
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceZnO vapor phase infiltration into photo-patternable polyacrylate networks for the microfabrication of hybrid organic–inorganic structures†
Lisanne
Demelius
 ab,
Li
Zhang
b,
Anna Maria
Coclite
*ac and
Mark D.
Losego
b
ab,
Li
Zhang
b,
Anna Maria
Coclite
*ac and
Mark D.
Losego
b
aInstitute of Solid State Physics, NAWI Graz, Graz University of Technology, Petersgasse 16, 8010 Graz, Austria
bSchool of Materials Science and Engineering, Georgia Institute of Technology, Atlanta, Georgia 30332, USA
cDepartment of Phjysics, University of Bari, via Amendola 173, Bari, Italy
First published on 1st October 2024
Abstract
Photopatterning of polymers enables the microfabrication of numerous microelectronic, micromechanical, and microchemical systems. The incorporation of inorganics into a patterned polymer material can generate many new interesting properties such as enhanced stability, optical performance, or electrical properties. Vapor phase infiltration (VPI) allows for the creation of hybrid organic–inorganic materials by infiltrating polymers with gaseous metalorganic precursors. This study seeks to explore the potential of integrating VPI with existing photopatterning techniques to achieve top-down hybridization and property modification of polymer structures of different complexity. For this, VPI of diethylzinc (DEZ) is studied for four highly crosslinked acrylate networks that can be patterned by photolithography and two-photon polymerization (2PP): pentaerythritol triacrylate (PETA), pentaerythritol tetraacrylate (PETeA), trimethylolpropane triacrylate (TMPTA) and ethoxylated trimethylolpropane triacrylate (ETPTA). The findings show that for highly crosslinked polymer networks, VPI can be limited by slow precursor diffusion. However, by introducing flexible segments (e.g., ethoxylated chains), the polymer's free volume can be increased, and infiltration is accelerated, leading to faster infiltration times and higher and more uniform inorganic loading. Finally, selective infiltration of ZnO into photolithographically patterned copolymer networks of TMPTA and ETPTA on non-infiltrating poly(methyl methacrylate) (PMMA) is demonstrated illustrating the potential of VPI for advanced maskless patterning strategies.
Introduction
Photolithographic patterning of photosensitive polymers is commonly used to fabricate many microdevices. For example, micropatterned polymers are important in fabricating micro electromechanical systems (MEMS), microfluidic systems, optics and flexible electronics, or as etch masks to define and transfer patterns onto other (inorganic) materials during microelectronics fabrication.1,2 Maskless photopatterning methods, such as e-beam lithography,3 direct laser writing (DLW)4,5 or two-photon polymerization (2PP),6,7 are especially interesting because they enable the creation of highly customizable nano- and microstructures of varying complexity.Incorporating inorganics into a polymer can introduce a variety of new synergistic properties. Such hybrid organic–inorganic materials8 have demonstrated applications in electronics,9 sensors,10 catalysis,11 biomaterials,12,13 and energy storage.14,15 Common synthesis routes rely on solution-based processes involving dispersions of organic matrices (e.g. polymers, monomers, etc.) and inorganic species (e.g. nanoparticles, metals, clusters, etc.).16 These processes pose challenges due to agglomeration of the inorganic particles, non-uniform distribution or poor miscibility of the components, as well as solvent-related safety concerns and substrate compatibility issues.
Vapor phase infiltration (VPI), also referred to as sequential infiltration synthesis (SIS) or atomic layer infiltration (ALI), is an emerging technique that allows for the hybridization of a polymer by infiltrating it with gaseous metalorganic precursors while preserving the polymer's original macroscale form and microstructure.17 In VPI, gas-phase inorganic precursors are sorbed into and diffuse throughout a polymer matrix, eventually becoming immobilized by either a chemical reaction with the polymer or loss of volatility, often due to reaction with a subsequently delivered co-reactant. Under ideal process conditions, a uniform distribution of inorganic species in the polymer matrix can be achieved that exhibits self-limiting saturation behavior akin to atomic layer deposition (ALD). Hybridization by VPI has shown huge potential in modifying material properties such as mechanical strength,18–20 chemical stability,21–23 optical emission,24,25 and triboelectric response.26
At present, the library of photo-patternable polymers that have been studied for modification by VPI is fairly limited. VPI has been investigated for AlOx,27–30 TiOx,31 ZnOx,32,33 and InOx34 into the epoxy-based photoresist SU-8,31–33 poly(methyl methacrylate) (PMMA)28,34 and bisphenol A-based dimethacrylate resins.29,30 VPI was mainly employed to enhance pattern transfer during EUV lithography,27,34,35 e-beam lithography,28 and UV-NIL29,30 by increasing photosensitivity or etch resistance of the polymer mask, or to fabricate inorganic nanostructures from lithographically patterned polymers by etching away the organic part after infiltration.31–33
Only a few rare examples have explored the integration of VPI with direct writing techniques to create hybrid organic–inorganic structures with novel properties. Li et al.36 infiltrated SU-8 nanopillars, patterned by e-beam lithography, with AlOx and demonstrated a polymer-like Young's modulus and a metal-like high yield strength, making the material interesting for applications in MEMS cantilever mass sensors. Subramanian et al.37 demonstrated that VPI of AlOx enables control of the bipolar switching characteristics and stochasticity of SU-8 used in resistive random-access memory (RRAM) devices. Another example was provided by Singhal et al.25 who used VPI of DEZ to increase the refractive index of photonic crystals printed by 2PP using the commercial photoresin IP-Dip (Nanoscribe).
VPI is also chemically selective and can be used to selectively modify certain polymer structures while leaving others unaltered. This ability is similar to area-selective deposition (ASD) where the chemistry present on a substrate surface dictates deposition.38 However, the mechanisms that can lead to volume-selective infiltration are more varied and include differences in solubility,39 reactivity40 or precursor kinetics.41 Selectivity in VPI has been extensively explored for self-assembled BCP structures, where, for certain copolymer combinations, the metalorganic precursor preferentially infiltrates into one polymer phase versus the other.42 The most widely studied system is probably TMA in PS-b-PMMA, where infiltration occurs only into the PMMA phase.43,44 McGuinness et al.39 demonstrated that selective VPI can also be achieved for larger volumes and structures by patterning a PS thin film on PMMA and showing that TMA infiltration was effectively blocked in regions covered by PS.
The present work aims to extend the VPI library of photo-patternable polymers by investigating the infiltration of ZnO (DEZ + H2O) into a set of highly crosslinked polyacrylate networks that are common components of 2PP photoresins.6 Within this system, we demonstrate that designing the polymer's flexibility and free volume is important to promoting precursor infiltration and inorganic loading. We also show the ability to selectively infiltrate a photo-patterned polyacrylate structure on a non-infiltrating PMMA supporting layer.
Methods
Preparation of crosslinked polyacrylate thin films
Crosslinked polyacrylate thin films were prepared from four different monomers: pentaerythritol triacrylate (PETA, Santa Cruz Biotechnology, Inc.), pentaerythritol tetraacrylate (PETeA, Sigma-Aldrich) with a PETA content of ≥30 to <50%, trimethylolpropane triacrylate (TMPTA, Sigma Aldrich), and ethoxylated trimethylolpropane triacrylate (ETPTA, average Mn ∼428, Sigma Aldrich). 2-(Dimethylamino)-2-(4-methylbenzyl)-1-(4-morpholinophenyl)butan-1-one (Omnirad 379, >95.0%, TCI), which has its main absorption at 233 and 320 nm, was used as photoinitiator for UV polymerization.Two different recipes were used for spin-coating thin films of the acrylate monomers. For Recipe A, 4 wt% monomer with 6 wt% photoinitiator with respect to the monomer were dissolved in 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 toluene (≥99.5%, Sigma Aldrich) and chloroform (>99.0%, stabilized with Ethanol, TCI) and spun-cast on single-side polished 〈100〉 silicon wafers (p-type, GlobiTech, Inc.) at 3000 rpm for 60 s with static dispensing. For Recipe B, an 8 wt%-solution of monomer in ethyl acetate (≥99.5%, Sigma Aldrich), again with 6 wt% photoinitiator with respect to the monomer, was prepared, which was then diluted with 10 wt% 1-butanol (≥99.4%, Sigma Aldrich) for better spin-coating results. 30 μl of the solution were spun-cast at 6000 rpm for 10 s with dynamic dispensing. Recipe B was developed as an optimization step to eliminate the use of the hazardous solvents toluene and chloroform. Both recipes produced thin films of comparable uniformity and thickness (between 100 and 250 nm depending on the monomer), as determined by eye and ellipsometry, and had the same chemical composition, as measured by FTIR. Copolymerized films of p(TMTPA-ETPTA) were prepared by mixing the two monomers in solution in a 1:3 weight ratio of TMPTA to ETPTA.
:50 toluene (≥99.5%, Sigma Aldrich) and chloroform (>99.0%, stabilized with Ethanol, TCI) and spun-cast on single-side polished 〈100〉 silicon wafers (p-type, GlobiTech, Inc.) at 3000 rpm for 60 s with static dispensing. For Recipe B, an 8 wt%-solution of monomer in ethyl acetate (≥99.5%, Sigma Aldrich), again with 6 wt% photoinitiator with respect to the monomer, was prepared, which was then diluted with 10 wt% 1-butanol (≥99.4%, Sigma Aldrich) for better spin-coating results. 30 μl of the solution were spun-cast at 6000 rpm for 10 s with dynamic dispensing. Recipe B was developed as an optimization step to eliminate the use of the hazardous solvents toluene and chloroform. Both recipes produced thin films of comparable uniformity and thickness (between 100 and 250 nm depending on the monomer), as determined by eye and ellipsometry, and had the same chemical composition, as measured by FTIR. Copolymerized films of p(TMTPA-ETPTA) were prepared by mixing the two monomers in solution in a 1:3 weight ratio of TMPTA to ETPTA.
The as-spun acrylate films were polymerized under a Dymax 2000-EC UV lamp with a 400 W Metal Halide flood bulb for 1 to 30 minutes at ambient conditions. Isopropyl alcohol (IPA, ≥99.5%, Sigma Aldrich) was used to test the stability of the polymerized films.
To pattern the copolymerized acrylate films, a lithographic mask was positioned on top of the spun-cast acrylate films (Recipe B with 15 wt% photoinitiator with respect to the monomer and a weight ratio of 1:3 TMPTA:ETPTA) which were then exposed to UV for 1 minute, followed by a quick rinse with isopropyl alcohol (>99.5%, Sigma Aldrich) to remove the unpolymerized parts of the film. To demonstrate selective infiltration, the polyacrylate patterns were created on a poly (methyl methacrylate) (PMMA, Polysciences Inc., 75 k MW) film that had been spun-cast on silicon substrates from a 5 wt% solution of PMMA in toluene (3000 rpm, 30 s, static dispensing).
Vapor phase infiltration of DEZ
Vapor phase infiltration (VPI) of the crosslinked polyacrylate thin films was performed in a custom-built system described elsewhere,45,46 using nitrogen (99.995% purified from air) as purging gas at a flow rate of 250 slpm, diethylzinc (DEZ, STREM chemicals, 95% purity, DANGER: pyrophoric) as the metalorganic precursor, and deionized water as the oxidizing co-reactant. The polyacrylate films were subjected to one VPI cycle at a process temperature of 120 °C. To remove any moisture from the polyacrylate films prior to infiltration, a 2-hour nitrogen purge followed by a 2-hour pump-down of the system to base pressure was performed. Then, the system was isolated from the pump and DEZ was dosed for 3 seconds, resulting in a chamber pressure of approximately 0.75 Torr. The reactor was held in this static DEZ atmosphere for 5 or 15 hours, and then pumped down to base pressure for 1 hour. For the water dose, the reactor was once again isolated from the pump, and water was dosed for 1 second resulting in a pressure of approximately 3.5 Torr. The system was then held in this static H2O atmosphere for 30 minutes before being pumped down for 1 hour.Characterization methods
FTIR spectra of the crosslinked polyacrylate films on silicon were measured in transmittance with a Thermo Scientific Nicolet iS5. A bare silicon wafer was used as background reference and each measurement consisted of 200 scans in a wavelength range from 400 to 4000 cm−1 at a resolution of 4 cm−1. All spectra were baseline corrected and normalized with respect to the film thickness measured by ellipsometry during post-processing.The thickness and the refractive index (n) of the crosslinked polyacrylate films were measured by spectroscopic ellipsometry (SE, J. A. Woollam Alpha-SE). Spectra were obtained in a wavelength range from 380 to 900 nm at a 70° angle of incidence and fitted to a model consisting of a silicon substrate, 1.7 nm native oxide, and a Cauchy layer using the CompleteEASE software (J. A. Woollam, Version 5.19). To achieve better fit results, thickness non-uniformity was included.
The zinc loading of the polyacrylate films after VPI was analyzed by X-ray fluorescence spectroscopy (XRF) using a Thermo Scientific Niton FXL FM-XRF with an Ag anode at a voltage of 50 kV and a current of 200 μA. Spectra were taken in general metals mode with a measurement time of 90 s. To get a measure of the zinc content, the spectra were baseline corrected by subtracting a spectrum measured before VPI, and the area of the Zn Kα peak at 8.637 keV was obtained by fitting it with a Gaussian.
A Thermo Scientific K-Alpha system equipped with a monochromatic Al-Kα X-ray source (1486.6 eV) was used to measure X-ray photoelectron spectroscopy (XPS). A pass energy of 200 eV, a dwell time of 50 ms and a step size of 1 eV were used for the survey scans, while high resolution scans were conducted with a pass energy of 50 eV, a dwell time of 50 ms and a step size of 0.1 eV. The crosslinked polyacrylate films were measured without any further surface treatment, and charge compensation was achieved using an in-built flood gun. The spot size was 400 μm. Depth profiles were obtained using a 2000 eV mid-current Ar ion beam with etch steps ranging from 35 to 60 s and a 5 s pause between etching and measuring. Data analysis and component fitting was performed with the software CasaXPS (Casa Software Ltd, Teignmouth, UK). All spectra were calibrated with respect to the C–C adventitious carbon peak at 284.8 eV. For the XPS depth profiles, the etch time was calibrated to etch depth using the film thickness obtained by ellipsometry. The position of the polyacrylate–Si interface was assumed to be at the point where the Si 2p peak reached 50 at%.
To analyze the topography of the patterned polyacrylate films, a Filmetric Profilm3D Optical Profilometer was used.
Energy-dispersive X-ray spectroscopy (EDX) elemental mapping was performed to demonstrate the selective infiltration of the patterned sample using a Phenom XL G2 scanning electron microscope (SEM) at 5 kV electron energy and a chamber pressure of 0.1 Pa in imaging mode. The atomic concentrations were calculated using the Phenom software.
Results and discussion
UV photopolymerization of acrylate monomers
Scheme 1 displays the chemical structures of the polyfunctional acrylate monomers (PETA, PETeA, TMPTA and ETPTA) investigated within the scope of this study along with the chemical structure of the photoinitiator Omnirad 379. While PETA, TMPTA and ETPTA are trifunctional, i.e. each monomer features three vinyl bonds that can be reacted to form covalent bonds to other monomers during polymerization, PETeA is tetrafunctional. Fig. 1 shows FTIR spectra collected from these four different chemistries upon UV polymerization. The black lines in Fig. 1(a)–(d) represent the FTIR spectra of the respective monomer films after spin-coating. The ester groups in these monomers show up in the FTIR spectra as a carbonyl stretch at approximately 1720 cm−1 and a series of peaks related to C–O bonding between 1010 and 1320 cm−1. The vinyl bonds are represented as absorption bands corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching (1625 cm−1) and C–H bending vibrations (1405 and 810 cm−1). The hydroxyl group of PETA produces a broad O–H peak at around 3500 cm−1. A weak O–H peak is also present for PETeA that is due to the fact that the used PETeA is not pure but contains 30–50% PETA. The methyl groups in TMPTA and ETPTA show up in the C–H stretch region at around 2970 cm−1. The ethoxy groups in ETPTA result in a shoulder peak in the C–H stretch region at a slightly lower wavenumber of approximately 2875 cm−1, and in an additional peak in the ether region at around 1110 cm−1.47,48
C stretching (1625 cm−1) and C–H bending vibrations (1405 and 810 cm−1). The hydroxyl group of PETA produces a broad O–H peak at around 3500 cm−1. A weak O–H peak is also present for PETeA that is due to the fact that the used PETeA is not pure but contains 30–50% PETA. The methyl groups in TMPTA and ETPTA show up in the C–H stretch region at around 2970 cm−1. The ethoxy groups in ETPTA result in a shoulder peak in the C–H stretch region at a slightly lower wavenumber of approximately 2875 cm−1, and in an additional peak in the ether region at around 1110 cm−1.47,48
 | ||
| Scheme 1 Chemical structures of the monomers (a) pentaerythritol triacrylate (PETA), (b) pentaerythritol tetraacrylate (PETeA), (c) trimethylolpropane triacrylate (TMPTA), (d) ethoxylated trimethylolpropane triacrylate (ETPTA), and (e) the photoinitiator 2-(dimethylamino)-2-(4-methylbenzyl)-1-(4-morpholinophenyl)butan-1-one (omnirad 379). The average molecular weight of ETPTA is 428, which corresponds to an average m, n, o = 1. | ||
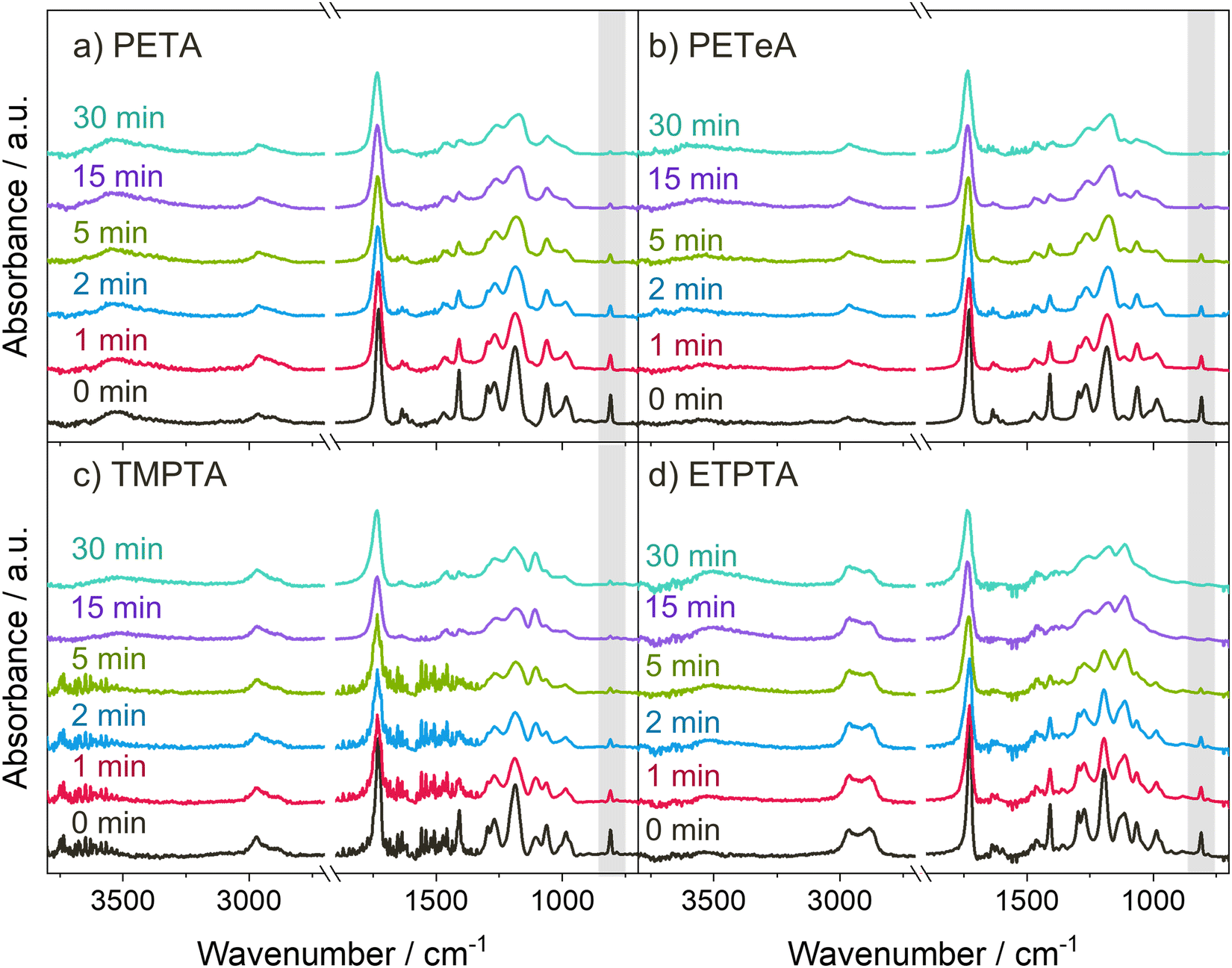 | ||
| Fig. 1 UV polymerization of the acrylate monomers. (a)–(d) FTIR spectra of the monomer thin films after different UV exposure times. The peak highlighted in gray corresponds to the C–H bending at 810 cm−1 used for calculation of the degree of polymerization (see Fig. 2). | ||
During photopolymerization, thin films of the four monomers are exposed to UV light for 0 to 30 minutes. The chemical changes are monitored by FTIR (Fig. 1), while film thickness (Fig. 2(a)) and refractive index n at a wavelength of 632.8 nm (Fig. 2(b)) are tracked by spectroscopic ellipsometry. Upon increasing UV exposure time, the FTIR spectra show a decrease in the absorption peaks related to the vinyl bonds (1625, 1405 and 810 cm−1), confirming successful reaction of these bonds through photopolymerization. For pTMPTA (Fig. 1(c)) and pETPTA (Fig. 1(d)), extended UV exposure also results in the emergence of a broad O–H absorption band around 3500 cm−1 which indicates that a certain degree of photooxidation of the acrylates takes place in addition to the polymerization reaction.49–51 The effect is stronger in pETPTA, where along with the appearance of an O–H band, the C–H peak at 2875 cm−1, corresponding to the ethoxy groups, sees a significant decrease for UV exposure times longer than 15 minutes.
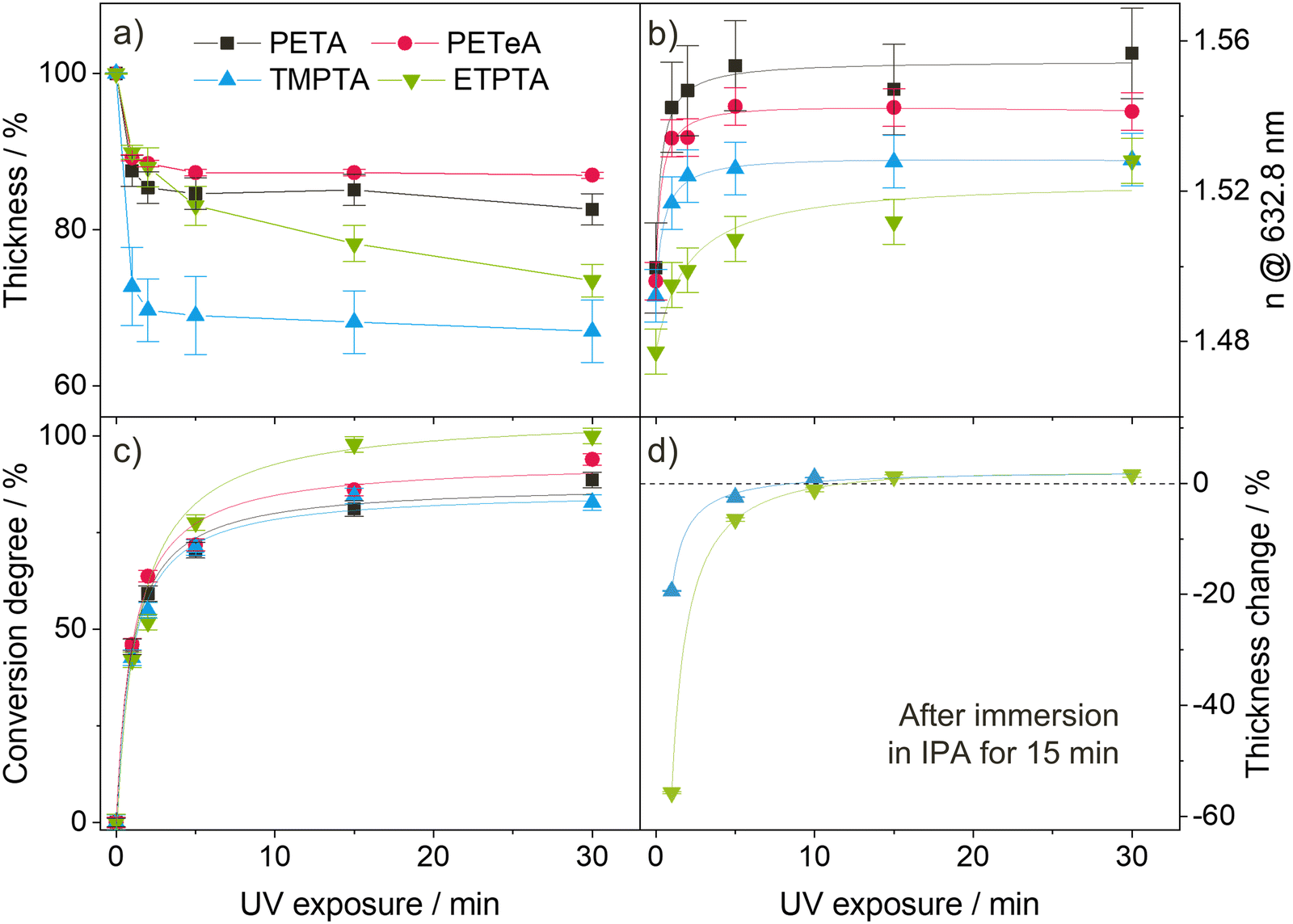 | ||
| Fig. 2 UV polymerization of the acrylate monomers. (a) Relative thickness change and (b) refractive index n at a wavelength of 632.8 nm measured by ellipsometry, (c) conversion degree calculated from the area change of the FTIR vinyl-bond peak at 810 cm−1, and (d) chemical stability of the UV-polymerized films determined by the change in thickness (measured by ellipsometry) after immersion in IPA for 15 minutes. The lines serve as a guide for the eye. Error bars in (a)–(c) derive from averaging over 2 samples. Error bars in (d) correspond to the error of the ellipsometry fit for 1 sample. | ||
Fig. 2(a) illustrates the films’ shrinkage upon UV photopolymerization. The decrease in film thickness is most pronounced during the first minute of irradiation, after which it quickly stabilizes. PETA and PETeA exhibit the least shrinkage (approximately 13–15%) while TMPTA and ETPTA shrink considerably more (up to 30%). The refractive index n (Fig. 2(b)) increases upon UV exposure and saturates after about 5 minutes to values ranging from 1.524 ± 0.011 for ETPTA, to 1.526 ± 0.002 for TMPTA, 1.542 ± 0.001 for PETeA and 1.560 ± 0.015 for PETA. To estimate the ratio of reacted vinyl bonds, the FTIR absorption band at 810 cm−1, which corresponds to the C–H bending vibration, was selected and the conversion degree (Fig. 2(c)) was calculated using eqn (1):47
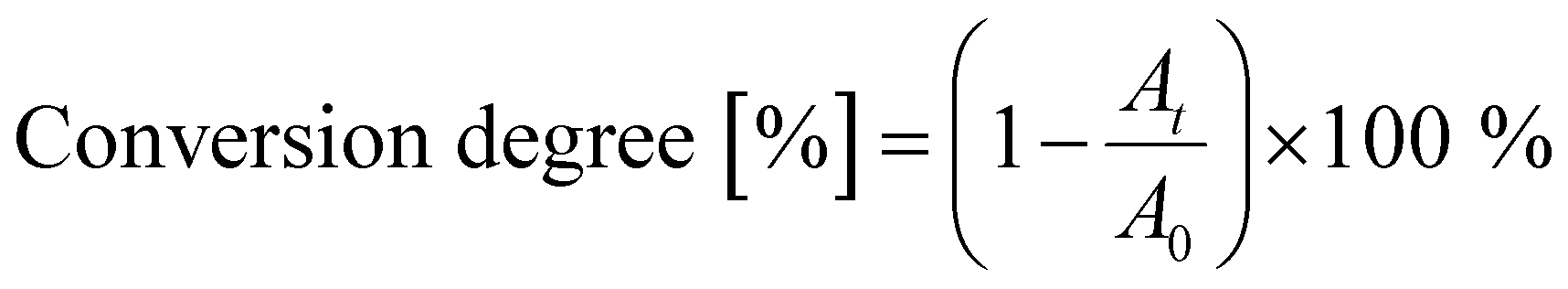 | (1) |
To account for potential material loss due to evaporation, the absorbance at 801 cm−1 was normalized by the absorbance of the carbonyl peak at 1735 cm−1. The results in Fig. 2(c) show that the conversion of the vinyl bonds begins to saturate after 5 minutes UV exposure, reaching very high degrees of conversion ranging from approximately 85% for PETA and TMPTA to 90% for PETeA, and almost 100% for ETPTA. To test the stability of the UV-exposed films, TMPTA and ETPTA UV cured for varying times were immersed in IPA (a good solvent for the pure monomers) for 15 minutes, and the change in film thickness was tracked by ellipsometry. The results (Fig. 2(d)) show that films exposed to UV light for 1 and 5 minutes still partially dissolve in IPA, indicating that they are insufficiently crosslinked. After 10 minutes of UV exposure, the film thickness remains stable, in good agreement with the saturation behavior observable in Fig. 2(a)–(c).
DEZ infiltration into crosslinked polyacrylate thin films
VPI of DEZ was first tested for the three acrylate monomers PETA, PETeA and TMPTA. Thin films of the monomers were polymerized and crosslinked under UV light for 30 min, then subjected to one cycle of VPI with a 5-hour DEZ hold at 120 °C. XRF was performed before and after VPI, and the area of the XRF Zn Kα peak was used as a measure for Zn loading (see Fig. S1a, ESI†). Both crosslinked PETA and PETeA (denoted in the following as pPETA and pPETeA) exhibit almost negligible inorganic loading after VPI, while the Zn signal for pTMPTA is significantly stronger (about 5-fold). To put the measured inorganic loading into perspective, a plane Si wafer, which is expected to grow a ZnO monolayer under ideal conditions, was subjected to the same VPI process. The resulting Zn signal is comparable to that of pTMPTA, indicating that infiltration, if it has occurred, is very limited. These results are corroborated by FTIR spectra (see Fig. S2 in the ESI†) that show no change in the absorbance for pPETA and pPETeA upon VPI and a minimal decrease in the carbonyl and ester bands for pTMPTA that might be an indication of precursor–polymer reactions.Based on the present understanding of VPI processes, a lack of infiltration can be due to a lack of miscibility (i.e. the precursor does not sorb into the polymer), a lack of or too slow precursor diffusion (e.g. because the polymer free volume is too small or the precursor molecule too big) or a lack of precursor–polymer reactions (i.e. infiltrated precursor molecules desorb during the pumping step). However, it has been demonstrated that when the density of reactive polymer groups is too high, reacted precursor molecules can form a barrier layer at the surface that inhibits diffusion of unreacted precursor molecules into the polymer bulk,23,52,53 thus limiting precursor uptake to the polymer surface.
In an attempt to improve infiltration of the crosslinked acrylates by allowing more time for the precursor to diffuse into the polymer and possibly react with its functional groups, the DEZ hold time was increased from 5 to 15 hours for subsequent experiments. Since pPETA and pPETeA showed similarly low Zn loading after the 5-hour DEZ hold, only Zn infiltration into pPETA was investigated further. Instead, ETPTA, an ethoxylated derivative of TMPTA, was included to investigate whether its longer side chains which are expected to increase the flexibility and free volume of the crosslinked network, enhance infiltration. The effect of varying UV polymerization time was studied as well. Fig. 3 shows the achieved Zn loading for the different polyacrylate networks after VPI with a 15-hour DEZ hold, measured by the area of the XRF Zn Kα peak. (The original spectra can be found in Fig. S1b and c, ESI†). Indeed, the longer DEZ hold time slightly increases the inorganic loading for both pTMPTA and pPETA, but the absolute value is still comparable to that on silicon. It is reducing the UV polymerization time from 30 minutes to 10 or 5 minutes that results in a quite significant increase in Zn loading for pTMPTA to approximately 4 times the amount measured on silicon indicating the presence of bulk infiltration. pETPTA exhibits an even higher overall Zn loading and reproduces the trend towards higher Zn loading for shorter UV crosslinking time. The inorganic loading of pPETA shows only a negligible increase with reduced UV time.
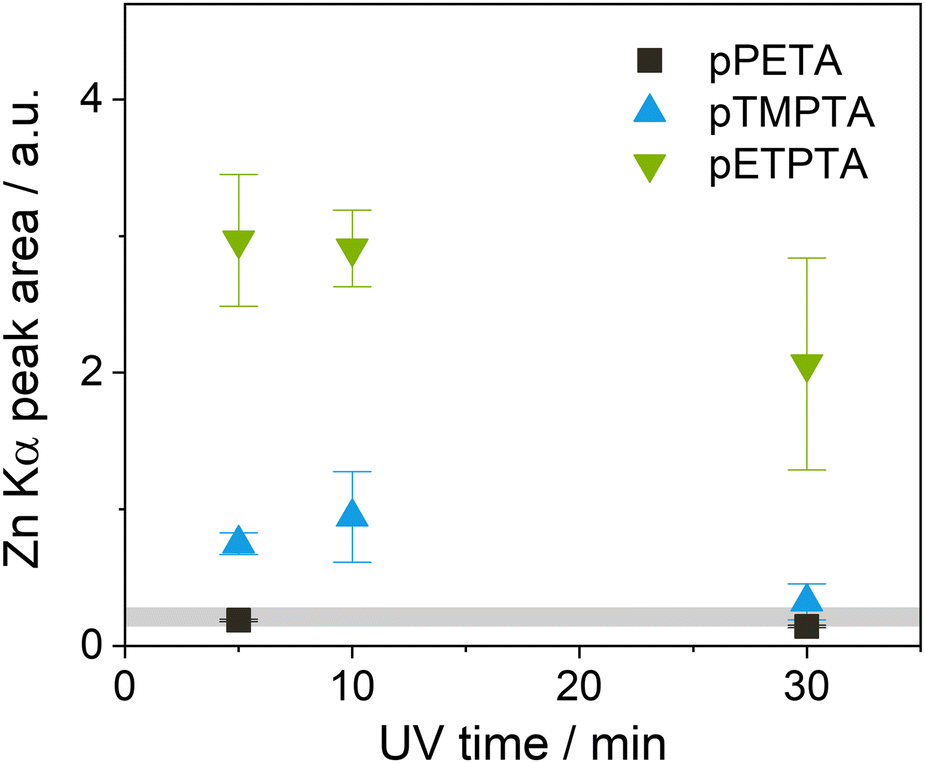 | ||
| Fig. 3 Zn loading of crosslinked acrylate films with different UV exposure times after VPI at 120 °C with a 15-hour DEZ hold based on the XRF Zn Kα peak area. The gray area indicates the average Zn loading of a Si wafer subjected to the same VPI process. | ||
The occurrence of bulk infiltration in pTMPTA and pETPTA is corroborated by FTIR spectra collected for films UV polymerized for 10 min before and after VPI with a 15-hour DEZ hold (see Fig. 4). Both polyacrylate networks show a significant decrease in the CO and C–O–C absorption bands, indicating the consumption of acrylate functional groups, as well as an increase in the O–H peak and the emergence of a new peak at 1650–1550 cm−1. The increase in the O–H band can be attributed to the water co-reaction that likely forms some kind of zinc hydroxide, while the new peak has previously been associated with C–O–Zn–R bonds in VPI literature.23,54–56 pETPTA shows larger spectral changes compared to pTMPTA, in agreement with the higher Zn loading measured by XRF. The FTIR spectrum of pPETA is not affected by VPI, indicating that the bulk of the material remains unaltered. The spectrum of the p(TMPTA-ETPTA) copolymer network will be discussed later. To ensure that the observed spectral changes result from precursor–polymer reactions rather than being thermally induced, FTIR spectra of crosslinked polyacrylate films heated to 120 °C in a vacuum chamber for 24 h were collected (see Fig. S3, ESI†). Although slight heat-induced losses in the CO and C–O–C regions can be observed, they cannot fully account for the much larger changes measured after VPI. It should be noted that for pETPTA, an additional CO shoulder peak emerges at around 1785 cm−1 upon heat exposure. This peak, which does not occur for pTMPTA, could correspond to the formation of γ-lactones, whose main absorption is at 1780 cm−1, or anhydrides, which absorb at around 1800 cm−1. Both structures have been frequently observed during degradation of aliphatic polymers.49,51,57
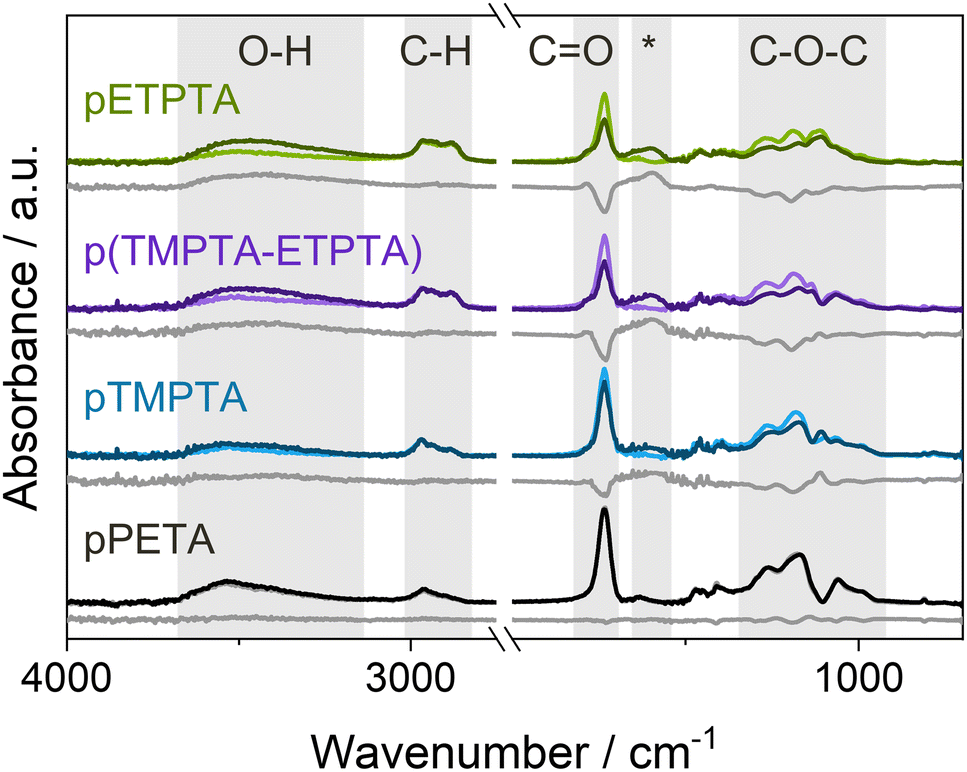 | ||
| Fig. 4 FTIR spectra of crosslinked acrylate films before (light color) and after (dark color) VPI with a 15-hour DEZ hold at 120 °C. The difference spectra are shown as gray lines. Relevant absorption bands are highlighted in gray. *marks a new peak that appears upon reaction with DEZ. pPETA was UV-polymerized for 5 min, pTMPTA, p(TMPTA-ETPTA) and pETPTA for 10 min. | ||
To gain insights into the spatial Zn distribution, XPS depth profiles were performed on infiltrated crosslinked polyacrylate films of PETA, TMPTA, ETPTA and a mixture of TMPTA-ETPTA, the latter of which will be discussed later. The resulting atomic concentrations as a function of film depth are shown in Fig. 5. It should be noted that during depth profiling, the crosslinked polyacrylate films were exposed to a high-energy Ar ion beam, which may cause chemical changes of the organic material.58,59 Preferential etching, such as faster removal of oxygen compared to carbon, as well as of organic matter compared to inorganic species, has also been observed. Therefore, the XPS measurements may not reflect the exact atomic composition of the undamaged polyacrylate–zinc complex, and sharp interfaces may not appear as abrupt changes in composition but as smeared and more gradual transitions. Despite these limitations, qualitatively investigating the in-depth distribution of zinc is still meaningful, as also previous studies have shown.18,60,61Fig. 5(a) shows that after 5 min UV polymerization and VPI with a 15-hour DEZ hold, Zn loading in pPETA is restricted to the surface of the polymer layer without significant infiltration into the bulk. (Note that the presence of a small amount of Cl is due to cross-contamination with other VPI processes and has not been shown to affect the Zn distribution.) pTMPTA, exposed to UV for 10 min, shows significant diffusion of DEZ into the polyacrylate layer (Fig. 5(b)), in agreement with the higher Zn loading measured with XRF and the chemical changes observed in FTIR. However, infiltration of pTMPTA after VPI with a 15-hour DEZ hold is only partial. The Zn atomic concentration drops to zero at approximately a third of the total film thickness. The shape of the depth profile suggests that infiltration is limited by precursor diffusion.61 The XPS depth profile for pETPTA shows an almost uniform Zn concentration throughout the entire film thickness indicating that the material has been fully infiltrated by DEZ. Again, the XPS result agrees with the XRF and FTIR data, both of which show that pETPTA achieves the highest Zn loading. When comparing the XRF results for the different polyacrylates, note that the absolute Zn loading is given without normalization to the film thickness. For pPETA and pTMPTA, where infiltration is non-existent or partial, the absolute Zn loading is independent of film thickness. However, once the layer is fully infiltrated (i.e. saturated), as assumed for pETPTA, the total Zn loading becomes dependent on the polyacrylate film thickness. A table with the thicknesses of all polyacrylate films studied in this work can be found in the ESI† (Table S1).
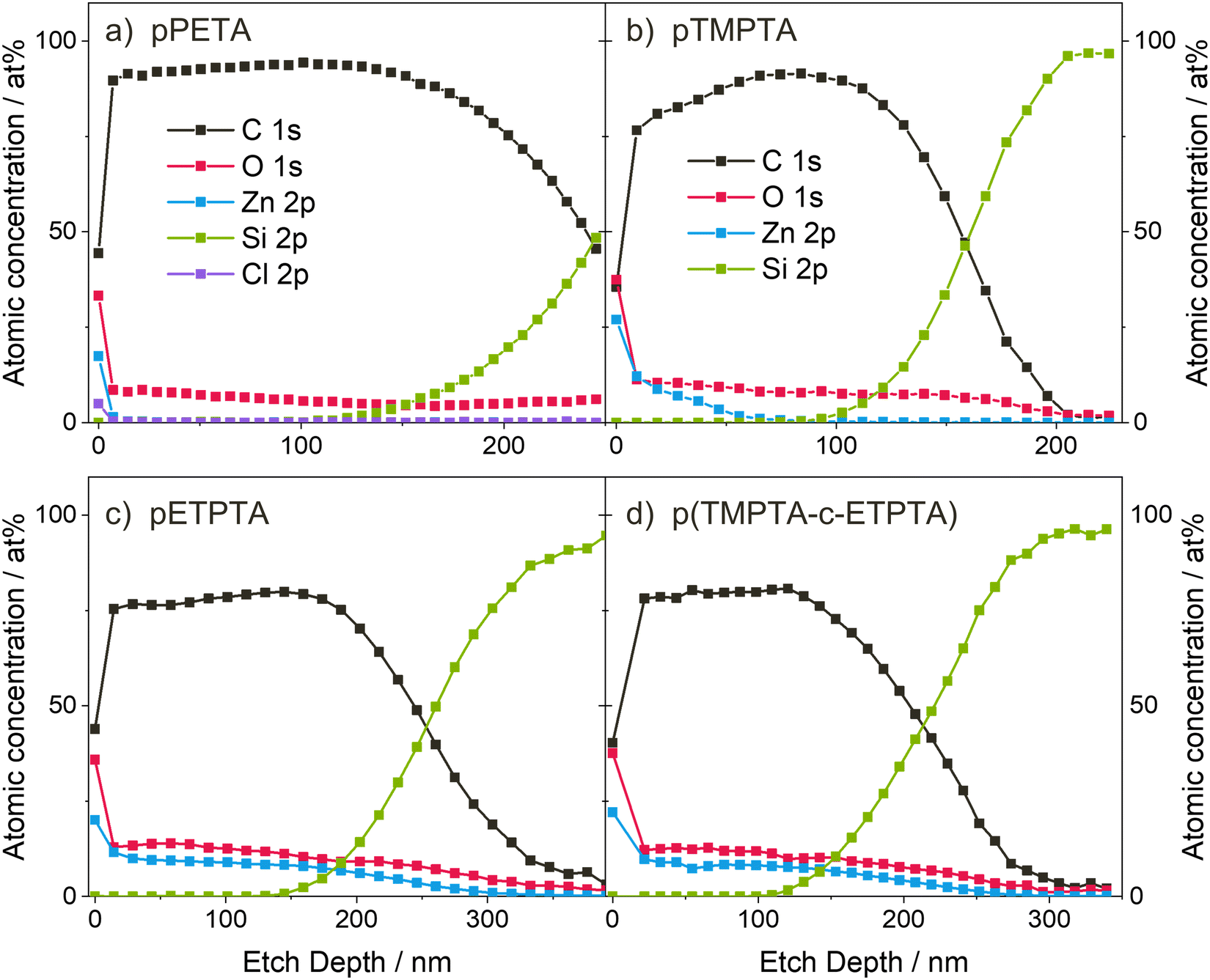 | ||
| Fig. 5 XPS depth profiles of (a) pPETA after 5 minutes UV exposure, and (b) pTMPTA, (c) pETPTA and (d) p(TMPTA-ETPTA) after 10 minutes UV exposure and VPI with a 15-hour DEZ hold at 120 °C. | ||
Due to the convolution of sorption, diffusion and reaction contributions, VPI process kinetics can be very complex, making it challenging to predict and understand why infiltration works for certain polymer–precursor combinations and process parameters and why it does not for others. Based on the data presented in this study, a plausible explanation for the differences in the infiltration behavior of the 4 acrylates can be found. These differences in infiltration behavior can be attributed to variations in the chemical structure and physical properties of the polyacrylate networks, such as their crosslinking density, the rigidity of the polymer network, and the presence of functional groups that can interact with DEZ. In terms of crosslinking density and network rigidity, pPETA and pTMPTA can be assumed to exhibit fairly similar properties, while pPETeA, due to its tetrafunctionality, can be expected to be more densely crosslinked. While a high crosslinking density or network rigidity may limit or inhibit precursor diffusion, more flexible acrylates, such as pETPTA, may enhance diffusivity, resulting in improved infiltration. In terms of reactivity towards the precursor, all 4 acrylates feature identical ester groups that have been shown to react with DEZ (see FTIR spectra for pTMPTA and pETPTA in Fig. 4). PETA exhibits an additional hydroxyl group, a functional species known to be highly reactive towards DEZ.62 In addition, all polyacrylate films contain 6 wt% photoinitiator with respect to the monomer. Due to the small quantity and because the photoinitiator exhibits functional groups similar to the acrylate monomers (i.e., CO and C–O–C bonds), it is unlikely that the presence of the photoinitiator affects the overall infiltration kinetics.
Keeping the just mentioned properties of the acrylates in mind, it can be concluded that the lack of infiltration into pPETA and pPETeA must be either due to a lack of diffusion or due to a lack of sorption, since both monomers have functional groups reactive towards DEZ. Since the chemistry of pPETA and pPETeA is quite similar to that of pTMPTA and pETPTA, it seems unlikely (though not impossible) that DEZ would only sorb into the latter two. A much more probable explanation is that the density of reactive groups in pPETA and pPETeA is too high, so that reacting precursor molecules form a barrier layer at the surface that hinders diffusion. The XPS depth profile of pTMPTA (see Fig. 5(b)) already showed that in these highly crosslinked acrylate networks, diffusion is the limiting factor (as opposed to a reaction-limited process, see ref. 61 and 63 for comparison). Bamford et al.52 have demonstrated that for TMA infiltration into poly(styrene-r-2-hydroxyethyl methacrylate) (PS-r-PHEMA) random copolymer thin films, a ratio of 20 molar % PHEMA, where each monomer unit has 1 hydroxyl group, results in the formation of a barrier layer and incomplete infiltration. This corresponds to a density of hydroxyl groups of roughly 0.0019 mol cm−3 (for details on the estimation see ESI†). Based on a similar ballpark estimation, the hydroxyl density of pPETA is 0.0033 mol cm−3 and thus significantly higher. Taking into account that DEZ is larger in size than TMA, that diffusion can be expected to be slower in the highly crosslinked acrylate networks compared to the linear (PS-r-PHEMA) copolymer, and that interactions between DEZ and the ester groups have not been considered, it seems reasonable that barrier layer formation could occur for PETA (and PETeA, which contains 30–50% PETA and is more densely crosslinked than PETA). In contrast, TMPTA's lack of hydroxyl groups may facilitate diffusion of DEZ into the polymer bulk. The simultaneous consumption of CO and C–O–C peaks and emergence of a new peak corresponding to an organic–Zn bond in the FTIR suggest that DEZ reacts with the ester groups but the reactivity appears to be low enough to still allow for substantial bulk diffusion. The FTIR spectra (Fig. 4) suggest that DEZ infiltration in pETPTA follows similar reaction pathways as in pTMPTA, since measured spectral changes mainly differ in magnitude. However, a more in-depth study of precursor–polymer interactions is necessary to be able to completely exclude the possibility of the additional ethoxy groups affecting the reaction mechanism or the precursor solubility. Assuming that the ethoxy groups in ETPTA do not substantially change the DEZ solubility and reactivity, the main difference between TMPTA and ETPTA lies in the length of the side chains. It is hypothesized that the longer side chains in ETPTA increase the flexibility and free volume of the crosslinked acrylate network and thus facilitate precursor diffusion. An indication for the change in flexibility and free volume with increasing ETPTA content is provided by the thermal expansion of the polymer networks, which is indeed more pronounced for pETPTA compared to pTMPTA (see Fig. S4 in the ESI†).
In addition to the monomer chemistry, UV crosslinking time was also shown to affect DEZ infiltration (see Fig. 3), with shorter UV exposure times (5–10 min) resulting in higher Zn loading and long UV times of 30 min resulting in lower Zn loading. According to Fig. 2(c), the crosslinking density, and thus the network's rigidity, increases from 5 min to 30 min of UV exposure, which may hinder DEZ diffusion. The FTIR spectra in Fig. 1 also show the emergence of a broad O–H absorption band upon prolonged UV exposure, which might be a result of photo-induced degradation.49–51,64 A higher density of hydroxyl groups may also contribute to reduced infiltration, and thus lower Zn loading. For efficient infiltration, it is therefore favorable to keep the UV exposure time as short as possible while still achieving sufficiently crosslinked, stable polyacrylate networks. According to the solvent stability tests in Fig. 1(d), the ideal UV crosslinking time for the given recipe and lamp set up is 10 min.
Overall, pETPTA was revealed to be most suitable for DEZ infiltration. However, FTIR spectra (Fig. 4) have shown that pETPTA is prone to heat-induced degradation. To enhance polyacrylate stability while at the same time maintaining efficient and complete DEZ infiltration, a crosslinked polyacrylate film consisting of a 1:3 weight ratio of TMPTA to ETPTA was created and tested. XRF reveals similarly high Zn loading in the copolymer network film as for pETPTA alone (see Fig. S1c and d, ESI†) after 10 minutes UV exposure and VPI with a 15-hour DEZ hold, and the XPS depth profile in Fig. 4(d) demonstrates that the p(TMTPA-ETPTA) film is fully infiltrated by DEZ after a 15-hour DEZ hold. The FTIR difference spectrum in Fig. S3 (ESI†) shows that the p(TMPTA-ETPTA) copolymer network is indeed thermally more stable than pETPTA, even though some changes in the CO peak can still be observed. The excellent infiltration characteristics of the p(TMPTA-ETPTA) film suggest that the ETPTA content in the copolymer network can be further reduced to further improve stability while still achieving efficient DEZ infiltration – a hypothesis that needs to be confirmed by future experiments.
Selective infiltration into patterned p(TMPTA-ETPTA) on PMMA
As a proof of concept of how VPI can be integrated with existing photopatterning techniques to selectively modify certain polymers’ properties in a top-down fashion, patterns of the well infiltrating p(TMPTA-ETPTA) copolymer network were created on pMMA by photolithographic masking. PMMA proves to be non-infiltrating by DEZ under the process conditions used in this work (see Fig. S1d, ESI†), a result consistent with previous reports.18,65 The patterns consist of circles and squares of different sizes. Fig. 6(a) shows the surface topography of a selected pattern prior to VPI, as measured by optical profilometry. The patterned shapes have a uniform thickness except for the outer edges, which form a kind of raised rim. It is hypothesized that this rim formation is a result of capillary forces pulling unpolymerized liquid monomer up along the edges of the shadow mask, an effect that is exploited in capillary force lithography66 but could likely be avoided using other methods such as direct laser writing. For this simple proof-of-concept, rim formation was minimized by increasing the amount of photoinitiator. This change in chemistry reduced the UV exposure time needed to obtain stable films to 1 minute, thus also reducing the time for rim formation.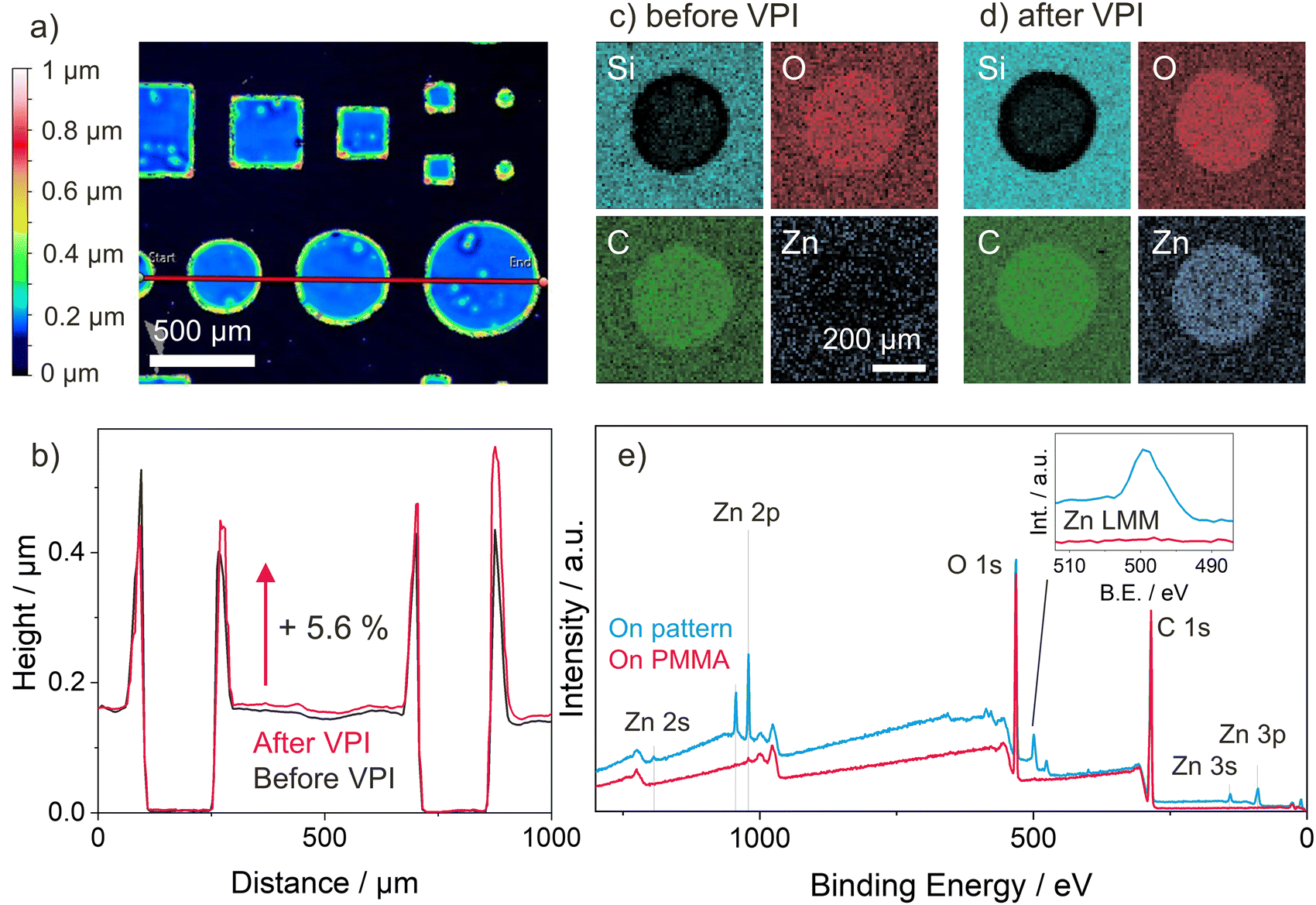 | ||
| Fig. 6 Selective Zn infiltration (5 h DEZ, 120 °C) of a p(TMPTA-ETPTA) pattern on PMMA. (a) Optical Profilometry of the pattern before VPI, (b) height profile before and after VPI cut along pink line in (a), (c) and (d) EDX elemental maps of a p(TMPTA-ETPTA) feature on PMMA (c) before and (d) after VPI. (e) XPS survey scan of samples on the p(TMPTA-ETPTA) pattern and on PMMA after VPI. The inset shows a zoom on the Zn L3M4,5M4,5 Auger peak. | ||
Height profiles for the red line shown in Fig. 6(a) were collected both before and after VPI and are reported in Fig. 6(b). The lateral dimensions of the patterned features remain unchanged after VPI, but their height increases by approximately 5.6%, consistent with the thickness increase upon VPI measured by ellipsometry for uniform p(TMPTA-ETPTA) films (see Table S1, ESI†). Note that an apparent thickness change of a few percent could also be caused by the change in optical properties upon VPI. EDX elemental mapping and XPS confirm the selective infiltration of Zn into p(TMPTA-ETPTA) while leaving PMMA unchanged. Results of the EDX elemental mapping are shown in Fig. 6(c) and (d) for one circular p(TMPTA-ETPTA) feature of 500 nm diameter before and after VPI, respectively. The contrast visible between the two materials, p(TMPTA-ETPTA) and PMMA, before VPI is caused by the limited penetration depth of the incident electrons. In areas with PMMA, electrons penetrate the entire polymer layer, and the detector collects signals from both the polymer and the underlying Si substrate. In areas where an approximately 150-nm thick layer of p(TMPTA-ETPTA) is patterned on top of the PMMA, the maximum depth of information lies inside the polymer layers, and no Si signal is detected. After VPI, the presence of Zn is observed only in the regions with p(TMPTA-ETPTA), but not on the surrounding PMMA. The EDX mapping data is corroborated by XPS survey scans (Fig. 6(e)) performed on both the pattern and pMMA, which exhibit characteristic Zn peaks on the former and no such peaks on the latter. An XPS depth profile performed in the region of a p(TMPTA-ETPTA) feature (see Fig. S5, ESI†) confirms that DEZ only infiltrated the p(TMPTA-ETPTA) layer but did not diffuse into the underlying PMMA.
Conclusion
The present study explores vapor phase infiltration (VPI) of DEZ into four different highly crosslinked polyacrylate networks based on the monomers pentaerythritol triacrylate (PETA), pentaerythritol tetraacrylate (PETeA), trimethylolpropane triacrylate (TMPTA) and ethoxylated trimethylolpropane triacrylate (ETPTA) that can be patterned by photolithography and two-photon polymerization (2PP). Thin films of the crosslinked polyacrylates were prepared by spin-coating and subsequent UV polymerization. The polymerization and crosslinking process was monitored by spectroscopic ellipsometry and FTIR, showing that stable films with a high degree of vinyl bond conversion could be achieved after 10 minutes UV exposure. The results of VPI with DEZ at 120 °C showed that for such highly crosslinked polyacrylate networks, infiltration is limited by the precursor diffusion. While for the hydroxyl-containing pPETA and pPETeA, the reaction with DEZ was restricted to the surface with no significant bulk infiltration, partial DEZ diffusion into the bulk could be achieved for pTMPTA after a 15-hour DEZ hold. For pETPTA, whose monomer has longer and more flexible side chains, complete and uniform infiltration by DEZ could be obtained, highlighting the impact of polymer flexibility and free volume on VPI kinetics. Copolymerizing ETPTA with 25 wt% TMPTA was shown to enhance the thermal stability of the material without affecting the inorganic loading.Finally, selective infiltration of DEZ into a photolithographically patterned p(TMPTA-ETPTA) film on non-infiltrating PMMA was demonstrated, illustrating the potential of integrating VPI with existing polymer patterning techniques to achieve selective material-specific top-down hybridization and property modification. Such volume-selective VPI processes open new avenues towards advanced maskless patterning strategies. The 2PP-processability of the investigated polyacrylate networks adds versatility and customizability, enabling the application of selective infiltration to printed 3D microstructures of varying complexity.
Author contributions
L. Demelius: conceptualization, methodology, investigation, formal analysis, writing – original draft, writing – review & editing, funding acquisition. L. Zhang: investigation, formal analysis. A. M. Coclite: supervision, writing – review & editing, funding acquisition. M. D. Losego: resources, supervision, writing – review & editing, funding acquisition.Data availability
Data for this article is available at Zenodo at https://doi.org/10.5281/zenodo.13310291.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement no. 899349. L. D. acknowledges personal funding from the Austrian Marshall Plan Foundation for funding her research stay at Georgia Institute of Technology. The experimental part of this work was performed at the Georgia Tech Institute for Electronics and Nanotechnology, a member of the National Nanotechnology Coordinated Infrastructure (NNCI), which is supported by the National Science Foundation (ECCS-2025462). Additional financial support was also supplied by the U.S. National Science Foundation under DMREF-1921873. Some measurements were also completed in Georgia Tech's Materials Innovation and Learning Laboratory (The MILL), an open-access research space for materials science and engineering. The authors acknowledge Shuaib Balogun for performing the XPS depth profile measurement on the patterned sample.References
- M. Qiu, W. Du, S. Zhou, P. Cai, Y. Luo, X. Wang, R. Yang and J. Zhao, Prog. Polym. Sci., 2023, 142, 101688 CrossRef CAS.
- Z. Nie and E. Kumacheva, Nat. Mater., 2008, 7, 277–290 CrossRef CAS PubMed.
- Y. Chen, Microelectron. Eng., 2015, 135, 57–72 CrossRef CAS.
- S. Diez, The next genergation of maskless lithography, ed. M. R. Douglass, P. S. King and B. L. Lee, San Francisco, California, USA, 2016, p. 976102 Search PubMed.
- Z.-L. Wu, Y.-N. Qi, X.-J. Yin, X. Yang, C.-M. Chen, J.-Y. Yu, J.-C. Yu, Y.-M. Lin, F. Hui, P.-L. Liu, Y.-X. Liang, Y. Zhang and M.-S. Zhao, Polymers, 2019, 11, 553 CrossRef CAS PubMed.
- V. Harinarayana and Y. C. Shin, Opt. Laser Technol., 2021, 142, 107180 CrossRef CAS.
- A. Selimis, V. Mironov and M. Farsari, Microelectron. Eng., 2015, 132, 83–89 CrossRef CAS.
- C. Sanchez, B. Julián, P. Belleville and M. Popall, J. Mater. Chem., 2005, 15, 3559 RSC.
- Y. Zhao and K. Zhu, Chem. Soc. Rev., 2016, 45, 655–689 RSC.
- S. Wang, Y. Kang, L. Wang, H. Zhang, Y. Wang and Y. Wang, Sens. Actuators, B, 2013, 182, 467–481 CrossRef CAS.
- S. Chongdar, S. Bhattacharjee, P. Bhanja and A. Bhaumik, Chem. Commun., 2022, 58, 3429–3460 RSC.
- W. Park, H. Shin, B. Choi, W.-K. Rhim, K. Na and D. Keun Han, Prog. Mater. Sci., 2020, 114, 100686 CrossRef CAS.
- S. Marcelja, L. Demelius, T. A. Ali, M. Aghito, F. Muralter, G. H. Rodriguez, M. Kräuter, K. Unger, L. Wolfsberger and A. M. Coclite, J. Phys. Mater., 2023, 6, 042001 CrossRef.
- P. Gómez-Romero, O. Ayyad, J. Suárez-Guevara and D. Muñoz-Rojas, J. Solid State Electrochem., 2010, 14, 1939–1945 CrossRef.
- Z. Liang, R. Zhao, T. Qiu, R. Zou and Q. Xu, EnergyChem, 2019, 1, 100001 CrossRef.
- K. Lu, Int. Mater. Rev., 2020, 65, 463–501 CrossRef CAS.
- C. Z. Leng and M. D. Losego, Mater. Horiz., 2017, 4, 747–771 RSC.
- M. Qiu, W. Du, X. Luo, S. Zhu, Y. Luo and J. Zhao, ACS Appl. Mater. Interfaces, 2021, 14, 22719–11722 CrossRef PubMed.
- S.-M. Lee, E. Pippel, O. Moutanabbir, I. Gunkel, T. Thurn-Albrecht and M. Knez, ACS Appl. Mater. Interfaces, 2010, 2, 2436–2441 CrossRef CAS PubMed.
- R. P. Padbury and J. S. Jur, J. Vac. Sci. Technol., A, 2015, 33, 01A112 CrossRef.
- V. P. Nguyen, J. Yoo, J. Y. Lee, J. J. Chung, J. H. Hwang, Y. Jung and S. M. Lee, ACS Appl. Mater. Interfaces, 2020, 12, 43501–43512 CrossRef CAS PubMed.
- N. A. Vogel, P. S. Williams, A. H. Brozena, D. Sen, S. Atanasov, G. N. Parsons and S. A. Khan, Adv. Mater. Interfaces, 2015, 2, 1500229 CrossRef.
- J. Pilz, A. M. Coclite and M. D. Losego, Mater. Adv., 2020, 1, 1695–1704 RSC.
- L. E. Ocola, D. J. Gosztola, A. Yanguas-Gil, H.-S. Suh and A. Connolly, in Proceedings Quantum Sensing and Nano Electronics and Photonics XIII, ed. M. Razeghi, San Francisco, California, USA, 2016, vol. 9755, p. 97552C Search PubMed.
- A. Singhal, R. Divan, A. Dalmiya, L. Stan, A. Ghiacy, P. T. Lynch and I. Paprotny, J. Vac. Sci. Technol., A, 2024, 42, 012404 CrossRef CAS.
- Y. Yu, Z. Li, Y. Wang, S. Gong and X. Wang, Adv. Mater., 2015, 27, 4938–4944 CrossRef CAS PubMed.
- M. Baryshnikova, D. De Simone, W. Knaepen, K. Kachel, B. Chan, S. Paolillo, J. Willem Maes, D. De Roest, P. Rincon Delgadillo and G. Vandenberghe, J. Photopolym. Sci. Technol., 2017, 30, 667–670 CrossRef CAS.
- Y. C. Tseng, Q. Peng, L. E. Ocola, D. A. Czaplewski, J. W. Elam and S. B. Darling, J. Mater. Chem., 2011, 21, 11722–11725 RSC.
- M. Nakagawa, T. Uehara, Y. Ozaki, T. Nakamura and S. Ito, J. Vac. Sci. Technol., B: Nanotechnol. Microelectron.: Mater., Process., Meas., Phenom., 2018, 36, 06JF02 Search PubMed.
- C. Miyajima, S. Ito and M. Nakagawa, J. Vac. Sci. Technol., B: Nanotechnol. Microelectron.: Mater., Process., Meas., Phenom., 2021, 39, 032603 CAS.
- C.-Y. Nam, A. Stein and K. Kisslinger, J. Vac. Sci. Technol., B: Nanotechnol. Microelectron.: Mater., Process., Meas., Phenom., 2015, 33, 06F201 Search PubMed.
- C. Y. Nam, A. Stein, K. Kisslinger and C. T. Black, Appl. Phys. Lett., 2015, 107, 1–4 Search PubMed.
- X. Ye, J. Kestell, K. Kisslinger, M. Liu, R. B. Grubbs, J. A. Boscoboinik and C. Y. Nam, Chem. Mater., 2017, 29, 4535–4545 CrossRef CAS.
- A. Subramanian, N. Tiwale, W. Lee, K. Kisslinger, M. Lu, A. Stein, J. Kim and C. Nam, Adv. Mater. Interfaces, 2023, 10, 2300420 CrossRef CAS.
- D. N. Le, T. Park, S. M. Hwang, J.-H. Kim, Y. C. Jung, N. Tiwale, A. Subramanian, W.-I. Lee, R. Choi, M. M. Sung, C.-Y. Nam and J. Kim, Jpn. J. Appl. Phys., 2023, 62, SG0812 CrossRef CAS.
- Z. Li, J. He, A. Subramanian, N. Tiwale, K. J. Dusoe, C.-Y. Nam, Y. Li and S.-W. Lee, Mater. Des., 2023, 227, 111770 CrossRef.
- A. Subramanian, N. Tiwale, K. Kisslinger and C. Nam, Adv. Electron. Mater., 2022, 8, 2200172 CrossRef CAS.
- G. N. Parsons and R. D. Clark, Chem. Mater., 2020, 32, 4920–4953 CrossRef CAS.
- E. K. McGuinness, Y. Liu, R. Ramprasad and M. D. Losego, Mater. Chem. Phys., 2023, 294, 127026 CrossRef CAS.
- J. Kamcev, D. S. Germack, D. Nykypanchuk, R. B. Grubbs, C. Y. Nam and C. T. Black, ACS Nano, 2013, 7, 339–346 CrossRef CAS PubMed.
- R. Azoulay, M. Barzilay, I. Weisbord, R. Avrahami, E. Zussman and T. Segal-Peretz, ACS Appl. Nano Mater., 2022, 5, 7228–7236 CrossRef CAS.
- R. Z. Waldman, D. J. Mandia, A. Yanguas-Gil, A. B. F. Martinson, J. W. Elam and S. B. Darling, J. Chem. Phys., 2019, 151, 190901 CrossRef PubMed.
- Q. Peng, Y. C. Tseng, S. B. Darling and J. W. Elam, Adv. Mater., 2010, 22, 5129–5133 CrossRef CAS PubMed.
- Y. C. Tseng, Q. Peng, L. E. Ocola, J. W. Elam and S. B. Darling, J. Phys. Chem. C, 2011, 115, 17725–17729 CrossRef CAS.
- E. K. McGuinness, F. Zhang, Y. Ma, R. P. Lively and M. D. Losego, Chem. Mater., 2019, 31, 5509–5518 CrossRef CAS.
- B. D. Piercy and M. D. Losego, J. Vac. Sci. Technol., B: Nanotechnol. Microelectron.: Mater., Process., Meas., Phenom., 2015, 33, 043201 Search PubMed.
- H. Kaczmarek and I. Vukovic-Kwiatkowska, eXPRESS Polym. Lett., 2012, 6, 78–94 CrossRef CAS.
- B. C. Smith, Spectroscopy, 2023, 10–14 Search PubMed.
- O. Chiantore, L. Trossarelli and M. Lazzari, Polymer, 2000, 41, 1657–1668 CrossRef CAS.
- G. G. Goourey, P. Wong-Wah-Chung, F. Delor-Jestin, B. Légeret, L. Balan and Y. Israëli, Polym. Degrad. Stab., 2015, 119, 208–216 CrossRef CAS.
- J. M. Chalmers, in Encyclopedia of Analytical Chemistry, ed. R. A. Meyers, Wiley, 1st edn, 2000 Search PubMed.
- J. T. Bamford, R. A. Smith, C. Z. Leng, W. R. Gutekunst and M. D. Losego, Macromolecules, 2021, 54, 6790–6798 CrossRef CAS.
- R. P. Padbury and J. S. Jur, Langmuir, 2014, 30, 9228–9238 CrossRef CAS PubMed.
- E. K. McGuinness, C. Z. Leng and M. D. Losego, ACS Appl. Polym. Mater., 2020, 2, 1335–1344 CrossRef CAS.
- G. T. Hill, D. T. Lee, P. S. Williams, C. D. Needham, E. C. Dandley, C. J. Oldham and G. N. Parsons, J. Phys. Chem. C, 2019, 123, 16146–16152 CrossRef CAS.
- W. Xie, S. Khan, O. J. Rojas and G. N. Parsons, ACS Sustainable Chem. Eng., 2018, 6, 13844–13853 CrossRef CAS.
- N. Grassie and J. D. Fortune, Makromol. Chem., 1973, 168, 13–18 CrossRef CAS.
- Y. J. Hofstetter and Y. Vaynzof, ACS Appl. Polym. Mater., 2019, 1, 1372–1381 CrossRef CAS.
- T. Miyayama, N. Sanada, S. R. Bryan, J. S. Hammond and M. Suzuki, Surf. Interface Anal., 2010, 42, 1453–1457 CrossRef CAS.
- N. Sasao, S. Sugimura and K. Asakawa, Jpn. J. Appl. Phys., 2021, 60, SCCC04 CrossRef CAS.
- S. A. Balogun, Y. Ren, R. P. Lively and M. D. Losego, Phys. Chem. Chem. Phys., 2023, 25, 14064–14073 RSC.
- A. Mameli, B. Karasulu, M. A. Verheijen, B. Barcones, B. Macco, A. J. M. Mackus, W. M. M. E. Kessels and F. Roozeboom, Chem. Mater., 2019, 31, 1250–1257 CrossRef CAS.
- Y. Ren, E. K. McGuinness, C. Huang, V. R. Joseph, R. P. Lively and M. D. Losego, Chem. Mater., 2021, 33, 5210–5222 CrossRef CAS.
- A. H. Soeriyadi, V. Trouillet, F. Bennet, M. Bruns, M. R. Whittaker, C. Boyer, P. J. Barker, T. P. Davis and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1801–1811 CrossRef CAS.
- Q. Peng, Y. C. Tseng, S. B. Darling and J. W. Elam, ACS Nano, 2011, 5, 4600–4606 CrossRef CAS PubMed.
- D. Ho, J. Zou, B. Zdyrko, K. S. Iyer and I. Luzinov, Nanoscale, 2015, 7, 401–414 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma00733f |
| This journal is © The Royal Society of Chemistry 2024 |