrossRef CAS PubMed.
- V. K. Maka, A. Mukhopadhyay, G. Savitha and J. N. Moorthy, Nanoscale, 2018, 10, 22389–22399 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma00811a |
| This journal is © The Royal Society of Chemistry 2024 |