
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTunable slow photon effect and local surface plasmon in Ag-immobilized TiO2 inverse opal films for enhancing pollutant photodegradation†
Thi Kim Ngan
Nguyen
 *a,
Fabien
Grasset
b,
Satoshi
Ishii
c,
Hiroshi
Fudouzi
d and
Tetsuo
Uchikoshi
bd
*a,
Fabien
Grasset
b,
Satoshi
Ishii
c,
Hiroshi
Fudouzi
d and
Tetsuo
Uchikoshi
bd
aInternational Center for Young Scientists, Global Networking Division, National Institute for Materials Science, 1-2-1 Sengen, Tsukuba, Ibaraki 305-0044, Japan. E-mail: ntkngan1912@gmail.com
bCNRS–Saint-Gobain–NIMS, IRL3629, Laboratory for Innovative Key Materials and Structures, National Institute for Materials Science, 1-1 Namiki, Tsukuba, Ibaraki 305-0044, Japan
cResearch Center for Materials Nanoarchitectonics (MANA), National Institute for Materials Science, 1-1 Namiki, Tsukuba, Ibaraki 305-0044, Japan
dResearch Center for Electronic and Optical Materials, National Institute for Materials Science, 1-2-1 Sengen, Tsukuba, Ibaraki 305-0044, Japan
First published on 26th September 2024
Abstract
Plasmonic silver-decorated TiO2 inverse opal has shown an interesting potential for photocatalysis owing to its physically tunable optical absorbance, highly active area, and flexible fabrication. In this study, electrophoretic deposition was used as a key technique to overcome the disadvantages of traditional inverse opal (IO)-fabricating methods, resulting in high reproducibility, chemical stability, and periodic area. The use of IO structural engineering, beneficially delocalizing and enhancing absorbed visible light, accounted for 46% of the total solar light, leading to the enhancement of the localized surface plasmonic resonance (LSPR) hot electrons of Ag NPs and an enhanced local electromagnetic (EM) field for the formation of photogenerated electrons on TiO2. These enhancements in Ag-deposited TiO2 IO promoted the excellent photocatalytic kinetic constant of methylene blue degradation around 17 × 10−3 min−1, responding to tunable optical absorption at the stopband edge of TiO2 IO containing 288-nm sized pores and low absorbance of Ag in the overlapped band. The explanation for the enhanced photocatalytic mechanism was studied based on high Ag deposition density, decrease in photocurrent, increase in electron lifetime in electrolytes, and the contribution of a slow photon effect to these characteristics. The proposed photocatalysis mechanism concerned the enhancement of EM-generated electrons on TiO2 that immigrate to the Ag surface for photoreduction while photooxidation occurred at the TiO2 surface by the holes. This study provides an interesting strategy to improve the photocatalysis of semiconductor–metal composite systems.
Introduction
Solar-driven photocatalysis has focused on the development of light-harvesting materials and structural designs to obtain the highest efficiency in utilizing solar energy for wastewater treatment, volatile organic carbon (VOC) removal, and photovoltaics.1–3 A heterogeneous photocatalyst based on semiconductors, such as TiO2, ZnO, and Fe2O3, possesses wide-band gaps that are photoactive in the ultraviolet-visible range and are renowned for efficiently initiating redox reactions.4–7 Among them, TiO2 has attracted the most interest owing to its low cost, high chemical stability, relative photocatalytic efficiency, flexible fabrication, and non-toxicity.8 However, the photocatalytic efficiency of TiO2 is restricted; photoinduced electrons are crucially created through excitation in the UV range, which is less than 5% of the total solar light and shows a high recombination rate of hole–electron pairs.5 Therefore, the combination of two or more semiconductors, such as Fe2O3 modified TiO2, ZnO/TiO2 nanocomposites, and Fe2O3/ZnO nanocomposites, have been revealed to undergo enhanced photocatalysis.9–11 In addition, nonmetal (N, C, S, Se, etc.) or transition metal (Fe and Cu) doped TiO2 can modify their bandgap belonging to the visible light range, which occupies almost 46% of the total solar light.12–16 The obtained chemical stability of doped elements is one of the key factors involved in tuning the photo-absorption of TiO2. Among the surface modifications of TiO2, the deposition of noble metal nanoparticles (NPs) (Ag, Au, Pt, etc.) can efficiently suppress electron–hole recombination and tune optical response in the visible light region.17,18 Localized surface plasmonic resonance (LSPR) on metal NPs allows for an increase in an electromagnetic wave (EM) field that enhances the light absorption of the band edge of TiO2, accelerating the generation of photoinduced electron/hole pairs.19,20 The light absorbance range, peak, and intensity of LSPR depend on the particle size, shape, structure, composition, and distance of metal NPs.21 The contact between TiO2 and the noble metal NPs was classified as an ohmic metal solution contact with Pt or Pd, forming an efficient catalyst for electron migration in solution, while non-ohmic interfaces (Ag, Au, Cu) tend to retain the photoinduced electron at the interface of both components.20,22,23 At the interface of the semiconductor and metal NPs, the equilibrium of the charge carrier enables the bending of the CB of the semiconductor, which aligns with the Fermi energy levels of the metal NPs, forming a Schottky barrier.24 The formation of the Schottky barrier and its height adjustability contribute to the efficient hot electron transferability from metals to semiconductors for optoelectronic devices and act as a barrier to prevent hole/electron recombination for photocatalysis.21,25 The purity of the metal phase and the distance between the metal and semiconductor affect the Schottky barrier energy the same as the charge transfer efficiency. Solving these problems becomes a motivating strategy for improving the photocatalytic activity of semiconductor–metal composite systems.Accompanying the chemical surface modifications, the morphology of TiO2 also properly contributes to the efficiency of the electron transfer to the interface and redox reactions, i.e., mesoporous nanoparticles, nanorods, and microporous IO structures provide a higher specific surface area than discrete particles. Moreover, the mesoporous morphology can suppress photogenerated hole/electron recombination. Among the interesting porous structures, photonic IO efficiently supplies a specific surface area and generates a slow photon effect that can broaden the light absorption range by amplifying light–matter interactions.26,27 The silver or gold-decorated TiO2 IO photocatalyst has attracted much interest due to its flexibility in preparation and high efficiency in separating photogenerated hole/electron pairs for photocatalytic reactions upon visible light irradiation.28–34 Most studies have confirmed the advantages of the specific surface area and injection of noble metal nanoparticles inside the pores. The efficiency of the slow photon effect has rarely been investigated regarding its photocatalytic properties due to the difficulty in IO structure fabrications on a large scale and low uniform reproducibility.
This study attempted to clarify the slow photon effect on the visible light absorbability of silver NPs inside microporous TiO2 IO to enhance the photodegradation of an organic dye. The use of electrophoretic deposition (EPD) shortens the fabricating time of opal films from several days (self-assembled deposition) to several minutes, controls the thickness, and provides high reproducibility on flexible scales. The key point for successful EPD is to control the relation between the template suspensions and the evaporation speed of the solvent to obtain a packed hexagonal structure after complete drying. Silver NPs were directly grown on the IO walls of TiO2via the sol immobilization method using chemical reduction and thermal treatment steps. The controlled size, hydrophilic properties, and high purification of the silver metal NPs introduced in TiO2 IO films using the naphthalene 2,6 disulfonate surfactant were investigated. The co-deposition of surfactants also improved the uniform adsorption of silver NPs on the IO walls, which radically utilized the enhanced absorbing light created by the slow photon effect. The purity of the Ag metal at the Schottky junction plays a role in promoting a high charge transfer and preventing electron/hole recombination, which is key to the improvement of photocatalytic reactions upon visible light irradiation.
Experimental
Preparation of TiO2 IO
The polystyrene spheres (PSs) were synthesized using standard emulsion polymerization methods with the participation of sodium dodecyl sulfate (SDS) surfactant.35 The synthesis process of PS was modified to adapt to the EPD conditions. The potassium persulfate (KPS, 10 g L−1) was prepared in distilled water at 70 °C for 1 hour and then mixed with a sodium dodecyl sulfate (SDS) solution of various concentrations. In the next step, 300 g of the PS monomers was added to the solution in a flask. The system used a nitrogen gas atmosphere and was stirred at 500 rpm and 70 °C. The reaction continued for 24 h, then cooled to room temperature and filtered to obtain a uniform PS size. The obtained average PS bead sizes were 316 ± 5, 338 ± 5, and 387 ± 5 nm, which were controlled by the amount of SDS. The opal PS film deposited on indium tin oxide (ITO) coated glass (Geomatec Co., Ltd, Tokyo, Japan; <5 Ohm sq−1) was prepared by EPD. Recently, EPD has been a good technique for fabricating PhCs; however, negatively charged PS beads and their suspension for EPD should be optimized.36 The high reproducibility and short time consumption for achieving the opal PS films using EPD have been confirmed (Fig. 1). Two electrodes were separated by 5 mm and connected to a Source Meter (Keithley Model 2400, Ohio, USA) used as the electric field generator. The as-synthesized 8 wt% aqueous PS slurry was diluted by ethanol to volume ratios of 1 and 4. The 2 × 3 cm2 ITO glass was first properly treated in ethanol and then coated with the PS beads by applying a voltage of 10 V for 1 minute. The PS opal film was dried at 50 °C for 24 hours to properly remove the solvent and increase the void volume.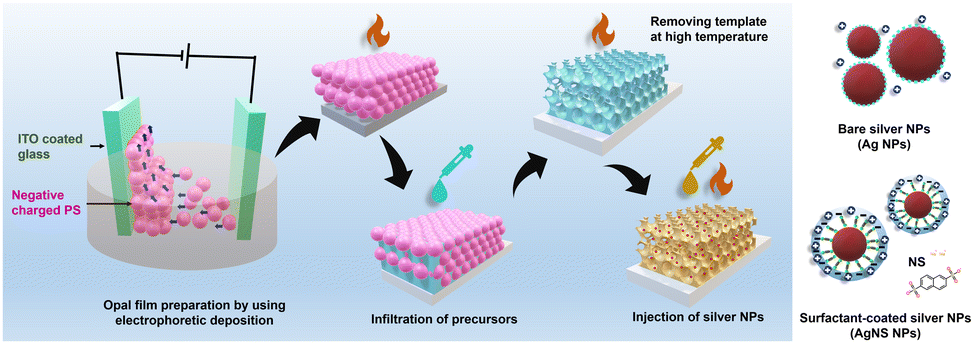 | ||
| Fig. 1 Schematic of the synthesis process of the TiO2 IO films, deposition of the Ag and NS-modified Ag NPs inside the IO walls, and different Ag NP structures. | ||
The TiO2 IO (TIO) film was fabricated by infiltrating the precursor of TiO2 and heating at high temperatures in air. The TiO2 precursor solution was composed of ethanol, water, and a TiCl4 solution (16.0–17.0% as Ti), corresponding to a volume ratio of 0.75, 0.125, and 0.125, respectively. The mixed solution was stirred at 70 °C for 15 minutes until a slightly opaque solution was obtained. The PS bead opal film was fixed at an angle of 20° using the drop-casting method; then, the residual solution on the surface was removed. The infiltrated PS opal film was heated to 550 °C at a speed of 1.8 °C per minute, remained at 550 °C for 2 hours, and then slowly cooled to room temperature. The TiO2 IO films with the abbreviations of TIOx (x = 250, 288, 318 nm as the pore sizes) were stored at 50 °C for the next characterizations and fabrications.
Preparation of Ag-decorated TiO2 IO
The Ag NPs were prepared by applying the chemical deposition method, i.e., the reduction of Ag+ cations from an AgNO3 aqueous solution using NaBH4 reagent to form Ag nuclei.37 The 0.023 M AgNO3 and 0.002 M NaBH4 aqueous solutions were separately prepared in water at a temperature of 5 °C for 1 hour. To prevent aggregation, 2, 4, 6, or 8 mL of AgNO3 solution was slowly dropped into 60 mL of the NaBH4 solution during strong stirring at a speed of 500 rpm for 5 minutes and then continually mixed for another 5 minutes. The mixed solutions were slowly heated to room temperature. Similarly, the Ag NPs were prepared with naphthalene 2,6 disodium sulfate (NS). The AgNO3 precursor solution (0.023 M) was well mixed with an aqueous NS solution (0.0125 M) before being reduced by NaBH4. Two kinds of Ag NPs were abbreviated Agy or AgNSy with y = 1, 2, 3, 4, which corresponded to 2, 4, 6, and 8 mL of the Ag precursor solution, respectively. The aqueous Ag and AgNS NP suspensions were stabilized for 1 hour and then deposited on the TIO films.To fabricate the Ag-deposited TIO films, 150 μL of an Ag or AgNS suspension was spread on the surface of TIOx (x = 250, 288, and 318) and then heated at 50 °C until a completely dried film was obtained. The same procedures were performed three times. In the last step, the Ag-decorated TIO films were continually heated at 100 °C for at least 24 hours. Fig. 1 shows a schematic diagram of the simple methods used to prepare the Ag-deposited TiO2 IO films.
Characterization
The diffuse absorbance and transmittance spectra of the samples were measured by UV-Vis-NIR spectroscopy (V570, Jasco Corp.) in the wavelength range of 200–1000 nm at a scan rate of 1000 nm min−1. The specular reflectance spectra of the IO films were measured at an incidence angle of 5 degrees in the wavelength range of 300–1000 nm using a UV-Vis-NIR spectrophotometer (Solidspec-3700, Shimadzu Corp.). The top view, pore size, and thickness characteristics were analyzed by field emission scanning electron microscopy (FE-SEM, SU8000, Hitachi High-Technologies Corp.) at 10 kV. High-resolution (HR) observations of the powder were performed by applying an HR-transmission electron microscope (HR-TEM) and STEM (JEOL JEM 2100F) equipped with an energy-dispersive X-ray (EDX) analysis device. The X-ray powder diffraction (XRD) patterns were recorded at room temperature at the 2-theta angle in the range of 20°–60° using a SmartLab diffractometer (RIGAKU, 40 kV and 30 mA) with Cu Kα radiation (λ = 1.54 Å), a step size of 0.02° and a scan speed of 1° min−1. The electron binding energy spectra were measured by X-ray photoelectron spectroscopy (XPS) (PHI Quantera SXM (ULVAC-PHI)) using Al Kα radiation at 20 kV and 5 mA and a take-off angle of 45°. All the binding energies were calibrated concerning the C 1s peak of the adventitious carbon at 285 eV. The typical chemical vibration of the powder was recorded by Fourier transform infrared spectroscopy (FTIR) (IRAffinity-1s, Shimadzu Corp.) in a wavenumber ranging from 4000 to 500 cm−1. The luminescent emitting spectra of the films were measured by high-performance fluorescence spectroscopy (JASCO FP8500) connected to a Xenon lamp at a scan rate of 1000 nm min−1.The photocurrent current and electrochemical impedance spectroscopy measurements were performed in a standard three-electrode configuration using a source meter (VSP300, Potentiostat, BioLogic). The TIO working electrode contained an active area of 2 × 2 cm2 and was fixed at the irradiating angle of 90°. The Ag/AgCl reference electrode in KCl (3 M) and the aqueous electrolyte of Na2SO4 (0.1 M) were applied. A bias voltage of 0.6 V referenced as the open voltage of TiO2 was applied in frequencies ranging from 0.1 Hz to 5 MHz with an oscillation amplitude of 10 mV. The electrochemical impedance spectroscopy measurements were measured in the dark and upon light irradiation of one sun (AM 1.5G illumination, HAL-320, Asahi Spectra USA, Inc.).
Photocatalytic degradation of organic pollutants
The efficiency of the photocatalytic degradation using bare TIO and Ag-decorated TIO films was evaluated by the decolorization of methylene blue (MB) in an aqueous solution at room temperature. The as-synthesized TIO films with an active area of 2 × 2 cm2 were immersed in 50 mL of an MB aqueous solution (5 × 10−4 g, 31 μM L−1) contained in a 100-mL beaker. The system was first stirred in the dark for 20 minutes to obtain the adsorption–desorption equilibrium among MB, TIO, and water. The surface of the TIO films was irradiated by the solar energy of one sun (AM 1.5G illumination, HAL-320, Asahi Spectra USA, Inc.) with a power density of 100 mW cm−2 at a 90° angle. MB decolorization was evaluated by the intensity of the absorbance at 664 nm using UV-Vis-NIR spectrometry (V570, Jasco Corp.). The concentration of MB corresponding to an intensity peak at 664 nm before irradiation was abbreviated as C0, while C(t) was the intensity after irradiating at the fixed time (t). The MB solution was collected at an interval time of 10 min for 60 or 120 minutes to calculate the kinetic photodegradation. The reaction was kept at room temperature to reduce the thermal effect. The MB photodegradation recycles were performed for 2 hours and repeated 4 times for every sample. The independence of the reaction kinetics vs. irradiance power was measured for 30 mL of MB aqueous solution (3 × 10−4 g, 31 μM L−1) for 40 minutes.Results and discussion
Characterization of Ag-deposited TIO films
In this study, three sizes of PS beads of 316 ± 5, 338 ± 5, and 387 ± 5 nm were successfully synthesized by controlling the SDS surfactant. Fig. 2a shows the top view of the TiO2 opal films prepared by EPD, having a hexagonal arrangement of the PS beads with a close-packed plane (111). Fig. 2b illustrates an IO structure with homogeneously 250 nm-sized pores in a large porous area after removing the template from the 316 nm-sized opal film. At higher magnification, a face-centered cubic (fcc) structure coming from the periodic arrangement of pores is observed, supplying the complete template removal. As a result of this structure, the film reflects a uniform blue color as depicted in the photograph in Fig. 2c. The volume of the pore size was reduced by an average of 20% compared to the PS template size due to the shrinkage of the PS beads during heat treatment before the TiO2 phase was formed. The cross-section of TIO250 also shows a face-centered cubic (fcc) structure with a thickness of 3.4 μm (Fig. 2d). Similarly, an approximately 20% shrinkage phenomenon also occurred for the 338 and 387-nm sized templates that corresponded to the achieved pore sizes of 288 and 318 nm (Fig. 2e and f). The photographs show the green and orange-colored TIO films corresponding to the 288 and 318 nm-sized pore structures. In addition, the thicknesses of TIO288 and TIO318 increase to 5.3 and 7.1 μm (Fig. S1, ESI†), respectively. To explain the realistic reflecting color of the TIO films with different pore sizes, the specular reflectance spectra of the TIO films were measured at an incidence angle of 5 degrees. The opal films containing 316, 338, and 387 nm-sized PS beads show a reflectance peak in the near-infrared range at 734, 818, and 921 nm (Fig. 2g and Table S1, ESI†), respectively. Similarly, the TIO films containing 250, 288, and 318 nm-sized pores have a reflectance peak at 474 (blue), 568 (green) and 613 (orange), respectively (Fig. 2f and Table S1, ESI†). The IO TiO2 films synthesized from the electrophoretic opal film show a stable structure with a large area and high reproducibility. In summary, the expected pore sizes of the TiO2 IO films are successfully obtained by controlling the PS bead size to generate a stopband that is tunable with the optical property of the depositing metal NPs. | ||
| Fig. 2 (a) HR-SEM images of the 316 nm PS opal film, (b) and (c) HR-SEM images of the top view and visual photographs of TIO250 at different magnifications, and (d) HR-SEM images of the cross-section view of TIO250. HR-SEM images of the top view of (e) TIO288 and (f) TIO318. (g) Specular reflectance spectra at the 5-degree incident angle of the PS opal (O) film and TIO films prepared using different PS bead sizes. | ||
The advantage of the IO structure is that it creates a photonic band gap that can delocalize the emitting and refracting light inside the pores and reduce the spontaneous scattering of light. Moreover, the IO structure can induce a slow photon effect to enhance light absorption, which is beneficial for the light-harvesting efficiency during photocatalysis.38 The approximate stopband position of the opal and IO can be calculated by a modification of Bragg's law, which is expressed by eqn (1) (Table S1, ESI†)39,40:
 | (1) |
However, the slow photon effect of the TIO films was evaluated for the photodegradation of organic pollutants in a water medium. In this case, the volume fraction (f) of water with a refractive index of 1.33 was considered similar to that of air with an f of 0.93. The incident angle (θ) of the light direction was set up at angles of 5° and 90° for the photocatalytic and electrochemical impedance spectroscopy measurements. Therefore, the real photonic stopband of the TIO films in a water medium depending on the light irradiating angles is recalculated, i.e., 460 (90°) and 438 nm (5°) for TIO250, 530 nm (90°) and 504 nm (5°) for TIO288, and 585 nm (90°) and 556 nm (5°) for TIO318 (Table S1, ESI†). It is suggested that the blue-shifting stopband edge created by TIO250 overlaps with the slow-photon absorbance of TiO2, and others induce slow photon light absorbance in the visible range (450–550 nm).
As is known, silver NPs broaden the photon absorbance in the visible band by utilizing free photoelectron coherent oscillation, which is called localized surface plasmon resonance (LSPR).41 In this study, the deposition of spherical Ag NPs on the IO walls of the macroporous TiO2 film was prepared by sol immobilization through the reduction and chemical deposition steps. In addition, LSPR strongly depended on the size, uniform deposition, concentration, and shape control of the Ag NPs, resulting in a different broadening of the Vis-NIR range and intensity of the light absorption.42 Broadening of the absorbance was expected in the range of 400–600 nm, which accounted for 46% of the total solar light corresponding to a metal size less than 100 nm. Therefore, the NS surfactant was employed for the first time to control the size, concentration, and hydrophilic properties of the Ag NPs. The objective of this study is to introduce small incomplete reduced Ag clusters on the TiO2 IO walls and then grow Ag NPs with the support of heat treatment. The role of NS is to slow down the aggregation of the Ag clusters in the solution before being introduced into the IO films. The mean average size (MA) and size distribution (SD) of the Ag NPs without and with NS measured after stopping the reaction for 1 hour are presented in Fig. 3a and Fig. S2 (ESI†). The mean average sizes of the NS-combined Ag NPs are recognized from 5 to 16 nm with a low size distribution, while they are larger than 115 nm for the bare Ag NPs. These small NS-combined Ag NPs in suspension were stabilized for 2 months that were not obtained in the bare Ag NPs (Fig. S3, ESI†). The amount of added Ag precursors also affected the size of the produced Ag NPs. The size distribution of the bare Ag NPs is greater in comparison to the NS-combined Ag NPs. The broad and large size of the Ag NPs directly affects their uniform growth on the IO walls. Most bare Ag NPs are bigger than 100 nm, and the pattern assigned to the (111) plane is confirmed in the HR-TEM images (Fig. 3b and c). This suggestion is supported by photographs of the Ag and AgNS4 NPs associated with methanol. To summarize, the NS molecules play a good role in preventing the aggregation of the Ag NPs, as observed in the HR-TEM images with a size smaller than 100 nm (Fig. 3d). Fig. 3e illustrates the confirmation of the Ag element in the metal NPs with the weak signal of S in the NS molecules. It is suggested that the NS will co-deposit with the Ag NPs on the TIO walls and support deposition inside the pores with a small size. The distributions of the bare Ag NPs and NS-combined Ag NPs on the IO wall of the TIO film were investigated based on the HR-TEM and STEM images (Fig. 5). The small white dots observed in Fig. 3f are assigned to the AgNS4 NPs with homogeneous small sizes. The NPs were also observed in the HR-TEM image as black dots measured at different magnifications (Fig. 3g and h). The hexagonal close-packed array structure of the TIO was successfully prepared. As illustrated in Fig. 3i, the patterns of the Ag and TiO2 lattices were observed in the (111) and (101) planes, respectively, as calculated in the HR-TEM images (Fig. S4, ESI†). The HR-TEM images assigned to the (101) plane of TiO2 were observed, which supported the performance of the anatase TiO2 pattern in the XRD. In addition, the growth of Ag NPs on the TiO2 lattice proves the creation of a heterojunction joint, which improves the plasmonic energy transfer from Ag near the band edge to generate the electron/hole pair on TiO2. Moreover, both the reduction and oxidation reactions were suggested to occur at least around the surface of the metal. Therefore, the short deposition distance of the Ag NPs is controlled to improve the charge transfer for photocatalysis.4,43
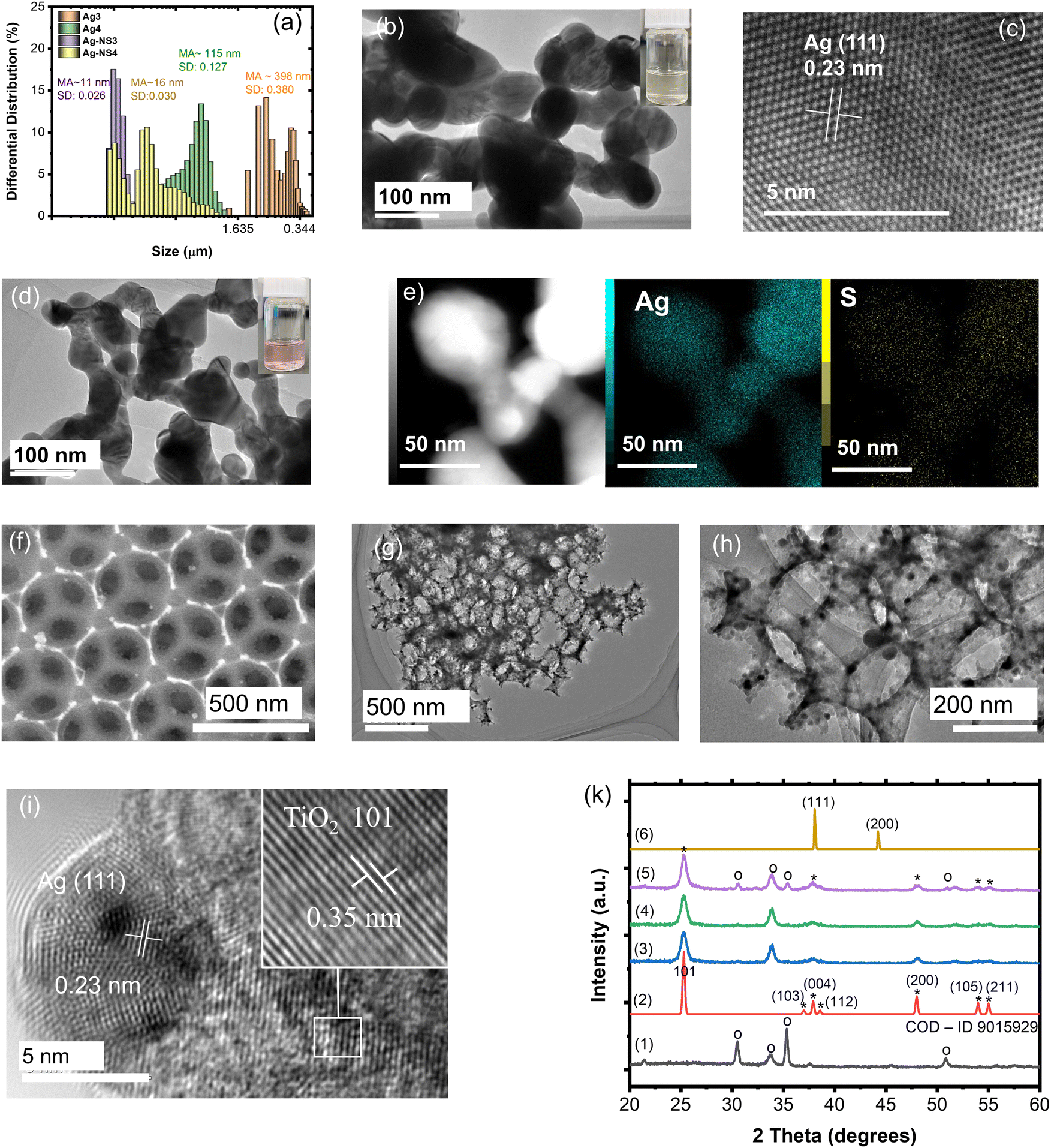 | ||
| Fig. 3 (a) Differential distribution and size distributions of the Ag3, Ag4, AgNS3, and AgNS4 NPs after synthesizing for 1 hour. (b) and (c) HR-TEM images of Ag4 NPs. (d) HR-TEM images and (e) STEM-EDX mapping of element compositions of AgNS4 NPs. (f) SEM and (g)–(i) HR-TEM images of TIO250 deposited AgNS4 at different magnifications. (k) XRD pattern of the ITO-coated glass (1), anatase TiO2 (reproduced using a file with COD–ID 9015929) (2), TIO250 (3), Ag4 deposited TIO250 (4), Ag-NS4 deposited TIO250 (5) and Ag4 NPs (6). *Photograph of the Ag4 and AgNS4 NPs associated in methanol. | ||
Fig. S5 (ESI†) illustrates the confirmation of the existence of the Ag-NS4 NPs on the IO pore wall and the element composition of silver. The silver NPs without stabilizers also succeeded in anchoring the TiO2 IO wall. However, the Ag element composition anchoring on the pore wall, which is calculated from 5 positions using EDX-STEM mapping, shows a difference between the two kinds of Ag NPs (Table S3, ESI†). The deposition of Ag with the support of the NS stabilizers shows a higher Ag concentration and better uniform distribution on the pore wall than that of Ag4 without stabilizers. This means that most Ag4 NPs deposited on the free cracking place the same as on the ITO surface and big-sized particles with a similar original concentration of the precursors. It can be suggested that NS will support the immobilization of the Ag nanoclusters on the TiO2 surface in the first stage. The sulfonate group with SO3− bonding on NS possibly forces the adsorption of the NS-combined Ag clusters at the Ti4+ site using hydrogen bonding with the OH group adopted of the TiO2 lattice and acid–base interaction occurs between S and O on the lattice.44,45 However, NS is suggested to adsorb directly on the TiO2 lattice rather than deposited on Ag NPs, as confirmed in the element spectra (Fig. S6, ESI†). This suggestion is also in agreement with the FT-IR spectra of the NS and AgNS-TIO films. The vibrational band assigned to C–C of the ring and S–O of the sulfonate group of NS appears in the spectrum of the NS-combined Ag-deposited TIO without changing the chemical bonding. The NS modification on the TiO2 surface is also beneficial for increasing the adsorption of the organic dye using sulfonate groups.
Fig. 3k shows the XRD pattern of the TiO2 IO films presented for TIO250 (line 3), TIO250 Ag4 (line 4), and TIO250 AgNS4 (line 5) with the observation of specific 2-theta angles centered at 25.3° (101), 37.9° (004), 48° (200), 54° (105), and 55.1° (211). These series of peaks completely overlapped with the anatase TiO2 pattern as observed in line (2) (COD – ID 9015929). The pattern peaks assigned to Ag NPs (line 6) are not observed in the Ag-decorated TIO films due to the low concentration of big crystals. Anatase TiO2 positively results in photocatalysis properties compared to the rutile or mixed phases. The IO TiO2 introduced Ag NPs were peeled off from the film, and the optical properties were measured to eliminate the effect of the slow photon effect. The increase in the amount of silver nitrate is proportional to the enhanced light absorbance of the metallic silver part and the saturated ratio obtained for the Ag4 and AgNS4 compounds (Fig. 4a). All the curves show an absorbance with a peak at around 450 nm and broadening to 650 nm caused by utilizing the LSPR of the Ag NPs, accompanying the absorbance of TiO2 in the ultraviolet ranging from 340 to 390 nm. A similar tendency was also obtained for the AgNS-deposited TiO2, as depicted in Fig. 4b. However, the absorption peak shifts from 477 to 465, 455, and 442 nm as a function of the increasing precursor amounts accompanied by a narrow range, as illustrated in Table S2 (ESI†). Consequently, the optical energy band gaps are also modified (Fig. S7, ESI†). Bare TIO films achieve an optical energy band gap of 3.17 eV, which decreases by tuning the visible absorbance of plasmonic Ag NPs, such as around 2.19 for TIO Ag4 and 2.35 for TIO AgNS4. As is expected, the as-synthesized silver NPs on the surface of TiO2 by chemical deposition produce a good absorbance in the visible range, which accounts for 46% of total solar light. In addition, the NS stabilizer is an efficient agent for controlling the small size of the Ag NPs and uniform-size distributions, which are properly attributed to the uniform deposition on the IO walls. The role of NS is clearly determined in the investigation of the slow photon effect.
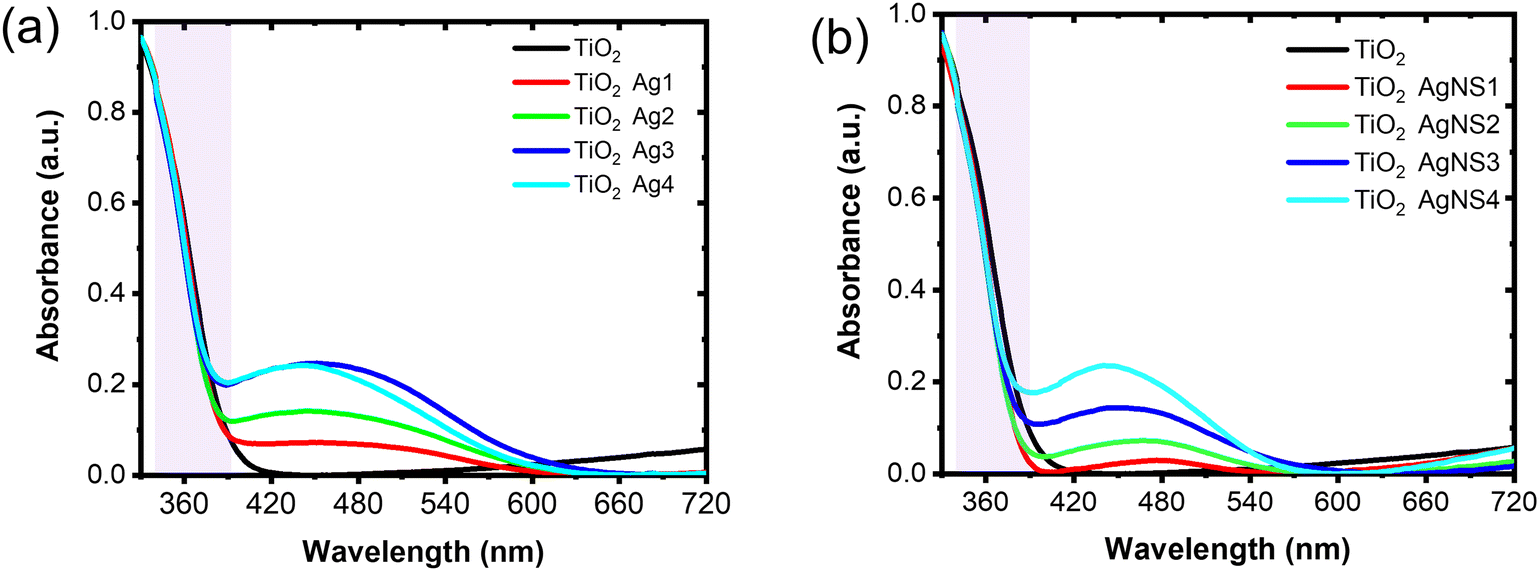 | ||
| Fig. 4 UV-vis absorption spectra of (a) disorder Ag-deposited TiO2 IO, and (b) disorder AgNS-deposited TiO2 IO. | ||
When plasmonic metal NPs contact a semiconductor, the Schottky barrier on the metal–semiconductor interface appears, affecting the charge carrier transfer efficiency to the surface of the TiO2 or Ag NPs. Therefore, the high purification of Ag0 nuclei at the interface can modify the Schottky barrier height to enhance the change in the transfer efficiency.46 The fabrication of pure Ag metal growth on the TiO2 lattice is important. The use of the NS stabilizer beneficially controls the existence of the impurity silver oxide, as proved by the XPS spectra. The XPS spectra assigned for the binding characteristics between elements were calibrated concerning the C1s peak of adventitious carbon at 285 eV (Fig. 5). NS increases the deposition of the Ag NPs inside the TIO pores, as confirmed by the Ti/Ag ratio of 100/8, while bare Ag NPs show that of 100/1 (Table S4, ESI†). The small size of the Ag NPs and high hydrophilic properties controlled by NS should be key factors for a uniform deposition. Fig. 5a shows the deconvolution of the C1s region assigned to NS containing C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonding centered at 284.6 eV, C–C, C–H bonding centered at 285.4 eV from the naphthalene rings, and C–S bonding centered at 286.1 eV from the sulfonate group (Table S5, ESI†). These characteristics also appear in the C1s spectrum of the TIO AgNS4 film at a low concentration.47 There is no difference in the spectrum of the Ti2p region for all the films (Fig. 5b). In great detail, the binding energy peaks centered at 459.4 eV and 465.2 eV are assigned to the splitting of the 2p3/2 and 2p1/2 regions, respectively, which come from the Ti4+ state in the TiO2 lattice.29 As depicted in Fig. 5c, the binding energy centered at 530.7 eV assigned to the O1s region of the O–Ti bonding was observed in TIO, TIOAg4, and TIO AgNS4. In addition, the binding energy centered at 531.8 eV was assigned to the SO bonding, and a small shoulder was attributed to a partially negative oxygen atom (–SO2–O−) on the sulfonate group.47 A deconvoluted peak centered at 531.3 eV in AgNS4 corresponds to oxygen in O–H bonding coming from bound water adsorbed on the surface of the Ag metal.48,49 This observation in AgNS4 is not observed in combination with TiO2. The deconvolution of the Ag3d region of TIO Ag4 and TIO AgNS4 or AgNS4 shows a significant difference in the peak positions (Fig. 5d). Two kinds of binding energies assigned to the Ag3d regions of TIO AgNS4, i.e., 368.1 eV (3d5/2) and 374.2 eV (3d3/2), were ascribed to metallic silver, and 367.6 eV (3d5/2) and 373.6 eV (3d3/2) ascribed to Ag–O in Ag2O metal oxide (Table S5, ESI†).49 The ratios of Ag and Ag2O in TIO Ag4 are 0.21 and 0.79, respectively, which are 0.94 and 0.06 in TIO AgNS4 or AgNS4. The synthesis of the Ag NPs combined with the NS stabilizer enhanced the purification of the silver metallic composition, which improved the charge transfer through the Schottky barrier. In general, the binding energy of the S2p region of AgNS4 shows a splitting of two peaks centered at 168.1 eV (2p1/2) and 169.3 eV (2p3/2), which is ascribed to the S–O bonding in the C–SO3− sulfonate groups of NS (Fig. 5e). However, the binding energy shifts to higher energies, corresponding to 168.6 eV (2p1/2) and 169.8 eV (2p3/2). It is suggested that the –C–SO3− sulfonate groups were chemically adsorbed on the surface of TiO2 and created a possible –C–SO2–O–Ti bonding. This result agrees with the previous report, i.e., the C–SOx–C (x = 1,2,3) bonding resulted in the binding energy of S2p centered at 168.4 eV.50 The adsorbed NS can enhance the hydrophilic interaction with organic dyes, which beneficially creates immobilization of the dye for catalytic reactions.
C bonding centered at 284.6 eV, C–C, C–H bonding centered at 285.4 eV from the naphthalene rings, and C–S bonding centered at 286.1 eV from the sulfonate group (Table S5, ESI†). These characteristics also appear in the C1s spectrum of the TIO AgNS4 film at a low concentration.47 There is no difference in the spectrum of the Ti2p region for all the films (Fig. 5b). In great detail, the binding energy peaks centered at 459.4 eV and 465.2 eV are assigned to the splitting of the 2p3/2 and 2p1/2 regions, respectively, which come from the Ti4+ state in the TiO2 lattice.29 As depicted in Fig. 5c, the binding energy centered at 530.7 eV assigned to the O1s region of the O–Ti bonding was observed in TIO, TIOAg4, and TIO AgNS4. In addition, the binding energy centered at 531.8 eV was assigned to the SO bonding, and a small shoulder was attributed to a partially negative oxygen atom (–SO2–O−) on the sulfonate group.47 A deconvoluted peak centered at 531.3 eV in AgNS4 corresponds to oxygen in O–H bonding coming from bound water adsorbed on the surface of the Ag metal.48,49 This observation in AgNS4 is not observed in combination with TiO2. The deconvolution of the Ag3d region of TIO Ag4 and TIO AgNS4 or AgNS4 shows a significant difference in the peak positions (Fig. 5d). Two kinds of binding energies assigned to the Ag3d regions of TIO AgNS4, i.e., 368.1 eV (3d5/2) and 374.2 eV (3d3/2), were ascribed to metallic silver, and 367.6 eV (3d5/2) and 373.6 eV (3d3/2) ascribed to Ag–O in Ag2O metal oxide (Table S5, ESI†).49 The ratios of Ag and Ag2O in TIO Ag4 are 0.21 and 0.79, respectively, which are 0.94 and 0.06 in TIO AgNS4 or AgNS4. The synthesis of the Ag NPs combined with the NS stabilizer enhanced the purification of the silver metallic composition, which improved the charge transfer through the Schottky barrier. In general, the binding energy of the S2p region of AgNS4 shows a splitting of two peaks centered at 168.1 eV (2p1/2) and 169.3 eV (2p3/2), which is ascribed to the S–O bonding in the C–SO3− sulfonate groups of NS (Fig. 5e). However, the binding energy shifts to higher energies, corresponding to 168.6 eV (2p1/2) and 169.8 eV (2p3/2). It is suggested that the –C–SO3− sulfonate groups were chemically adsorbed on the surface of TiO2 and created a possible –C–SO2–O–Ti bonding. This result agrees with the previous report, i.e., the C–SOx–C (x = 1,2,3) bonding resulted in the binding energy of S2p centered at 168.4 eV.50 The adsorbed NS can enhance the hydrophilic interaction with organic dyes, which beneficially creates immobilization of the dye for catalytic reactions.
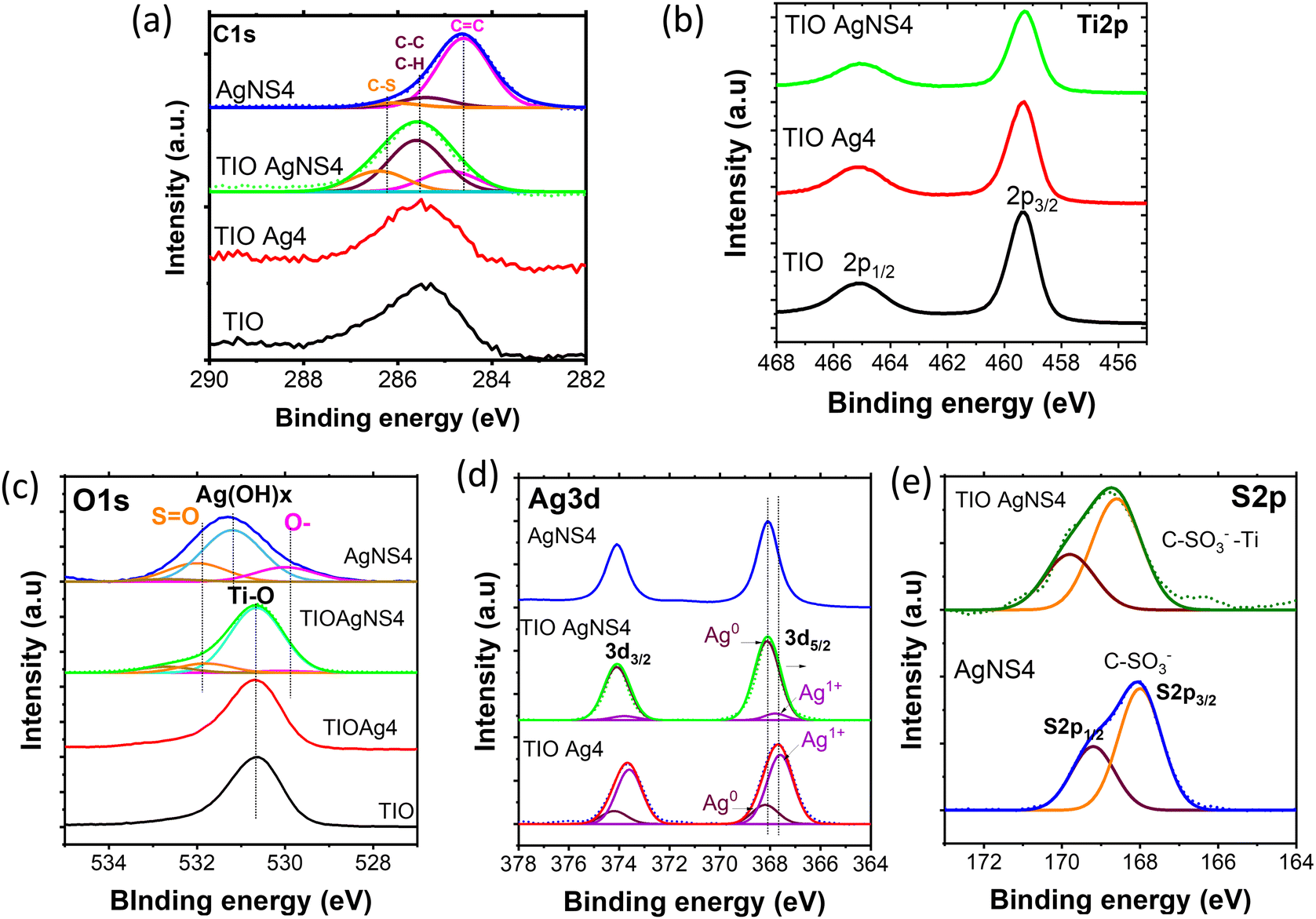 | ||
| Fig. 5 XPS and its deconvolution spectra of (a) C1s and (b) Ti2p, (c) O1s, (d) Ag3d, and (e) S2p regions of the TIO, TIO Ag4, TIO AgNS4 and AgNS4. | ||
Slow photon effect and enhanced photocatalytic efficiency of Ag-deposited TIO films
Fig. 6a shows the specular reflectance spectra at the 5-degree incident angle of TIOx (x = 250, 288, and 318), correspondingly observing the reflectance peaks at 474, 568, and 613 nm, respectively. The AgNS-deposited TIO powder shows an absorbance band narrower than that of the Ag-deposited TIO, agreeing better with the homogeneous sizes of the particles. Bare TIO does not show absorbance in the visible band. As depicted in Fig. 6b, the trapped refractive light is scattered inside the pores of the IO structure, which physically enhances the absorbed photon energy at the stopband edges. TIO250 has a stopband that almost overlapped with the strong visible absorption of Ag NPs, as observed by the dotted lines of the disordered TIO Ag4 and TIO AgNS4. The edges of the stopbands of TIO288 and TIO318 overlapped with the low absorption range caused by the Ag NPs. Consequently, the specular absorption spectra show a difference in absorbance caused by the Ag NPs, i.e., a reduction of the absorbance at about 478 nm, while an enhancement of the absorbance at 493 nm for TIO288 and 502 nm for TIO318 was observed, as illustrated in Fig. 6c–e. In addition, the absorbance of trapped light centered at 405 nm is achieved for all the ITO films. The slow photon effect properly occurs in the three IO structures, which agrees with the literature.38 The diffuse transparency spectra provide more evidence of the slow photon effect, as illustrated in Fig. 6f–h. The visible light-absorbing enhancement is proportional to the trapped light when the Ag NPs are deposited inside the IO pores. All diffuse absorption spectra confirm the homogeneous deposition and high density of the AgNS NP inside the IO pores, corresponding to better absorption for the same concentration powder spectra (Fig. 6a). Moreover, the diffuse absorbance of TIO is broadened in the range overlapped with the enhanced specular absorbance, i.e., enhancement at 405 nm and reduction at 480 nm for TIO250, and enhancements at 405 and 480 for ITO288 and ITO318, respectively. Interestingly, TIO288 shows an absorbance increase of 8% at 480 nm, and it is 15% for TIO318 in comparison to the absorbance at 480 nm for the disordered TIO (Table S6, ESI†).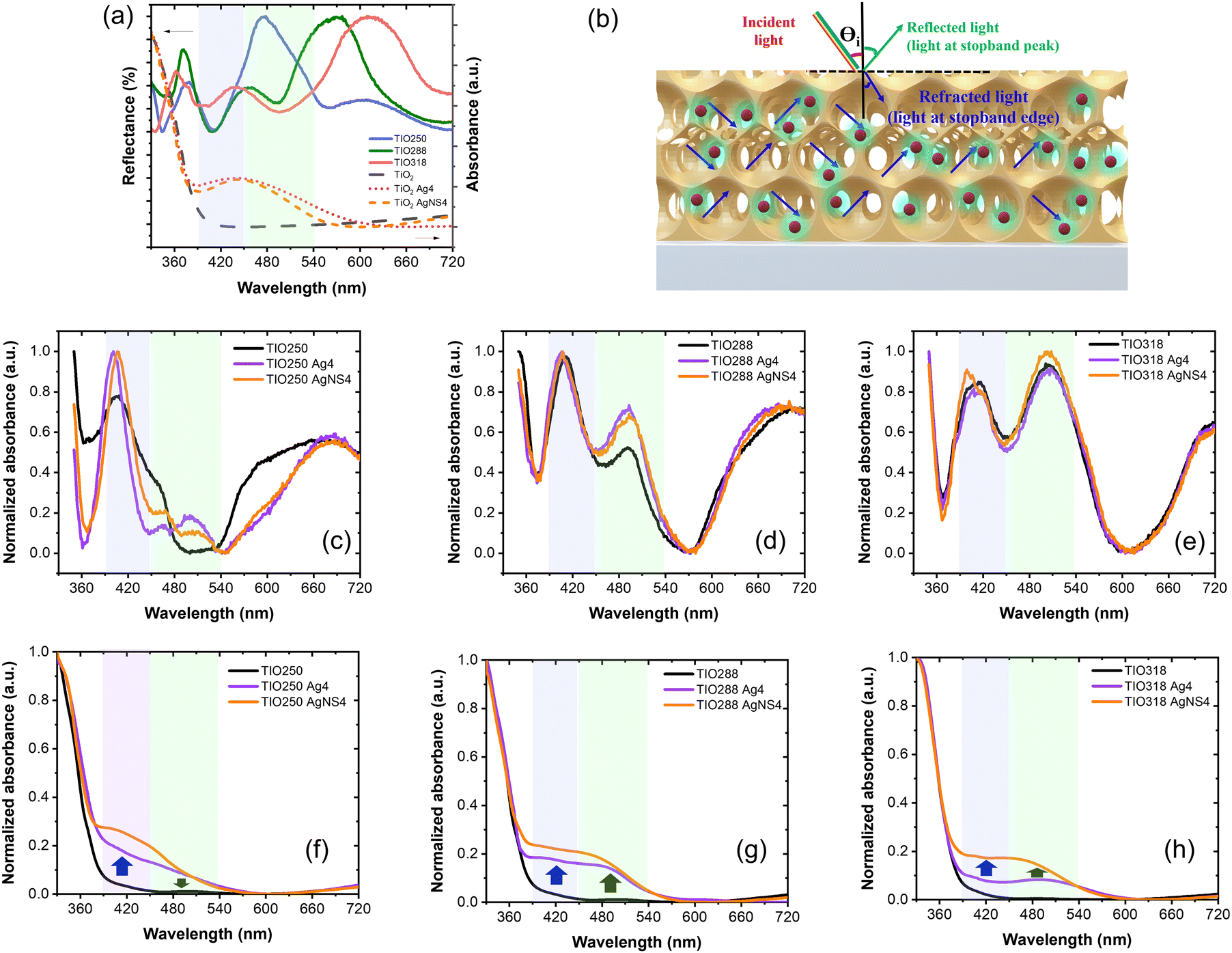 | ||
| Fig. 6 (a) UV-vis absorption of disorder TIO and specular reflectance spectra at the 5-degree incident angle of TIOx (x = 250, 288, and 318). (b) Schematic of the distribution of the Ag NPs (red) and the absorbed light scattered by the IO structure. Normalized specular absorbance spectra at the 5-degree incident angle and transparency spectra of (c) and (f) TIO250, (d) and (g) TIO288, and (e) and (h) TIO318 deposited Ag NPs. | ||
The efficiency of the slow photon effect was properly investigated by the decolorization of methylene blue upon irradiation by simulated solar light, in which a visible band from 400 to 600 nm accounted for 46%. The purpose of utilizing the IO is to enhance the optimal absorbance of solar energy in this range. Fig. 7a shows the photocatalytic reactivity of the Ag NP-deposited TIOx (x = 250, 288, 318 nm). The film was immersed in an aqueous MB solution for 20 minutes in the dark to obtain the adsorption–desorption equilibrium. The Ag NP-deposited TIO accelerates the MB photodegradation twice in comparison to the bare TIO for the first hour (Fig. 7a). Similarly, the photoreaction kinetics are faster in the case of the deposition of Ag metal; thus, the group of TIO AgNS4 shows the fastest photodegradation (kTIO288 = 17.3 min−1), while TIO shows low kinetic constants of kTIO250 = 9.3 min−1 (Table S7 and Fig. 7b). The lower photodegradation efficiency in the case of the Ag-deposited TIO (kTIO288 = 15.3 min−1) is due to the coagulation of big particles on the ITO glass and low distribution of the Ag NPs inside the IO pores even at the same concentration as the AgNS NPs. The photodegradation rate was performed three times for different series of samples, as illustrated in Fig. 7c. The bare TIO250 with the 3.4 μm thickened film and 250 nm-sized pores is the best photocatalyst upon excitation in the ultraviolet range. It is interesting to realize that the thickness of the films is not an essential factor attributed to the photodegradation efficiency. TIO288 (5.3 μm) and TIO318 (7.1 μm) show a lower photocatalysis even when they achieve a thicker film the same as the larger active areas. It can be explained that the contribution of the slow photon effect at the stopband of 405 nm in IO250 leads to the enhancement of the photo absorbance to create photo-excited electron/hole pairs.
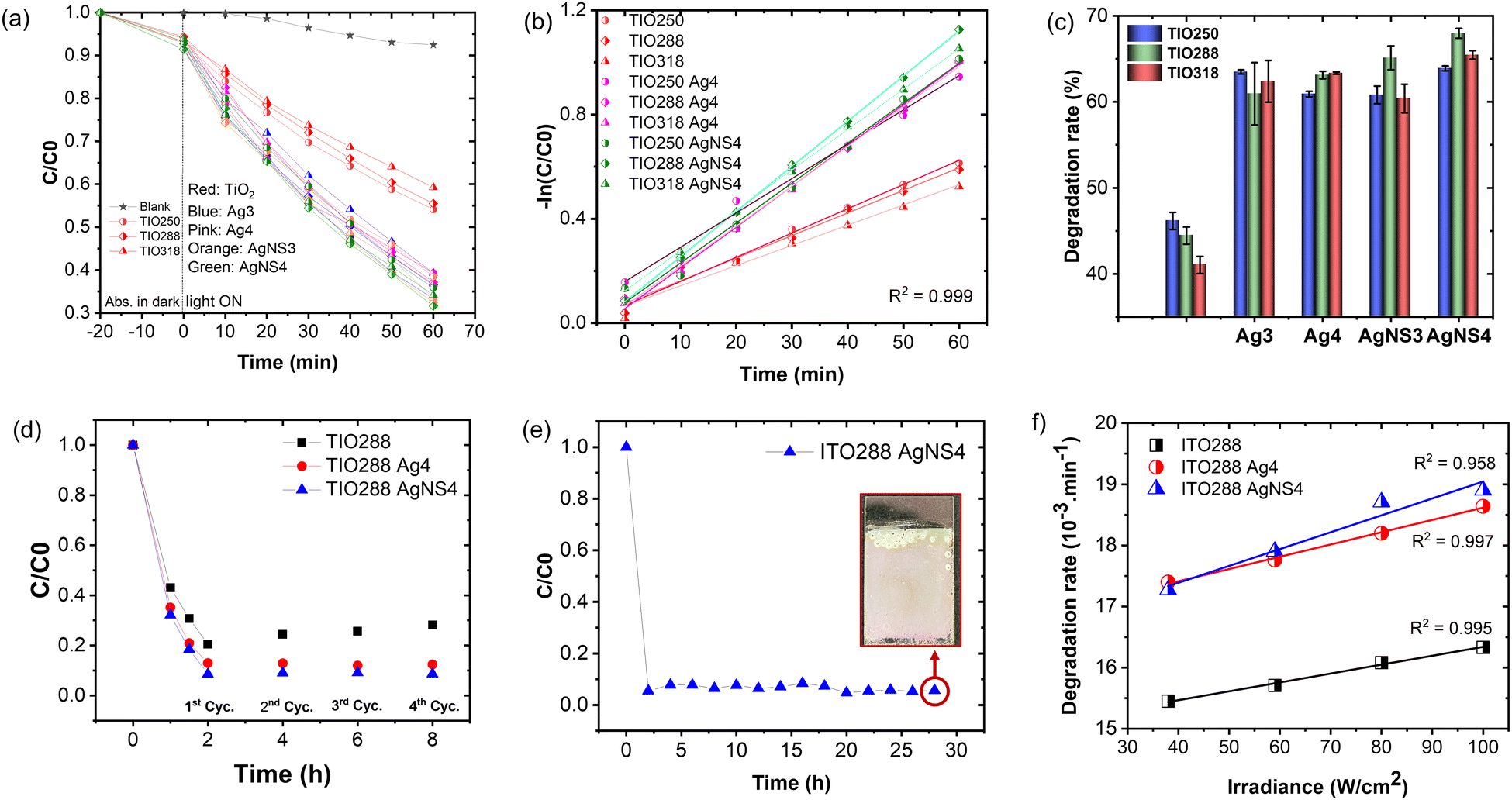 | ||
| Fig. 7 (a) Photocatalytic degradation efficiency of MB (50 mL of 31 μM L−1) using different samples under the illumination of an AM 1.5G standard solar simulator for 1 hour. (b) Chemical reaction kinetics calculated from photodegradation. (c) Degradation rate and (d) reproducibility of the photocatalytic degradation of different samples. (e) Reproducibility of the photocatalytic degradation of ITO288 AgNS4 after 14 cycles, and (f) photodegradation rate of MB (30 mL of 31 μM L−1) as a function of light solar irradiance fitted using a linear law. | ||
In the sample series of the Ag NP-deposited TIO, the photodegradation changes the tendency, i.e., the increase in the photocatalytic efficiency obtained for IO288 and IO318 due to the enhanced photon absorption occurring in the visible range of exciting the Ag NPs. The metallic NPs present two crucial factors to accelerate the reaction: (i) limiting the electron/hole recombination in TiO2 by generating the Schottky junction, and (ii) absorbing photon energy in the visible light range to create the plasmon photoexcited electron or called “hot carriers”, which increase the local electromagnetic field.21,46,51 The generated hole and hot charge electron on silver can create a local non-equilibrium charge and activate the redox reaction.52 The absorbed photon energy is proportional to the plasmonic hot electrons, similar to the electromagnetic field at the interface for exciting the electron at the edge band of TiO2 and increasing the redox ability to degrade the dye. The uniform size, high density, and distribution of the NS-combined Ag NPs inside the IO structure support the interaction between the slow photon effect and the absorption of visible light. Therefore, a higher photocatalyst efficiency is recorded for these TIO AgNS films with high reproducibility. Both the Ag and AgNS-deposited TIO288 show photocatalytic stability after the 4th cycle, while the ability of the bare TIO288 gradually decreases (Fig. 7d and Table S7, ESI†). The reproducibility of film fabrication and high photocatalytic efficiency provide huge potential for practical applications. Impressively, the ITO288 AgNS4 film can prove the stabilization of the MB photodegradation rate after 14 cycles, with an average value of 94% for every cycle (Fig. 7e). The film structure is retained so that the porous film fabricating technique in this study can provide a good strategy for scaling up in practical applications.
The plasmonic silver NPs accelerate the photocatalytic reaction via the generated hot-charge carriers and heat generation.21,42,53 To investigate the dependence of the photodegradation reaction on heat generation, an experiment was performed to determine the change in the MB degradation rate as a function of the irradiance power (Fig. 7f). This result confirms the linear dependence of the chemical transformation rate on the increase in the solar light powers for the bare TIO288 and Ag-deposited TIO288 films.53 The photothermal effect was less attributed to the photodegradation.
The photocurrents of the bare TIO and Ag-deposited TIO films are presented in Fig. 8a and Fig. S8 (ESI†). In general, the plasmonic hot carriers of the Ag NPs due to visible light absorbance are injected into the conducting band (CB) of TiO2, increasing the photocurrent by a transfer to an external circuit. The injection into the CB of TiO2 occurs when the energy of the hot plasmonic carriers is higher than the Schottky barrier energy generated by metal contact with TiO2.42,51 Under a small bias, the high resistance of the Schottky barrier can limit the transfer of hot carriers to the conduction band of TiO2. This could explain the decrease in the photocurrent after the deposition of Ag NPs in this study (Fig. 8a). Gross and his colleagues also revealed that the positive or negative effect on the photocurrent depended on the illumination, distance, concentration of the Ag NPs, and applied bias.54 Following their conclusion, at a high Ag deposited density and a short distance between the Ag NPs, the photogenerated electron/hole separation is more efficient. Therefore, the strengthened hole strongly photo-oxidizes H2O to form O2, which makes the evolution of the O2 concentration on the electrolyte/metal interface. The high catalytic activity of Ag then preferably utilizes the captured electrons to reduce the generated oxygen, thus reducing the migration of captured electrons into the external circuit. Another study also reported that an increase in the metal deposition amount led to an increase in the Schottky barrier height, thus limiting the primary hot electron transfer to the CB of TiO2.55 This clarification possibly explains the decrease in the photocurrent after the deposition of Ag NPs on TiO2 IO films in this study. Depending on the photocurrent at 90s, the Ag-NS deposited TIO films show a lower value, which indicates the better covering of Ag NPs the same as the homogeneous distribution and size. In summary, the chemical immobilization method in this study is efficient for growing small silver NPs with a high concentration and density on the IO walls, which promotes the active sites for photocatalytic reactions occurring on the metal interface. In addition, the photocurrent shows interesting different trends in the two groups of TIO films, i.e., bare TIO films result in a higher current vs. the smallest pore size of 250 nm with a stopband at 460 nm in water (Table S1, ESI†). In contrast, the Ag-deposited TIO films obtained a better photocurrent for TIO288 and ITO318 nm, showing the proper contribution of the slow photo effect at the blue shifting shoulder of the stopband at 530 and 585 nm, respectively, as depicted in Fig. 6.
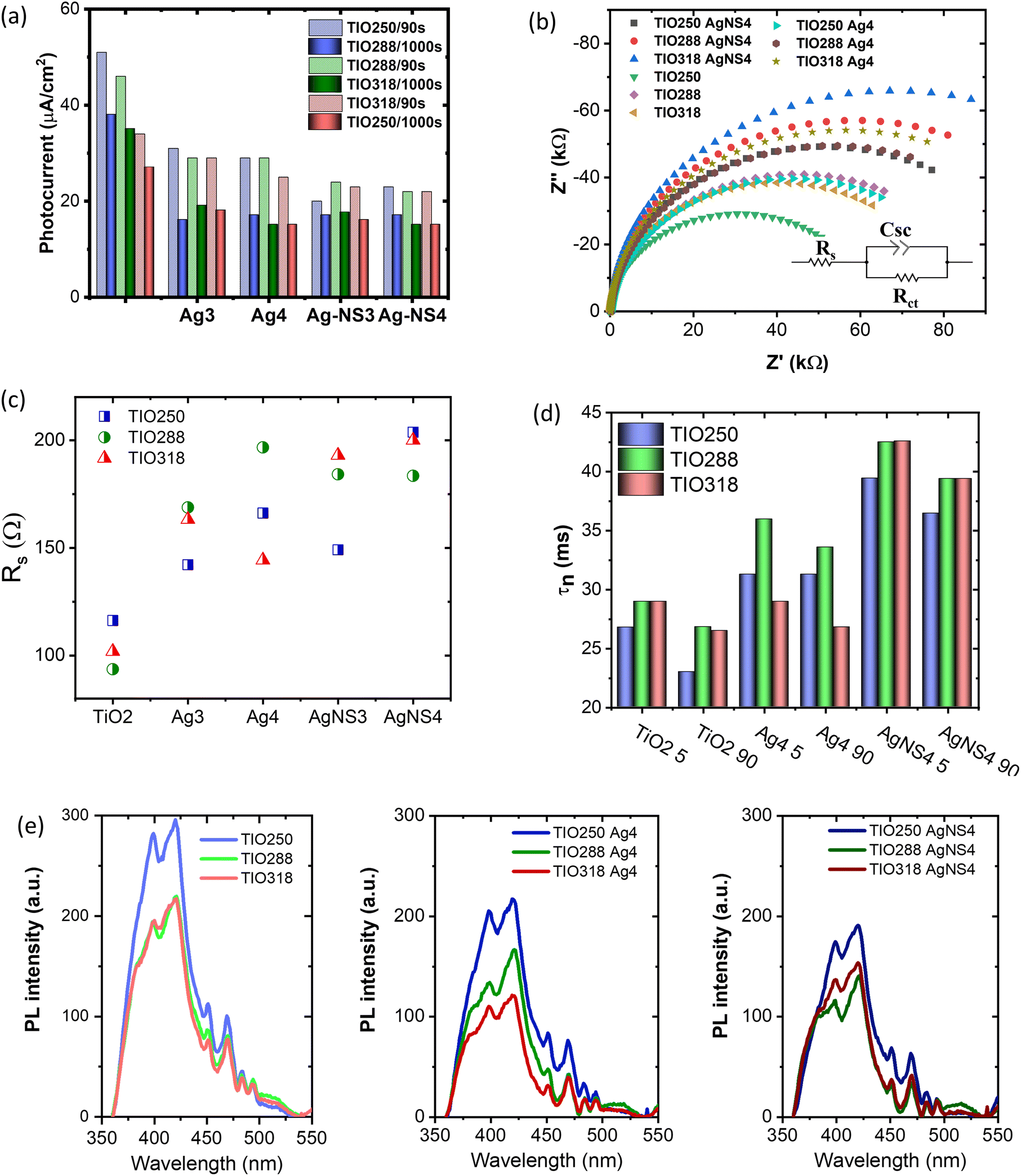 | ||
| Fig. 8 (a) Transient photocurrent after applying a bias of 0.6 V after 90s and 1000s under ambient conditions. (b) Nyquist plots and equivalent circuits used for fitting cell impedance, (c) electrode resistance calculated from the fitting of equivalent circuits using ZView software of the different ITO films, and (d) electron lifetime of the charge carrier calculated from the frequency maximum in Bode plots. (e) Photoluminescence emission of the TIO films without and with the deposition of the Ag NPs. | ||
It has been reported that the rate of hot electron transfer depends on the intensity of the generation of high-energy electrons, net surface area, plasmon wavelength, and plasmonic enhancement factor.21 As already mentioned, the enhancement of the photon absorbance as well as the electromagnetic (EM) field on the Ag NPs contributes to an increase in the light absorption in the near-band edge of TiO2 to create charge carriers and accelerate the number of primary hot carriers. When the plasmonic and photogenerated charge density increases, they tend to inject a conduction band of TiO2 and immigrate to an external circuit to increase the photocurrent. Consequently, all the Ag3 or AgNS3 deposited TIO288 and TIO318 films show a higher photocurrent in comparison to the Ag or AgNS deposited on TIO250, which did not have the slow photon effect. At a higher Ag concentration and density of Ag4 or AgNS4, the photocurrent is slightly reduced due to the electron utilized for redox reactions on the interface, as already discussed.
To understand the photocurrent result deeply, electrochemical impedance spectroscopy was done to characterize the charge separation at the electrolyte/semiconductor and the lifetime of the photogenerated electron/hole pairs. This measurement was performed using a three-electrode configuration in a neutral aqueous solution to stimulate the charge transfer for photodegrading a dye in an aqueous medium. Fig. 8b shows the Nyquist plots of the bare TIO and Ag-deposited TIO films, which are fitted to an equivalent electrical analogue using ZView software. Table S8 (ESI†) presents the charge-transfer resistance (Rct) and space charge capacitance (Csc) in the dark and under illumination. In the dark, the Ag-deposited TIO films show a lower Rct and Csc after introducing the Ag NPs. The lower Rct and Csc suggested a lower electron/hole recombination, i.e., allowing the fast electron/hole transfer to the electrolyte/semiconductor interface to be photoreactive with the dye.56 The sheet resistance (Rs) properly increased due to the deposition of Ag NPs, limiting the electron transfer between the electrode and electrolyte (Fig. 8c). As discussed, the covering of TIO on ITO glass is incomplete, and the Ag concentration is simultaneously deposited in cracks and IO wall positions. The Rs resistance is proportional to the deposited Ag concentration covering the ITO glass, thus achieving a lower photocurrent. This result agrees with the reduction in the total photocurrent of the Ag-deposited TIO, as shown in Fig. 8a.
Upon light irradiation, the lower Rct and higher double-layer Csc are obtained for all the films due to the assumption of photo-excited electrons and holes. This result is in agreement with other reports.57 The electron lifetime Tn = 1/(2πf) is calculated from the maximum frequency (ωmax) (Fig. S9 and Table S8, ESI†). The different electron lifetimes were recorded at two irradiating angles of the light of 5° and 90°. Several points should be clarified in this result, i.e., increasing the photoinduced electron lifetime caused by deposition of the Ag NPs and a slow photon effect. First, the slow photon effect-induced absorption creates a higher number of hot electrons on the Ag NPs and photoexcited electrons at the conduction band of TiO2 due to the enhanced local EM field, leading to a reduction of Rct upon illumination.53 Second, the increase in the hot electron density on a metal led to the low potential created by the equilibrium of the Fermi levels of Ag and TiO2.5 Consequently, the photoexcited electron on TiO2 in the conduction band is forced to transfer and be trapped in the metal NPs, increasing the electron lifetime and the interfacial charge transfer efficiency (Fig. 8c). The amount of the slow photon effect-induced energy is controlled by the stopband position. As mentioned in Fig. 6 and Table S1 (ESI†), the stopband edges of TIO288 and TIO318 enhance the photon energy caused by the Ag NPs at the irradiation angles of 5° and 90°, respectively. The stopband created by TIO250 at these angles overlapped with the strong absorbance of the Ag NPs, so it did not work to increase light absorption. The increased irradiation angle leads to a red shift in the stopband position, which is far from the low absorption band of the Ag NPs. Consequently, a stronger enhanced photon absorbance is obtained at an illumination angle of 5°. In relation to the stopband positions vs. electron lifetime, the enhanced photon energy is proportional to the photogenerated lifetime, as shown in Fig. 8d. The electron lifetime increases by 1.5 times after introducing Ag NPs and obtains high reproducibility with a film containing the NS-combined Ag NPs. These results are in agreement with the photoluminescence (PL) emission, the intensity of which is associated with the charge carrier recombination rate (Fig. 8e). The PL emission of TiO2 shows the main different contributions, i.e. 380, 398, and 420 nm assigned to the indirect band-to-band recombination between the gap, and 451 and 470 are assigned to the surface oxygen vacancies and defects, respectively.55,58 The average intensity of the PL emission decreases, followed by the tendency of TIO > TIO Ag > TIO AgNS for similar thickness and Ag amount. The introduction of the AgNS NPs is more efficient in slowing down the combination of the electron/hole pairs between the band gaps of TiO2.
The photocatalytic mechanism of the plasmonic metal/semiconductor is a complex process that is contributed by many compositions, i.e., (i) UV-absorption efficiency of TiO2 for photogenerated electrons (UV intensity accounted for 5% of total solar light), (ii) localized surface plasmon resonance caused by metal NPs for generating hot carriers (46% of visible light), and (iii) plasmon-induced resonance energy transfer (PRET) and enhanced local EM field from the plasmonic Ag to increase light absorption of TiO2, and (iv) light scattering and photothermal heat.21,32 The photothermal heat insignificantly affects the photocatalytic efficiency of MB degradation, as illustrated in Fig. 7f. Following the standard influence of the Schottky barrier (φSB), the hot plasmonic carriers from Ag with the appropriate energy can transfer to the CB of TiO2, separating the photogenerated holes and electrons. However, the SPR mechanism in the photocatalytic enhancement is still unclear, supposing the migration of electrons on the metal surface for the photocatalysis.59 In this study, the use of photonic IO engineering enables us to improve the effect of the LSPR and local EM field caused by the Ag NPs, leading to a strong distribution of the photogenerated electrons of TiO2.4,41,54 The IO structure can create a stopband whose edge can enhance the photon energy at the low absorption range of light-harvesting materials. The high density and homogeneous silver NPs were immobilized on the surface of the TiO2 IO wall with expectations, i.e., broadening the visible-light absorbance for Ag–TiO2 and enhancing the photon absorbance in the visible range by the slow photon effect. In summary, TIO288 and TIO318 have a stopband with the blue-shifting edge overlapped with the low absorbance band of the Ag NPs, resulting in an increase in their absorbance by about 15% at a wavelength of 480 nm. As shown, the AgNS-deposited TIO288 and TIO318 showed better MB photodegradation with a maximum photoreaction kinetic constant of 17. 10−3 min−1, responding to a longer photoexcited electron lifetime in an electrolyte medium.
In addition, the high photocatalytic activity of the Ag-deposited TIO films was achieved only when the samples were irradiated on both the UV and visible regions with a broader wavelength range, as shown in Fig. S10 (ESI†). The results present low photocatalytic efficiency when separately irradiating upon a single wavelength of 370 nm (UV) or a multiwavelength greater than 400 nm (Vis), showing a similar photodegradation rate for TIO and Ag-deposited TIO films. The photocatalytic result confirms the distinct role of UV-light adsorbing TiO2 and visible light-adsorbing LSPR Ag NPs in the catalytic mechanisms that support the suggestion of enhancement of LSPR-generated electrons on TiO2 more active than the “hot carriers” generated on Ag NPs. To summarize, the enhanced photocatalysis of the Ag-deposited TiO2 comes from the additional electron/hole pairs of TiO2 that are generated by an electromagnetic (EM) field in the LSPR of Ag NPs.32,60,61 The local EM field enhanced by LSPR, which is lower than the activation energy, excites TiO2 by resonance energy transfer. The local EM field depends on two factors, i.e., the intensity of LSPR is improved by the slow photon effect of the IO structure; the short distance between high-deposited Ag metal and TiO2 which is favorable for transferring plasmonic energy, followed by enhancement of charge carries and lifetime, as demonstrated in Fig. 8d. These electrons continue to transfer to Ti3+ defect states and then further to Ag NPs. The total photogenerated electron on the conduction of TiO2 tends to accumulate within the depletion layer at the Schottky junction at the interface of Ag, resulting in the prevention of hole/electron recombination. This hypothesis agrees with a previous report.32 The second abundant “hot carrier” excited by LSPR on the Ag NPs under visible light absorption is enhanced by the slow photon effect of IO.62,63 With the decrease in photocurrent, the plasmonic-induced hot carriers are preferably generated, and LSPR-generated electrons are suggested to immigrate to the surface of the metal for initiating the reduction of O2 to form oxygen species radicals (−O2˙, ˙OOH, and ˙OH) (Fig. 9). Simultaneously, the holes in TiO2 photo-oxidize H2O to form hydroxyl radicals (˙OH).4,51 The active oxygen and hydroxyl radical species degrade MB.29,32 The high density of the Ag NPs efficiently prevents the hole/electron recombination, resulting in the evolution of oxygen on the interface and promoting the electron transfer to the Ag metal. Consequently, the MB photodegradation kinetics of Ag–TiO2 IO is properly 2 times faster in comparison to that of bare TiO2 IO, resulting in impressive photocatalytic properties and high catalyst stability even after the 14th cycle (Fig. 7e).
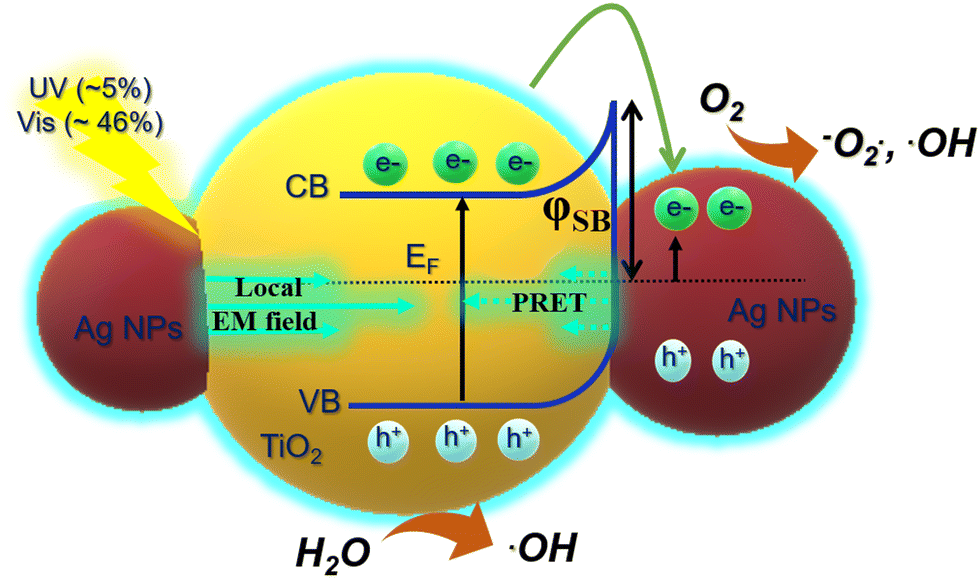 | ||
| Fig. 9 Scheme of the effect of LSPR and slow photon effect on the generation of electron/hole pairs on Ag-deposited TiO2 by absorbing UV-Vis light. | ||
Conclusions
Electrophoretic deposition is an efficient technique for the fabrication of a periodic IO structure with a flexible thickness and pore sizes in a very short time and high reproducibility. The investigation of the slow photon effect coming from these IO structures on the photocatalytic effect of the Ag-deposited TiO2 was studied. The growth of the Ag NPs with a high Ag chemical composition and their homogeneous distribution on the IO walls was strongly supported by the co-deposition of the naphthalene 2,6 disulfonate molecules. TIO288 and TIO318 showed strong slow photon effects during the LSPR absorbance of the Ag NPs, resulting in high photocatalytic kinetics of the MB photodegradation and a longer photogenerated charge lifetime in electrolytes. The decrease in the photocurrent of the Ag–TIO confirms the tendency of the photogenerated electron on TiO2, i.e., the photoexcited electrons on the CB of TiO2 are transferred to the surface of the Ag NPs for redox reactions due to the high concentration of the Ag NPs on the IO walls. The photocatalytic mechanism was also clarified with an impressive point. The slow photon effect in IO structures enhance the LSPR on Ag metal and enhance the local EM field on TiO2, which is mainly attributed to electron/hole pair generation used for photocatalytic reactions. This study provides a good strategy for improving the photocatalytic effect of semiconductor–metal composite systems for water splitting and CO2 reduction.Author contributions
Thi Kim Ngan Nguyen conceived and designed the experiments, performed the experiments, analyzed and interpreted the data, and drafted the article. Fabien Grasset, Satoshi Ishii, Hiroshi Fudouzi, and Tetsuo Uchikoshi critically reviewed the paper to improve the intellectual content.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
These studies were carried out under the continued financial support of the International Center for Young Scientists (ICYS-Sengen), NIMS, and the France-Japan International Collaboration Framework (IRL3629 LINK, ANR-22-CE09-0015). T.K.N Nguyen would like to thank Dr. Suzuki Tohru and Dr. Naoto Shirahata for the laboratory supplies and useful advice. This study was supported by the “Advanced Research Infrastructure for Materials and Nanotechnology in Japan (ARIM)” of the Ministry of Education, Culture, Sports, Science and Technology (MEXT); Proposal Number JPMXP1224NM5061.Notes and references
- P. She, J. S. Qin, H. Rao, B. Guana and J. Yu, Mater. Chem. Front., 2020, 4, 1671–1678 RSC.
- Y. Goto, K. Yamanaka, M. Ohashi, Y. Maegawa and S. Inagaki, Appl. Catal., B, 2021, 287, 119965 CrossRef.
- Y. Guo, J. Sun, T. Guo, Y. Liu and Z. Yao, Angew. Chem., 2023, 36, 19664 Search PubMed.
- J. Schneider, M. Matsuoka, M. Takeuchi, J. Zhang and Y. Horiuchi, Chem. Rev., 2014, 114, 9919–9986 CrossRef.
- H. H. Mohamed and D. W. Bahnemann, Appl. Catal., B, 2012, 128, 91–104 CrossRef.
- C. B. Ong, L. Yong and A. W. Mohammad, Renewable Sustainable Energy Rev., 2018, 81, 536–551 CrossRef.
- Z. Lin, C. Du, B. Yana and G. Yang, Catal. Sci. Technol., 2019, 9, 5582–5592 RSC.
- Y. Sari, P. L. Gareso, B. Armynah and D. Tahir, J. Hydrogen Energy, 2024, 55, 984–996 CrossRef CAS.
- Y. Q. Cao, T. Q. Zi, X.-R. Zhao, C. Liu, Q. Ren, J. B. Fang, W. M. Li and A. D. Li, Sci. Rep., 2020, 10, 13437 CrossRef CAS.
- P. Akhter, S. Nawaz, I. Shafiq, A. Nazir, S. Shafique, F. Jamil, Y.-K. Park and M. Hussain, Mol. Catal., 2023, 535, 112896 CrossRef CAS.
- P. C. Quero-Jiménez, A. Hernández-Ramírez, J. Luis Guzmán-Mar, M. Villanueva-Rodríguez, D. Alejandro Pino-Sandoval and L. Hinojosa-Reyes, J. Photochem. Photobiol., A, 2024, 446, 115154 CrossRef.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef PubMed.
- F. Grasset, G. Starukh, L. Spanhel, S. Ababou-Girard, D. S. Su and A. Klein, Adv. Mater., 2005, 17, 294–297 CrossRef.
- P. Birnal, M. C. Marco de Lucas, I. Pochard, B. Domenichini and L. Imhoff, Appl. Surface Sci., 2020, 512, 145639 CrossRef.
- H. An, L. Cao, R. Cheng, X. Zhang, S. Zhang, Y. Sun, L. Zhao, B. Wang and Z. Yin, CrystEngComm, 2022, 24, 657–666 RSC.
- D. Zhang, J. Chen, Q. Xiang, Y. Li, M. Liu and Y. Liao, Inorg. Chem., 2019, 58, 12511–12515 CrossRef.
- Y. Chen, L. Soler, C. Cazorla, J. Oliveras, N. G. Bastús, V. F. Puntes and J. Llorca, Nat. Commun., 2023, 14, 6165 CrossRef CAS PubMed.
- H. Zhang, P. Sun, X. Fei, X. Wu, Z. Huang, W. Zhong, Q. Gong, Y. Zheng, Q. Zhang, S. Xie, G. Fu and Y. Wang, Nat. Commun., 2024, 15, 4453 CrossRef CAS PubMed.
- M. L. de Souza, D. P. dos Santos and P. Corio, RSC Adv., 2018, 8, 28753–28762 RSC.
- G.-Y. Yao, Z.-Y. Zhao, Q.-L. Liu, X.-D. Dong and Q.-M. Zhao, Sol. Energy Mater. Sol. Cells, 2020, 208, 110385 CrossRef.
- A. Kumar, P. Choudhary, A. Kumar, P. H. C. Camargo and V. Krishnan, Small, 2022, 18, 2101638 CrossRef.
- M. C. Herrera-Beurnio, F. J. López-Tenllado, J. Hidalgo-Carrillo, J. Martín-Gómez, R. Estévez, M. Castillo-Rodríguez, G. de Miguel, F. J. Urbano and A. Marinas, Catal. Today, 2023, 413–415, 113967 CrossRef.
- T. Cai, Y. Liu, L. Wang, S. Zhang, J. Ma, W. Dong, Y. Zeng, J. Yuan, C. Liu and S. Luo, ACS Appl. Mater. Interfaces, 2018, 10, 25350–25359 CrossRef.
- K. Ozawa, M. Emori, S. Yamamoto, R. Yukawa, S. Yamamoto and R. Hobara, Phys. Chem. Lett., 2014, 5, 1953–1957 CrossRef.
- Z. Sun and Y. Fang, Sci. Rep., 2021, 11, 338 CrossRef.
- J. Yu, J. Lei, L. Wang, J. Zhang and Y. Liu, J. Alloys Compd., 2018, 769, 740–757 CrossRef.
- E. Armstrong and C. O'Dwyer, J. Mater. Chem. C, 2015, 3, 6109–6143 RSC.
- A. Zulfiqar, F. Temerov and J. J. Saarinen, ACS Omega, 2020, 5, 11595–11604 CrossRef.
- Z. Chen, L. Fang, W. Dong, F. Zheng, M. Shena and J. Wang, J. Mater. Chem. A, 2014, 2, 824–832 RSC.
- K. Pham, H. Ali-Löytty, J. Saari, M. Zubair, M. Valden, K. Lahtonen, N. Kinnunen, M. Gunell and J. J. Saarinen, Opt. Mater., 2022, 131, 112695 CrossRef.
- S. Loukopoulos, E. Sakellis, M. G. Kostakis, D. Triantafyllos Gerokonstantis, P. Tsipas, S. Gardelis, A. G. Kontos, F. K. Katsaros, Z. Sideratou, G. E. Romanos, A. Dimoulas, N. S. Thomaidis and V. Likodimos, ACS Omega, 2023, 8, 33639–33650 CrossRef PubMed.
- F. Temerov, K. Pham, P. Juuti, J. M. Mäkelä, E. V. Grachova, S. Kumar, S. Eslava and J. J. Saarinen, ACS Appl. Mater. Interfaces, 2020, 12, 41200–41210 CrossRef.
- M. Bijl, K. R. G. Lim, S. Garg, N. J. Nicolas, N. L. Visser, M. Aizenberg, J. E. S. van der Hoeven and J. Aizenberg, Nanoscale, 2024, 16, 13867–13873 RSC.
- P. Birnal, M. C. Marco de Lucas, I. Pochard, F. Herbst, O. Heintz, L. Saviot, B. Domenichini and L. Imhoff, Appl. Surface Sci., 2023, 609, 155213 CrossRef.
- S. Furumi, H. Fudouzia and T. Sawadaa, J. Mater. Chem., 2012, 22, 21519–21528 RSC.
- T. K. N. Nguyen, F. Grasset, S. Cordier, N. Dumait, S. Ishii, H. Fudouzi and T. Uchikoshi, Mater. Today Chem., 2023, 27, 101351 CrossRef.
- L. Mulfinger, S. D. Solomon, M. Bahadory, A. V. Jeyarajasingam, S. A. Rutkowsky and C. Boritz, J. Chem. Educ., 2007, 84, 322 CrossRef.
- M. Curti, J. Schneider, D. W. Bahnemann and C. B. Mendive, J. Phys. Chem. Lett., 2015, 6, 3903–3910 CrossRef PubMed.
- A. Lonergan, C. Hu and C. O’Dwyer, Phys. Rev. Mater., 2020, 4, 065201 CrossRef.
- A. Richel, N. P. Johnson and D. W. McComb, Appl. Phys. Lett., 2000, 76, 1816–1818 CrossRef.
- J. Zhao, S. Xue, R. Ji, B. Lia and J. Li, Chem. Soc. Rev., 2021, 50, 12070–12097 RSC.
- E. A. Coronado, E. R. Encinaa and F. D. Stefani, Nanoscale, 2011, 3, 4042–4059 RSC.
- R. Khana, S. WooKima,T., J. Kima and C.-M. Namb, Mater. Chem. Phys., 2008, 112, 167–172 CrossRef.
- G. Horányi, J. Colloid Interface Sci., 2003, 261, 580–583 CrossRef.
- U. Suzuki, T. Kameda, S. Kumagai, Y. Saito and T. Yoshioka, J. Water Process Eng., 2022, 49, 103081 CrossRef.
- G. B. Helene, P. de Souza Ono and D. S. Pellosi, ChemistrySelect, 2023, 8, 202302767 CrossRef.
- L. Ruangchuaya, J. Schwankb and A. Sirivata, Appl. Surface Sci., 2002, 199, 128–137 CrossRef.
- Q. Q. Liu, K. F. Yue, X. J. Wenga and Y. Y. Wang, CrystEngComm, 2019, 21, 6186–6195 RSC.
- N. J. Firet, M. A. Blommaert, T. Burdyny, A. Venugopal, D. Bohra, A. Longo and W. A. Smith, J. Mater. Chem. A, 2019, 7, 2597–2607 RSC.
- S. Cheng, C. Li, Z. Yu, Y. Sun, L. Lia and J. Yanga, RSC Adv., 2020, 10, 9814–9823 RSC.
- C. César, Nat. Photonics, 2014, 8, 95–103 CrossRef.
- Y. Tian and T. Tatsuma, Chem. Commun., 2004, 1810–1811 RSC.
- G. Baffou, I. Bordacchini, A. Baldi and R. Quidant, Light: Sci. Appl., 2020, 9, 108 CrossRef PubMed.
- P. A. Gross, S. N. Pronkin, T. Cottineau, N. Keller, V. Keller and E. R. Savinova, Catal. Today, 2012, 189, 93–100 CrossRef.
- G. Zerjav, M. Roskaric, J. Zavasnik, J. Kovac and A. Pintar, Appl. Surf. Sci., 2022, 579, 152196 CrossRef.
- J. Ângelo, P. Magalhães, L. Andrade and A. Mendes, Appl. Surf. Sci., 2016, 387, 183–189 CrossRef.
- Z. Hosseini, N. Taghavinia, N. Sharifi, M. Chavoshi and M. Rahman, J. Phys. Chem. C, 2008, 112, 18686–18689 CrossRef.
- A. Saha, A. Moya, A. Kahnt, D. Iglesias, S. Marchesan, R. Wannemacher, M. Prato, J. J. Vilatela and D. M. Guldi, Nanoscale, 2017, 9, 7911–7921 RSC.
- P. Kumari, N. Bahadur, L. Kong, L. A. Odell, A. Merenda and L. F. Dumee, Mater. Adv., 2022, 3, 2309–2323 RSC.
- T. Kong, A. Liao, Y. Xu, X. Quao, H. Zhang, L. Zhang and C. Zhang, RSC Adv., 2024, 14, 17041–17050 RSC.
- V. Amendola, R. Pilot, M. Frasconi, O. M. Marago and M. A. J. Lati, Phys. Condens. Matter, 2017, 29, 203002 CrossRef PubMed.
- R. Kamarudheen, G. W. Castellanos, L. P. J. Kamp, H. J. H. Clercx and A. Baldi, ACS Nano, 2018, 12, 8447–8455 CrossRef PubMed.
- K. Awazu, M. Fujimaki, C. Rockstuhl, J. Tominaga, H. Murakami, Y. Ohki, N. Yoshida and T. A. Watanabe, J. Am. Chem. Soc., 2008, 130, 1676–1680 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma00807c |
| This journal is © The Royal Society of Chemistry 2024 |