
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulti-electron redox reactions with iron and vanadium ions at a mixed phosphate–sulfate electrode during sodium intercalation†
Violeta
Koleva
 ab,
Trajche
Tushev
a,
Sonya
Harizanova
a,
Rositsa
Kukeva
a,
Maria
Shipochka
a,
Pavel
Markov
a and
Radostina
Stoyanova
*ab
ab,
Trajche
Tushev
a,
Sonya
Harizanova
a,
Rositsa
Kukeva
a,
Maria
Shipochka
a,
Pavel
Markov
a and
Radostina
Stoyanova
*ab
aInstitute of General and Inorganic Chemistry, Bulgarian Academy of Sciences, Acad. G. Bonchev Str., Bldg. 11, 1113 Sofia, Bulgaria. E-mail: radstoy@svr.igic.bas.bg
bNational Center of Excellence Mehatronics and Clean Technologies, Bldg. 29, 1113 Sofia, Bulgaria
First published on 27th September 2024
Abstract
In order to improve the specific capacity of intercalation electrodes for sodium-ion batteries, it is necessary to identify materials capable of storing Na+ ions by activating multi-electron redox reactions. Herein, we report a NaFeVPO4(SO4)2 compound as a multi-electron electrode that combines the most abundant Fe and V ions, having multiple oxidation states, with a stable mixed phosphate–sulfate matrix. NaxFeVPO4(SO4)2 reversibly intercalates a total of 3 moles of Na+ ions (corresponding to a specific capacity of 175 mA h g−1) within a potential range of 1.5–4.2 V, which is concomitant with a limited variation in the lattice volume (up to 5.2%). NaFeVPO4(SO4)2 interacts with rGO, resulting in rGO covering the phosphate–sulphate particles, and the thickness of the covering varies between 5 and 10 nm. The NaFeVPO4(SO4)2/rGO composite stores Na+ ions via a hybrid mechanism involving faradaic and capacitive reactions. In sodium half-cells, the NaFeVPO4(SO4)2/rGO composite displays high capacity (about 90 mA h g−1), and it exhibits an excellent long-term cycling stability at elevated temperatures (about 96–97% after 100 cycles at 20 °C, followed by the next 100 cycles at 40 °C). The improved electrochemical performance is discussed based on the structural robustness of NaFeVPO4(SO4)2 and the surface interaction of NaFeVPO4(SO4)2/rGO with an electrolyte salt and electrolyte solvent. The information from this study will be relevant to the design of high energy polyanionic electrodes for practical application in sodium-ion batteries.
1. Introduction
Intercalation of sodium ions into solid state materials is a reaction that underlies the design of sodium-ion batteries.1–5 This is a consequence of the unique interplay between the structure and redox capability of transition metal ions that are integrated inside a given material.1–5 Although the crystal structure determines the pathways of solid-state Na+ diffusion (which is of significance for the rate capability of cathodes), the transition metal ions ensure the compensation of Na+ charges during the intercalation, thus contributing to the cathode's specific capacity.1–5 Most materials exhibit a one-electron redox reaction during Na+ intercalation, which is the main factor limiting their theoretical specific capacities.6–8 To improve specific capacity, the state-of-the-art research work is nowadays directed towards the design of materials in which multi-electron redox reactions are activated.9 This can be achieved through selective substitution of material constituents with transition metal ions or anionic groups.9In this aspect, polyanionic electrode materials fulfil the above requirements to a great extent: they possess a diversity of structures, which are prone to accommodate a variety of transition metal ions.10–13 The replacement of vanadium by chromium in NASICON-type phosphates leads to the activation of multi-electron redox reactions, including V2+/V3+, V3+/V4+ and V4+/V5+ couples.14–16 The anionic substitution enables the tuning of the redox capability of the polyanionic electrode.17–20 Owing to the different electronegativities of the X cation in XO4n−, the covalent character of X–O bonds is weakened or strengthened, thereby resulting in respective strengthening or weakening of the covalency of M–O bonds.17–20 The stronger the X–O bonding, the weaker the M–O bonding and hence the higher the redox potentials for the Mn+/M(n−1)+ couple (inductive effect).17–20 The pioneering experimental studies of Goodenough et al. clearly demonstrated that the SO4 substitution for PO4 effectively enhances the redox voltage of the Fe3+/Fe2+ pair from 2.8 V in Li3Fe2(PO4)3 up to 3.3–3.4 V in LiFe2PO4(SO4)2.17 This approach opened a great perspective to design new types of electrode materials having exciting electrochemical properties both for Li-ion and Na-ion batteries.18–20 The concept of the inductive effect has successfully been applied to develop a diversity of mixed polyanionic electrode materials via combinations of anions in three different ways: (i) two different oxyanionic units (e.g. PO4–SO4, PO4–SiO4, PO4–CO3, etc.); (ii) oxyanionic units with single anions (e.g. PO4–F, SO4–F, PO4–OH, PO4–N, etc.); (iii) different structural units of the same polyanion unit (PO4–P2O7, BO3–B2O5, etc.).18,21 Besides the effect on the working voltage the anion substitution has been found to increase the ionic conductivity of the NASICON compounds. The study of Geng et al.22 has demonstrated that VO43− substitution for PO43− in Li3Sc2(PO4)3 enhances the conductivity by two orders of magnitude in comparison to the non-substituted sample.
For Fe-based NASICON type AxFe2(XO4)3 materials, it has been shown that the increase in the redox potentials follows the increase in the electronegativity of X in XO4n−: for instance, VPO4 < VMoO4 ∼ VWO4 < VSO4.23 Hence, the combination of PO4 with highly electronegative SO4 ions into the NASICON framework is expected to increase considerably the operating voltage without any loss of gravimetric capacity, because of the close atomic weights of S and P (both are lighter than Mo and W). By analogy with LiFe2PO4(SO4)2, a NASICON-type analogue, NaFe2PO4(SO4)2, with a theoretical capacity of 127 mA h g−1 (for 2e− reaction) has been proposed.24 The electrode fabricated to have a high amount of carbon black additive (44![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :55 weight ratio) operates at an average voltage of about 3 V vs. Na+/Na and delivers an initial discharge capacity of about 80 mA h g−1 at a rate of C/10.24 Yahia et al.25 prepared the same compound as a single powder phase by solid state reaction and they established that the material without any carbon coating is able to insert 1.4 Na+ ions during the first discharge up to 2 V, which corresponds to 70% of the theoretical specific capacity. During the cycling, the specific capacity decreases, reaching a value of 56 mA h g−1 after the 30th cycle. Wu et al. further optimized this material by carbon decoration.26 Using the sol–gel self-propagating combustion process a sponge-like NaFe2PO4(SO4)2@rGO material (rGO denotes reduced graphene oxide) is obtained.26 The NaFe2PO4(SO4)2@rGO displays a discharge capacity of 92 mA h g−1 under 25 mA g−1, while under high current loading (i.e. 50 mA g−1) a capacity of 40 mA h g−1 is reached after 300 cycles.26 Through partial substitution of Fe for V, Essehli et al.27 obtained a series of compositions NaFe2−xVx(PO4)(SO4)2 (0 ≤ x ≤ 2) and they found that pure NASICON phases can be only formed at a vanadium amount within the range of 0 < x ≤ 1. The maximum reversible capacity was reported for the sample NaFe1.6V0.4(PO4)(SO4)2, reaching a capacity of 90 mA h g−1 after 50 cycles at a rate of C/10.27 Very recently, Pati and Dhaka28 reported data on the same composition mixed with CNT as an electrode in sodium-ion batteries. Surprisingly, the authors claimed that this electrode NaFe1.6V0.4(PO4)(SO4)2@CNT operates at a working voltage of ∼3 V only due to the Fe3+/Fe2+ couple, which is opposite to the data of Essehli et al.,27 indicating a simultaneous function of iron and vanadium ions. Moreover, Pati and Dhaka reported that in this composition vanadium is present as V5+ and it dopes the Fe3+ sites, thus producing oxygen vacancies.28 These statements raise serious doubts about the stoichiometry of NaFe1.6V0.4(PO4)(SO4)2, as well as the appearance of oxygen vacancies in the crystal framework where oxygen atoms are covalently bonded in PO4 and SO4 groups. Recently, Kumar et al. proposed Na3Fe2PO4(SO4)2 composition as another 3 V-insertion electrode.29 It exhibited a single-phase Na de-intercalation with a stable specific capacity of 57 mA h g−1 at a rate of C/10 after 500 cycles.29 Based on these data, one can expect that coupling the multi-electron reaction with the mixed phosphate–sulfate matrix will enable the design of an electrode material with enhanced specific capacity and cycling stability.
:55 weight ratio) operates at an average voltage of about 3 V vs. Na+/Na and delivers an initial discharge capacity of about 80 mA h g−1 at a rate of C/10.24 Yahia et al.25 prepared the same compound as a single powder phase by solid state reaction and they established that the material without any carbon coating is able to insert 1.4 Na+ ions during the first discharge up to 2 V, which corresponds to 70% of the theoretical specific capacity. During the cycling, the specific capacity decreases, reaching a value of 56 mA h g−1 after the 30th cycle. Wu et al. further optimized this material by carbon decoration.26 Using the sol–gel self-propagating combustion process a sponge-like NaFe2PO4(SO4)2@rGO material (rGO denotes reduced graphene oxide) is obtained.26 The NaFe2PO4(SO4)2@rGO displays a discharge capacity of 92 mA h g−1 under 25 mA g−1, while under high current loading (i.e. 50 mA g−1) a capacity of 40 mA h g−1 is reached after 300 cycles.26 Through partial substitution of Fe for V, Essehli et al.27 obtained a series of compositions NaFe2−xVx(PO4)(SO4)2 (0 ≤ x ≤ 2) and they found that pure NASICON phases can be only formed at a vanadium amount within the range of 0 < x ≤ 1. The maximum reversible capacity was reported for the sample NaFe1.6V0.4(PO4)(SO4)2, reaching a capacity of 90 mA h g−1 after 50 cycles at a rate of C/10.27 Very recently, Pati and Dhaka28 reported data on the same composition mixed with CNT as an electrode in sodium-ion batteries. Surprisingly, the authors claimed that this electrode NaFe1.6V0.4(PO4)(SO4)2@CNT operates at a working voltage of ∼3 V only due to the Fe3+/Fe2+ couple, which is opposite to the data of Essehli et al.,27 indicating a simultaneous function of iron and vanadium ions. Moreover, Pati and Dhaka reported that in this composition vanadium is present as V5+ and it dopes the Fe3+ sites, thus producing oxygen vacancies.28 These statements raise serious doubts about the stoichiometry of NaFe1.6V0.4(PO4)(SO4)2, as well as the appearance of oxygen vacancies in the crystal framework where oxygen atoms are covalently bonded in PO4 and SO4 groups. Recently, Kumar et al. proposed Na3Fe2PO4(SO4)2 composition as another 3 V-insertion electrode.29 It exhibited a single-phase Na de-intercalation with a stable specific capacity of 57 mA h g−1 at a rate of C/10 after 500 cycles.29 Based on these data, one can expect that coupling the multi-electron reaction with the mixed phosphate–sulfate matrix will enable the design of an electrode material with enhanced specific capacity and cycling stability.
Herein, we report a NaFeVPO4(SO4)2 compound as a multi-electron electrode, which combines the most abundant Fe and V ions having multiple oxidation states with a stable mixed phosphate–sulfate matrix. In order to enhance the specific capacity and the long-term cycling stability, a composite between NaFeVPO4(SO4)2 and rGO is obtained by a ball-milling treatment. The electrochemical performance at room and elevated temperatures is discussed on the basis of the structural robustness of NaFeVPO4(SO4)2 and its interaction with the electrolyte. First data on the formation and on identification of the electrode surface layers are reported by means of ex situ XPS and EPR spectroscopy. The new knowledge will help in further electrode optimization and it will also be of importance for elaboration of polyanionic electrodes for practical application in sodium ion batteries.
2. Experimental
2.1. Synthesis
NaFeVPO4(SO4)2 (denoted as NFVPS) was synthesized via a two-step process involving freeze-drying of solutions and their thermal decomposition at 400 °C. The solution subjected to freeze-drying contained NaH2PO4, Fe(NO3)3·9H2O, NH4VO3 and (NH4)2SO4 at a ratio of 1:1:1:2 mole. This solution was obtained by mixing of separate aqueous solutions in a definite sequence to avoid precipitation processes before freezing. First, NaH2PO4 and (NH4)2SO4 were mixed (Solution 1). Then a solution of NH4VO3 in citric acid (1:2 mole ratio) was prepared (Solution 2) upon heating at about 50–60 °C, which caused colour transformations from yellow to blue and then to green, indicating changes in the V oxidation states: V5+ to V4+ to V3+. This green solution was added to Solution 3, containing Fe(NO3)3·9H2O acidified with HNO3 to about pH = 2 (Solution 4). Finally, Solution 1 was added to Solution 4 and the as-prepared complex solution was frozen. Freeze-drying was performed for 18 h at 0.15 mbar and then for 4 h at 0.1 mbar (−100 °C) using a Christ Alpha 3–4 LSCbasic freeze dryer. The resulting powder precursor was thermally treated at 400 °C under Ar flow: a pre-decomposition for 3 h to release the decomposition products such as NOx, CO2, and NH3, and further annealing in a tube furnace for 10 h. All the chemicals used were of ACS or reagent grade. The composite between NFVPS and 15% rGO (denoted as NFVPS/rGO) was prepared by simple ball-milling treatment for 4 h at a speed of 300 rpm using agate balls having a diameter of 10 mm (planetary mill “Pulverisette 6” from Fritsch GmbH). The powder to ball mass ratio was 1:10. The as-obtained composite NFVPS/rGO was annealed at 400 °C for 3 h under Ar flow to eliminate all the adsorbed species. The rGO material was provided by Graphit Kropfmühl GmbH (Hauzenberg, Germany). As a reference in the CV measurements a composite between NFVPS and 15% carbon black being a traditional additive (Super C65, TIMCAL Ltd) was prepared in the same way (denoted as NVFPS/C).
2.2. Material characterization
In order to determine the purity and structure of NFVPS, NFVPS/rGO and cycled electrodes, powder X-ray diffraction (XRD) was applied using a Bruker Advance D8 diffractometer with a LynxEye detector (CuKα radiation). The lattice parameters were calculated by the WinPLOTR program. An X-ray fluorescence (XRF) spectrometer (Bruker S2 PICOFOX 400) was employed for the analysis of the chemical composition of the samples after dissolving them in dilute nitric acid. The porosity of the samples was studied based on the low-temperature nitrogen adsorption–desorption isotherms (77.4 K) measured with a NOVA 1200e device (Quantachrome, Boynton Beach, FL, USA). The specific surface areas were calculated by the BET method, the total pore volumes were obtained according to Gurwitsch's rule at p/p0 = 0.99 and the pore size distributions were estimated according to the Barrett–Joyner–Halenda (BJH) method. Electron paramagnetic resonance (EPR) spectra were recorded within the X-band frequency region (9.4 GHz) on a Bruker EMXplus EPR spectrometer within a temperature range of 100–400 K. To probe the chemical and electronic state of the elements on the electrode surfaces X-ray photoelectron spectroscopy (XPS) analysis was carried out (VG Escalab II system with Al Kα radiation, Thermo Fisher Scientific, Waltham, MA, USA). The C1s line of adventitious carbon at 285.0 eV was used as an internal standard to calibrate the binding energies. The photoelectron spectra were corrected by subtracting a Shirley-type background and they were quantified using the peak area and Scofield's photo-ionization cross-section. The accuracy of the binding energy measured was ±0.1 eV. Scanning electron microscopy (SEM) was used to observe the morphology of the powder samples (JEOL JSM 6390 microscope, Japan). HR-TEM analysis was performed using a JEOL 2100 microscope (Tokyo, Japan) with a GATAN Orius 832 SC1000 camera (Plesantan, CA, USA). The optical images of the electrodes were collected using a ZEISS Stemi 508 stereo microscope (Carl Zeiss, Germany).2.3. Electrochemical characterization
The electrochemical tests were performed in Swagelok type sodium half-cells by means of a 32 channel Biologic VMP-3e battery cycler. The electrochemical cycling was carried out under galvanostatic and potentiostatic regimes at 20 and 40 °C (KB-53 incubator, Binder GmbH, Germany). The positive electrodes consisted of NFVPS/rGO, Super C65 carbon and polyvinylidene fluoride (PVDF) (Sigma-Aldrich) at a 80:10:10 mass ratio. This mixture together with N-methyl-2-pyrrolidone was homogenized using a planetary centrifugal mixer ARE-250 CE (THINKY, Japan) for 10 min at a speed of 2000 rpm, and then the as-prepared slurry was cast onto carbon-coated aluminium foil (Goodfellow, Cambridge Ltd, UK) with a doctor blade film coater (ZAA 2600.A, Proceq SA). The electrode film having a thickness of 200 μm was dried at 120 °C overnight under vacuum. Discs of 10 mm diameter were cut, pressed and then additionally dried at 120 °C under vacuum for 1 h, and then further kept in a glove box (MBrawn Unilab Pro SP). The electrode loading was around 3 mg cm−2. A sodium metallic disc was used as a negative electrode. 1 M NaPF6 in PC was used as an electrolyte and a Whatman GF/D glass fibre as a separator. The cyclic voltammetry (CV) curves were recorded at scanning rates of 0.1 and 1 mV s−1 within the potential window between 1.5 and 5.0 V vs. Na+/Na at 20 and 40 °C. The galvanostatic tests (C/20 rate) and the long cycling experiments (C/2 rate) were also performed at the two temperatures (20 and 40 °C) over the range 1.5–4.5 V vs. Na+/Na (1C = 128 mA g−1 for 2 e− reaction). The specific capacity was calculated based on the mass of the active NFVPS phase in the electrodes. All the experiments were repeated at least twice to avoid the experimental errors and a high reproducibility was established. For ex situ analyses (XRD, EPR, XPS and optical microscopy), the electrodes were switched off at different potentials (e.g. 1.5, 3.25, 4.5 V, etc.), the electrochemical cells were disassembled inside the glove-box, and the electrodes were covered by a plastic film and then subjected to different analyses.
3. Results and discussion
3.1. Characterization of NFVPS and NFVPS/rGO composite
The two-step synthetic method enables the preparation of NFVPS composition at relatively low temperatures (i.e. at 400 °C). The atomic ratio between all elements, Fe:V:P:S, in the target product coincides well with the nominal values in the freeze-dried solution (1:1:1:2). The NFVPS compound crystallizes in the NASICON-type structure (space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) ), in agreement with the previous data of Essehli et al.27 (Fig. 1). The lattice parameters (Table S1, ESI†) are calculated using the structural model for NaFe1.6V0.4PO4(SO4)227 with a fixed Fe:V ratio of 1. The structure of NaFeVPO4(SO4)2 is built by corner shared (Fe,V)O6 octahedra and (P,S)O4 tetrahedra, thus arranging three-dimensional channels for sodium ion diffusion27 (Fig. 1(a)). The Fe3+ and V3+ ions reside randomly in the two octahedral 6c sites (at a ratio of 0.5:0.5), while P5+ and S6+ at a ratio of 0.333:0.667 are statistically distributed over one tetrahedral 18f site. Sodium ions, Na1 and Na2, occupy 3a and 3b sites, forming triangular NaO6 prisms. It should be mentioned that the lattice parameters for NFVPS slightly deviate from those reported by Essehli et al. for the same composition: a = 8.4580(1) Å, c = 22.0286(1) Å, V = 1364.74(12) Å3vs. a = 8.4019 Å, c = 22.0562 Å, V = 1.348.40 Å327 (the difference in the cell volume is 1.2%).
), in agreement with the previous data of Essehli et al.27 (Fig. 1). The lattice parameters (Table S1, ESI†) are calculated using the structural model for NaFe1.6V0.4PO4(SO4)227 with a fixed Fe:V ratio of 1. The structure of NaFeVPO4(SO4)2 is built by corner shared (Fe,V)O6 octahedra and (P,S)O4 tetrahedra, thus arranging three-dimensional channels for sodium ion diffusion27 (Fig. 1(a)). The Fe3+ and V3+ ions reside randomly in the two octahedral 6c sites (at a ratio of 0.5:0.5), while P5+ and S6+ at a ratio of 0.333:0.667 are statistically distributed over one tetrahedral 18f site. Sodium ions, Na1 and Na2, occupy 3a and 3b sites, forming triangular NaO6 prisms. It should be mentioned that the lattice parameters for NFVPS slightly deviate from those reported by Essehli et al. for the same composition: a = 8.4580(1) Å, c = 22.0286(1) Å, V = 1364.74(12) Å3vs. a = 8.4019 Å, c = 22.0562 Å, V = 1.348.40 Å327 (the difference in the cell volume is 1.2%).
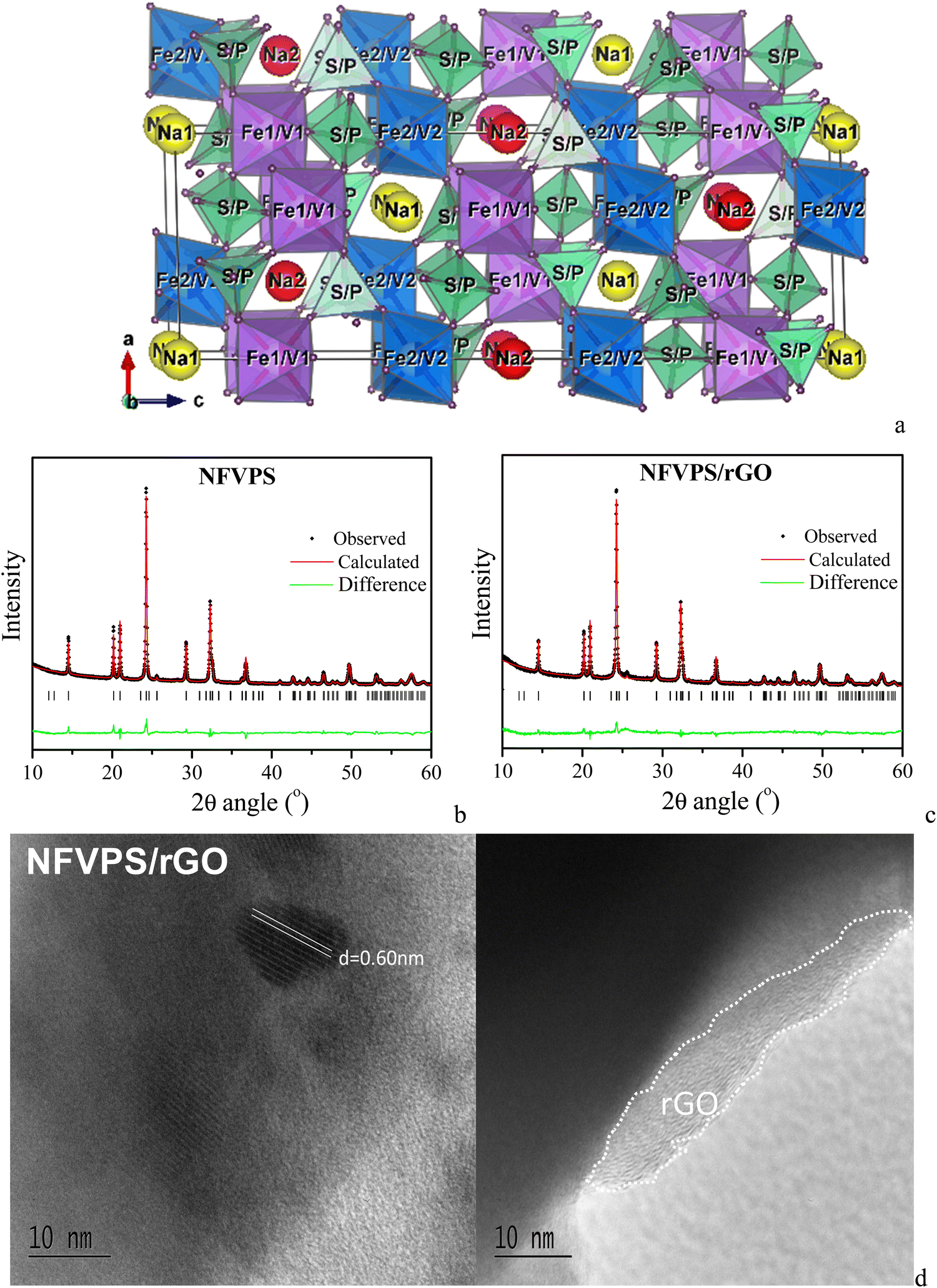 | ||
| Fig. 1 Crystal structure (a) and Rietveld refinement plots of NFVPS (b) and NFVPS/rGO (c); HR-TEM images of NFVPS/rGO (d). | ||
In the composite of NFVPS with rGO, the atomic ratio between Fe:V:P:S elements remains the same. The NFVPS component in the composite NFVPS/rGO also adopts the NASICON-type crystal structure (Fig. 1(c)). Moreover, the lattice parameters also retain their values (Table S1, ESI†). The average crystallite size of NFVPS in the composite varies around 53 nm as is the case of pure NFVPS. The crystal structure data disclose the structural stability of NFVPS during the composite formation by ball-milling mixing.
The rGO additive in the composite is detected by HR-TEM (Fig. 1(d)). The images show that the rGO additive covers the phosphate–sulfate particles, and the layer thickness varies between 5 and 10 nm (Fig. 1(d)). This gives evidence for a good interphase contact between rGO and phosphate–sulfate particles in the composite. Furthermore, well resolved lattice fringes, corresponding to the (012) plane of NFVPS (d-value of 0.60 nm), are observed. This is another evidence for the preservation of the crystal structure and crystallinity of the phosphate–sulfate phase in the composite with rGO.
Fig. 2 compares the morphology and texture peculiarities of the individual NFVPS and NFVPS/rGO composites.
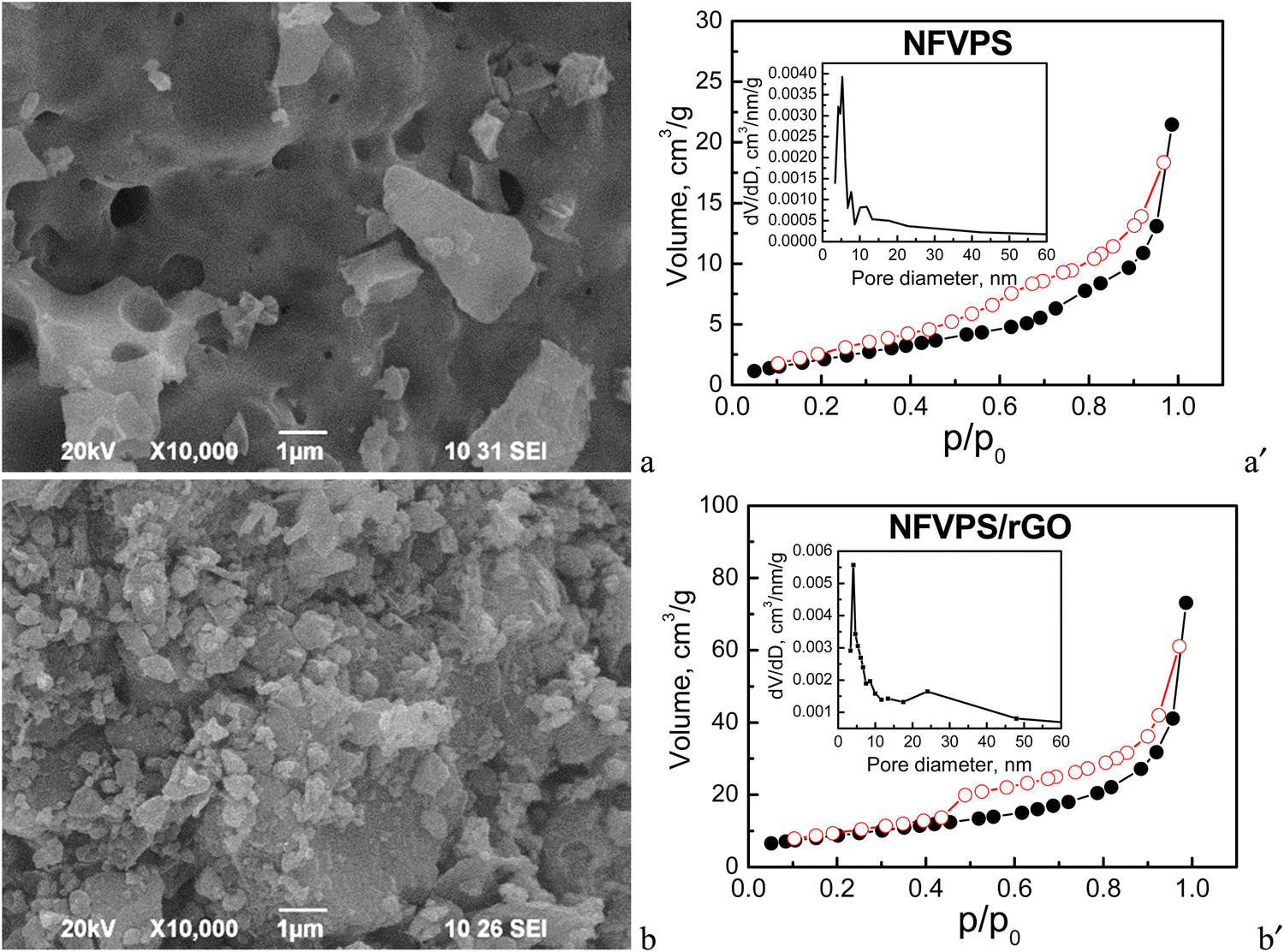 | ||
| Fig. 2 (left) SEM images and (right) nitrogen adsorption/desorption isotherms (full/open symbols) with pore size distributions (insets) of NFVPS (a, a′) and NFVPS/rGO (b, b′). | ||
The morphology of NFVPS consists of large aggregates (above 5–10 μm size) with various shapes containing big holes on their surface (Fig. 2). This is a result from the synthetic method, applied for the preparation of NFVPS: during the thermal decomposition of the freeze-dried salts several gases such as CO2, NOx and NH3 are released, generating the holes inside the aggregates. After the ball milling of NFVPS with rGO additive the aggregates break into smaller particles with dimensions below 1 μm.
Contrary to the SEM analysis, the nitrogen adsorption isotherms reveal a significant difference in the porosity of the two samples (Fig. 2). According to the adsorption/desorption isotherms NFVPS can be regarded as a mesoporous material (pseudo-type II isotherm, H3 hysteresis) with a specific surface area of 9 cm2 g−1 and comparatively narrow pore size distribution between 3 and 25 nm (average pore diameter is 15 nm), with the predominant mesopores having diameters between 3 and 9 nm. The formation of the composite with rGO leads to considerable changes in the textural characteristics with preservation of the pseudo-type II isotherm: (i) the specific surface area increases threefold up to 31 cm2 g−1; (ii) the total pore volume is enhanced four times (0.12 cm3 g−1 for NFVPS/rGO vs. 0.03 cm3 g−1 for individual NFVPS); (iii) the pore size distribution is changed from unimodal to bimodal ones in the composite (Fig. 2). These textural changes are a result of the rGO additive, which is characterized by much higher specific surface area (363 cm2 g−1) and total pore volume (1.58 cm3 g−1) previously reported by us.30 At the same time, the total pore volume of NFVPS/rGO appears to be lower than expected, considering the much larger total pore volume of pure rGO. We established a similar finding in studying the porous structure of the composite between Na4Fe3(PO4)2P2O7 and the rGO additive30 and it was explained by a partial blocking of the small mesopores of the rGO material having diameters between 3.5 and 6.5 nm by the larger Na4Fe3(PO4)2P2O7 particles.30 An analogous process could be supposed in the case of the NFVPS/rGO composite considering the crystallite sizes of the NFVPS phase (about 53 nm). All these results indicate that the rGO additive modifies effectively the texture of phosphate-based electrodes, which is of importance for their sodium storage performance.
3.2. Structural stability of NFVPS during Na+ intercalation
Fig. 3 compares the CV curves of individual NFVPS and NFVPS/rGO composites. It is worth noting that the electrode based on individual NFVPS is fabricated by a standard procedure including mixing with carbon black additive, 15 wt%. The CV curve of NFVPS contains several oxidation- and reduction-peaks between 1.5 and 5.0 V. In order to couple the oxidation peaks with reduction peaks, the CV curves are recorded keeping the lower potential limit constant (i.e. at 1.5 V) and increasing the upper voltage limit from 3.5 up to 5.0 V via 4.0 V (Fig. 3(c)). This allows identifying at least three types of redox couples which are located at 2.30/1.88 V, 3.31/2.74 V and 4.04/3.71 V. By increasing the cell temperature from 20 °C up to 40 °C the redox peaks become more pronounced and additional high-voltage peaks are observed at 4.04 and 4.51 V (Fig. 3(a)).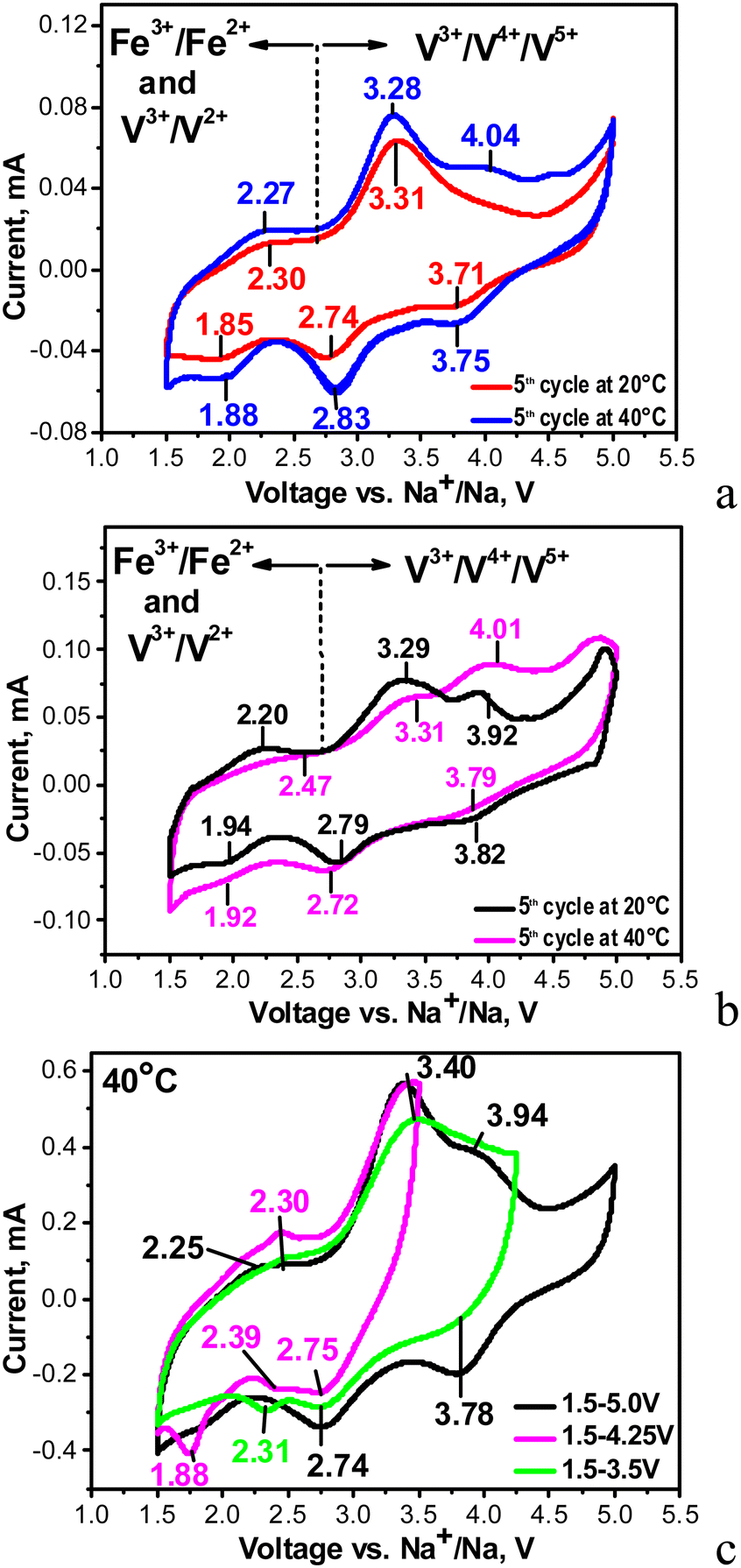 | ||
| Fig. 3 CV curves in sodium-half cells at a scanning rate of 0.1 mV s−1 for NFVPS/C (a) and NFVPS/rGO (b) at 20 and 40 °C within a voltage window of 1.5–5.0 V; CV curves at a scanning rate of 1 mV s−1 for NFVPS/C at 40 °C within different voltage windows (c). | ||
Based on previous electrochemical data for iron and vanadium phosphates,12,31–35 the low-voltage redox peaks (2.30/1.88 V) could be assigned to V3+/V2+ redox couples, while the intermediate and the high-voltage redox peaks originate from V3+/V4+ and V4+/V5+ couples. Thus, for NASICON structured compounds Na3+xFeV(PO4)3 (x = 0 and 0.4) and Na4MnV(PO4)3 the V3+/V4+ peaks are observed in the range of 3.3–3.5 V12,31,32,35 similar to the pure Na3V2(PO4)3 compound (3.4 V). In the same materials an electrochemical oxidation of V4+ to V5+ ions is reported to occur at about 4 V.12,31,32,35 The presence of the V5+ ions in the charged state at 4 V has been verified by XPS spectra.12 Regarding the iron ions, we consider that the Fe2+/Fe3+ couple operates at 3.31/2.74 V rather than at 2.30/1.88 V. This assignment is related to the inductive effect of the SO4 groups on the redox potential of Fe2+/Fe3+ in the mixed phosphate–sulfate frameworks.24,25,29,36 It is worth mentioning that in the mixed phosphate–sulfate systems the Fe2+/Fe3+ and V3+/V4+ ion couples are difficult to distinguish, in contrast to the individual phosphate systems.12,31,32 In this aspect, sodium deficient phosphate, Na3.41£0.59FeV(PO4)3, is an excellent example for consecutive oxidation of Fe2+ to Fe3+, V3+ to V4+ and V4+ to V5+ occurring at 2.55 V, 3.46 V and around 4 V during Na+ deintercalation.32 These values are close to those observed by us, especially for the high-voltage peaks associated with V3+/V4+ and V4+/V5+ couples (i.e. at 3.31 V and 4.04 V, respectively). In our case the low-voltage peak centred at 2.30 V is relatively broad and hardly visible, which gives us the reason to associate this peak mainly to the V3+/V2+ redox couple instead of the two couples V3+/V2+ and Fe3+/Fe2+. Herein, the new finding is that the elevated temperature favours the occurrence of V4+/V5+ redox processes and thus also contributes to the activation of the multi-electron reactions in NFVPS. This is an interesting observation that deserves future investigation.
In comparison with the individual NFVPS, the composite NFVPS/rGO demonstrates the same redox peaks which are associated with Fe and V multi-electron reactions (Fig. 3(b)). However, the CV curve of NFVPS/rGO brings also the electrochemical feature of the rGO additive (Fig. S1, ESI†). Between 1.5 and 5.0 V, the CV curve profile of rGO shows that it stores sodium only by a capacitive reaction, which is in contrast to NFVPS accommodating Na+ by an intercalation reaction (i.e. faradaic reaction, Fig. S1, ESI†). The comparison of the CV curve profiles for NFVPS and rGO reveals that both the intercalation and capacitive reactions are responsible for the sodium storage properties of the composite NFVPS/rGO (Fig. S1, ESI†). This is of importance for the electrochemical performance of NFVPS/rGO in sodium-ion cells. It should also be mentioned that the presence of the rGO additive in the composite results in broadening of all redox peaks in the CV curve of NFVPS/rGO in comparison with NFVPS/C (Fig. 3(a), (b) and Fig. S1, ESI†). Due to this broadening the two high-voltage peaks above 4 V observed in NFVPS/C cannot be distinguished in NFVPS/rGO and most probably, the highest-voltage peak at 4.5 V is hidden by the broad peak at 4.01 V (Fig. 3(a) and (b)).
It is worth comparing the NaFeVPO4(SO4)2/rGO electrode with the data provided by Pati and Dhaka on NaFe1.6V0.4PO4(SO4)2@CNT.28 Although NaFeVPO4(SO4)2/rGO displays CV with several redox peaks between 1.5 and 4.5 V, only one peak at 3.3/2.8 V has been detected for NaFe1.6V0.4PO4(SO4)2@CNT (the same scan rate of 0.1 mV s−1 is used in the two studies). This comparison demonstrates once again that the multi-electron reactions are responsible for the electrochemical activity of NaFeVPO4(SO4)2/rGO.
To get insight into the structural peculiarities of NFVPS during the Na+ intercalation, ex situ XRD experiments were undertaken (Fig. 4).
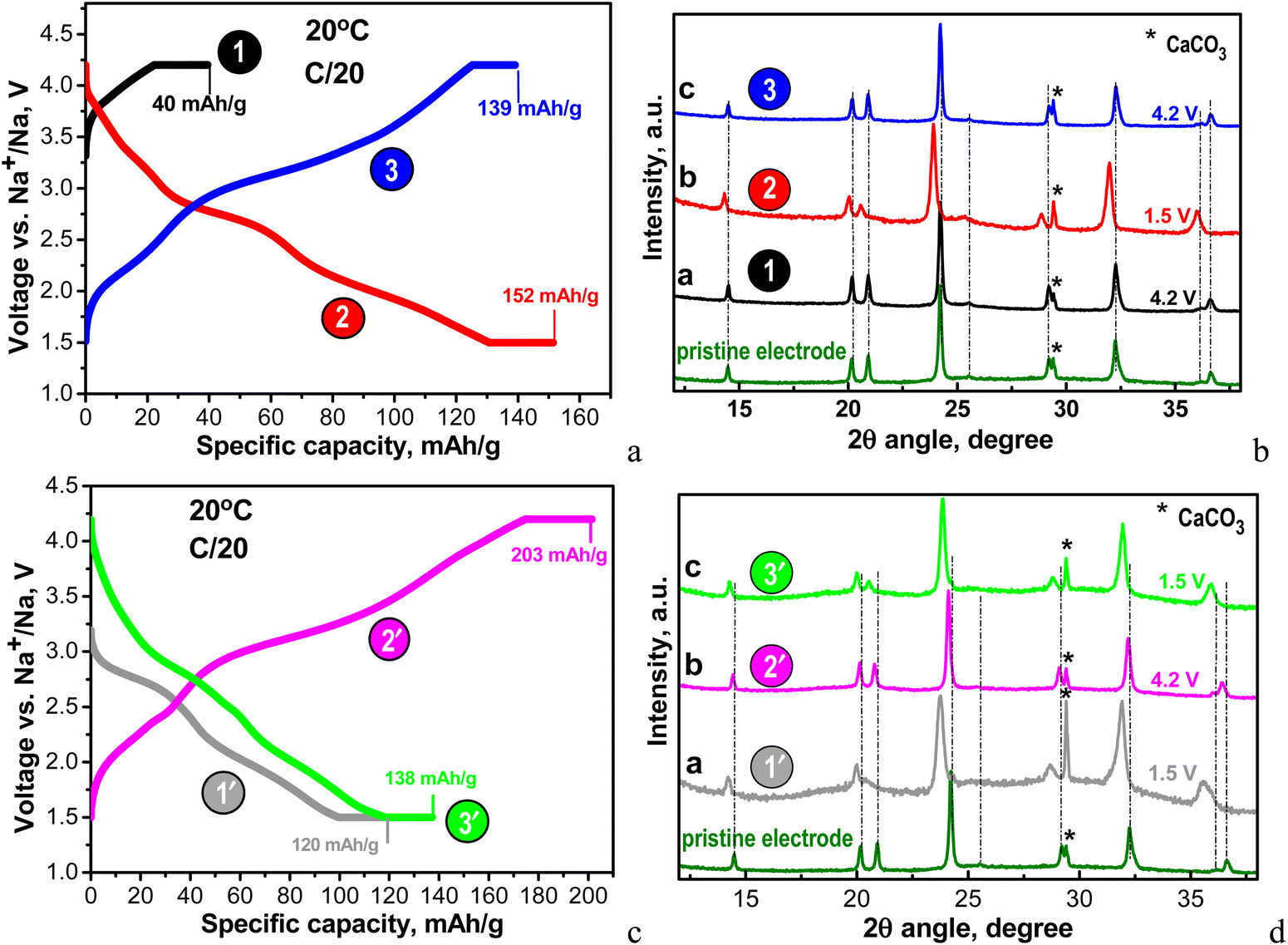 | ||
| Fig. 4 (a) Charge–discharge curves of NFVPS/rGO at 20 °C with a C/20 rate starting with a charge mode: 1st charge to 4.2 V (point 1), followed by 1st discharge to 1.5 V (point 2) and 2nd charge to 4.2 V (point 3); (b) ex situ XRD patterns of the cycled electrodes stopped at points 1, 2 and 3; (c) charge–discharge curves of NFVPS/rGO at 20 °C with C/20 rate starting with a discharge mode: 1st discharge to 1.5 V (point 1′), followed by 1st charge to 4.2 V (point 2′) and 2nd discharge to 1.5 V (point 3′); the numbers in (a) and (c) correspond to the specific capacities obtained at charged and discharged states before and after holding for 10 h. (d) Ex situ XRD patterns of the cycled electrodes stopped at points 1′, 2′ and 3′. The asterisk (*) symbol in the XRD patterns denotes the peaks due to calcite CaCO3 from plastic holder (see ESI,† Fig. S2). | ||
Because of the multi-electron redox reactions, the phosphate–sulfate electrode can operate in two manners: first, by charging the cell from OCV to 4.2 V and when keeping the voltage constant for 10 hours (curve charge 1, Fig. 4(a)) and second, by discharging the cell from OCV to 1.5 V and then keeping the voltage constant for 10 hours (discharge 1′, Fig. 4(c)). Then, the galvanostatic experiments (i.e. CC/CV) continue by subsequent cycles, with the total being 3 half-cycles. These experiments were performed at a rate of C/20 (current of 6.4 mA g−1). In Table S1 (ESI†) the Na amount in the cycled electrodes is calculated based on the capacity achieved. After the first charging, NFVPS delivers a capacity of 40 mA h g−1 (Fig. 4(a), curve 1) corresponding to the extraction of 0.6 moles of Na owing to the oxidation of the V3+ ions only, while the first discharge capacity reaches a much higher magnitude (i.e. of 120 mA h g−1, Fig. 4(c), curve 1′) corresponding to the insertion of almost 2 moles of Na, giving a composition of Na2.8FeVPO4(SO4)2 (Table S1, ESI†). The electrochemical processes during the first discharge are related to both Fe3+/Fe2+ and V3+/V2+ couples. The next two half-cycles yield specific capacities between 140 and 200 mA h g−1 depending on whether the cell begins with charging or discharging mode (Fig. 4(a) and (c)). Irrespective of the manner of the cell testing, the XRD patterns of electrodes display the same NASICON-type structure as in the case of the pristine NFVPS (Fig. 4(b) and (d)). The comparison of the XRD patterns shows small shifts in the peak positions, thus reflecting the lattice “breathing” during the Na+ intercalation. Based on the XRD patterns, the lattice parameters for charged and discharged electrodes are calculated and these are listed in Table S1 (ESI†). As one expects, the insertion of Na+ could produce a cell extension, while the Na+ extraction leads to a cell contraction. The question is what the degree of the lattice volume variation is during the Na+ intercalation.
Assuming a lack of side reactions between the electrode and electrolyte, the total specific capacity enables us to calculate the amount of inserted/deinserted sodium ions (Fig. 5(a) and Fig. S3, ESI†).
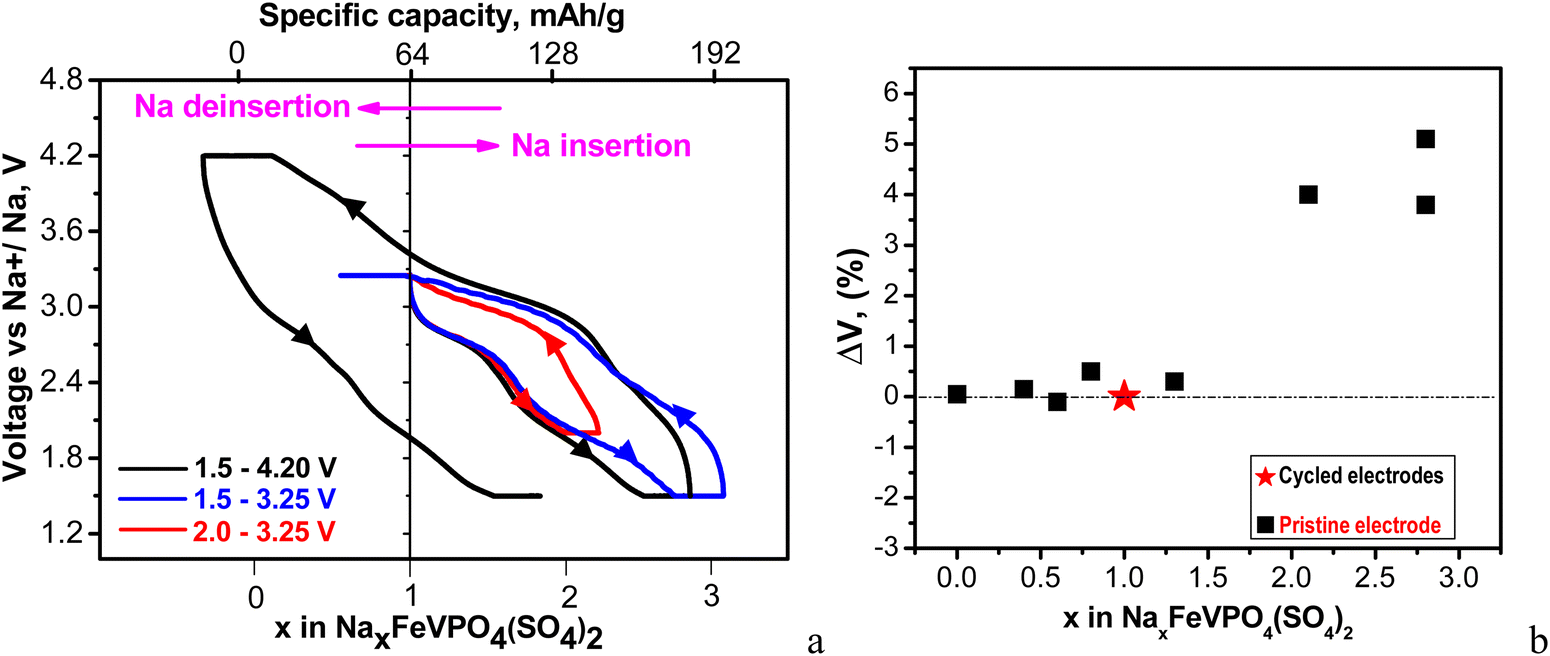 | ||
| Fig. 5 (a) First charge–discharge curves between 2.0 and 3.25 V (red curves) and between 1.5 and 3.25 V (blue curves); first charge–discharge and second discharge curves between 1.5 and 4.2 V (black curves). The cells start with a discharge mode, and they are kept at discharged and charged states for 10 h. All curves are recorded at a C/20 rate at 20 °C. The intercalation of 1 Na per formula unit corresponds to a capacity of 64 mA h g−1; (b) changes in the unit-cell volumes of cycled electrodes with respect to the pristine electrode depending on Na amount. The cells are stopped at different potentials (discharged and charged states are described in detail in Table S1, ESI†). | ||
The calculation reveals that up to 3 moles of Na+ could be intercalated into NFVPS between 1.5 and 4.2 V. It is worth mentioning that this amount of intercalated Na+ into NFVPS (i.e. 3 moles) is among the highest values found for phosphate-based electrodes (for example, 3 and 2.95 moles of Na+ in Na3V2(PO4)2F337 and Na3.41£0.59FeV(PO4)3,32 respectively). This gives evidence for the capability of NFVPS to intercalate sodium in higher amount. The amount of intercalated Na+ ions was controlled by changing the voltage limits from 4.2–1.5 V to 3.25–1.5 and 3.25–2.0 V, respectively. Within the shorter voltage range between 3.25 and 2.0 V the main working couple is Fe2+/Fe3+, whereas between 3.25 V and 1.5 V, Fe2+/Fe3+ and V2+/V3+ operate. Within the broader voltage range between 4.2 V and 1.5 V three couples Fe2+/Fe3+, V2+/V3+ and V3+/V4+ work. Thus, the amount of intercalated Na+ progressively increases from 1 to 2 and 3 moles, respectively, upon broadening of the voltage limits (Fig. 5(a) and Fig. S3, ESI†). The highly oxidized vanadium couple V4+/V5 appears to be inactive up to 4.2 V.
Fig. 5(b) shows the amount of the intercalated Na+ ions versus the lattice volume variation of the electrodes at charged and discharged states (data are given in Table S1, ESI†). The data show that the Na extraction from NaFeVPO4(SO4)2 does not lead to essential changes in the lattice volume. Depending on the Na amount in NaxFeVPO4(SO4)2 (i.e. x between 0 and 0.6) the lattice volume changes between −0.1 and +0.15% (Fig. 5(b) and Table S1, ESI†). The insertion of ≈2 moles of Na+ into NaFeVPO4(SO4)2 to a composition of Na2.8FeVPO4(SO4)2 yields a 5.1% lattice expansion (Table S1, ESI†). This means that the Na+ intercalation into NaFeVPO4(SO4)2 even in high amount is concomitant with limited overall lattice volume variation up to 5.2% (calculated as the sum of the maximal absolute values of the volume changes in the desodiated and sodiated states). It is considered that the slight change in the lattice volume during alkali ion intercalation is the main prerequisite for prolonged cycling stability of the electrodes.38 Among the intercalation electrodes for sodium ion batteries, there are few examples for electrodes with minimal lattice volume change (called electrodes with zero-strain characteristics), some of them being layered oxides with lithium dual-site-substitution,39 for example, Na4Fe7(PO4)6 with a tiny volume change of only ∼0.24%,40 nickel ferricyanide,41 Na2FeSiO4,42 and Na3V2(PO4)2F3@rGO/CNT composite.43 For mixed Fe–V phosphates and mixed phosphate–sulfate electrodes, the reported lattice volume variations are slightly higher. The insertion of 1.4 Na+ ions into the structure of NaFe2PO4(SO4)2 is found to induce a 4.6% lattice volume change.25 In the case of Na3.4VFe0.6Mn0.4(PO4)3 the lattice expansion is 5.25% (fully discharged state) and the lattice contraction is 0.37% (fully charged state).44 Hadouchi et al. reported that the extraction of about 2.16 Na+ from Na3.41£0.59FeV(PO4)3 causes a lattice volume change of 2.36%.32 The ex situ XRD data demonstrate that NaFeVPO4(SO4)2 can also be classified as a positive electrode having low-strain characteristics.
3.3. Electrochemical performance of NFVPS/rGO at elevated temperatures
The cycling stability of the NFVPS/rGO composite is monitored in sodium half-cells with CC-mode (i.e. a current of 64 mA g−1 corresponding to C/2 rate). The electrochemical experiments begin in two ways: by cell discharging and cell charging from OCV (Fig. 6(a) and (b)). | ||
| Fig. 6 Charge–discharge curves with a C/2 rate between 1.5 and 4.5 V in sodium half-cells starting with a discharge mode (a) and charge mode (b). The cells operate firstly at 20 °C (cycles from 1 to 100) and then at 40 °C (cycles from 101 to 200); cycling stability and Coulombic efficiency with a C/2 rate at 20 and 40 °C (c); (d) ex situ XRD patterns of electrodes cycled between 1.5 and 4.5 V at a C/2 rate: (a) pristine electrode; (b) discharged electrode cycled for 200 cycles (100 cycles at 20 °C and subsequent 100 cycles at 40 °C), the cell beginning with a charge mode; (c) charged electrode cycled for 200 cycles (100 cycles at 20 °C and subsequent 100 cycles at 40 °C), the cell beginning with a discharge mode; and (d) discharged electrode cycled for 100 cycles at 40 °C, the cell beginning with a charge mode. | ||
The first 100 cycles were carried out at 20 °C, and then the cell temperature was increased up to 40 °C for the next 100 cycles. The charge/discharge curves and corresponding capacities are compared in Fig. 6. In general, at fast rates the individual plateaus for the multi-electron reactions merge, giving sloping shapes between 1.5 and 4.5 V, while at slow rates well-defined plateaus are clearly observed (Fig. 4). When the cell begins with a discharge mode, Coulombic efficiency (CE) is about 93%, which increases during cycling to around 98%. In comparison, the cell starting with a charge mode displays a stable Coulombic efficiency of around 96–97%. After 100 cycles at 20 °C, the specific capacity of NFVPS/rGO reaches a magnitude of 62 mA h g−1 with a capacity retention of 66%, irrespective of the starting mode (charging or discharging). This fact indicates that the starting mode affects mainly the performance of NFVPS/rGO during the first few cycles, while during the cycling the performance becomes comparable. This suggests that during the first cycles, a surface layer resulting from the interaction of NFVPS with the electrolyte grows, and its formation is dependent on whether the experiment starts with a discharging or charging mode. During cycling, the surface layers lose their identities and then become comparable. In this aspect, the variation of Coulombic efficiency in the first cycles can be used as conditioning cycles to achieve a high CE value.
The temperature raise from 20 up to 40 °C causes the enhancement in the specific capacity (reaching a value of about 90 mA h g−1) without changing the curve profile. At the same time, the Coulombic efficiency slightly decreases down to around 93–94%. It is of importance that the capacity retention increases dramatically from 65 up to 95% going from 20 up to 40 °C. After the next 100 cycles at 40 °C, the specific capacity reaches a value of 92 mA h g−1. The improved performance of NFVPS at 40 °C can be related to an already formed surface layer at 20 °C, which appears to prevent the further interaction of the electrode with the electrolyte at 40 °C. In order to check this suggestion, we perform another experiment where the galvanostatic testing begins directly at 40 °C and all of the rest of the experimental parameters remain the same (Fig. 7).
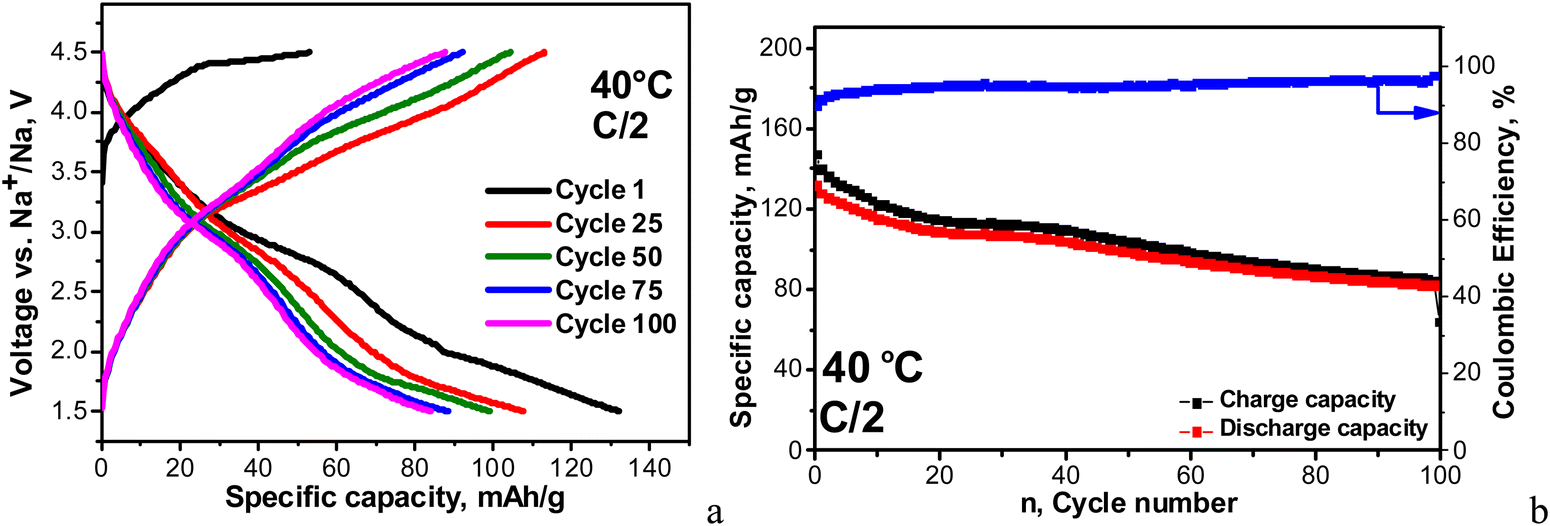 | ||
| Fig. 7 Charge–discharge curves with a C/2 rate at 40 °C between 1.5 and 4.5 V in a sodium half-cell staring with a charge mode (a). Cycling stability and Coulombic efficiency (b). | ||
In this case, the first discharge capacity increases strongly: 135 mA h g−1 for the 1st cycle at 40 °C (Fig. 7) versus 88 mA h g−1 after 101 cycles at 40 °C (i.e. immediately after the test at 20 °C, Fig. 6). In the same order, the Coulombic efficiency is lower: 84% versus 93%. However, the Coulombic efficiency increases during cycling, reaching a value of 96–97%, which is comparable with that for the long-term experiment at 20 °C, followed by 40 °C. The most important difference between the two experiments comes from the capacity retention: for the long-term experiment at 20 °C followed by 40 °C, the capacity retention varies around 95%, while for the direct cycling at 40 °C, the capacity retention decreases down to 64%. This is a next sign for the role of the surface layer in the electrochemical performance of NFVPS/rGO.
3.4. Ex situ surface analysis of NFVPS/rGO electrodes
The excellent electrochemical performance of NFVPS/rGO is related to its structural and morphological stability (Fig. S4, ESI†). After 200 cycles at 20 °C and 40 °C, the NASICON-type structure is preserved, with the lattice volume being varied no more than 1.5% (i.e. −1.2% for the charged state and +0.3% for the discharged state, Table S1, ESI†). The morphological integrity also remains intact after cycling (Fig. S4, ESI†). The parameter which undergoes changes during the cycling is the surface interaction between the electrode and electrolyte.In order to identify the chemical composition of the surface layer formed on NFVPS/rGO, ex situ XPS was undertaken. The XPS spectra of pristine NFVPS/rGO and corresponding electrodes are collected in Fig. 8 and Fig. S5 (ESI†). The XPS survey of the pristine NFVPS/rGO electrode (Fig. 8(a)) displays the specific features of NaFeVPO4(SO4)2 and the rGO components. Within the range of the Na1s binding energy, there is one band centered at 1071.7 eV, which could be assigned to Na+ ions bounded to PO43− groups.45 This corroborates with the P2p spectrum, where a band centered at 133.8 eV comes from the P element of the PO43− group.32 Within the range of binding energy for the V2p core level, there are two broad bands at 517 and 524 eV, which correspond to V2p3/2 and V2p1/2 transitions, respectively. In general, these values can be assigned to V3+ ions.46 The close inspection of the V2p3/2 band shows that this band is composed of at least two components at 516.8 eV and 517.7 eV, while V2p1/2 remains too broad that prevents distinguishing any splitting. This implies that highly oxidized V ions (i.e. V4+/V5+) in addition to V3+ are also present on the surface of the NFVPS/rGO composite.47 The XPS spectrum of S2p shows two peaks having binding energies at 169.1 eV and 170.3 eV, which are due to S2p3/2 and S2p1/2 transitions.48 Both the peak positions and the spin orbital splitting (i.e. ∼1.2 eV) are typical for metal sulfates.49 The core level spectra of Fe2p are not well resolved, which makes their clear assignment difficult (Fig. S5, ESI†).
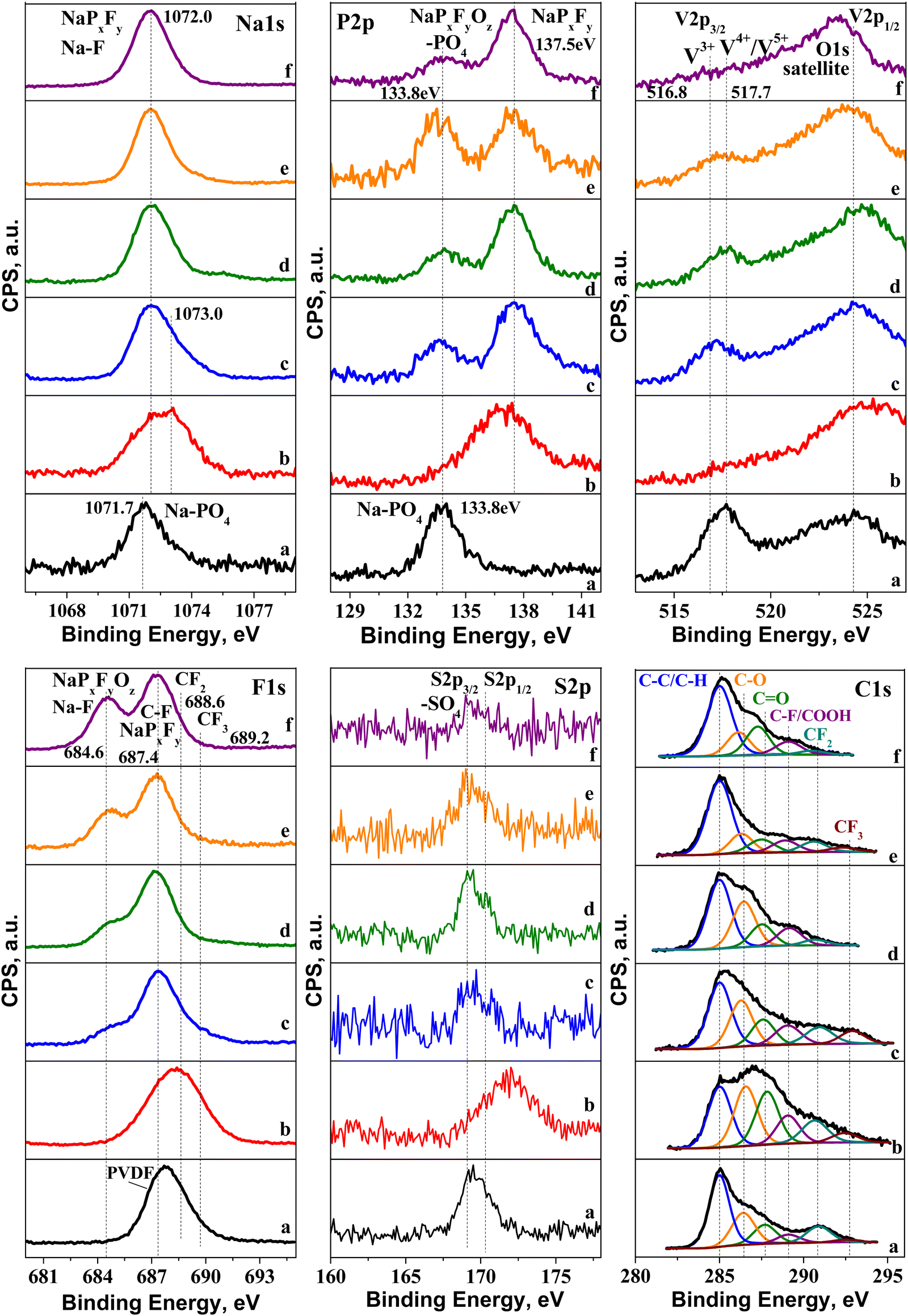 | ||
| Fig. 8 XPS spectra of electrodes cycled between 1.5 and 4.2 V and stopped at the discharged or charged state: (a) pristine electrode; (b) 1st charge to 4.2 V at 20 °C (point 1 in Fig. 4(a)); (c) 1st discharge to 1.5 V at 20 °C (point 1′ in Fig. 4(c)); (d) charged electrode after 1st discharge at 20 °C (point 2′ in Fig. 4(c)); (e) 2nd discharge at 20 °C (point 3′ in Fig. 4(c)); (f) charged electrode after 1st discharge at 40 °C. | ||
The features of the rGO component are visible within the range of the C1s binding energy: in addition to the main peak due to carbon (285 eV), there are two low intensity peaks at 286.4 and 287.7 eV coming from the functional oxygen groups, such as C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups.50
O groups.50
The highly shifted peaks (i.e. above 289 eV) are related to C atoms, bounded to 1F, 2F and 3F atoms.51 Along with this, the F1s spectrum consists of a band at 687.4 eV corresponding to F in PVDF. Given that PVDF is used as a binder, the observation of CFx-signals can be regarded as a consequence of the method of the electrode fabrication: it appears that PVDF partially reacts with rGO, yielding CFx-products.
The O1s spectrum (Fig. S5, ESI†) displays broad and asymmetric bands, where several oxygen species can be identified: oxygen in the metal lattice of NFVPS (529.8 eV), oxygen bonded to phosphates and to carbonyl groups (∼531.6 eV), COOH (∼532.1 eV) and C–O bonds (533.5 eV).52,53 The set of all XPS spectra reveal that the surface of NFVPS/rGO is composed mainly of PO43−, SO42−, Na+ and V3+ species as it is expected from the nominal composition of NaFeVPO4(SO4)2, as well as of O-based functional groups coming from the rGO component.
After the electrochemical reaction, the XPS spectra undergo different changes depending on whether the cell begins with charging or discharging mode (Fig. 8). To quantify this process, Table S2 (ESI†) shows the calculated element concentrations in the electrodes. The first charge (i.e. extraction of 0.6 mole Na+, Fig. 8(b)) yields an enhancement of the surface content of Na, P and F elements, while V and C elements decrease more than two times, and the S content remains relatively unchanged. The variations in the surface element content correlate with a change in their spectral profiles. The Na1s band is shifted from 1071.7 to around 1073 eV, which indicates a decomposition of Na–PO4 bonds and formation of Na–F and/or Na–PxFy species.54,55 In the same order, in the P2p spectrum the band due to PO43− disappears at the expense of the growth of a new band at 137.5 eV. This band can be assigned to NaPxFy species.55,56 Supporting the above assignment, the F1s signal becomes very broad. The ratios of F-to-P and F-to-Na are 6.7 and 7.4, respectively, and these values approach the stoichiometric ones for the NaPF6 salt used in sodium electrolyte. This means that NaPF6 salt is strongly adsorbed on the electrode surface without undergoing any significant decomposition. The surface film is so thick and dense (i.e. roughly up to 5 nm), thus preventing the observation of the signal due to V2p3/2.
Further information on the film composition is provided by the C1s spectrum. The comparison shows a drastic raise in the bands due to C–O and CO bonds, more than 2 times (Table S3, ESI†). This suggests that the electrolyte solvent (i.e. PC) is partially decomposed on the electrode surface, contrary to that observed for the electrolyte salt (NaPF6). The partial decomposition of PC on the electrode surface at 4.2 V can be related to the rGO component rather than NFVPS. It is recognized that the sodium electrolyte containing 1 M NaPF6 in PC is stable at high potentials with respect to the polyanionic electrodes such as Na2FePO4F.57 Thus, it appears that the O-functional groups of rGO preferentially react with PC solvent. Based on XPS studies, one can conclude that the first charge leads to a formation of a thick and dense film, composed of adsorbed NaPF6 electrolyte salt and solvent decomposition products on NFVPS/rGO.
In contrast to the first cell charge, the first cell discharge provokes the decomposition of the electrolyte salt instead of the electrolyte solvent (Fig. 8(c)). Within the range of the Na1s binding energy, the band becomes broad and asymmetric. However, it is possible to distinguish at least two overlapped bands: the band at 1071.7 corresponds to Na atoms bounded to PO43− (as in the pristine electrode), while the band at 1073 eV can be assigned taking into account the P2p and F1s spectra. The low-energy band at 133.8 eV is due both to PO43− groups and to oxidized PxFyOz species.58 In addition, the P2p spectrum displays a high-energy band at 137.5 eV assigned to NaPxFy species. The F1s spectrum supports the presence of PxFyOz species by the band at 684.6 eV.59 The latter band is also associated with F bonded to Na.59 The deposition of NaF can be related to the strong increase in the total Na amount (more than one order, Table S2, ESI†). On the other hand, the ratios of F-to-P and F-to-Na (4.2 and 1.3, respectively) are smaller than that expected for NaPF6 salt, which is a next indication for the deposition of oxidized PxFyOz species on the surface of NFVPS/rGO. Based on Na1s, F1s and P2p spectra, it is found that the electrolyte NaPF6 salt decomposes on the electrode surface, forming mainly NaF and PxFyOz products. Unlike PO43− groups, the SO4 groups remain unchanged after the electrochemical reaction. Here, the surface film is not so dense, so that the signals due to the pristine PO43−, V3+ and SO42− species are still visible on the electrode surface (Fig. 8(c)). Contrary to the electrolyte salt, the electrolyte solvent remains inert towards the electrode surface: the bands due to C–O and CO bonds are well visible and their ratios to the main C signal remain relatively similar to that of the pristine electrode (Fig. 8 and Table S3, ESI†). Thus, one can conclude that after the first cell discharge, the electrolyte salt reacts with the surface of NFVPS/rGO, leading to the deposition of a non-dense film containing NaF and PxFyOz products.
After the first discharge, the next charge and discharge half-cycles (Fig. 8(d) and (e)) produce the densification of the surface film, which is established through the decrease in the surface content of V and S (Table S2, ESI†). The F1s peak, due to NaF, grows in intensity after each half-cycle, thus indicating an increasing amount of deposed NaF inside the surface film. However, the total Na content changes depending on whether charging or discharging is applied: after the cell charging the total Na content decreases in respect to that found after the first discharge (Fig. 8(c), (d) and Table S2, ESI†), while after the second discharge (i.e. 3 half-cycles) the Na content increases (Fig. 8(c)–(e) and Table S2, ESI†). This can be related to a transformation of the surface composition leading to a deposition of NaF after each half-cycle.
After the cell discharging and next charging at 40 °C (Fig. 8(f)), the XPS spectra in all element energy regions bring the same features as those observed for the electrodes cycled at 20 °C. However, the close comparison of the spectra evidences an increase in the NaF content, concomitant with a disappearance of signals due to vanadium. This means that at elevated temperatures the surface film is denser than that at 20 °C.
The EPR spectroscopy in addition to the XPS method aims to access paramagnetic V and Fe ions (Fig. 9).
 | ||
| Fig. 9 (left) EPR spectra at 100 K of short-cycled electrodes (up to 3 half-cycles) between 1.5 and 4.2 V and stopped at the discharged or charged state: (a) pristine electrode; (b) 1st charge to 4.2 V at 20 °C (point 1 in Fig. 4(a)); (c) 1st discharge to 1.5 V at 20 °C (point 1′ in Fig. 4(c)); (d) charged electrode after 1st discharge at 20 °C (point 2′ in Fig. 4(c)); (e) 2nd discharge at 20 °C (point 3′ in Fig. 4(c)); (f) charged electrode after 1st discharge at 40 °C. (right) EPR spectra at 100 K of long-term cycled electrodes between 1.5 and 4.2 V and stopped at the discharged or charged state: (a) pristine electrode; (g) step-wise cycled electrode (20 and 40 °C for 200 cycles in total) stopped at the charged state; (h) step-wise cycled electrode (20 and 40 °C for 200 cycles in total) stopped at the discharged state; (i) electrode directly cycled at 40 °C for 100 cycles and stopped at the discharged state. | ||
The EPR spectrum of pristine NFVPS/rGO (Fig. 9(a)) is dominated by a narrow signal with a g-value of 2.003. This signal is assigned to paramagnetic defects in the carbon network, which, in turn, is associated with C65 binder or/and rGO.60 The EPR signal from NFVPS is not registered due to the availability of non-identical paramagnetic V3+ (spin state of S = 1) and Fe3+ (spin state of S = 5/2) ions, which are coupled between themselves by exchange interactions. After the first cell charge (i.e. after deinsertion of 0.6 moles Na+), two overlapped signals contribute to the EPR spectrum of NFVPS/rGO: a multiplet signal having a simulated g-tensor of g‖ = 1.930 and gc = 1.975 and hyperfine structure of A‖ = 200 G and A⊥ = 80 G (Fig. 9(b) and Fig. S6, ESI†) as well as a single line having a g-value of 2.004 and line width of 50 mT. Based on previous EPR studies on V-compounds the multiplet signal can be assigned to VO2+ ions (Table S4, ESI† and references therein). The observed g-values and the hyperfine structure allow the identification of the coordination of VO2+ ions, namely, they are coordinated by F ions, forming complexes such as VOF5 and/or VOF4.61,62 This means that the VO2+-complexes are outside the structure of NFVPS and they are formed as a result of side reactions of the electrode–electrolyte interaction. Given that the VO2+-complexes are detectable only by EPR and they remain undetectable by XPS, it appears that VO2+-complexes are located in the deeper surface layers. The single EPR signal could be attributed to Fe3+ ions. Contrary to VO2+-complexes, Fe3+ ions reside, most probably, in the structure of the charged Na0.4FeVPO4(SO4)2 composition. On the other hand, the observation of Fe3+ ions in charged NFVPS can be considered as an indirect sign for a complete oxidation of paramagnetic V3+ to diamagnetic V5+ ions, which magnetically dilute the identical paramagnetic Fe3+ ions.
When the cell begins with a discharge mode (i.e. after insertion of 1.8 Na+), there are no signals due to VO2+-based complexes and Fe3+ ions (Fig. 9(c)). This provides indirect evidence for a reduction of V3+ and Fe3+ to V2+ and Fe2+ ions. Both V2+ (spin state of S = 3/2) and Fe2+ (spin state of S = 2) are also paramagnetic and they are responsible for the lack of EPR signals due to the exchange interactions between them. The next charging half-cycle (Fig. 9(d)) produces a growth in the intensities of signals due to VO2+ and Fe3+ ions, thus confirming once again the ion oxidation at 4.2 V together with a formation of VO2+-complexes. This process is observed at 20 and at 40 °C (Fig. 9(d) and (f)). After the subsequent discharging half-cycle (Fig. 9(e)), the signal due to Fe3+ disappears as one could expect, but the signal due to VO2+ is still visible. This discloses the fact that the formation of the VO2+-complexes is an irreversible process.
In order to analyse cycle-induced changes in materials, the EPR spectra of the long-term cycled electrodes at 20 °C and at 40 °C are given in Fig. 9 (right). The analysis of EPR spectra of these long-term electrodes is based on the data for the short-term cycled charged and discharged NFVPS/rGO electrodes (i.e. up to one half-cycle, Fig. 9(b) and (c)) used as references. For NFVPS/rGO cycled 100 cycles at 20 °C, followed by next 100 cycles at 40 °C and switched off at 1.5 V (i.e. at a discharged state), the EPR spectrum displays only the signal due to the VO2+-complexes (Fig. 9(h)). When the same electrode is switched off at 4.2 V (i.e. at a charged state), both signals due to VO2+-complexes and Fe3+ ions are clearly distinguished (Fig. 9(g)). This implies that the redox reaction with the Fe2+/Fe3+ couple is a reversible process, while VO2+-complexes are irreversibly formed.
It is interesting to compare the EPR spectra of the long-term cycled electrodes after step-wise cycling at 20 °C and then at 40 °C with a long-term cycled electrode cycled directly at 40 °C (Fig. 9(h) and (i)). Both electrodes are switched off at 1.5 V (i.e. they are in a discharged state). The comparison shows that the EPR spectrum of NFVPS/rGO cycled directly at 40 °C contains signals due to VO2+-complexes and Fe3+ ions (Fig. 9(i)), which is in contrast to NFVPS/rGO cycled step-wise at 20 and 40 °C, where only the signal due to VO2+-complexes is detected (Fig. 9(h)). This evidences a partial irreversibility of the redox reaction of Fe3+/Fe2+ couples when it proceeds directly at 40 °C. On the other hand, this observation can be related to the role of the surface layer in the electrochemical performance of NFVPS/rGO (Fig. 6 and 7): the cycling stability of NFVPS/rGO becomes worse, when the reaction is carried out directly at 40 °C.
3.5. Electrochemical performance of NFVPS/rGO electrodes, compared to other mixed Fe–V phosphates
To highlight the electrochemical performance of NFVPS/rGO, Table 1 summarizes the available electrochemical data for other mixed Fe–V phosphates, used as electrodes in sodium half-cells.| Electrode | Theoretical capacity (mA h g−1) | Electrolyte | Voltage window vs. Na+/Na (V) | Discharge capacity (mA h g−1) | Cycles number | Capacity retention (%) |
|---|---|---|---|---|---|---|
| a These data are estimated from Fig. 2(g) in ref. 28. | ||||||
| NaFeVPO4(SO4)2/rGO [present work] | 128 for 2 e− reaction | 1 M NaPF6 in PC | 1.5–4.5 | |||
| – Start with charge, 20 °C | 90 at C/2 | 100 | 67 | |||
| 40 °C | 88 at C/2 | 100 | 99 | |||
| – Start with discharge, 20 °C | 96 at C/2 | 100 | 65 | |||
| 40 °C | 98 at C/2 | 100 | 93 | |||
| NaFe1.6V0.4PO4(SO4)227 | 128 for 2 e− reaction | 1 M NaPF6 in EC/PC | 1.5–4.5 | ≈90 at C/10 | 50 | 96 |
| NaFeVPO4(SO4)227 | 128 for 2 e− reaction | 1 M NaPF6 in EC/PC | 1.5–4.5 | 65 at C/10 | 50 | 58 |
| NaFe1.6V0.4PO4(SO4)2@CNT28 | 102 for 1.6 e− reaction | 1 M NaPF6 in EC/DEC | 1.5–4.5 | 88.3 at C/2 | 6 cycles | |
| ≈80 mA h g−1 at 2Ca | 1000 | 48 | ||||
| ≈65 mA h h−1 at 5Ca | 1000 | 53 | ||||
| ≈ 55 mA h g−1 at 10Ca | 1000 | 57 | ||||
| 82.2 mA h g−1 at 5C at 45 °C | 100 | 80 | ||||
| NaFe2PO4(SO4)2@rGO26 | 127 | 1 M NaPF6 in PC + FEC | 2.0–4.4 | 65 mA h g−1 at 50 mA g−1 (C/2.5) | 300 | 59.9 |
| NaFe2PO4(SO4)226 | 127 | 1 M NaPF6 in PC + FEC | 2.0–4.4 | 50 mA h g−1 at 50 mA g−1 (C/2.5) | 100 | 34.7 |
| NaFe2PO4(SO4)225 | 127 | 1 M NaPF6 in PC + FEC | 2.0–4.5 | 56 at C/5 | 30 | 96 |
| Na3Fe2PO4(SO4)229 | — | 1 M NaPF6 in EC/PC | 1.5–4.2 | 92 at C/10 | 500 | ≈60 |
| Na3FeV(PO4)312 | 115.8 | 1 M NaClO4 in PC/FEC | 2.0–3.8 | 103 at 1C | 1000 | 95 |
| Na4FeV(PO4)331 (electrochemical sodiation of Na3FeV(PO4)3) | 58.2 for 1 e− reaction | 1 M NaPF6 in EC/DMC + 2% FEC. | 1.3–4.3 | 137.8 at C/20 | 20 | 78 |
| Na3.41£0.59FeV(PO4)332 | — | 1 M NaClO4 in PC + 5% FEC | 2.0–3.8 | 108 at 5C | 300 | 99.4 |
| 99 at 10C | 700 | 94.7 | ||||
| 81.4 at 20C | 400 | 98.4 | ||||
At similar rates of charging, the comparison reveals that NFVPS/rGO outperforms the analogue NaFe2PO4(SO4)2@rGO26 and it is among the best performing phosphates such as Na3.41£0.59FeV(PO4)3.32 Recent data demonstrate that the NaFe1.6V0.4PO4(SO4)2@CNT exhibits a good performance at high rate of charging, delivering a specific capacity of ≈25 mA h g−1 at 5C and ≈ 20 mA h h−1 at 10 C after 2000 cycles (data are estimated based on Fig. 2(g) in ref. 28).
At intermediate charging rate (C/2), NFVPS/rGO studied by us exhibits a specific capacity which is slightly higher than that for NaFe1.6V0.4PO4(SO4)2@CNT: 120 mA h g−1versus 88.3 after 6 cycles.28 Irrespective of the significance of the electrode performance at elevated temperatures, there are few data on phosphate-based electrodes in the current literature (see Table 1). This study reveals that the capacity retention of NFVPS/rGO is dramatically enhanced at 40 °C, when the first cycles are performed at 20 °C: the capacity retention is above 93% for step-wise cycling versus 64% for direct cycling at 40 °C. The worse cycling stability for direct cycling at 45 °C is also observed for NaFe1.6V0.4PO4(SO4)2@CNT (i.e. 80% capacity retention at 45 °C after 100 cycles,27Table 1). However, the comparison should be carefully taken into account since the electrolyte composition and voltage windows used for long-term electrochemical testing are quite different (Table 1). Moreover, our studies clearly demonstrate a dependence of the storage performance of NFVPS/rGO on its interactions with the electrolyte. Thus, we expect to achieve further improvement in the electrochemical performance of NFVPS/rGO when we replace the standard electrolyte, 1 M NaPF6 in PC, with some more advanced ones, as well as to use SEI-forming additives in electrolytes.
For practical application of the electrode materials for sodium ion batteries the achievement of high energy density is one of the main criteria. In general, for Fe and V based phosphate electrodes the theoretical energy density varies in the range of 300–420 W h kg−1.63,64 For instance, for Na3Fe2(PO4)3 and Na3V2(PO4)3 the energy densities are 326 and 400 W h kg−1, respectively.64,65 For NaFeVPO4(SO4)2 the energy density calculated at C/2 rate (mean operating voltage is about 3 V) is 384 W h kg−1 and this value is reasonable for practical application which is another advantage of this electrode material.
4. Conclusions
A simple and reproducible synthetic method was elaborated for the preparation of a robust mixed polyanionic electrode for sodium-ion batteries. The method includes two steps: first, the preparation of NaFeVPO4(SO4)2 through thermal decomposition at 400 °C of freeze-dried solutions containing NaH2PO4, Fe(NO3)3·9H2O, NH4VO3 and (NH4)2SO4 and then the formation of composites between NaFeVPO4(SO4)2 and rGO additive by ball-milling mixing. This method enables covering the phosphate–sulfate particles by rGO, and the thickness of the covering varies between 5 and 10 nm. The rGO plays an effective role in increasing the specific surface area and the total pore volume more than threefold.The composite NFVPS/rGO stores Na+ by faradaic (i.e. intercalation) and capacitive reactions. The intercalation of Na+ into NaFeVPO4(SO4)2 proceeds owing to the multiple redox couples of iron and vanadium: between 1.5 and 3.0 V mainly V2+/V3+ participates in the charge compensation of Na+ in NaxFeVPO4(SO4)2, while between 3.0 V and 4.5 V the iron and highly oxidized vanadium ions (Fe3+/Fe2+, V3+/V4+ and V4+/V5+) are responsible for the intercalation process. Thus, NaxFeVPO4(SO4)2 is capable of reversibly intercalating a total of 3 moles of Na+ (which corresponds to a specific capacity of 175 mA h g−1 in a potential range of 1.5–4.2 V due to the operation of all Fe and V couples). Irrespective of the high amount of intercalated Na+, the lattice volume of NaxFeVPO4(SO4)2 undergoes a limited variation (up to 5.2%), which allows classifying it as a low-strain material. The capacitive reaction of the composite NFVPS/rGO is associated with the rGO component.
The electrochemical performance correlates with the structural robustness of NaFeVPO4(SO4)2 and its interaction with the electrolyte. Through ex situ XPS and EPR studies, surface layers on NFVPS/rGO are detected and it is established that their composition and thickness depend on the voltage applied and on the cell temperature. After the first cell charging up to 4.2 V, NaPF6 electrolyte salt is adsorbed on the surface of NFVPS/rGO together with the electrolyte solvent decomposition. In deeper surface layers, VO2+-complexes are formed. In contrast to it, after the first cell discharging down to 1.5 V, the electrolyte salt, NaPF6, decomposes, forming mainly NaF and PxFyOz products on the electrode surface. The subsequent charge and discharge half-cycles lead to unification of the surface layer composition on NFVPS/rGO. When the electrochemical reaction proceeds at 40 °C, a denser surface film is formed in comparison with that formed at 20 °C. The effect of the surface layer formation on the electrochemical performance is a new finding in mixed polyanionic compounds, which deserves further investigation.
The storage performance of NFVPS/rGO is among the best performing Fe–V phosphates. After 100 cycles at 20 °C under a current load of 64 mA g−1, NFVPS/rGO delivers a specific capacity of 62 mA h g−1 with a capacity retention of 66%, irrespective of the starting mode of charging or discharging. Upon temperature raise from 20 to 40 °C, in the next 100 cycles, it shows an increase in the specific capacity to about 92 mA h g−1 and a capacity retention to 95%. The improved performance of NFVPS at 40 °C is associated with an already formed surface layer at 20 °C, which prevents further interaction of the electrode surface with the electrolyte. Given the role of the surface layer in the performance of NFVPS/rGO, further improvement in the cycling stability at elevated temperatures could be achieved by replacement of the standard electrolyte (1 M NaPF6 in PC) with some advanced ones, as well as by using SEI-forming additives in the electrolytes.
Data availability statements
The data supporting this article have been included as part of the ESI.† Software used and code: (a) OriginPro 8.6 (64 bit) Sr3, b99 (Academic). (b) EC-Lab Software, Biologic. (c) ESCAppe™1.2.0.1325, Kratos Analytical A Shimadzu.Conflicts of interest
The authors declare no competing interest.Acknowledgements
The authors thank to the financial support of project “Master“ (KΠ-06-DO02/3 dated 18.05.2023) and project “National center of excellence mechatronics and clean technologies“ (BG16RFPR002-1.014-0006) under “Research Innovation and Digitization for Smart Transformation” program 2021–2027. The authors are sincerely grateful to Robert Feher from Graphit Kropfmühl GmbH (Hauzenberg, Germany) for providing rGO additive.References
- B. L. Ellis and L. F. Nazar, Curr. Opin. Solid State Mater. Sci., 2012, 16, 168, DOI:10.1016/j.cossms.2012.04.002.
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114, 11636, DOI:10.1021/cr500192f.
- P. Gupta, S. Pushpakanth, M. Ali Haider and S. Basu, ACS Omega, 2022, 7, 5605, DOI:10.1021/acsomega.1c05794.
- J.-Y. Hwang, S.-T. Myung and Y.-K. Sun, Chem. Soc. Rev., 2017, 46, 3529, 10.1039/C6CS00776G.
- V. Koleva, M. Kalapsazova, D. Marinova, S. Harizanova and R. Stoyanova, ChemSusChem, 2023, 16, e202201442, DOI:10.1002/cssc.202201442.
- N. Nitta, F. Wu, J. T. Lee and G. Yushin, Mater. Today, 2015, 18, 252, DOI:10.1016/j.mattod.2014.10.040.
- R. J. Clément, P. G. Bruce and C. P. Grey, J. Electrochem. Soc., 2015, 162, A2589, DOI:10.1149/2.0201514jes.
- S. Kumakura, Y. Tahara, K. Kubota, K. Chihara and S. Komaba, Angew. Chem., Int. Ed., 2016, 55(41), 12760, DOI:10.1002/anie.201606415.
- F. Wu, C. Zhao, S. Chen, Y. Lu, Y. Hou, Y.-S. Hu, J. Maier and Y. Yu, Mater. Today, 2018, 21, 960, DOI:10.1016/j.mattod.2018.03.004.
- P. Barpanda, L. Lander, S. Nishimura and A. Yamada, Adv. Energy Mater., 2018, 8(17), 1703055, DOI:10.1002/aenm.201703055.
- T. Jin, H. Li, K. Zhu, P.-F. Wang, P. Liu and L. Jiao, Chem. Soc. Rev., 2020, 49, 2342, 10.1039/C9CS00846B.
- W. Zhou, L. Xue, X. Lu, H. Gao, Y. Li, S. Xin, G. Fu, Z. Cui, Y. Zhu and J. B. Goodenough, Nano Lett., 2016, 16, 7836, DOI:10.1021/acs.nanolett.6b04044.
- W. Zhang, Z. Zhang, H. Li, D. Wang, T. Wang, X. Sun, J. Zheng and Y. Lai, ACS Appl. Mater. Interfaces, 2019, 11, 35746, DOI:10.1021/acsami.9b12214.
- M. J. Aragón, P. Lavela, G. Ortiz and J. L. Tirado, ChemElectroChem, 2015, 2, 995, DOI:10.1002/celc.201500052.
- R. Liu, G. Xu, Q. Li, S. Zheng, G. Zheng, Z. Gong, Y. Li, E. Kruskop, R. Fu, Z. Chen, K. Amine and Y. Yang, ACS Appl. Mater. Interfaces, 2017, 9, 43632, DOI:10.1021/acsami.7b13018.
- P. Lavela, R. Klee and J. L. Tirado, J. Power Sources, 2021, 495, 229811, DOI:10.1016/j.jpowsour.2021.229811.
- A. K. Padhi, V. Manivannan and J. B. Goodenough, J. Electrochem. Soc., 1998, 145, 1518, DOI:10.1149/1.1838513.
- B. Senthilkumar, C. Murugesan, L. Sharma, S. Lochab and P. Barpanda, Small Methods, 2018, 1800253, DOI:10.1002/smtd.201800253.
- A. Zhao, Y. Fang, X. Ai, H. Yang and Y. Cao, J. Energy Chem., 2021, 60, 635, DOI:10.1016/j.jechem.2021.01.014.
- C. Pérez-Vicente and R. Alcántara, Phys. Chem. Chem. Phys., 2023, 25, 15600, 10.1039/D2CP06044B.
- M.-Y. Wang, J.-Z. Guo, Z.-W. Wang, Z.-Y. Gu, X.-J. Nie, X. Yang and X.-L. Wu, Small, 2020, 1907645, DOI:10.1002/smll.201907645.
- S. Geng, Y. Yang, Y. Zhang, W. Ding, X. Wang, H. Peng and Z. Bakenov, Electrochim. Acta, 2015, 176, 327, DOI:10.1016/j.electacta.2015.06.047.
- C. Masquelier and L. Croguennec, Chem. Rev., 2013, 113, 6552, DOI:10.1021/cr3001862.
- K. Shiva, P. Singh, W. Zhou and J. B. Goodenough, Energy Environ. Sci., 2016, 9, 3103, 10.1039/C6EE01093H.
- H. B. Yahia, R. Essehli, R. Amin, K. Boulahya, T. Okumura and I. Belharouak, J. Power Sources, 2018, 382, 144, DOI:10.1016/j.jpowsour.2018.02.021.
- S.-F. Li, X.-K. Hou, Z.-Y. Gu, Y.-F. Meng, C.-D. Zhao, H.-X. Zhang and X.-L. Wu, New J. Chem., 2021, 45, 4854, 10.1039/d1nj00262g.
- R. Essehli, A. Alkhateeb, A. Mahmoud, F. Boschini, H. Ben Yahia, R. Amin and I. Belharouak, J. Power Sources, 2020, 469, 228417, DOI:10.1016/j.jpowsour.2020.228417.
- J. Pati and R. S. Dhaka, J. Power Sources, 2024, 609, 234646, DOI:10.1016/j.jpowsour.2024.234646.
- S. Kumar, R. Ranjeeth, N. K. Mishra, R. Prakash and P. Singh, Dalton Trans., 2022, 51, 5834, 10.1039/D2DT00780K.
- S. Harizanova, T. Tushev, V. Koleva and R. Stoyanova, Materials, 2023, 16, 6546, DOI:10.3390/ma16196546.
- S. Park, J.-N. Chotard, D. Carlier, I. Moog, M. Courty, M. Duttine, F. Fauth, A. Iadecola, L. Croguennec and C. Masquelier, Chem. Mater., 2021, 33, 5355, DOI:10.1021/acs.chemmater.1c01457.
- M. Hadouchi, N. Yaqoob, P. Kaghazchi, M. Tang, J. Liu, P. Sang, Y. Fu, Y. Huang and J. Ma, Energy Storage Mater., 2021, 35, 192, DOI:10.1016/j.ensm.2020.11.010.
- G. Li, D. Jiang, H. Wang, X. Lan, H. Zhong and Y. Jiang, J. Power Sources, 2014, 265, 325, DOI:10.1016/j.jpowsour.2014.04.054.
- S. Mirza, Z. Song, H. Zhang, A. Hussain, H. Zhang and X. Li, J. Mater. Chem. A, 2020, 8, 23368, 10.1039/D0TA08186H.
- F. Chen, V. M. Kovrugin, R. David, O. Mentré, F. Fauth, J.-N. Chotard and C. Masquelier, Small Methods, 2018, 3, 1800218, DOI:10.1002/smtd.201800218.
- J. Lu, S. Nishimura and A. Yamada, Chem. Mater., 2017, 29, 3597, DOI:10.1021/acs.chemmater.7b00226.
- G. Yan, S. Mariyappan, G. Rousse, Q. Jacquet, M. Deschamps, R. David, B. Mirvaux, J. W. Freeland and J.-M. Tarascon, Nat. Commun., 2019, 10, 585, DOI:10.1038/s41467-019-08359-y.
- P. Yan, J. Zheng, M. Gu, J. Xiao, J.-G. Zhang and C.-M. Wang, Nat. Commun., 2017, 8, 14101, DOI:10.1038/ncomms14101.
- Z. Wu, Y. Ni, S. Tan, E. Hu, L. He, J. Liu, M. Hou, P. Jiao, K. Zhang, F. Cheng and J. Chen, J. Am. Chem. Soc., 2023, 145, 9596, DOI:10.1021/jacs.3c00117.
- X. Pu, C. Rong, S. Tang, H. Wang, S. Cao, Y. Ding, Y. Cao and Z. Chen Zero-strain, Chem. Commun., 2019, 55, 9043, 10.1039/C9CC04136B.
- Y. You, X.-L. Wu, Y.-X. Yin and Y.-G. Guo, J. Mater. Chem. A, 2013, 1, 14061, 10.1039/C3TA13223D.
- S. Li, J. Guo, Z. Ye, X. Zhao, S. G. Wu, J.-X. Mi, C.-Z. Wang, Z. Gong, M. J. McDonald, Z. Zhu, K.-M. Ho and Y. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 17233, DOI:10.1021/acsami.6b03969.
- C. Shi, J. Xu, T. Tao, X. Lu, G. Liu, F. Xie, S. Wu, Y. Wu and Z. Sun, Small Methods, 2024, 8, 2301277, DOI:10.1002/smtd.202301277.
- P. Lavela, R. Klee, M. A. Hidalgo and J. L. Tirado, J. Electroanal. Chem., 2021, 895, 115533, DOI:10.1016/j.jelechem.2021.115533.
- M. Xu, F. Zhang, Y. Zhang, C. Wu, X. Zhou, X. Ai and J. Qian, Chem. Sci., 2023, 14, 12570M, 10.1039/D3SC03498D.
- M. J. Aragón, P. Lavela, G. F. Ortiz and J. L. Tirado, J. Electrochem. Soc., 2015, 162, A3077, DOI:10.1149/2.0151502jes.
- C. Wu, J. Tong, J. Gao, J. Li, X. Li, J. Zhu, M. Gu and W. Zhou, ACS Appl. Energy Mater., 2021, 4, 1120, DOI:10.1021/acsaem.0c02203.
- V. Sharova, A. Moretti, T. Diemant, A. Varzi, R. Behm and S. Passerini, J. Power Sources, 2018, 375, 43, DOI:10.1016/j.jpowsour.2017.11.045.
- M. A. Naghmash and M. M. Ibrahim, Mater. Chem. Phys., 2022, 283, 126036, DOI:10.1016/j.matchemphys.2022.126036.
- Y. Wu, X. Feng, M. Yang, C.-Z. Zhao, X. Liu, D. Ren, Z. Ma, L. Lu, L. Wang, G.-L. Xu, X. He, K. Amine and M. Ouyang, Adv. Sci., 2022, 9, 2204059, DOI:10.1002/advs.202204059.
- N. Schulz, R. Hausbrand, C. Wittich, L. Dimesso and W. Jaegermann, J. Electrochem. Soc., 2018, 165, A833, DOI:10.1149/2.0881803jes.
- S. Park, S. Y. Jeong, T. K. Lee, M. W. Park, H. Y. Lim, J. Sung, J. Cho, S. K. Kwak, S. Y. Hong and N.-S. Choi, Nat. Commun., 2021, 12, 838, DOI:10.1038/s41467-021-21106-6.
- A. Moretti, D. V. Carvalho, N. Ehteshami, E. Paillard, W. Porcher, D. Brun-Buisson, J.-B. Ducros, I. de Meatza, A. Eguia-Barrio, K. Trad and S. Passerini, Batteries, 2019, 5, 45, DOI:10.3390/batteries5020045.
- M. Ma, H. Cai, C. Xu, R. Huang, S. Wang, H. Pan and Y.-S. Hu, Adv. Funct. Mater., 2021, 31, 2100278, DOI:10.1002/adfm.202100278.
- V. Shipitsyn, N. Antrasian, V. Soni, L. Mu and L. Ma, Energy Mater., 2023, 3, 300038, DOI:10.20517/energymater.2023.22.
- Y. Jin, P. M. L. Le, P. Gao, Y. Xu, B. Xiao, M. H. Engelhard, X. Cao, T. D. Vo, J. Hu, L. Zhong, B. E. Matthews, R. Yi, C. Wang, X. Li, J. Liu and J.-G. Zhang, Nat. Energy, 2022, 7, 718, DOI:10.1038/s41560-022-01055-0.
- C. Vidal-Abarca, P. Lavela, J. L. Tirado, A. V. Chadwick, M. Alfredsson and E. Kelde, J. Power Sources, 2012, 197, 314, DOI:10.1016/j.jpowsour.2011.09.008.
- R. Aukstakojyte, J. Gaidukevic, G. Niaura, M. Skapas, V. Bukauskas and J. Barkauskas, Inorganics, 2022, 10, 142, DOI:10.3390/inorganics10090142.
- M. Kalapsazova, K. Kostov, E. Zhecheva and R. Stoyanova, Front. Chem., 2020, 8, 600140, DOI:10.3389/fchem.2020.600140.
- E. Zhecheva, R. Stoyanova, J. M. Jiménez-Mateos, R. Alcántara, P. Lavela and J. L. Tirado, Carbon, 2002, 40, 2301, DOI:10.1016/S0008-6223(02)00121-5.
- G. Jayaram and V. G. Krishnan, Z. Naturforsch., 1995, 50a, 953, DOI:10.1515/zna-1995-1008.
- P. T. Manoharam and M. T. Rogers, J. Chem. Phys., 1968, 49, 3912, DOI:10.1063/1.1670700.
- X. Wang, S. Roy, Q. Shi, Y. Li, Y. Zhao and J. Zhang, J. Mater. Chem. A, 2021, 9, 193, 10.1039/D0TA10610K.
- H. Wu, Y. Chen, T. Wen, L. Chen, X. Pu and Z. Chen, Batteries, 2023, 9, 56, DOI:10.3390/batteries9010056.
- R. Rajagopalan, B. Chen, Z. Zhang, X.-L. Wu, Y. Du, Y. Huang, B. Li, Y. Zong, J. Wang, G.-H. Nam, M. Sindoro, S. X. Dou, H. K. Liu and H. Zhang, Adv. Mater., 2017, 1605694, DOI:10.1002/adma.201605694.
Footnote |
| † Electronic supplementary information (ESI) available: Cyclic voltammetry curves of NaFeVPO4(SO4)2 and rGO at a scanning rate of 1 mV s−1 in Na half-cells; XRD pattern of the carbon-coated Al foil used; charge–discharge curves of NaFeVPO4(SO4)2/rGO with C/20 rate within different voltage windows; lattice parameters of NaFeVPO4(SO4)2/rGO and electrodes cycled in Na half-cells and stopped at different states; ex situ XRD patterns and optical images of long-term cycled electrodes; XPS spectra in the Fe2p and O1s regions of cycled electrodes; element content of the electrodes determined from XPS spectra; percentages of the different components of the C peaks; EPR spectrum of the step-wise cycled electrode after the subtraction of the signal due to Fe3+ ions; Spin-Hamiltonian parameters of VO2+ ions in various crystal lattices. See DOI: https://doi.org/10.1039/d4ma00754a |
| This journal is © The Royal Society of Chemistry 2024 |