
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBifunctional molybdenum and vanadium materials: semiconductor properties for advanced electronics and catalytic efficiency in linalool oxidation†
Josipa
Sarjanović
 a,
Mateja
Cader
a,
Edi
Topić
a,
Marta
Razum
b,
Dominique
Agustin
cd,
Mirta
Rubčić
a,
Luka
Pavić
*b and
Jana
Pisk
*a
a,
Mateja
Cader
a,
Edi
Topić
a,
Marta
Razum
b,
Dominique
Agustin
cd,
Mirta
Rubčić
a,
Luka
Pavić
*b and
Jana
Pisk
*a
aDepartment of Chemistry, Faculty of Science, University of Zagreb, Horvatovac 102a, 10000 Zagreb, Croatia. E-mail: jana.pisk@chem.pmf.hr
bDivision of Materials Chemistry, Ruđer Bošković Institute, Bijenička 54, 10000 Zagreb, Croatia. E-mail: luka.pavic@irb.hr
cLCC-CNRS (Laboratoire de Chimie de Coordination), 205 Route de Narbonne, BP44099, CEDEX 4, 31077 Toulouse, France
dDepartment of Chemistry, IUT Paul Sabatier, Université Paul Sabatier, University of Toulouse, Av. G. Pompidou, CS20258, 81104 Castres, France
First published on 28th October 2024
Abstract
Polynuclear and mononuclear molybdenum(VI) complexes, coordinated with water or methanol, were synthesized using acyl-hydrazone ligands, derived from the reactions of 2-hydroxy-3-methoxybenzaldehyde with formic – (H2L1) or acetic acid hydrazide (H2L2). Characterization of the complexes was conducted utilizing advanced spectroscopic techniques and elemental analysis. Crystal and molecular structures of ligand H2L2, and complexes [MoO2(L1)(H2O)], [MoO2(L2)(MeOH)], together with (Hpy)2Mo8O26 were determined by single crystal X-ray diffraction. The thermogravimetry provided insights into the thermal stability and decomposition patterns of the complexes. In situ solid-state impedance spectroscopy was employed, revealing correlations between the electrical properties and the thermal and structural transformations of Mo complexes. This multifaceted approach enabled a profound understanding of the interplay between structure, thermal behaviour, and electrical characteristics. The polynuclear complex [MoO2(L1)]n exhibited remarkable conductivity, achieving values up to 10−8 (Ω cm)−1 at room temperature. This performance, compared to previously reported vanadium-based analogues, highlights its considerable potential for integration into electronic device manufacturing. Additionally, the catalytic efficiency of these newly synthesized molybdenum complexes was evaluated in linalool oxidation, alongside previously reported vanadium compounds, further demonstrating their promising applications in catalysis.
Introduction
Molybdenum(VI) and vanadium(V, IV) coordination complexes are renowned for their versatile applications, primarily due to their biological significance and potential as antitumor and antiviral agents.1,2 These complexes also have extensive industrial applications, capitalising on catalytic capabilities in sulfoxidation, epoxidation, and oxidation processes.3–5 Additionally, materials containing molybdenum, due to good electrical properties, are suitable for semiconductor use, with DC conductivity on the order of magnitude up to 10−9–10−12 (Ω cm)−1.6,7 Furthermore, vanadium(IV) complexes with tetradentate Schiff bases and vanadium(IV) indanone derivatives show semiconductor characteristics, making them suitable for use in manufacturing electronic devices such as light-emitting diodes, rechargeable batteries, sensors, and other electronic devices.8,9 In our recent publication,10 we investigated electrical properties of oxo- and dioxo vanadium(V) complexes coordinated by ligands derived from 2-hydroxy-3-methoxybenzaldehyde and acetic acid hydrazide, Scheme 1, ligand H2L2, with general formulas [VO(L2)(OMe)(MeOH)], [VO(L2)(OEt)(H2O)], and [VO2(HL2)]2·2H2O. Furthermore, we studied similar vanadium coordination compounds: (NH4)[VO2(L1*)], [VO2(HL2*)], and [VO(L1*)(OMe)(MeOH)], where the ligands were derivatives of acetic acid hydrazide (ESI,† Scheme S1).10 One of the investigated compounds, [VO(L2)(OEt)(H2O)], exhibited high conductivity being 10−7 (Ω cm)−1 at 120 °C, while others from the study are in the range of 10−11–10−13 (Ω cm)−1.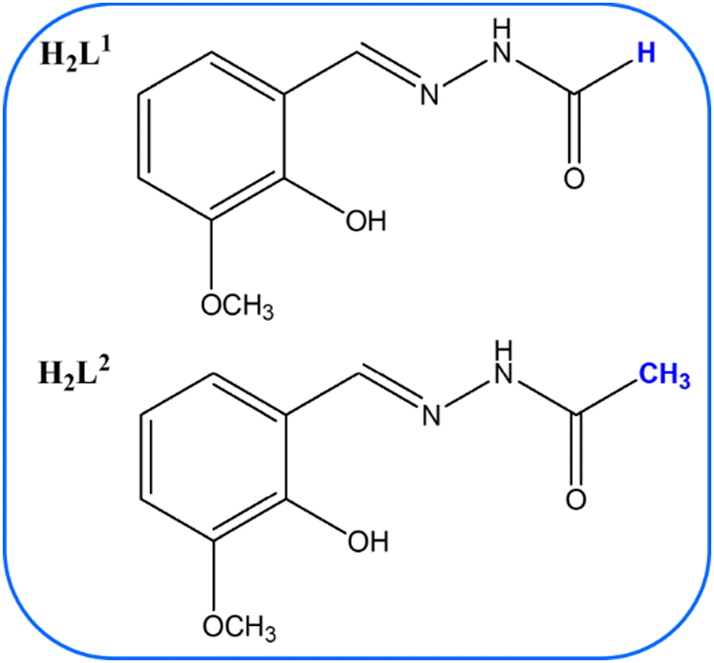 | ||
| Scheme 1 Ligands used for the presented investigation. | ||
Previously, we studied the variation of the aldehyde component of the acyl hydrazine.10 In this study, we focused on the hydrazide part of the acyl hydrazone and examined how the presence of hydrogen (H) or methyl (CH3) group at the end of the chain affected the structural and consequently electrical properties of Mo coordination complexes (Schemes 1 and 2). Therefore, solid-state impedance spectroscopy was employed to conduct an in-depth analysis of the in situ transformation of molybdenum compounds.
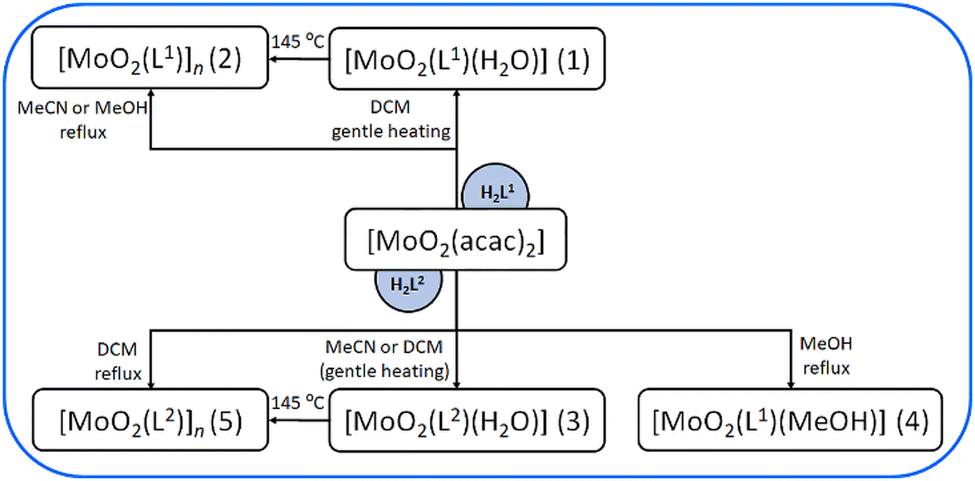 | ||
| Scheme 2 The solution-based synthetic route towards Mo complexes. MeCN = acetonitrile, DCM = dichloromethane (gente heating), MeOH = methanol. | ||
Mo complexes derived from aroyl-hydrazones and thiosemicarbazones are well-studied in the literature.11–15 On the contrary, it is worth noting that molybdenum (Mo) compounds derived from acyl-hydrazone ligands are to the best of our knowledge rarely reported in the literature, presenting challenges for material production.16 To understand the full potential of these systems, catalytic studies on linalool oxidation were explored and further broadened towards previously characterised vanadium coordination compounds. Linalool is an interesting substrate due to its biological origin,17,18 while its oxidation products (pyranoid and furanoid) are essential in biochemistry, medicine, and the food industry, due to their structural properties and biological activities.19–21 Further, the number of publications reporting linalool oxidation employing transition metal-based catalysts is quite limited.7,22–25 Our approach includes a horizontal comparison between Mo and V catalysts, and a vertical comparison among Mo catalysts featuring two types of ligands and coordination complexes. This dual comparative viewpoint provides a deeper understanding of the catalytic and electrical properties and potential applications of these compounds. While a direct correlation between these properties cannot be established, given that electrical properties and semiconducting potential are assessed in the solid state, while catalytic reactions occur in solution, indirect correlations are possible. Namely, obvious correlations exist between structural and electrical properties, as well as between structural and catalytic performance. Ultimately, our goal is to develop materials exhibiting dual functionalities, capable of serving simultaneously as catalysts and semiconductors.
Results and discussion
Synthesis, IR characterisation and thermal behaviour of molybdenum(VI) compounds
Both ligands were synthesized using standard solution-based methods as well as mechanochemical techniques. While the solution-based method yielded high product quantities, the mechanochemical approach minimized solvent usage, aligning with green chemistry principles. IR characterisation and DSC data are presented in Table 1 and ESI,† Fig. S1, S2, S8 and S9. Further, the reactions of the prepared ligands H2L1,2 with the starting molybdenum(VI) precursor ([MoO2(acac)2]) were carried out in different solvents (methanol, acetonitrile, dichloromethane), in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio (Scheme 2).
:1 molar ratio (Scheme 2).
| Compound | IR bands/cm−1 | |||||
|---|---|---|---|---|---|---|
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N N |
C–Ohydr | C–Ophe | MoO |
H2O | MeOH | |
| [MoO2(L1)]n (2) | 1595 | 1308 | 1259 | 922, 877 | — | — |
| [MoO2(L2)]n (5) | 1608 | 1310 | 1265 | 922, 834 | — | — |
| [MoO2(L1)(H2O)] (1) | 1599 | 1319 | 1264 | 895, 861 | 3343 | — |
| [MoO2(L2)(H2O)] (3) | 1596 | 1328 | 1254 | 906, 866 | 3244 | — |
| [MoO2(L2)(MeOH)] (4) | 1612 | 1337 | 1259 | 906, 868 | — | 1028 |
| Ligand | CN |
C–OH | CO |
|||
| H2L1 | 1605 | 3195 | 1688 | |||
| H2L2 | 1609 | 3185 | 1672 | |||
The reaction between [MoO2(acac)2] and H2L1 in methanol and acetonitrile yielded a polynuclear complex [MoO2(L1)]n (2), while with H2L2 in dichloromethane, under vigorous heating, produced [MoO2(L2)]n (5). Both brown-coloured complexes were analysed via IR-ATR spectroscopy (Tables 1, 2 and ESI,† Fig. S3 and S4). Broad bands at 922 cm−1 and 877 cm−1 ([MoO2(L1)]n (2)) and 834 cm−1 ([MoO2(L2)]n (5)) were attributed to intramolecular stretching of MoO⋯Mo bonds.26,27 On the other hand, gentle heating in DCM, yielded mononuclear complexes of general formula [MoO2(L1or2)(H2O)] (1) and (3), respectively. Interestingly, the complex [MoO2(L2)(H2O)] (3) could be obtained if the reaction of [MoO2(acac)2] and H2L2 was provided in acetonitrile, also under gentle heating, suggesting the moisture from the solvent facilitates the coordination of a water molecule at the sixth coordination site of Mo. IR analysis (ESI,† Fig. S5 and S6) showed that bands at 895 and 861 cm−1 ([MoO2(L1)(H2O)] (1)) and 906 and 866 cm−1 ([MoO2(L2)(H2O)] (3)) are characteristic of stretching of the OMoO group, while the band around 3250–3350 cm−1 corresponds to O–H stretching of the water molecule.7 Additionally, the reaction with H2L2, in methanol, produced the mononuclear complex [MoO2(L2)(MeOH)] (4), in which the methanol molecule occupied the sixth coordination site. Bands at 906 and 868 cm−1 were observed, indicating stretching of the OMoO bond, while a band at 1028 cm−1 was characteristic for MeOH (ESI,† Fig. S7).26,27 In all the complexes, the coordination of the ligand to the Mo centre is confirmed through the absence of the band characterizing CO groups of the neutral ligand at 1688 (H2L1) and 1672 cm−1 (H2L2), implying keto–enol tautomerization and consequential appearance of the band at ∼1250 cm−1 characteristic for C–O.26,27
| Compound | TGA analysis/°C | ||
|---|---|---|---|
| H2O | MeOH | Complex decomposition | |
| [MoO2(L1)]n (2) | — | — | 275–475 |
| [MoO2(L2)]n (5) | — | — | 280–545 |
| [MoO2(L1)(H2O)] (1) | 110–125 | — | 270–520 |
| [MoO2(L2)(H2O)] (3) | 120–140 | — | 280–535 |
| [MoO2(L2)(MeOH)] (4) | — | 100–140 | 265–525 |
Upon heating in an oxygen atmosphere, both polynuclear complexes [MoO2(L1)]n (2) and [MoO2(L2)]n (5) exhibited a single-step mass loss, with decomposition occurring within distinct temperature ranges: 275–475 °C for [MoO2(L1)]n (2) and 280–545 °C for [MoO2(L2)]n (5), ESI,† Fig. S10 and S11. Decomposition of [MoO2(L1)(H2O)] (1), ESI,† Fig. S12, occurred in two steps: first, the release of a water molecule between 110 and 125 °C, followed by the complex decomposition between 270 and 520 °C. A similar analysis was conducted for [MoO2(L2)(H2O)] (3), ESI,† Fig. S13, revealing analogous decomposition steps in the temperature range of 120–140 °C and 280–535 °C, respectively. TGA for [MoO2(L2)(MeOH)] (4), revealed a two-step decomposition process, ESI,† Fig. S14, involving the release of coordinated methanol 100–140 °C, followed by the complex decomposition between 265 and 525 °C. All the resulting white residues were identified as MoO3 through IR-ATR and PXRD comparison with commercially available molybdenum(VI) oxide. The experimentally obtained amounts of MoO3 corresponded to the theoretically calculated ones.
When the mononuclear complexes [MoO2(L1or2)(H2O)] were heated at 145 °C for one hour, transformation to polynuclear species [MoO2(L1or2)]n occurred. While [MoO2(L1)]n (2) remained stable and did not convert to the mononuclear complex upon exposure to water vapours, [MoO2(L2)]n (5) transformed into [MoO2(L2)(H2O)] (3) after 24 hours of exposure. The transitions were confirmed via IR-ATR spectra comparison.
Furthermore, the mononuclear complex [MoO2(L1)(H2O)] (1) and [MoO2(L2)(MeOH)] (4) could also be obtained via mechanochemical synthesis using methanol as solvent. At the same time, no reaction occurred when methanol was substituted by dichloromethane or acetonitrile, as evidenced by IR spectra comparison. The unexpected formation of [MoO2(L1)(H2O)] (1), assisted by MeOH, can likely be explained by the ball milling energy and the presence of water in the MeOH solvent, which together promote the formation of a water-coordinated species.
Crystal and molecular structure
In this work a total of six crystal structures were determined (ESI,† Table S1), that of side-product (pyH)4(Mo8O26), ligands H2L1, H2L2, and complexes [MoO2(L1)(H2O)] (1), [MoO2(L2)(H2O)] (3) and [MoO2(L2)(MeOH)] (4). The ligand H2L1 and complex [MoO2(L2)(H2O)] (3) were known in the database as references CCDC 69881228 and 2001210,16 respectively. Surprisingly, and to the best of our knowledge, the crystal structure of (pyH)4(Mo8O26) (Fig. 1a and ESI,† Fig. S15) was not reported before, even though a significant number of pyridinium polyoxomolybdates are known. The structure features a centrosymmetric β-octa molybdate anion and pyridinium cations that engage in linear and bifurcated hydrogen bonds with the axial oxygen atoms. | ||
| Fig. 1 (Supra)molecular structures of (a) pyridinium octamolybdate, (b) H2L1, (c) [MoO2(L2)(H2O)] and (d) [MoO2(L2)(MeOH)], showcasing differences in hydrogen bonding. | ||
H2L1 and H2L2 hydrazones (Fig. 1b and ESI,† Fig. S16 and S17) are in the expected keto-amino tautomeric form as evidenced by relevant bond lengths (ESI,† Table S2) and anti-conformation. Both form supramolecular homodimers through amide (CO)–NH synthons. Significant differences however exist in the arrangement of the dimers. While in H2L1 the dimers pack in parallel stacks forming a zig-zag arrangement, the presence of an auxiliary methyl group in H2L2 causes the dimers to extend into sheets through (CO)–CH3 short contacts (ESI,† Table S3).
Similarly, the dioxomolybdenum(VI) complexes (Fig. 1c and d) derived from H2L1 and H2L2 also show features well-established in the related works. The dianion L2− is in an enol-imino tautomeric form (ESI,† Table S2) in all three cases, forming an ONO chelating pocket. The sixth coordination site of {MoO2}2+ core is occupied by the solvent molecule. However, the packing of the molecules in the crystal structures differs greatly between the three complexes.
The complex [MoO2(L1)(H2O)] (1), ESI,† Fig. S18, is noteworthy for reassessment as it presents the sturdiest supramolecular architecture in the presented set of crystal structures. The molecules form unusually complex R66(24) and larger supramolecular rings through Ow–H⋯OMo and Ow–H⋯Namide hydrogen bonds. Additional hydrogen bonds from the water ligand, not involved in a ring formation, interact with neighbouring complex molecules, creating an extended supramolecular network.
In contrast, the [MoO2(L2)(H2O)] (3) complex, previously reported,16 (Fig. 1c and ESI,† Fig. S19) forms somewhat less intertwined supramolecular chains via Ow–H⋯Ophen, Ow–H⋯Omethoxy, and Ow–H⋯Namide hydrogen bonds. These chains are weakly interconnected through numerous MoOax⋯H–C contacts, differing from the L1 counterpart by exhibiting a linear rather than a net-like arrangement. The structure of [MoO2(L2)(MeOH)] (4) (Fig. 1d and ESI,† Fig. S20) lacks a ditopical hydrogen bond donor, resulting in the formation of non-interacting homodimers through OMeOH–H⋯Namide hydrogen bonds. These dimers, formed between symmetrically equivalent molecules, produce two geometrically distinct but similar types, stabilized by various weak C–H⋯O contacts. The differences in supramolecular architectures can be correlated with observed distinctions in electrical measurements obliterated with impedance spectroscopy, discussed in detail vide infra, paragraph on electrical properties.
Catalytic properties of molybdenum- and vanadium-based materials for linalool oxidation
Five molybdenum complexes [MoO2(L1)]n (2), [MoO2(L2)]n (5), [MoO2(L1)(H2O)] (1), [MoO2(L2)(H2O)] (3), [MoO2(L2)(MeOH)] (4), and five vanadium complexes (NH4)[VO2(L1*)], [VO2(HL2*)], [VO(L1*)(OMe)(MeOH)], [VO(L2)(OEt)(H2O)], [VO(L2)(OMe)(MeOH)] were used as catalysts for linalool oxidation. The initial idea was the comparison of the catalytic activity based on (i) different metal centres (Mo vs. V), (ii) the use of oxidant in different solvents (TBHP in decane vs. TBHP in water), and (iii) the addition of organic solvent to the reaction media (H2O2vs. H2O2 and MeCN). The obtained data are presented in Table 3 and ESI,† Fig. S22–S25, and Table S4. Generally, Mo compounds are superior catalysts for linalool epoxidation, exhibiting higher activity and selectivity towards the formation of furanoid and pyranoid products. For Mo catalysts, TBHP in decane or water as a solvent did not affect the maximum conversion rate. However, the furanoid yield remained consistent, regardless of the TBHP used, ranging from 55% to 70%, with the highest yield achieved by [MoO2(L2)(MeOH)] (4). The pyranoid yield was approximately 10% higher when TBHP in water was used, resulting in around 30% yield compared to around 20% with TBHP in decane, and this trend was similar across all the Mo catalysts tested. The highest TOF value, 598, was achieved with TBHP in decane in the presence of the catalyst [MoO2(L2)(H2O)] (3), indicating the fastest conversion of the complex into the pentacoordinated active species that was proven to be the first step of linalool oxidation,3 since it was proven that those oxacycles are obtained from a transient epoxide.22 This was proven by TOF values for [MoO2(L1)(H2O)] (1), [MoO2(L2)]n (5), and [MoO2(L1)]n (2), in descending order. Unlike TBHP, using H2O2 as an oxidant did not enhance activity or selectivity towards furanoid and pyranoid products. Adding MeCN to the reaction mixture significantly increased linalool conversion and tremendously boosted the TOF parameter. However, the furanoid yield was three times lower compared to TBHP. The pyranoid yield was generally lower with H2O2 than with TBHP, and surprisingly, the addition of MeCN to H2O2 further reduced the catalytic efficiency for pyranoid formation, negatively impacting its yield.| Catalyst | Conversiona/% | TOF20minb | TONc | Furanoid yield/% | Pyranoid yield/% | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| TBHPdec | TBHPaq | TBHPdec | TBHPaq | TBHPdec | TBHPaq | TBHPdec | TBHPaq | TBHPdec | TBHPaq | |
| * TBHP stands for test-butyl hydroperoxide, H2O2 for hydrogen peroxide, MeCN acetonitrile, dec for decane and aq for aqueous.a Linalool consumed at the end of reaction.b n(linalool) transformed/n(catalyst)/time(h) at 20 min.c n(linalool) transformed/n(catalyst) at the end of reaction. | ||||||||||
| [MoO2(L1)(H2O)] (1) | 547 | 270 | 61 | 57 | 20 | 28 | ||||
| [MoO2(L1)]n (2) | >99 | 511 | 435 | 200 | 56 | 57 | 18 | 29 | ||
| [MoO2(L2)(H2O)] (3) | 598 | 234 | 57 | 58 | 18 | 29 | ||||
| [MoO2(L2)(MeOH)] (4) | 437 | 280 | 71 | 61 | 18 | 29 | ||||
| [MoO2(L2)]n (5) | 523 | 405 | 59 | 57 | 18 | 33 | ||||
| H2O2 | H2O2 MeCN | H2O2 | H2O2 + MeCN | H2O2 | H2O2 + MeCN | H2O2 | H2O2 + MeCN | H2O2 | H2O2 + MeCN | |
| [MoO2(L1)(H2O)] (1) | 39 | 79 | 21 | 104 | 78 | 157 | 21 | 25 | 16 | 14 |
| [MoO2(L1)]n (2) | 50 | 85 | 71 | 301 | 100 | 167 | 21 | 18 | 16 | 8 |
| [MoO2(L2)(H2O)] (3) | 45 | 84 | 21 | 187 | 91 | 169 | 23 | 17 | 15 | 7 |
| [MoO2(L2)(MeOH)] (4) | 59 | 85 | 15 | 429 | 119 | 167 | 19 | 19 | 13 | 9 |
| [MoO2(L2)]n (5) | 69 | 83 | 87 | 357 | 138 | 165 | 11 | 23 | 5 | 10 |
| TBHPdec | TBHPaq | TBHPdec | TBHPaq | TBHPdec | TBHPaq | TBHPdec | TBHPaq | TBHPdec | TBHPaq | |
| [VO(L2)(OEt)(H2O)] | >99 | >99 | 351 | 345 | 198 | 200 | 12 | 20 | 4 | 14 |
| [VO(L2)(OMe)(MeOH)] | 325 | 349 | 200 | 12 | 19 | 2 | 14 | |||
| H2O2 | H2O2 MeCN | H2O2 | H2O2 + MeCN | H2O2 | H2O2 + MeCN | H2O2 | H2O2 + MeCN | H2O2 | H2O2 + MeCN | |
| [VO(L2)(OEt)(H2O)] | 24 | 81 | 63 | 469 | 48 | 162 | 6 | 27 | 3 | 33 |
| [VO(L2)(OMe)(MeOH)] | 22 | 81 | 60 | 479 | 44 | 161 | 7 | 27 | 4 | 15 |
Linalool oxidation reactions were also tested with V compounds. The most interesting comparison is between catalysts [VO(L2)(OMe)(MeOH)] with the [MoO2(L2)(MeOH)] (4). As shown in Table 3 and ESI,† Table S4, the V catalyst achieves the highest conversion and TON in reactions with TBHP. However, its TOF is lower compared to [MoO2(L2)(MeOH)] (4). Additionally, the yields of desired pyranoid and furanoid products are significantly lower with the V catalyst than with [MoO2(L2)(MeOH)] (4). Furthermore, reactions utilizing H2O2 as the oxidizing agent are less effective than those using TBHP. Detailed discussion about linalool oxidation assisted by vanadium catalysts can be found in ESI.† When comparing to the previously published results, where the Mo loading was 0.25 mol%, and an oxidising agent was TBHP in water, the furanoid yield was 44% for the [MoO2(L)(H2O)] and 27% for [MoO2(L)(MeOH)] (L stands for the doubly deprotonated form of the ligand obtained by the synthesis of 2-hydroxy-5-nitrobenzaldehyde and benzhydrazide) while pyranoid was 18 and 11%, respectively.7 On the other hand, [MoO2(SAP)]2 (SAP is the doubly deprotonated form of salicylideneaminophenol) catalyst,22 operating under the same conditions provided a furanoid yield of 27% and pyranoid yield of 11%. Significant improvement was noticed when employing TBHP in decane, since the furanoid yield was 52%, and the pyranoid 13%.
Electrical properties of molybdenum-based materials and comparison with vanadium-based ones
The electrical properties of molybdenum complexes were investigated employing solid-state impedance spectroscopy (ss-IS), a powerful technique applied to various materials, amorphous and/or crystalline ones.29–32 Five distinct complexes [MoO2(L1)]n (2), [MoO2(L2)]n (5), [MoO2(L1)(H2O)] (1), [MoO2(L2)(H2O)] (3), [MoO2(L2)(MeOH)] (4), utilizing two ligands (H2L1 and H2L2) and synthesized from methanol, acetonitrile, and dichloromethane, were studied over a broad frequency and temperature range. Due to the potential decomposition of these complexes with temperature increase (loss of solvent or amorphization of the complex itself), results of thermal analysis were carefully evaluated to guide the planning of electrical measurements, thereby ensuring accuracy and reliability. All IS measurements incorporated a thermal cycling process, where the temperature was ramped from 30 °C to 200 °C (heating run) and subsequently back to 30 °C (cooling run) across a wide frequency spectrum.Fig. 2(a) and (b) illustrates the conductivity spectra for polynuclear compounds [MoO2(L1)]n (2) and [MoO2(L2)]n (5), respectively, during the cooling cycle. These spectra are characteristic of all measured samples and exhibit two distinct regions. The first region is the frequency-dependent part (dispersion), which is prominent in the high-frequency domain and is more significant at lower temperatures. As the temperature rises, the frequency-dependent region shifts beyond the measurement frequency range, giving way to the second frequency-independent section (i.e. plateau), known as DC (direct current) conductivity, which becomes dominant part of the spectrum. The extent of this plateau, indicative of the material's electrical properties, varies among samples. Accordingly, it can be observed that in [MoO2(L1)]n (2), the DC plateau spans a larger frequency range compared to [MoO2(L2)]n (5) one.
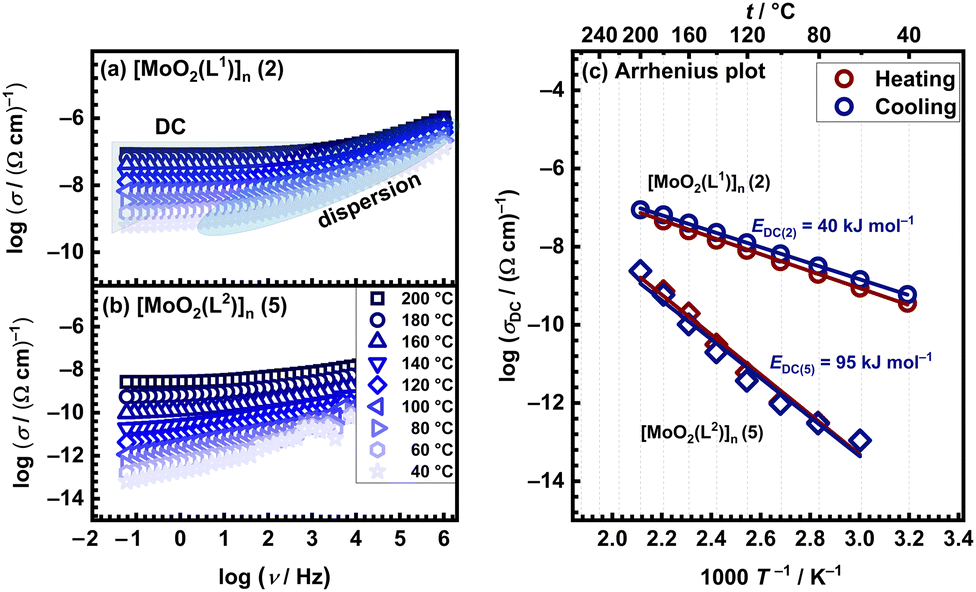 | ||
| Fig. 2 Conductivity spectra for polynuclear (a) [MoO2(L1)]n (2) compound and (b) [MoO2(L2)]n (5) compound in the cooling run, along with (c) Arrhenius plots for DC conductivity in both runs for both polynuclear complexes (red circle – heating, blue circle – cooling). | ||
Furthermore, it is noteworthy that both [MoO2(L1or2)]n compounds exhibited no significant changes during the heating/cooling run. This observation aligns with the TG analysis, which indicates a single-step decomposition occurring at 275–280 °C, Table 2. Such thermal stability is anticipated, given that these compounds lack coordinated or crystalline solvent molecules that could be lost during the heating run.
All studied compounds exhibit semiconductive behaviour, characterized by an Arrhenius-type dependence of DC conductivity, from which the characteristic activation energy can be derived.
The activation energy for DC conductivity, EDC, is determined from the slope of log(σDC) vs. 1000/T using the following equation:
| σDC = σ0*exp(–EDC/kBT), | (1) |
| Compound | σ DC /(Ω cm)−1 | logσ*0/(Ω cm)−1 |
E DC/kJ mol−1 (h) | E DC/kJ mol−1 (c) |
|---|---|---|---|---|
| a Measured at 200 °C. | ||||
| [MoO2(L1)(H2O)] (1) | 1.72 × 10−11 | –4.29 | — | 60 |
| [MoO2(L1)]n (2) | 8.77 × 10−8 | –2.69 | 42 | 40 |
| [MoO2(L2)(H2O)] (3) | 1.80 × 10−10 | –0.78 | — | 84 |
| [MoO2(L2)(MeOH)] (4) | 8.96 × 10−11 | –1.32 | — | 81 |
| [MoO2(L2)]n (5) | 2.66 × 10−9 | 1.58 | 97 | 95 |
This range aligns with those seen in a variety of semiconductive materials,6,7,10,33–39 both amorphous and crystalline, indicating in our case dominant electronic transport mechanisms. Furthermore, considering low conductivity values at lower temperatures, and the absence of a well-defined DC plateau in conductivity spectra, the DC conductivity values were determined by analysing the impedance spectra using equivalent electrical circuit (EEC) modelling, specifically through the complex nonlinear least squares (CNLLS) method. The employed simple EEC model is based on the R-CPE parallel circuit, where R denotes the resistor corresponding to the sample resistance, and the constant phase element, CPE, determines the sample capacitance.
For polynuclear compounds [MoO2(L1or2)]n (2 and 5, respectively) there are negligible differences in different runs (heating vs. cooling) which is an indicator of temperature stability and absence of compound transformations in the studied temperature range, Fig. 2(c). For [MoO2(L1)]n (2) the EDC value is determined to be ∼40 kJ mol−1, whereas for [MoO2(L2)]n (5) is ∼95 kJ mol−1 correlating with their differences in conductivity.
At 200 °C during the cooling run polynuclear compound [MoO2(L1)]n (2) exhibits a DC conductivity of 8.77 × 10−8 (Ω cm)−1, which is almost two orders of magnitude higher than polynuclear compound [MoO2(L2)]n (5), with DC conductivity being 2.66 × 10−9 (Ω cm)−1. In both cases, the dominant transport is electronic, while EDC and higher DC conductivity of the polynuclear compound [MoO2(L1)]n (2) suggest a more facile and continuous transfer of charge carriers through its structure. Despite the only structural difference between ligands H2L1 and H2L2 being a single methyl group, this small variation seems to significantly impact conductivity and activation energy. Typically, an electron-donating group like methyl (CH3) is expected to enhance conductivity compared to a hydrogen atom (H). However, in this case, replacing the methyl group with a hydrogen atom results in higher conductivity values comparing the [MoO2(L1or2)]n (2 and 5) compounds. The hydrogen atom is smaller than the methyl group, which may lead to better planarity of the molecule. Enhanced planarity can facilitate better π–π stacking interactions between molecules, creating more efficient charge transport pathways. Additionally, the absence of the methyl group reduces steric hindrance, allowing for tighter molecular packing, which should strengthen intermolecular interactions, and potentially improve conductivity.
Conversely, the mononuclear compounds [MoO2(L1)(H2O)] (1), [MoO2(L2)(H2O)] (3) and [MoO2(L2)(MeOH)] (4), exhibit discrepancies during measuring cycles. Representative spectra of the heating vs. cooling run for compound [MoO2(L1)(H2O)] (1) are shown in Fig. 3(a) and (b), while those for [MoO2(L2)(H2O)] (3) and [MoO2(L2)(MeOH)] (4) are in the ESI,† Fig. S26 and S27, respectively. For all three mononuclear compounds, in both runs, one can observe the predominance of the frequency-dependent component in the conductivity spectra, consequently having lower DC values compared to the polynuclear ones [MoO2(L1or2)]n (2 and 5, respectively), see Table 4. The observed differences in heating and cooling conductivity spectra could be related to structural changes associated with the release of coordinated solvent, i.e. H2O and MeOH, during IS measurements from 100 to 140 °C, Table 2. Furthermore, the changes in DC conductivity values in the different runs are more pronounced in the Arrhenius plots, Fig. 3(c) and ESI,† Fig. S26 and S27(c). Notably, the heating run exhibits non-monotonic DC conductivity changes, while the cooling shows a monotonic change, suggesting structural transformations in heating while being stable in cooling. Activation energies obtained from cooling run for mononuclear compounds after transformation, are listed in Table 4.
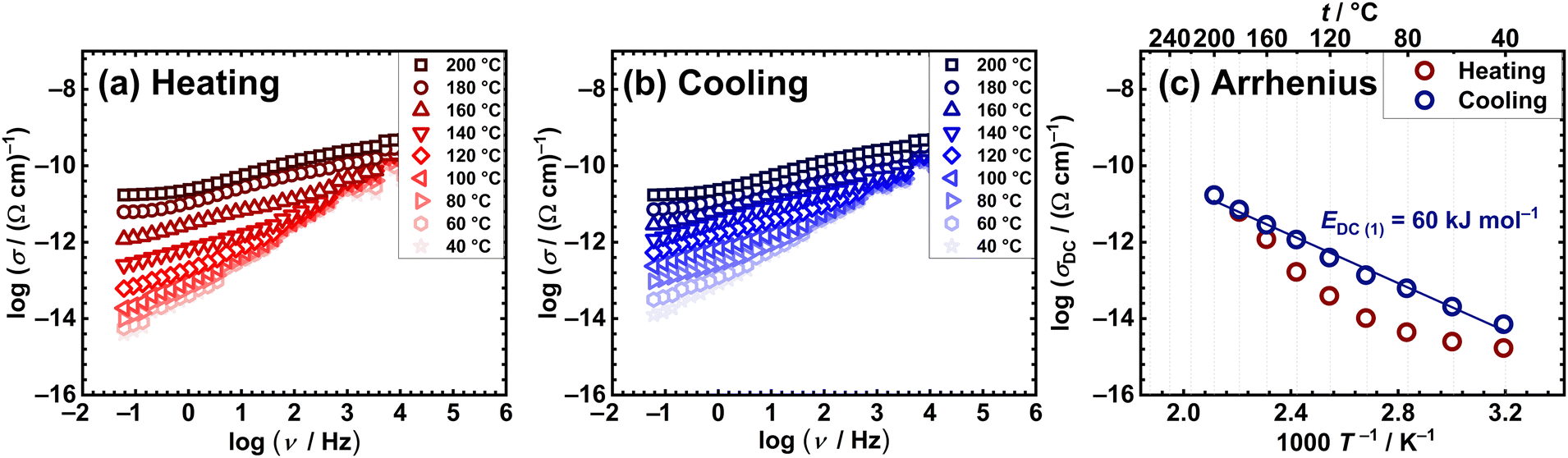 | ||
| Fig. 3 Conductivity spectra for mononuclear [MoO2(L1)(H2O)] (1) compound in heating (a) and cooling (b) runs, along with (c) Arrhenius plots for DC conductivity for both runs (red circle – heating, blue circle – cooling). | ||
Comparison of complex [MoO2(L1)(H2O)] (1) with [MoO2(L2)(H2O)] (3) and [MoO2(L2)(MeOH)] (4) before structural transformations, reveals interesting trends which can be correlated to structural features. One can conjecture that the more complex arrangement of [MoO2(L1)(H2O)] (1) with an extended supramolecular network present, has a positive effect on conductivity at RT (1.45 × 10−15 (Ω cm)−1 for (1) vs. 1.07 × 10−15 Ω cm)−1 (3) and 6.92 × 10−16(4)), but more interestingly, shows an impact on temperature dependence. At RT, complex [MoO2(L1)(H2O)] (1) shows slightly higher DC conductivity, whereas temperature-dependent measurements reveal moderate differences between the mentioned complexes (up to one order of magnitude at 120 °C, 4.17 × 10−14 (Ω cm)−1 for (1) vs. 3.98 × 10−15 (Ω cm)−1 (3) and 5.50 × 10−15 (Ω cm)−1 (4).
Correlation between electrical properties of all five complexes in this study, based on different ligands (H2L1 and H2L2) and type of Mo-complex (poly- vs. mononuclear and coordinated solvent, H2O vs. MeOH, for mononuclear complexes), can be done, Fig. 4. One can see that transformed complexes (mono/polynuclear) with the H2L1 ligand exhibit significantly lower activation energy values (60 and 40 kJ mol−1, respectively) compared to those with the H2L2 ligand (81–95 kJ mol−1). As mentioned above, this indicates that the H2L1 ligand facilitates easier electron transfer, resulting in lower energy barriers for conduction processes.
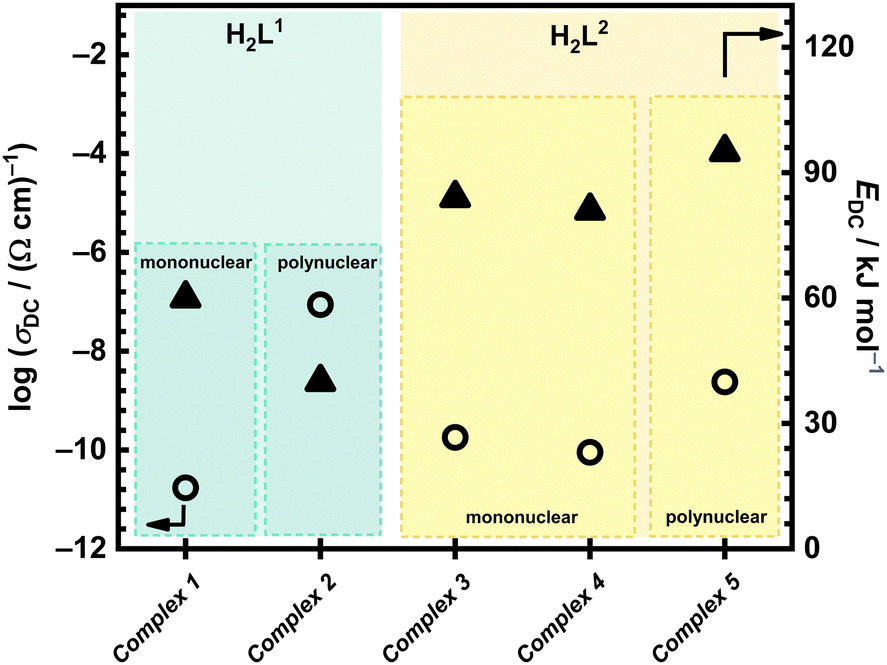 | ||
| Fig. 4 Conductivity at 200 °C and activation energy in the cooling run after transformation from IS analysis for all compounds in this study. | ||
The mononuclear complexes with H2L2, namely [MoO2(L2)(H2O)] (3) and [MoO2(L2)(MeOH)] (4), after structural transformation demonstrate similar activation energies, as shown in Table 4 and Fig. 4, which suggests that even though different solvent molecules (H2O and MeOH) are present before transformation, the resulting transformed compound is the same. This transformation likely involves the loss of the solvent molecules in complexes [MoO2(L2)(H2O)] (3) and [MoO2(L2)(MeOH)] (4) above 140 °C, and subsequent polymerization to [MoO2(L2)]n (5) species, leading to changes in their electrical properties. In the cooling run, we do not observe non-monotonic changes, indicating the stability of the transformed complexes. The DC conductivity at 200 °C, and EDC values of the transformed mononuclear complexes [MoO2(L2)(H2O)] (3) and [MoO2(L2)(MeOH)] (4) and the polynuclear complex [MoO2(L2)]n (5) are comparable (84 and 81 vs. 95 kJ mol−1), Table 4., and this consistency suggests the polymerisation process. The intrinsic factors, such as the ligand structure and the coordination environment around the molybdenum centre, play a crucial role and affect the electrical properties of these compounds. The ligand structure influences the electronic distribution and steric effects, while molybdenum centre's coordination environment affects the overall stability and conductivity of the complexes.
On the other hand, in the case of complexes with H2L1 ligand, mononuclear complex [MoO2(L1)(H2O)] (1) and polynuclear complex [MoO2(L1)]n (2) exhibit noticeable differences in their electrical properties, see Fig. 4. Compound [MoO2(L1)(H2O)] (1), which includes a coordinated water molecule, exhibits after transformation DC conductivity of 1.72 × 10−11 (Ω cm)−1 and EDC of 60 kJ mol−1, which is following the previously reported Mo compounds with the similar type of ligands.6 In contrast, we should emphasize here the interesting behaviour observed for the polynuclear [MoO2(L1)]n (2) that shows significantly higher DC conductivity of 8.77 × 10−8 (Ω cm)−1 (an increase of 4 order of magnitude) and lower activation energy (40 kJ mol−1) in comparison to complex [MoO2(L1)(H2O)] (1) where polymerization is expected upon heating. This pronounced difference, with DC conductivity being the highest while at the same time the lowest activation energy, observed among the five studied complexes, is both intriguing and promising for potential tuning of the material's properties. Further investigation into this effect is warranted to understand better and exploit these properties.
Here, it is noteworthy to make the correlation between the electrical properties of molybdenum complexes in this study and corresponding vanadium complexes with the same H2L2 ligand, from our previous work,10Table 5 and ESI,† Table S5. Comparison between mononuclear [MoO2(L2)(H2O)] (3) and [VO(L2)(OEt)(H2O)], as well as dinuclear [VO2(L2)]2 × 2H2O, reveals that [VO(L2)(OEt)(H2O) shows the highest conductivity. This suggests a strong positive effect of the vanadium centre in conjunction with water on electrical properties, being both much higher (3–4 orders of magnitude) in comparison to Mo-based one with H2L2 ligand.
| Compound | σ DC /(Ω cm)−1 |
|---|---|
| a Measured at 200 °C. | |
| [MoO2(L2)(H2O)] (3) | 2.43 × 10−15 |
| [VO(L2)(OEt)(H2O)] | 1.53 × 10−11 |
| [VO2(L2)]2 × 2H2O | 3.00 × 10−12 |
| [MoO2(L2)(MeOH)] (4) | 5.91 × 10−15 |
| [VO(L2)(OMe)(MeOH)] | 7.02 × 10−14 |
On the other, a comparison for complexes containing coordinated MeOH and different metal centres (Mo/V), namely [MoO2(L2)(MeOH)] (4) and [VO(L2)(OMe)(MeOH)], reveals that the latter shows one order of magnitude higher conductivity, thereby, one more time, reinforcing the improved electrical properties of vanadium over molybdenum. The presence of methoxy ligand in [VO(L2)(OMe)(MeOH)], in addition to methanol in both complexes, exhibit a positive effect on conductivity also.
Overall, the study underscores the critical role of the metal centre in the resulting electrical properties of the complexes. The coordination environment, including the type of ligand and coordinated solvent, additionally plays a significant role in modulating electrical characteristics, albeit to a lesser extent compared to the influence of the metal centre. In molybdenum complexes, the coordinated solvent shows minimal effect compared to vanadium complexes, where coordinated water results in complexes with improved conductivity.
Experimental
Materials
Solution-based synthesis. Yellow 2-hydroxy-3-methoxybenzaldehyde (0.5292 g, 3.478 mmol) was dissolved in 30 mL of methanol in a 100 mL round bottom flask. White formic or acetic acid hydrazide (0.2090 g, 3.478 mmol) was added to the colourless solution. The reaction was refluxed for two hours and left at room temperature. The obtained yellow crystals were separated by filtration and analysed.
Mechanochemically-based synthesis. 2-Hydroxy-3-methoxybenzaldehyde was placed in a stainless-steel crucible (14.5 mg, 0.0954 mmol), formic acid hydrazide (5.7 mg, 0.0954 mmol) and 30 μL of methanol. The mechanochemical synthesis was carried out for 30 minutes at a frequency of 3 Hz. Mechanochemical synthesis provides yields of >99%.
H2L1. Colour: white. Yield for solution-based synthesis: 0.5372 g, 79.5%. IR–ATR ν/cm−1: 3300, 3195 ν O–H, 2943 ν C–H (CH3), 2859 νas C–H (CH2), 2845 νs C–H (CH2), 1688 ν C
O, 1605 CN.
DSC: Te = 174.97 °C, E = 32.01 kJ mol−1.
1H NMR (DMSO) δ/ppm: 3,81 (d, 3H, C–H, 2, 3, 4) J = 1.96 Hz, 6,83 (m, 1H, H–Ar, 6) J1 = 15.28 Hz J2 = J3 = 5.2 Hz, 6,99 (dd, 1H, N–H, 9) J1 = 5.2 Hz J2 = 0.9 Hz, 7,03 (dd, 1H, H–Ar, 5) J1 = 5.2 Hz J2 = 0.9 Hz, 7,16 (dd, 1H, H–Ar, 7) J1 = 5.2 Hz J2 = 0.9 Hz, 8,37 (s, 1H, C–H, 10), 8,69 (d, 1H, C–H, 8) J = 4.16 Hz, 11,68 (s, 1H, O–H, 1).
13C NMR (DMSO) δ/ppm: 56,30 (b), 113,47 (d), 114,29 (e), 119,59 (g), 120,76 (f), 143,30 (h), 147,94 (c), 157,39 (a), 165,22 (i).
H2L2. Colour: white. Yield for solution-based synthesis: 0.5911 g, 83.6%. IR–ATR: ν/cm−1: 3317 i 3185 ν O–H, 2939 ν C–H (CH3), 2868 νas C–H (CH2), 2838 νs C–H (CH2), 1672 ν C
O, 1609 CN.
DSC: Te = 179.98 °C, E = 33.24 kJ mol−1.
1H NMR (DMSO) δ/ppm: 1,97 (s, 3H, C–H, 10, 11, 12) 3,81 (d, 3H, C–H, 2, 3, 4) J1 = 5.2 Hz, 6,83 (m, 1H, H–Ar, 6) J1 = 11.32 Hz J2 = J3 = 5.2 Hz, 7,01 (dd, 1H, H–Ar, 5) J1 = 5.2 Hz J2 = 0.9 Hz, 7,10 (dd, 1H, H–Ar, 7) J1 = 5.2 Hz J2 = 0.9 Hz, 8,33 (d, 1H, C–H, 8) J = 18.52 Hz, 11,24 (s, 1H, N–H, 9), 11,59 (s, 1H, O–H, 1).
13C NMR (DMSO) δ/ppm: 20,78 (j), 56,29 (b), 113,26 (d), 114,13 (e), 119,51 (g), 120,10 (f), 141,05 (h), 147,47 (c), 165,84 (a), 172,00 (i).
Mononuclear complexes.
Water-coordinated complexes [MoO2(L)(H2O)]. In a 100 mL round bottom flask, the ligand H2L1 (53.0 mg, 0.2728 mmol) was dissolved in 30 mL of dichloromethane with heating. [MoO2(acac)2] (88.9 mg, 0.2728 mmol) was then added. The reaction was refluxed for two hours. Precipitate appears immediately. The products were analyzed using IR–ATR spectroscopy, TGA/DSC analysis and NMR spectroscopy.
[MoO 2 (L 1 )(H 2 O)] ( 1 ). Colour: orange. Yield for solution-based synthesis: 48.8%
IR–ATR: ν/cm−1: 3343 ν O–H, 2958 ν C–H (CH3), 2934 νas C–H (CH2), 2839 νs C–H (CH2), 1599 CN (imin), 1264 C–O, 895 i 861 ν OMoO.
TGA: wt (H2O) = 5.30%, wexp (H2O) = 5.77%, wt (MoO3) = 42.32%, wexp (MoO3) = 42.27%.
EA: Ct: 31.97, Cexp: 31.02, Ht: 2.98, Hexp: 2.81, Nt: 8.28, Nexp: 7.87.
1H NMR (DMSO) δ/ppm: 3,80 (d, 3H, C–H, 1, 2, 3) J = 5.2 Hz, 7,01 (t, 1H, H–Ar, 5) J = 5.2 Hz, 7,24 (dd, 1H, H–Ar, 4) J1 = 5.2 Hz J2 = 0.9 Hz, 7,29 (dd, 1H, H–Ar, 6) J1 = 5.2 Hz J2 = 0.9 Hz, 7,76 (s, 1H, C–H, 8), 8,82 (s, 1H, C–H, 7).
13C NMR (DMSO) δ/ppm: 56,30 (b), 117,66 (e), 120,48 (d), 121,88 (g), 125,88 (f), 148,97 (a), 149,71 (c), 157,55 (i), 163,15 (h).
The same complex can be obtained by mechanochemical synthetic approach: [MoO2(acac)2] (19,0 mg, 0,5186 μmol), and ligand H2L1 (11,3 mg, 0,5186 μmol) are put in the stainless–steel crucible. 45 μL methanol was added and milled for 60 min at 25 Hz. Mechanochemical synthesis provides yields of >99%.
[MoO 2 (L 2 )(H 2 O)] ( 3 ). Colour: yellow. Yield: 59.6%.
IR–ATR: ν/cm−1: 3440 ν N–H, 3244 ν O–H, 2951 ν C–H (CH3), 2848 ν C–H (CH2), 1596 CN (imin), 1254 C–O, 906 i 866 ν OMoO.
TGA: wt (H2O) = 5.09%, wexp (H2O) = 5.79%, wt (MoO3) = 40.64%, wexp (MoO3) = 39.29%.
EA: Ct: 34.11, Cexp: 34.05, Ht: 3.43, Hexp: 3.11, Nt: 7.95, Nexp: 7.15.
1H NMR (DMSO) δ/ppm: 2,06 (s, 3H, C–H, 8, 9, 10), 3,79 (d, 3H, C–H, 1, 2, 3) J = 5.2 Hz, 6,99 (t, 1H, H–Ar, 5) J = 4.72 Hz, 7,21 (dd, 1H, H–Ar, 4) J1 = 5.2 Hz J2 = 0.9 Hz, 7,24 (dd, 1H, H–Ar, 6) J1 = 5.2 Hz J2 = 0.9 Hz, 8,70 (s, 1H, C–H, 7).
13C NMR (DMSO) δ/ppm: 16,79 (j), 55,19 (b), 116,20 (e), 119,67 (d), 120,66 (g), 124,53 (f), 147,82 (a), 148,40 (c), 154,19 (h), 171,90 (i).
Methanol-coordinated complex [MoO2(L2)(MeOH)]. In a 100 mL round bottom flask, the ligand H2L2 (49.5 mg, 0.2376 mmol) was dissolved in 30 mL of methanol. [MoO2(acac)2] (77.6 mg, 0.2376 mmol) was then added. The reaction was, refluxed for two hours and left until the appearance of a precipitate at room temperature. The resulting yellow crystals were analyzed using IR–ATR spectroscopy, TGA/DSC analysis and single-crystal X-ray diffraction.
[MoO 2 (L 2 )(MeOH)] ( 4 ). Colour: orange. Yield for solution-based synthesis: 25.0%.
IR–ATR: ν/cm−1: 2964 ν C–H (CH3), 2933 ν C–H (CH2), 1612 CN (imin), 1259 C–O, 906 i 868 ν OMoO.
TGA: wt (MeOH) = 8.70%, wexp (MeOH) = 8.97%, wt (MoO3) = 39.09%, wexp (MoO3) = 38.40%.
EA: Ct: 36.08, Cexp: 35.57, Ht: 3.85, Hexp: 3.21, Nt: 7.68, Nexp: 7.16.
The same complex can be obtained by mechanochemical synthetic approach: [MoO2(acac)2] (14.4 mg, 0.0442 mmol), and ligand H2L2 (9.2 mg, 0.0442 mmol) are put in the stainless-steel jar. 45 μL methanol was added and milled for 60 min at 25 Hz. Mechanochemical synthesis provides yields of >99%.
Polynuclear complexes [MoO2(L1or2)]n. Ligand H2L1 (49.5 mg, 0.2548 mmol) was dissolved in 30 mL of methanol or acetonitrile with heating. [MoO2(acac)2] (83.1 mg, 0.2548 mmol) was added to the reaction mixture. The reaction was refluxed for two hours and left at room temperature until a precipitate appeared. The resulting brown precipitate was filtered off and analyzed using IR–ATR spectroscopy and TGA/DSC analysis.
In a 100 mL round bottom flask, the ligand H2L2 (44.8 mg, 0.2151 mmol) was dissolved in 30 mL of dichloromethane with heating. [MoO2(acac)2] (70.16 mg, 0.2151 mmol) was then added. The reaction was refluxed for two hours. Precipitate appears immediately. The products were analyzed using IR–ATR spectroscopy and TGA/DSC analysis.
[MoO 2 (L 1 )] n ( 2 ). Colour: brown. Yield: 36.1%.
IR–ATR: ν/cm−1: 2938 ν C–H (CH3), 2839 ν C–H (CH2), 1595 CN, 1259 C–O, 922 i 877 ν OMoO.
TGA: wt (MoO3) = 44,.8%, wexp (MoO3) = 48.70%.
EA: Ct: 33.77, Cexp: 33.23, Ht: 2.52, Hexp: 2.12, Nt: 8.75, Nexp: 8.23.
[MoO 2 (L 2 )] n ( 5 ). Colour: brown. Yield: 39.5%.
IR–ATR: ν/cm−1: 2965 ν C–H (CH3), 2935 νas C–H (CH2), 2843 νs C–H (CH2), 1618 CN, 1265 C–O, 834 ν OMoO.
TGA: wt (MoO3) = 42.82%, wexp (MoO3) = 42.66%.
EA: Ct: 35.95, Cexp: 35.16, Ht: 3.02, Hexp: 3.23, Nt: 8.38, Nexp: 8.60.
Methods
The prepared compounds were characterized and identified using elemental analysis (EA), infrared spectroscopy (IR–ATR, Infrared spectroscopy – Attenuated Total Reflectance), nuclear magnetic resonance (NMR), thermogravimetric analysis (TGA), differential scanning calorimetry (DSC), Single Crystal X-ray Diffraction (SCXRD), and impedance spectroscopy (IS).IR–ATR spectroscopic analysis was performed on a PerkinElmer Spectrum Two spectrometer equipped with a diamond ATR attachment. The recording was carried out in the 4000–400 cm−1 range with four scans. The spectra were processed and analysed with the Excel program. Nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Avance III HD 400 spectrometer operating at 400 MHz. Compounds were dissolved in dmso-d6 and measured in 5 mm NMR tubes at 298 K with TMS as an internal standard. The proton and carbon NMR chemical shifts of all compounds in dmso-d6 solution were deduced by combined use of one (1H, 13C APT) and two-dimensional NMR techniques (COSY, HMQC and HMBC). NMR spectra are available in ESI,† Scheme S2 and Fig. S28–S35.
Differential scanning calorimetry (DSC) analysis was performed on a Mettler–Toledo DSC823e instrument in the range from 25 °C to 300 °C in an inert nitrogen atmosphere with a flow rate of 50 mL min−1 and heating of 10 °C min−1. The samples were recorded in closed aluminium containers. The measurement results were processed with the Mettler STARe Evaluation Software v17.00 program.
Thermogravimetric (TGA) analyses were performed using a Mettler–Toledo TGA/DSC 3+ in closed aluminium oxide containers. All experiments were performed in an oxygen atmosphere with a flow rate of 200 cm3 min−1 and heating at 10 °C min−1. The recording was carried out in the temperature range from 25 °C to 600 °C. The measurement results were processed with the Mettler STARe Evaluation Software v17.00 program.
Complex impedance was measured across a broad frequency range (0.01 Hz to 1 MHz) and temperature range (30–200 °C, in 10 °C increments) using a Novocontrol Alpha–AN dielectric spectrometer. Temperature control was maintained within ±0.2 °C. Measurements were conducted on powder samples pressed into cylindrical disks with a diameter of 5 mm and a thickness of 1 mm under a uniform load of 2 × 103 kg using a hydraulic press. Gold electrodes, 3.8 mm in diameter, were sputtered on both sides of the disk using a Quorum Technologies SC7620 magnetron for electrical contact. The sample was then positioned between the two electrodes of the BDS cell in a sandwich configuration. Impedance spectra were analyzed using equivalent electrical circuits (EEC) modelling, with parameters obtained through complex non-linear least square (CNLLSQ) fitting with WinFit software.40 The system shows resistance (R) and capacitance (C) related to the sample and can be modelled with an RC element in a sandwich configuration. The impedance spectrum exhibited a single semicircle related to bulk effects for all studied complexes. The complex impedance plots typically consisted of a single depressed semicircle with the center below the real axis. Therefore, a constant-phase element (CPE) was used in the equivalent circuits rather than a simple capacitor. The empirical function defines the CPE: 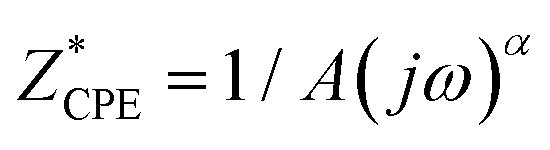 , where A and a are the constants, 0 ≤ α ≤ 1, a being 1 for the ideal capacitor, and 0 for the ideal resistor. In a rough approximation, the parameter A can be equated with the value of the “real” capacity. From the resistance values obtained through the fitting procedures, and the electrode dimensions (sample thickness d and electrode area A), the DC conductivity was calculated.
, where A and a are the constants, 0 ≤ α ≤ 1, a being 1 for the ideal capacitor, and 0 for the ideal resistor. In a rough approximation, the parameter A can be equated with the value of the “real” capacity. From the resistance values obtained through the fitting procedures, and the electrode dimensions (sample thickness d and electrode area A), the DC conductivity was calculated.
The catalytic reactions were followed by GC techniques on Agilent 6890A chromatograph equipped with an FID detector and Agilent CP7495 (25 m × 0.25 mm × 0.12 μm). The GC parameters were quantified using authentic samples of the reactants and products. The conversion of linalool and formation of 2-(5-methyl-5-vinyl-tetrahydrofuran-2-yl propan-2-ol) and 2,2,6-trimethyl-6-vinyltetrahydropyran-3-ol were calculated from calibration curves using authentic samples of the studied species relative to dodecane or ethylbenzene as an internal standard (r2 = 0.999).
Linalool oxidation
A mixture of linalool (1.54 g, 10 mmol), ethylbenzene (0.3 mL) as internal standard, vanadium catalyst (0.05 mmol), and acetonitrile (5 mL) was mixed and heated at 70 °C before adding H2O2 in water (30% w/w) (2.06 mL, 20 mmol). The reaction was monitored for five hours by taking and analyzing aliquots of the organic phase (0.6 mL) at defined time points (0, 20, 60, 120, 180, 240, and 300 minutes). Each sample taken was mixed with 0.4 mL of MeCN and analyzed using GC.
Crystallography
Single crystals of (pyH)4(Mo8O26), H2L1, H2L2, [MoO2(L1)(H2O)] (1), [MoO2(L2)(H2O)] (3) and [MoO2(L2)(MeOH)] (4) of appropriate quality were selected for the diffraction experiments. Data were compiled using a Rigaku XtaLAB Synergy-S diffractometer equipped with a Dualflex source (Cu Kα radiation, λ = 1.54184 Å) and a HyPix detector. Data were gathered via ω-scans at 170 K and data were processed with the CrysAlis program package.41 A summary of the general crystallographic data is presented in Table S1 (ESI†). The structures were solved by dual-space methods with SHELXT.42 The refinement was done via full-matrix least-squares methods based on F2 values against all reflections, including the anisotropic displacement parameters for all non-H atoms. Hydrogen atoms attached to carbon atoms were placed in geometrically idealized positions and refined by using the riding model, with Uiso = 1.2Ueq of the connected carbon atom, or as ideal CH3 groups, with Uiso = 1.5Ueq. Hydrogen atoms attached to heteroatoms were located in the difference Fourier map in the final stages of the refinement procedure. All refinements were conducted using SHELXL.43 The SHELX programs were operated within the Olex2 suite.44 Geometrical calculations were performed by Platon45 and molecular graphics were produced using Mercury.46 CCDC 2367284–2367289† contains the supplementary crystallographic data for this paper.Conclusions
In the synthesis of molybdenum complexes, two distinct ligands characterized by variations in substituent groups on the hydrazide chain end were employed. Five novel molybdenum complexes were prepared from different solvents and subsequently characterised via different analytical techniques. These compounds encompassed the following entities: [MoO2(L1)]n (2), [MoO2(L2)]n (5), [MoO2(L1)(H2O)] (1), [MoO2(L2)(H2O)] (3), [MoO2(L2)(MeOH)] (4). In all instances, the ligand established coordination with the metal centre through the ONO atom, resulting in mononuclear or polynuclear Mo moiety.Building on our previous work with vanadium coordination compounds, we extended our research to molybdenum complexes, investigating them using impedance spectroscopy. We recognized the complexity associated with the liberation of coordinated solvents and the decomposition stages of the complexes, which we correlated with thermogravimetric analysis. This approach provided valuable insights into optimizing ss-IS measurements, determining appropriate heating temperatures, and identifying potential sample transformations (e.g., from mononuclear to polynuclear forms). All examined molybdenum complexes exhibited a temperature-dependent increase in conductivity, characteristic of semiconducting behaviour. Notably, the polynuclear samples [MoO2(L1)]n (2) and [MoO2(L2)]n (5) demonstrated high conductivities of 8.77 × 10−8 (Ω cm)−1 and 2.39 × 10−9 (Ω cm)−1, respectively. These values surpass those of previously published vanadium coordination complexes with acylhydrazone ligands, suggesting potential applications in various electronic devices, such as light-emitting diodes, rechargeable batteries, sensors, and other electronic components.
Furthermore, this research highlights the dual functionality of the synthesized molybdenum complexes, which, along with recently reported vanadium complexes, show promise not only for electronic applications but also for catalytic activities. The catalytic potential was evaluated using linalool as a bio-based substrate, revealing the significant efficacy of molybdenum catalysts in producing furanoid and pyranoid compounds when employing TBHP as an oxidant.
Bifunctionality is a pivotal factor in the evolution of next-generation materials, critically enabling innovative applications for future technologies. This principle underpins the objectives of our research investigations.
Author contributions
Conceptualization: D. A., L. P., J. P. Data curation: J. S., M. C., E. T., M. R., L. P., J. P. Formal analysis: J. S., M. C., E. T., M. R., L. P., J. P. Funding acquisition: D.A., L. P., J. P. Investigation: J. S., E. T., J. P., L. P. Methodology: E. T., D. A., L. P., J. P. Project administration: D. A., J. P., L. P. Resources: M. R., D. A., L. P., J. P. Software: J. S., E. T., L. P. Supervision: D. A., L. P., J. P. Validation: J. S., M. C. Visualization: J. S., E. T., L. P., J. P. Writing – original draft: L. P., J. P. Writing – review & editing: J. S., E. T., L. P., J. P.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded in part by COGITO 2023–2024: Sustainable polyesters using Mo and V catalysts (SusPolMoV) and CNRS: International Emerging Actions 2023–2024: Solvent-sober Processes-Organic solvent-free polyterpenes oxidation assisted by mechanochemically synthesized catalysts (SoSoPro). E. T., M. R., and J. P. acknowledge the support of project CIuK (grant KK.01.1.1.02.0016) co-financed by the Croatian Government and the European Union through the European Regional Development Fund-Competitiveness and Cohesion Operational Programme. LCC CNRS and IUT Chem Dpt are acknowledged for all the facilities needed during catalytic experiments.Notes and references
- D. C. Crans, J. J. Smee, E. Graidamauskas and L. Yang, The chemistry and biochemistry of vanadium and the biological activities exerted by vanadium compounds, Chem. Rev., 2004, 104(2), 849–902 CrossRef CAS PubMed.
- A. Galani, V. Tsitsias, D. Stellas, V. Psycharis, C. P. Raptopoulou and A. Karaliota, Two novel compounds of vanadium and molybdenum with carnitine exhibiting potential pharmacological use, J. Inorg. Biochem., 2015, 142, 109–117 CrossRef CAS PubMed.
- J. Morlot, N. Uyttebroeck, D. Agustin and R. Poli, Solvent-Free Epoxidation of Olefins Catalyzed by “[MoO2(SAP)]”: A New Mode of tert-Butylhydroperoxide Activation, ChemCatChem, 2013, 5, 601–611 CrossRef CAS.
- J. Pisk, D. Agustin, V. Vrdoljak and R. Poli, Epoxidation Processes by Pyridoxal Dioxomolybdenum(VI) (Pre)Catalysts Without Organic Solvent, Adv. Synth. Catal., 2011, 353, 2910–2914 CrossRef CAS.
- M. Sutradhar and A. J. L. Pombeiro, Reference Module in Chemistry, Molecular Sciences and Chemical Engineering, 2017 Search PubMed.
- J. Sarjanović, M. Stojić, M. Rubčić, L. Pavić and J. Pisk, Impedance Spectroscopy as a Powerful Tool for Researching Molybdenum-Based Materials with Schiff Base Hydrazones, Materials, 2023, 16, 1064 CrossRef PubMed.
- J. Pisk, M. Šušković, E. Topić, D. Agustin, N. Judaš and L. Pavić, Molybdenum Complexes Derived from 2-Hydroxy-5-nitrobenzaldehyde and Benzhydrazide as Potential Oxidation Catalysts and Semiconductors, Int. J. Mol. Sci., 2024, 25, 4859 CrossRef CAS PubMed.
- S. Sarkar, Y. Aydogdu, F. Dagdelen, B. B. Bhaumik and K. Dey, X-ray diffraction studies, thermal, electrical and optical properties of oxovanadium(IV) complexes with quadridentate Schiff bases, Mater. Chem. Phys., 2004, 88, 357 CrossRef CAS.
- M. Lozano-González, M. E. Sánchez-Vergara, I. Alvarado-Beltrán, M. Leyva-Esqueda, M. Rivera and C. Álvarez-Toledano, Synthesis and Evaluation of the Semiconductor Behavior in Vanadium Indanone Derivatives Thin Films, Adv. Mater. Phys. Chem., 2017, 7(2), 70–83 CrossRef.
- J. Sarjanović, E. Topić, M. Rubčić, L. Androš Dubraja, L. Pavić and J. Pisk, Evaluation of vanadium coordination compounds derived from simple acetic acid hydrazide as non-conventional semiconductors, J. Mater. Chem. C, 2024, 12, 4013–4025 RSC.
- M. A. Hussein, T. S. Guan, R. A. Haque, M. B. K. Ahamed and A. M. S. A. Majid, Structures, DNA Binding, DNA Cleavage, and Antitumor Investigations of a Series of Molybdenum(VI) Complexes with Some N(4) Methyl and Ethyl Thiosemicarbazone Ligands, J. Coord. Chem., 2014, 67(4), 714–727 CrossRef CAS.
- A. Fuior, D. Cebotari, M. Haouas, J. Marrot, G. M. Espallargas, V. Guérineau, D. Touboul, R. V. Rusnac, A. Gulea and S. Floquet, Synthesis, Structures, and Solution Studies of a New Class of [Mo2O2S2]-Based Thiosemicarbazone Coordination Complexes, ACS Omega, 2022, 7(19), 16547–16560 CrossRef CAS PubMed.
- S. Roy, Saswati, S. Lima, S. Dhaka, M. R. Maurya, R. Acharyya, C. Eagle and R. Dinda, Synthesis, structural studies and catalytic activity of a series of dioxidomolybdenum(VI)-thiosemicarbazone complexes, Inorg. Chim. Acta, 2018, 474, 134–143 CrossRef CAS.
- J. Pisk, B. Prugovečki, D. Matković-Čalogović, R. Poli, D. Agustin and V. Vrdoljak, Charged dioxomolybdenum(VI) complexes with pyridoxal thiosemicarbazone ligands as molybdenum(V) precursors in oxygen atom transfer process and epoxidation (pre)catalysts, Polyhedron, 2012, 33(1), 441–449 CrossRef CAS.
- V. Vrdoljak, M. Mandarić, T. Hrenar, I. Đilović, J. Pisk, G. Pavlović, M. Cindrić and D. Agustin, Cryst. Growth Des., 2019, 19(5), 3000–3011 CrossRef CAS.
- M. Maiti, S. Thakurta, G. Pilet, A. Bauzá and A. Frontera, Two new hydrogen-bonded supramolecular dioxo-molybdenum(VI) complexes based on acetyl-hydrazone ligands: Synthesis, crystal structure and DFT studies, J. Mol. Struct., 2021, 1226, 129346 CrossRef CAS.
- C. S. Letizia, J. Cocchiara, J. Lalko and A. M. Api, Fragrance material review on linalool, Food Chem. Toxicol., 2003, 41, 41943–41964 Search PubMed.
- K. A. D. Swift, Catalytic Transformations of the Major Terpene Feedstocks, Top. Catal., 2004, 27, 143–155 CrossRef CAS.
- M.-A. Mirata, M. Wust, A. Mosandl and J. Schrader, Fungal Biotransformation of (±)-Linalool, J. Agric. Food Chem., 2008, 56, 3287–3296 CrossRef CAS PubMed.
- T. Hofmann, P. Schieberle and W. Grosch, Model studies on the oxidative stability of odor-active thiols occurring in food flavors, J. Agric. Food Chem., 1996, 44(1), 251–255 CrossRef CAS.
- D. Voet, J. G. Voet and C. W. Pratt, Fundamentals of Biochemistry: Life at the Molecular Level, 5th edn, Wiley, 2016 Search PubMed.
- A. A. H. Haidar and D. Agustin, Tetrahedron Green Chem, 2023, 2, 100029 CrossRef.
- M. G. Speziali, P. A. Robles-Dutenhefner and E. V. Gusevskaya, Palladium-Catalyzed Oxidation of Monoterpenes: Novel Aerobic Pd(II)/Cu(II)-Catalyzed Oxidation of Linalool under Chloride-Free Nonacidic Conditions, Organometallics, 2007, 26, 4003–4009 CrossRef CAS.
- M. G. Speziali, V. V. Costa, P. A. Robles-Dutenhefner and E. V. Gusevskaya, Aerobic Palladium(II)/Copper(II)-Catalyzed Oxidation of Olefins under Chloride-Free Nonacidic Conditions, Organometallics, 2009, 28, 3186–3192 CrossRef CAS.
- C. B. Vilanculo, M. J. Da Silva, M. G. Teixeira and J. A. Villarreal, One-pot synthesis at room temperature of epoxides and linalool derivative pyrans in monolacunary Na7PW11O39-catalyzed oxidation reactions by hydrogen peroxide, RSC Adv., 2020, 10, 7691–7697 RSC.
- M. Cindrić, V. Vrdoljak, N. Strukan and B. Kamenar, Synthesis and characterization of some mono- and dinuclear molybdenum(VI) thiosemicarbazonato complexes, Polyhedron, 2005, 24, 369–376 CrossRef.
- V. Vrdoljak, M. Cindrić, D. Milić, D. Matković-Čalogović, P. Novak and B. Kamenar, Synthesis of five new molybdenum(VI) thiosemicarbazonato complexes. Crystal structures of salicylaldehyde and 3-methoxy-salicylaldehyde 4-methylthiosemicarbazones and their molybdenum(VI) complexes, Polyhedron, 2005, 24, 1717–1726 CrossRef CAS.
- A. Meetsma, CSD Commun., 2016, private communication Search PubMed.
- J. R. Macdonald and W. B. Johnson, Fundamentals of Impedance Spectroscopy, John Wiley & Sons, Inc., 2018 Search PubMed.
- N. Bonanos, P. Pissis and J. R. Macdonald, Impedance Spectroscopy of Dielectrics and Electronic Conductors, John Wiley & Sons, Inc., 2012 Search PubMed.
- J. R. Macdonald, Impedance spectroscopy, Ann. Biomed. Eng., 1992, 20, 289 CrossRef CAS PubMed.
- E. Barsoukov and J. R. Macdonald, Impedance Spectroscopy: Theory, Experiment, and Applications, John Wiley & Sons, Inc., 2018 Search PubMed.
- A. Bafti, V. Mandić, I. Panžić, L. Pavić and V. Špada, CdSe QDs modified cellulose microfibrils for enhanced humidity sensing properties, Appl. Surf. Sci., 2023, 612, 155894 CrossRef CAS.
- V. Kojić, M. Bohač, A. Bafti, L. Pavić, K. Salamon, T. Čižmar, D. Gracin, K. Juraić, M. Leskovac and I. Capan, Formamidinium Lead Iodide Perovskite Films with Polyvinylpyrrolidone Additive for Active Layer in Perovskite Solar Cells, Enhanced Stability and Electrical Conductivity, Materials, 2021, 14, 4594 CrossRef PubMed.
- M. Razum, L. Pavić, D. Pajić, J. Pisk, P. Mošner, L. Koudelka and A. Šantić, Structure–polaronic conductivity relationship in vanadate–phosphate glasses, J. Am. Ceram. Soc., 2024, 107, 5866–5880 CrossRef CAS.
- L. Pavić, J. Nikolić, M. P. F. Graça, B. F. Costa, M. A. Valente, Ž. Skoko, A. Šantić and A. Moguš-Milanković, Effect of controlled crystallization on polaronic transport in phosphate-based glass-ceramics, Int. J. Appl. Glass Sci., 2020, 11, 97–111 CrossRef.
- J. E. Garbarczyk, P. Jozwiak, M. Wasiucionek and J. L. Nowinski, Nanocrystallization as a method of improvement of electrical properties and thermal stability of V2O5-rich glasses, J. Power Sources, 2007, 173, 743–747 CrossRef CAS.
- T. K. Pietrzak, M. Maciaszek, J. L. Nowiński, W. Ślubowska, S. Ferrari, P. Mustarelli, M. Wasiucionek, M. Wzorek and J. E. Garbarczyk, Electrical properties of V2O5 nanomaterials prepared by twin rollers technique, Solid State Ion., 2012, 225, 658–662 CrossRef CAS.
- T. K. Pietrzak, L. Pawliszak, P. P. Michalski, M. Wasiucionek and J. E. Garbarczyk, Highly Conductive 90V2O5·10P2O5 Nanocrystalline Cathode Materials for Lithium-ion Batteries, Procedia Eng., 2014, 98, 28–35 CrossRef CAS.
- WinFIT software (version 3.2, Novocontrol Technologies GmbH & Co. KG, Hundsangen, Germany).
- Rigaku Oxford Diffraction, CrysAlisPro Software System; Versions 1.171.42.49, 1.171.41.92a, 1.171.41.93a and 1.171.42.53a; Rigaku Oxford Diffraction: Oxford, UK, 2020.
- G. M. Sheldrick, SHELXT – Integrated space-group and crystal-structure determination, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, OLEX2: a complete structure solution, refinement and analysis program, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- A. L. Spek, Structure validation in chemical crystallography, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, D65, 148–155 CrossRef PubMed.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, Mercury 4.0: from visualization to analysis, design and prediction, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2367284–2367289. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ma00790e |
| This journal is © The Royal Society of Chemistry 2024 |