
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceROS-releasing PVA sub-micron antimicrobial dressing with enhanced aqueous stability and mechanical properties†
Joel Yupanqui
Mieles
*a,
Cian
Vyas
 *ab,
Gavin
Humphreys
c,
Carl
Diver
d and
Paulo
Bartolo
*ab
*ab,
Gavin
Humphreys
c,
Carl
Diver
d and
Paulo
Bartolo
*ab
aDepartment of Mechanical, Aerospace and Civil Engineering, University of Manchester, UK. E-mail: joel.yupanquimieles@manchester.ac.uk
bSingapore Centre for 3D Printing, School of Mechanical and Aerospace Engineering, Nanyang Technological University, Singapore 639798, Singapore. E-mail: cian.vyas@ntu.edu.sg; pbartolo@ntu.edu.sg
cDivision of Pharmacy & Optometry, University of Manchester, UK
dDepartment of Engineering, Manchester Metropolitan University, UK
First published on 8th November 2024
Abstract
This study aimed to develop a biocompatible nanofibrous mesh for wound healing applications that is stable in aqueous environments. The mesh was produced by electrospinning RO-101-loaded polyvinyl alcohol (PVA) fibres and crosslinking them using glutaraldehyde (GA) vapour exposure. RO-101™ is a wound gel that produces therapeutic levels of hydrogen peroxide (H2O2). The results of Fourier-transform infrared spectroscopy (FTIR) and scanning electron microscopy (SEM) confirmed successful incorporation of RO-101 wound gel and crosslinking of the mesh, with average fibre diameters of 400 nm. The vapour crosslinking process resulted in enhanced mechanical strength and flexibility, improved aqueous stability, and an increase in contact angle compared to the uncrosslinked mesh whilst maintaining hydrophilicity. The vapour-crosslinked mesh also demonstrated sustained release of H2O2 at similar concentrations (1103 ± 199 μM g−1 mL−1) to the uncrosslinked mesh, but with a more gradual release. The developed mesh showed antimicrobial activity against S. aureus and its released H2O2 presented no cytotoxicity in human adipose-derived stem cells (hADSCs) metabolic activity. Overall, the developed mesh has potential for wound healing applications, providing a barrier against infection and promoting tissue regeneration.
1. Introduction
Human host systems have evolved complex methods to provide barriers against disease-causing microorganisms, where the skin is the first and largest defence against infection. Still, these microbes can enter the host system in a variety of ways: via the respiratory tract, via food, along with foreign objects entering the body, or through wounds in the skin.1 When wounds occur, the body is usually able to regenerate skin integrity via a complex and interactive process divided into a sequence of four time-dependent phases: coagulation and haemostasis, inflammation, proliferation, and remodelling.2,3 However, the natural healing response can be interrupted due to local factors (wound infection, lack of oxygenation, wound area and depth, local pressure, and venous insufficiency) and systemic factors (age, ischaemia, diabetes and other underlying diseases or immunocompromised conditions, obesity, medications, smoking and alcoholism, and nutrition).2 In these cases, it is imperative that medical treatment is provided-normally in the form of wound dressings- to offer an environment that favours skin regeneration.2,3 Ideally, wound dressings should be engineered to: provide an antimicrobial barrier against infection, absorb wound exudates, support oxygenation, sustain a moist environment, be noncytotoxic, and mimic the structural morphology of the extracellular matrix (ECM) with adequate mechanical properties to promote cell migration, adhesion, and proliferation.2–4Electrospun meshes are an excellent approach for wound management and tissue regeneration, as they present high surface-to-volume ratio which enhances exudate absorption.5 They also have porosity with suitable pore size for ultrafiltration which acts as a barrier against infection whilst aiding oxygenation and exudate drainage.6,7 Moreover, sub-micron electrospun fibres assemble a three-dimensional organisation that supports cell growth by mimicking the structure of native ECM, which is made of proteins and polysaccharides fibres with diameters ranging from tens to hundreds of nanometers.5 Furthermore, meshes can be loaded with bioactive molecules and drugs that can further aid the healing process through targeted and controlled delivery to the wound site.3,5 Honey —a traditional antimicrobial and wound healing biomaterial8—has been successfully incorporated into electrospun fibres to harness their enzymatic activity.9–12
RO-101® is a honey-inspired polymeric wound gel that includes glucose oxidase (GOx) and glucose—its substrate—for in situ generation of hydrogen peroxide (H2O2) via an enzymatic reaction. Reactive oxygen species (ROS), such as H2O2, act as a powerful antimicrobial and can support the different stages of wound healing through their active role in tissue regeneration mechanisms such as: recruitment of immunocytes, regulation of matrix metalloproteinases (MMPs), fibroblast proliferation with increased mitogenic rates, and neutrophil stimulation for clearing of damaged tissue, simplifying debridement.8,13 However, their use as a therapeutic agent is limited by their short duration of activity.14 To overcome this limitation, RO-101 was engineered to release low concentrations of ROS to a target site and sustain them for a prolonged period of time.14,15 This patented technology is the antimicrobial agent used for this research.
Electrospun biopolymeric sub-micron fibres will act as the ROS delivery platform, further supporting the controlled release of therapeutic levels of H2O2in situ. Meshes fabricated in this manner have shown excellent physicochemical and biological properties, and successful encapsulation and release of bioactive compounds.16,17 Furthermore, the electrospinning process favours enzyme immobilisation, enhances thermal stabilisation, and boosts the efficiency of the enzyme's catalytic reactions with substrates from the surrounding environment by diffusion.17
Honey has been previously encapsulated in electrospun polyvinyl alcohol (PVA) fibres,9,10,18,19 and since RO-101's is physiochemically similar to honey, PVA was considered as a viable option to encapsulate the wound gel for this research. PVA is a non-toxic biocompatible and biodegradable polymer with suitable spinnability and physical properties, making it ideal for a range of biomedical applications such as wound dressings, drug delivery systems, and temporary tissue engineering scaffolds.3,9,16 Similar to RO-101, PVA can be dissolved in water, which favours a less hazardous and more cost-effective fabrication process when compared to meshes electrospun using organic solvents. Nevertheless, that same nature can be disadvantageous for electrospun PVA RO meshes in aqueous experiments and in wounds with heavy exudate production when the dressing's psychochemical properties are required to be stable for over 24 h. To enhance the stability and mechanical properties of the meshes crosslinking is required, commonly achieved using chemical reagents such as glutaraldehyde (GA), glyoxal, or boric acid.20 Among these, GA is a well-investigated and effective approach, especially in its vapour phase as it has low cytotoxic effect when exposed to electrospun sub-micron meshes.21
The present work aims to fabricate a PVA/RO bioactive wound dressing with improved physicochemical properties by crosslinking the electrospun sub-micron meshes using GA vapour. To observe the impact of crosslinking, the morphology and chemical structure of the sub-micron fibres were investigated by scanning electron microscopy (SEM) and nuclear magnetic resonance spectroscopy (NMR), respectively. Wettability, swelling ratio, and weight loss of the nanofibrous membranes were studied. The enzymatic activity was evaluated for meshes with different crosslinking processes through H2O2 release assays. The antibacterial activity against Staphylococcus aureus was investigated by disc diffusion assay and time-kill assays. The cell viability of the proposed wound dressing material was evaluated by Alamar Blue assay. This preliminary study has developed a crosslinked PVA/RO-101 electrospun mesh which shows promise as a wound dressing and in other clinical applications such as surgical device coating.
2. Materials and methods
2.1 Materials
PVA (Mw: 89![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–98000, 99 + % hydrolysed), Triton-X 100 (TX), GA, hydrochloric acid (HCl), phosphate-buffered saline (PBS), Mueller Hinton Broth (MHB) and Mueller Hinton Agar (MHA) were all purchased from Sigma-Aldrich (Gillingham, UK). Amplex® Red (AR) assay kit, StemPro™ human adipose-derived stem cells (hADSCs), MesenPRO RS™ medium, glutamine, penicillin/streptomycin, trypsin-EDTA solution and alamarBlue™ (AB) reagent were all obtained from Thermo Fisher Scientific (Loughborough, UK). RO-101® antimicrobial gel was given by Matoke Holdings Ltd. (Abingdon, UK).
000–98000, 99 + % hydrolysed), Triton-X 100 (TX), GA, hydrochloric acid (HCl), phosphate-buffered saline (PBS), Mueller Hinton Broth (MHB) and Mueller Hinton Agar (MHA) were all purchased from Sigma-Aldrich (Gillingham, UK). Amplex® Red (AR) assay kit, StemPro™ human adipose-derived stem cells (hADSCs), MesenPRO RS™ medium, glutamine, penicillin/streptomycin, trypsin-EDTA solution and alamarBlue™ (AB) reagent were all obtained from Thermo Fisher Scientific (Loughborough, UK). RO-101® antimicrobial gel was given by Matoke Holdings Ltd. (Abingdon, UK).
2.2 Fabrication of electrospun PVA RO nanofibres
A 12% w/v PVA solution was prepared in deionised water at 90 °C and 800 rpm overnight. RO-101® antimicrobial gel (0.01% GOx) was then added to the PVA solution at room temperature and vortexed. TX was introduced into the polymeric solution (0.5% w/w) to improve fibre homogeneity and avoid bead formation. PVA and RO-101 concentrations in water were kept constant at 12% and 8% w/v, respectively. To minimise the reaction time and production of H2O2 during solution preparation, the RO-101 gel was added to the polymeric solution immediately prior to electrospinning and the total volume was restricted to 3 mL per batch. Once electrospun the reaction is effectively paused due to the lack of free water. The name of the samples represents the theoretical weight concentration that will be present in the electrospun mesh after the solvent has evaporated. A 100 mg PVA RO40 mesh consists of 40 mg RO-101 (40% w/w) and 60 mg PVA (60% w/w).A vertical electrospinning system (Spraybase, Maynooth, Ireland) with a 21G needle was used with a voltage of 17.5 kV, a flow rate of 0.75 mL h−1, and a spinneret distance of 13 cm. The collector was an aluminium foil sheet covering a stainless-steel plate.
Vapour crosslinking (Vap CL). an uncrosslinked mesh is attached to the inside of the lid of a Petri dish, where the container portion of the dish enclosed 2M GA – the chemical crosslinking reagent – and HCl – the catalyst – in a 3
:1 ratio of GA:HCl.22 The closed and sealed Petri dish was left inside an incubator at 35 °C for 2, 6 or 48 h. The crosslinked samples were then removed from the lid and placed inside a desiccator for 48 h.
In situ crosslinking (in situ CL). 220 μL GA and 10 μL HCl, were added directly into the syringe containing the PVA/RO solution at room temperature and vortexed briefly. The final PVA/RO/GA/HCl solution was electrospun immediately.
2.3 Fourier-transform infrared spectroscopy
To confirm the incorporation of RO-101 gel and to verify if there are any GA residuals after crosslinking, the material chemistry of PVA RO40 Vap CL electrospun meshes was investigated using Fourier-transform infrared spectroscopy (FTIR, Alpha-P System, Bruker, Coventry, UK). The FTIR transmittance spectra was collected between 4000 cm−1 to 400 cm−1 using a 24-scan resolution at 2 cm−1.2.4 Scanning electron microscopy
A scanning electron microscope (SEM) (Quanta 200, FEI Company, USA) at an accelerating voltage of 20 kV was used to study the morphology and diameter of the meshes. Samples were sputter-coated with a 6 nm thick gold/palladium (80:20) layer (Q150T ES, Quorum Technologies, UK) prior to observation. Three images per sample group were collected (n = 3), and the diameter of at least 50 fibres from each mesh were assessed quantitatively using ImageJ (Fiji, v1.53c) image processing package.24
2.5 Wettability, water absorption capacity and weight loss behaviour
To assess the wettability of crosslinked meshes (n = 5, 19 mm diameter), water contact angle measurements were taken via sessile drop method using DSA100 drop shape analyser (Kruss Scientific, Germany) and deionised water at room temperature.A water absorption study was carried out on PVA RO40 samples (n = 3) with increasing exposure time to GA vapour (uncrosslinked, 6 h, 48 h) by measuring the swelling percentage. Meshes were submerged in 1 mL of 1× PBS for 1 h and 24 h. Soaked meshes were flipped gently on filter paper until no excess water was visible. Meshes were then weighed in triplicate. To measure weight loss, swollen meshes were dried in an oven at 37 °C for 24 h.
Swelling and weight loss abilities were calculated with the following equations:
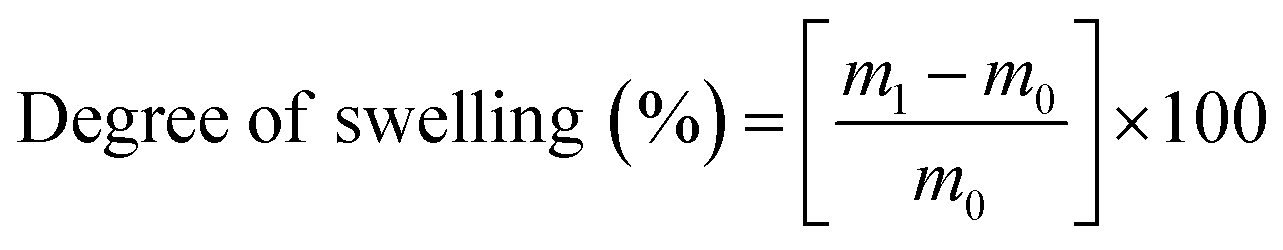 | (1) |
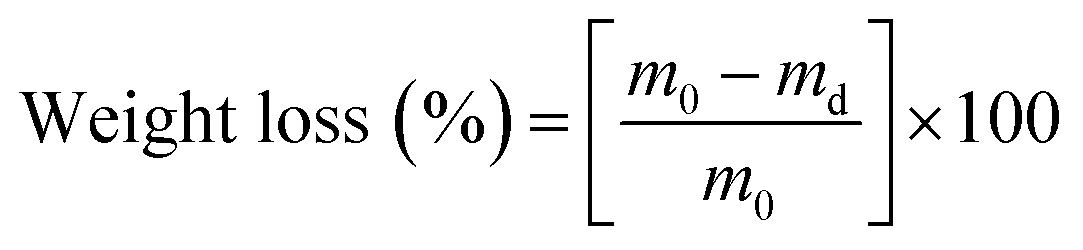 | (2) |
2.6 Mechanical properties: tensile testing
Tensile testing was used to evaluate the mechanical properties of the electrospun meshes (Instron 3344 and Bluehill Universal Software, Instron, Norwood, MA, USA) following ISO-527-3/2/5 standard. Meshes (n = 6) were cut into strips (5 × 50 mm), clamped with an initial distance between grips of 25 mm, and stretched at a deformation rate of 5 mm min−1 using a 10N load cell until break. Young's moduli were determined using the slope of the linear portion of the stress–strain curves within the range of 1–2% strain.2.7 Hydrogen peroxide production
AR assay kit was used to determine H2O2 release in the samples following the manufacturers protocol. In this study, PVA RO40 electrospun meshes (n = 5) with different crosslinking procedures were considered at 0, 0.25, 1, 2, 3, 6, 24, 48, and 72 h. Meshes (100 mg) were placed in 6-well plates and 500 μL of 1X PBS were added for activation of the enzymatic action; plates were kept at room temperature and protected from light. At each timepoint 10 μL were taken from each well and serially diluted five-fold. 50 μL of standards and diluted samples were loaded into a 96-well plate containing 50 μL of AR working solution. Fluorescence was measured for each dilution at an emission detection of 590/20 nm using a microplate reader with excitation in the range 530/30 nm (CLARIOstar, BMG Labtech, Germany). The results were normalised for dilution and mass, and presented as μM mL−1 g per H2O2 (molarity detected as a result of placing 1 g of material in 1 mL of PBS).2.8 Determination of antimicrobial susceptibility
Meshes were then removed and placed into 9 mL sterile MHB with glass beads. Meshes were vortexed to detach cells from their surface and serially diluted (1:10) in fresh MHB. 200 μL from each dilution were inoculated onto agar plates in technical duplicates and incubated overnight. A digital SLR camera (D3200, Nikon) was used to take pictures of every plate, and colonies were counted using ImageJ (Fiji, v1.53c).24 The number of colony forming units (CFU) was recorded for each dilution and results were expressed as log10(CFU count per mm2).
2.9 Cell metabolic activity
hADSCs cells were cultured with MesenPRO RS™ medium containing 2% (v/v) growth supplement, 1% (v/v) glutamine, and 1% (v/v) penicillin/streptomycin until 80% confluence and harvested using 0.05% trypsin-EDTA at passage 7. A total of 25000 cells in 600 μL of medium were seeded onto each mesh (n = 6, 19 mm diameter) and incubated in a cell culture incubator (37 °C, 5% CO2, and 95% humidity).
The metabolic activity of cells, indicating cell proliferation, was evaluated using AB resazurin assay. Fluorescence intensity was measured on days 1, 3, 7, and 14 after cell seeding. On day 1, all samples were transferred to a new 12-well plate to prevent unattached cells from influencing the result. At each time point, 10% of the total medium by volume (60 μL) of AB solution were added to each well and incubated for 4 h. After incubation, 150 μL of each sample were transferred to a 96-well plate and fluorescence was measured (540 nm excitation/590 nm emission wavelength) using a plate reader (CLARIOstar, BMG Labtech, Germany). Samples were washed twice in sterile PBS to remove the AB solution before the addition of fresh media. Cell culture media was changed every 2 days.
2.10 Statistical analysis
Statistical analysis was performed using one-way analysis of variance (ANOVA) and post hoc Tukey test using GraphPad Prism software v. 9.4.0 (Graphpad Software Inc., San Diego, CA, USA). A p value < 0.05 (* = p < 0.05, ** = p < 0.01, *** = p < 0.001, **** = p < 0.0001) was considered statistically significant and results are reported as mean ± standard deviation (SD).3. Results and discussion
3.1. Fourier-transform infrared spectroscopy (FTIR)
Fig. 1 displays the FTIR spectra obtained from electrospun meshes of PVA and PVA RO40, crosslinked with GA vapor for 6 and 48 h. The PVA Vap CL 6 h mesh exhibits two minor peaks around 2940 cm−1 (region B, Fig. 1), whereas a third band attributed to C–H stretching related to aldehydes appears around 2830 cm−125 when the exposure time is 48 h. This peak is indicative of the formation of crosslinks between PVA and GA25 and suggests that longer exposure times result in better crosslinking. Another characteristic band of crosslinked PVA resulting from the carbonyl group (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) is observed at around 1735 cm−121,25 (region C, Fig. 1), which further validates the vapour crosslinking method and is particularly prominent after 48 h of GA exposure.
O) is observed at around 1735 cm−121,25 (region C, Fig. 1), which further validates the vapour crosslinking method and is particularly prominent after 48 h of GA exposure.
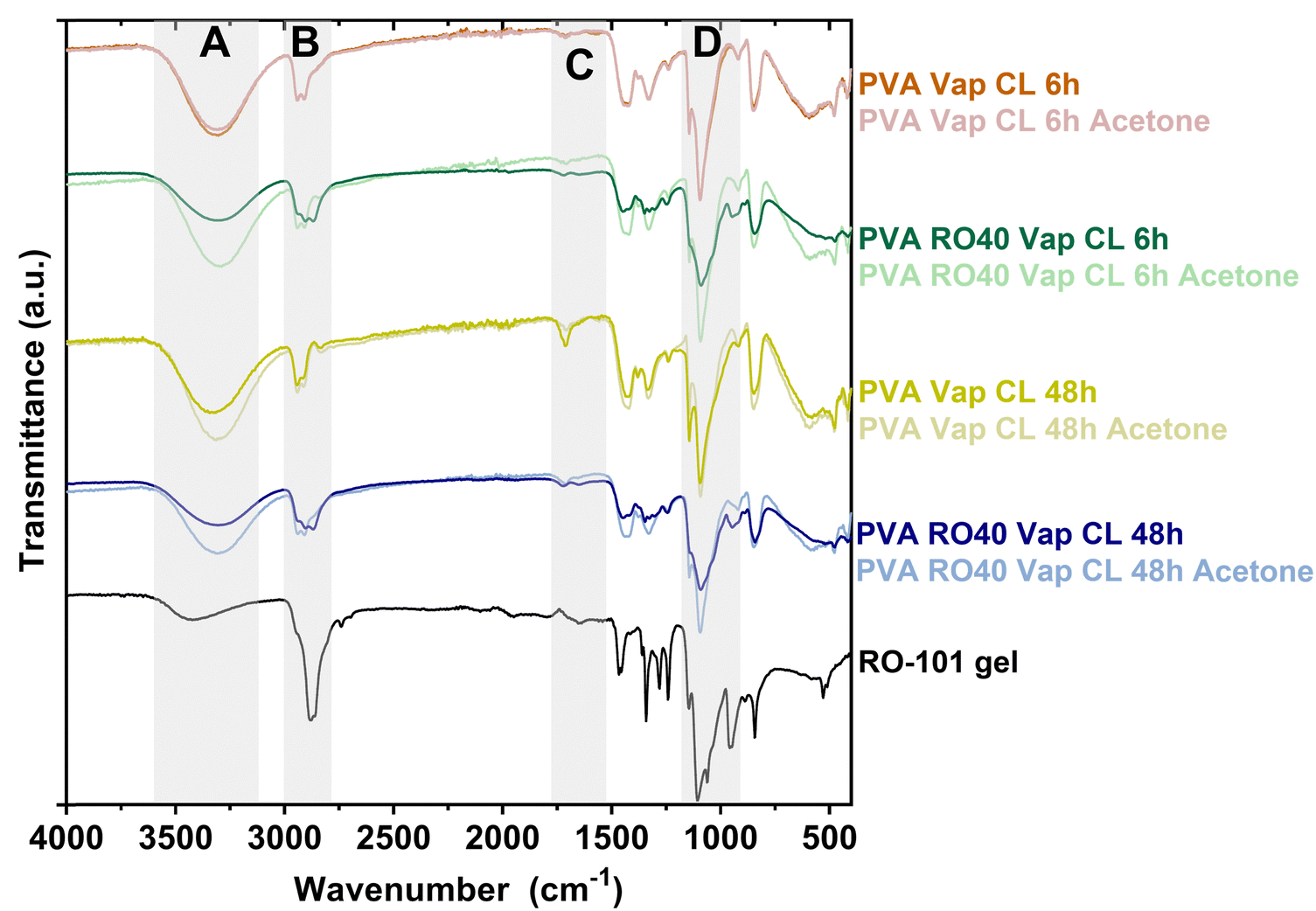 | ||
| Fig. 1 ATR-FTIR spectra of PVA and PVA RO40 electrospun meshes vapour crosslinked for either 6 h or 48 h exposure to GA. | ||
The FTIR spectra also provided confirmation of the successful encapsulation of RO-101 in the electrospun meshes and evidenced its presence after crosslinking for 6 and 48 h (Fig. 1). The incorporation of RO-101 in the samples was marked by broadening of the 2940 cm−1 peaks (C–H from alkyl groups), which are a characteristic feature of crosslinked PVA,25 leading to a wider peak structure (region B in Fig. 1). Furthermore, samples that contained RO-101 caused broadening and reduction in the intensity of the 1087 cm−1 peak (C–O–C bond) characteristic of crosslinked PVA25 (region D in Fig. 1). Interestingly, washing the samples containing RO-101 with acetone resulted in a spectrum that was more similar to pure PVA, which suggests the loss of RO-101 during the washing process.
The O–H stretching vibration peak (region A in Fig. 1, 3330–3350 cm−1) of all the samples exhibited lower intensity when compared to uncrosslinked PVA,21,25 providing strong evidence of the reaction between PVA and GA, which results in the formation of acetal bridges among the pendant hydroxyl groups of the PVA chains.21,25 Furthermore, the intensity of this band was significantly lower in the samples crosslinked for 48 h compared to those crosslinked for 6 h, indicating more efficient crosslinking and less unreacted GA. This observation was also apparent when comparing PVA RO40 samples to PVA exposed for 6 and 48 h, suggesting that the incorporation of RO-101 improves the effectiveness of the PVA–GA reaction. Moreover, samples containing RO-101 exhibited lower intensity in the peaks corresponding to unreacted ends of GA (regions A and C in Fig. 1), which can be attributed to the bi-functional nature of GA, where one aldehyde group reacts with the hydroxyl groups of the PVA polymer chain to form a hemi-acetal structure,25 while the other aldehyde group reacts with the components of RO-101 polymeric gel, facilitating the immobilization of GOx enzyme present in the gel.17,26,27
Results indicate that the electrospun meshes incorporating RO-101 are sufficiently crosslinked. Furthermore, exposure of PVA RO40 meshes to GA vapour for 48 h appears to result in a greater degree of crosslinking, potentially with reduced GA residuals compared to exposure for 6 h. Acetone washes resulted in the partial removal of unreacted GA in PVA RO40 samples, but also in the loss of encapsulated gel.
3.2 Electrospun mesh morphology
Morphology studies of uncrosslinked and crosslinked PVA RO40 submicron fibres are shown in Fig. 2. Regardless of crosslinking mode, all electrospun membranes containing RO-101 remained in the submicrometric range could mimic the three-dimensional structure of the natural extracellular matrix.9 Uncrosslinked meshes were smooth and beadles (Fig. 2(a)), with fibres averaging 378 ± 94 nm in diameter. However, these meshes immediately degrade in aqueous environments, with behaviour that resembles that of hydrogels. This presents a practical challenge for any biological studies and raises concerns for the application of the product in wound care, especially in cases with heavy exudate. Hence, it was aimed to fabricate more stable meshes with enhanced mechanical properties via different modes of crosslinking: in situ CL (GA added to the polymeric solution right before electrospinning) and vapour CL (exposure to GA vapour for 2 h, 6 h and 48 h).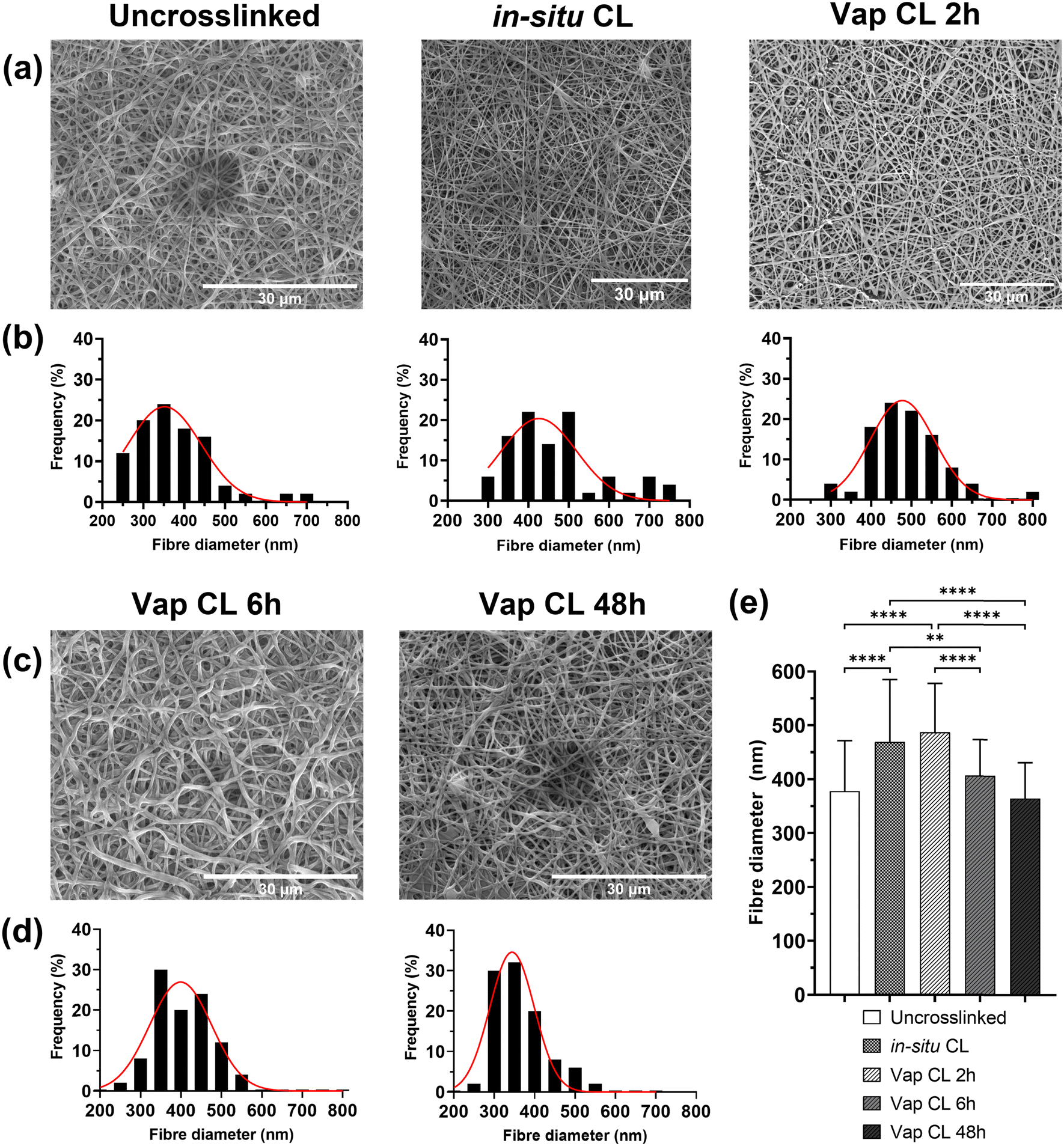 | ||
| Fig. 2 Morphology of uncrosslinked, vapour and in situ crosslinked electrospun fibres. (a), (c) SEM images, (b), (d) fibre diameter distribution, and (e) average fibre diameter of uncrosslinked, in situ CL, and vapour CL (at different GA exposure times) PVA RO40 electrospun samples. | ||
It was observed that In situ CL meshes’ diameters had significantly increased (Fig. 2(e)) to an average of 469 ± 116 nm and with a wider distribution (Fig. 2(b)). The increase in fibre diameter could be the product of the increase in viscosity in the polymeric solution due to its reaction with GA.28 This may lead to an initial lower occurrence of defects in the mesh when it reaches a threshold in solution cohesiveness.29 However, as time progresses viscosity continues to increment in the syringe, which leads to thicker fibres and subsequent flow instability,29 and practical difficulties in fabrication after 45 min. Moreover, the pH of PVA RO40 solutions decreases as time progresses (ESI,† Fig. S1), possibly due to the formation of gluconic acid by GOx,8 which contributes to an increase in conductivity without affecting viscosity and surface tension.30 This may be counteracting the effect of the increase in viscosity and preventing in situ CL fibres to have even thicker diameters. Still, this process was not considered viable for large-scale fabrication of wound dressings due to the practical inconsistencies of this time-dependent crosslinking mode.
In the case of vapour CL samples, an exposure of 2 h also resulted in a significantly larger fibre diameter (487 ± 90 nm) when compared to the uncrosslinked case. Moreover, unusual dark lumpy regions were observed in fibre intersections which could be GA residuals on the mesh, and some fibres started to merge (Fig. 2(a)). This has been previously observed in meshes crosslinked using lower concentrations of GA vapour for the same exposure time.31 This represents a major challenge to this research, as it was already anticipated that the encapsulation of the GOx enzyme caused by the exposure to GA could decrease the enzymatic production of H2O2,17,26,27 but the residuals could also have a cytotoxic effect on cells which could negatively impact proliferation, and thus the healing process.32,33 Hence, if vapour crosslinking is employed, it is imperative to adequately remove all GA residuals from the meshes, without removing the encapsulated RO gel in the process. SEM was used to assess the potential of a polar (isopropyl alcohol ∼100%, IPA) and a non-polar solvent (acetone) as rinsing medium for extraction of GA in vapour-crosslinked samples by observing their effect on fibre morphology (ESI,† Fig. S2). ESI,† Fig. S2b–e show that rinsing Vap CL 2 h meshes for 30 s in either acetone or IPA may reduce or eliminate the observed dark lumpy regions. Both rinses significantly reduced the fibre diameter (ESI,† Fig. S2a), which could be an indicator of removal of either RO-101 or residual GA on the fibres, as opposed to the reported swelling for longer washes.31 Nevertheless, these washes could be also removing the encapsulated gel, which is why H2O2 production assays will be carried out for washed samples. Out of the two, acetone wash will be further studied as it achieved a slightly greater fibre diameter reduction, a narrower fibre distribution, and a more uniform submicron structure was obtained which resembles the morphology of the uncrosslinked mesh.
Vap CL 6 h samples showed a considerable increase in the number of fused fibres compared to 2 h exposure (Fig. 2(c)). This bonding between fibres could be explained by their softening and swelling when in continued contact with the aqueous GA solution.21 Conversely, fibre diameter was significantly reduced to 407 ± 67 nm when exposure was increased from 2 to 6 h (Fig. 2(e)). Further reductions were observed at an exposure of 48 h, and the distribution continued to narrow. Both 6 and 48 h exposure achieved fibre diameters that were not significantly different to the uncrosslinked mesh.
Since Vap CL with a 2 h exposure may lead to inconsistent morphology with a potentially high presence of unreacted aldehyde groups, and in situ CL causes difficulties with the fabrication process, only 6 h and 48 h vapour CL exposure times were studied further.
To simulate fibre behaviour in aqueous environments, SEM images of Vap CL 6 h and 48 h samples immersed in PBS for different times were studied (ESI,† Fig. S3). It was evident that CL for 48 h resulted in improved stability in water compared to a 6 h exposure. After only 1 h of immersion, fibres in Vap CL 6 h samples were merged and flattened (ESI,† Fig. S3a). Vap CL 48 h samples showed no significant change in fibre diameter and distribution after being immersed in PBS for 1 h (ESI,† Fig. S3c–e). When submerged in PBS for 24 h there was observable swelling, represented by an increase in diameter and a wider fibre distribution. In the case of Vap CL 6 h samples immersed for the same time, morphology was severely affected, with the majority of fibres merging and a visible decrease in porosity. This was caused due to softening and degradation of the fibres which contributed to a high rate of inter-fibre bonding (ESI,† Fig. S3a). The differences in the samples after being immersed in 24 h were visible to the naked eye (ESI,† Fig. S4). Vap CL 6 h samples were similar to uncrosslinked ones, providing insufficient improvements in aqueous stability (supported by swelling results, Fig. 2(b)). The reduced porosity, along with the hydrogel-like texture of these samples when in contact with water, may be beneficial for certain drug delivery and tissue engineering applications. However, since this study aims for a mesh that can be stable for up to 72 h in a wound setting, morphological results suggest that Vap CL 48 h samples may be promising candidates as dressings.
3.3 Wettability, water absorption capacity and weight loss behaviour
Measurements of water contact angles on uncrosslinked and vapour-crosslinked (6 h and 48 h exposure times) PVA RO40 meshes, along with swelling and weight loss studies provided valuable information about their hydrophilicity, their ability to absorb exudate, and mechanical stability in water.Uncrosslinked samples showed an average water contact angle of 20 ± 2°. Meshes showed a gradual increase in hydrophobicity with crosslinking (Fig. 3(a)). Vap CL 6 h samples averaged 27 ± 3°, and longer exposure time to GA resulted in a significantly higher contact angle (31 ± 10° for Vap CL 48 h). The highest contact angle measured was 45° for a Vap CL 48 h sample, which indicates that all samples were still hydrophilic, and demonstrated potential for high absorption rates since the majority of the drops were distributed over the meshes’ surface area within the first 30 s.
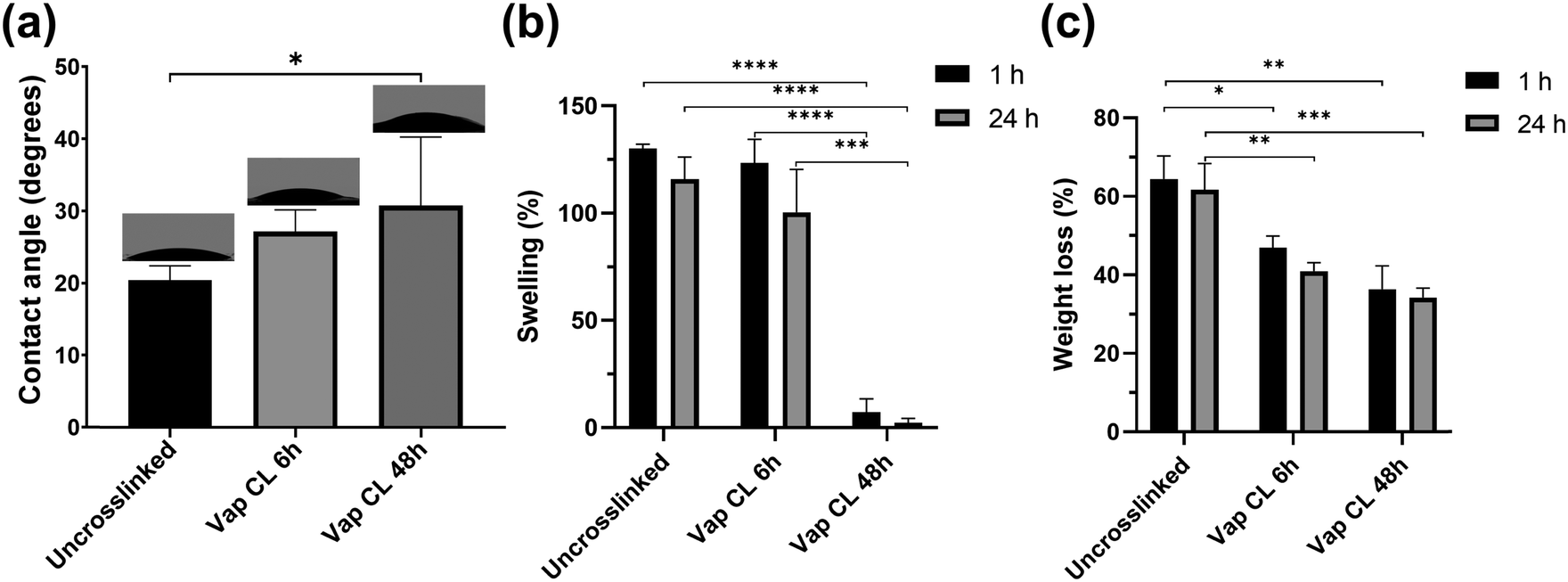 | ||
| Fig. 3 Wettability, swelling, and degradation characteristics. (a) Contact angle measurements, (b) percentage swelling and (c) percentage weight loss of electrospun PVA RO40 samples after immersion in PBS (pH 7.4) for 1 and 24 h. | ||
From the water absorption studies, it was found that the degree of swelling was not affected with 6 h vapour crosslinking time, but significantly reduced when the exposure time was 48 h (Fig. 3(b)). Average swellings after 24 h immersion were 116 ± 10% and 100 ± 20% for uncrosslinked and Vap CL 6 h PVA RO40 meshes, respectively. On the contrary, Vap CL 48 h samples averaged only 2 ± 2% swelling after the same time. Even though wettability data demonstrated that this sample was still hydrophilic, the swelling study proved there is no water retention by Vap CL 48 h fibres, which suggests that they are capable of accumulating fluids and spreading them over their large surface area without absorbing it into the fibres. These results correlate to the unaffected porosity observed in SEM studies (Fig. 2(c)) and denote a stable permeability in an aqueous environment which is beneficial to allow oxygen into a wound bed, and could facilitate debridement and exudate removal.
Moreover, there was no significant difference in any sample's swelling when immersed in PBS for 1 h when compared to 24 h, which supports the claim that samples provide a fast fluid collection within the first seconds of application. Still, all samples show a slightly higher swelling percentage with shorter immersion time. This may be due to the larger reduction in mass resulting from the release and enzymatic activity of the encapsulated gel in PVA RO40 fibres when the immersion time is 24 h. Weight loss studies support this, as there is proportionally larger loss in mass when the immersion time is 24 h when compared to 1 h (Fig. 3(c)).
Weight loss percentages were also directly correlated with exposure time to GA. Immersions in PBS for both 1 and 24 h resulted in significantly lower weight losses when crosslinking time increased. Uncrosslinked meshes lost an average of 62 ± 7% of their mass after 24 h in PBS, whereas Vap CL 6 h and Vap CL 48 h samples lost 41 ± 2% and 34 ± 2%, respectively. It is worth highlighting that Vap CL 6 h meshes swell as much as uncrosslinked ones but retained approximately 20% more mass, which demonstrates less fibre degradation whilst retaining PVA's characteristic hydrophilicity and their ability to absorb exudate. The vapour crosslinking method employed in this study improved meshes’ mechanical stability in aqueous environments without affecting the interaction of the encapsulated RO gel with water molecules, even with an exposure of 48 h.
3.4 Mechanical properties: tensile testing
Tensile mechanical properties of uncrosslinked and crosslinked PVA RO40 meshes were tested to compare the impact of GA vapour exposure and evaluate their suitability for wound dressings and other tissue engineering applications (Fig. 4). It was expected that crosslinking would enhance the meshes’ mechanical properties.21Fig. 4(a)–(c) illustrates strain–stress curves before and after vapour crosslinking for exposures of 6 h and 48 h. | ||
| Fig. 4 Mechanical properties of uncrosslinked, vapour and in situ crosslinked electrospun meshes. (a)–(c) Stress–strain curves of uncrosslinked, Vap CL 6 h and Vap CL 48 h, (d) Young's modulus, (e) ultimate tensile strength and (f) ductility of the same meshes. | ||
Fig. 4(d) shows that uncrosslinked samples averaged a Young's modulus of 2.37 ± 0.42 MPa. This started to decrease with 6 h GA exposure (1.88 ± 0.34 MPa), and it was significantly diminished when exposure time was increased to 48 h (1.56 ± 0.38 MPa). On the other hand, the uncrosslinked meshes’ ultimate tensile strength (UTS) was increased from 9.43 ± 0.82 MPa to 12.72 ± 1.25 MPa after crosslinking for 6 h (Fig. 4(e)). Further exposure (48 h) boosted it to 14.23 ± 4.17 MPa. It is important to note that while Fig. 4(e) shows a slightly higher tensile stress for the 48 h crosslinked samples, no significant difference is observed compared to the 6 h crosslinking, the primary difference is between crosslinked and uncrosslinked samples. SEM studies (Fig. 2) had previously demonstrated there was no significant difference in fibre diameter between uncrosslinked and Vap CL 6 h and 48 h samples. Hence, this increase in strength could be attributed to the rigid inter-fibre bonding that occurred at intersection points with vapour CL.21 The variation in the 48 h crosslinked sample stress–strain curves (Fig. 4(c)) and higher standard deviation (Fig. 4(e)) may be attributed to the variability in the electrospinning and vapour crosslinking process (i.e., precisely controlling thickness, fibre size distribution, fibre orientation, and homogenous vapour exposure), highlighting the challenge in manufacturing consistent meshes.
Fig. 4(f) shows that uncrosslinked meshes’ fracture strain was significantly incremented when GA exposure time was 6 h (from 136 ± 15% to 166 ± 27%). It was observed that half of Vap CL 6 h samples (Fig. 3(b)) have a much larger elastic region than any other sample tested. However, further increase in exposure time to 48 h resulted in a decrease in fracture strain (151 ± 14%), but this was still slightly larger than uncrosslinked meshes.
Based on the results, it is possible to conclude that increasing the crosslinking exposure time significantly increases the maximum stress that PVA RO meshes can withstand before breaking, confirming improved strength and stability. Moreover, the lower Young's moduli confirm that meshes become more flexible in the elastic region when they are exposed to GA vapour for longer. Conversely, increasing the exposure time from 6 to 48 h reduces the material's ductility (Fig. 4(f)). The crosslinking process presented in this study provides a way to control the mechanical properties of the electrospun wound dressing, which could allow to better mimic the natural skin environment for scarless regeneration by favouring differentiation.34
3.5 Hydrogen peroxide production
H2O2 release studies over 72 h were carried out on PVA RO40 samples to verify if exposure to GA, either added as a pre-processing step to the polymeric solution (in situ CL) or as a vapour post-processing step for electrospun meshes, had a negative impact on GOx activity. Also, since acetone wash was considered as a method to remove unreacted GA remaining in vapour CL meshes, washed samples were compared to their unwashed counterparts to check if the wash would nullify H2O2 production. This assay was extended to 5 days to discover for how long Vap CL 6 h samples were able to support H2O2 production.Fig. 5(a) shows H2O2 production for uncrosslinked, in situ CL and Vap CL 6 h samples for 72 h. It was evident that uncrosslinked samples displayed the highest release, with a peak H2O2 concentration of 1403 ± 143 μM g−1 mL−1 at 72 h. These samples demonstrated rapid enzymatic activity in aqueous environment, releasing 1341 ± 105 μM g−1 mL−1 within the first 15 min of activation.
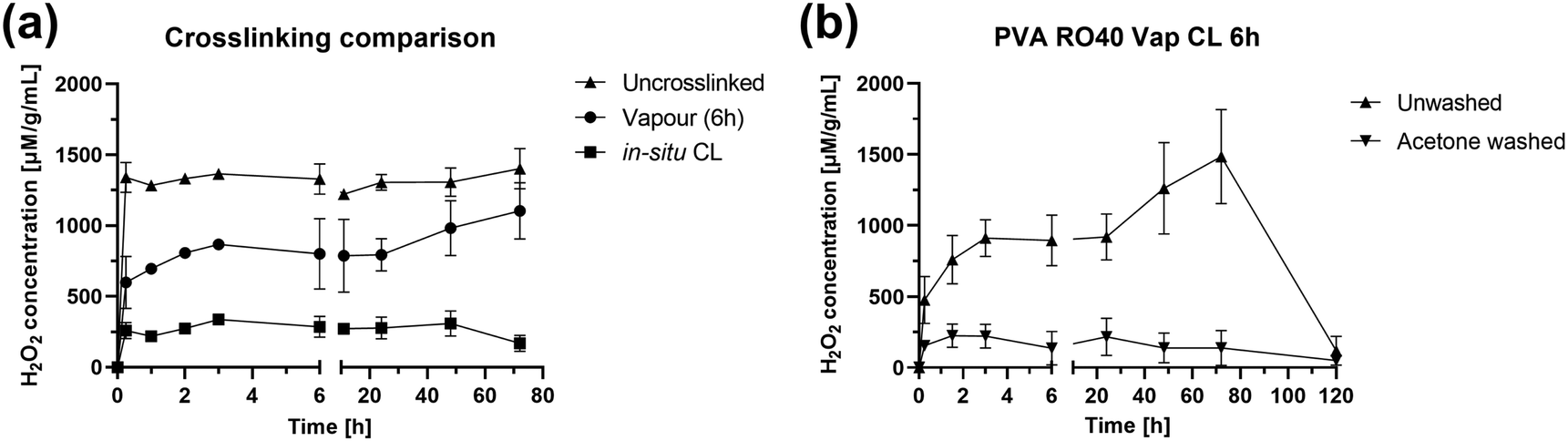 | ||
| Fig. 5 H2O2 production over 72 h for PVA RO40 samples showing: (a) the effect of crosslinking method, and (b) the effect of acetone wash in enzymatic activity. | ||
On the contrary, in situ CL showed the lowest release out of the samples studied, with a maximum of 309 ± 89 μM g−1 mL−1, which represents an approximate 78% reduction in maximum H2O2 production compared to uncrosslinked samples. In situ CL process required the presence of HCl in the polymeric solution to provide the acid catalyst for the crosslinking reaction.21 Lowering the pH of the solution to 3 as suggested by the literature,21 could render GOx inactive since this enzyme presents optimum activity at pH of 6.1, then rapidly decreasing with pH below 5.5, and could become inactive at pH 4.8 Hence, only 10 μL of HCl were added to the solution, enough to lower the pH of the solution to ∼5. However, the pH of PVA RO40 solutions continuously decreases as time progresses (ESI,† Fig. S1), possibly due to the formation of gluconic acid by GOx,8 reaching a pH of 4.4 after 1 h. This could explain the limited enzymatic activity observed by these samples, which may result in a reduced antimicrobial action in a wound setting.
It was expected that the encapsulation of the GOx enzyme caused by GA vapour crosslinking could decrease the production of H2O2.17,26,27 Vapour CL samples exposed to GA for 6 h achieved a H2O2 peak of 1103 ± 199 μM g−1 mL−1 after 72 h, which is comparable to uncrosslinked samples (21% lower). Both samples reached their peak after 3 days (Fig. 5(a)), but Vap CL 6 h samples displayed a more gradual release. The steady-state portion of the H2O2 release profile is given by the rate at which H2O2 is produced balanced by the rate at which H2O2 is consumed. Over time, H2O2 will start to oxidise the glucose oxidase rendering it inactive.35 Release profiles suggest that the enzyme immobilisation caused by GA vapours allows a more gradual H2O2 production compared to uncrosslinked,17,26,27 and consequently, a later inactivation of GOx. This explains why Vap CL 6 h achieved the highest productions in the last two time points, suggesting potential release even beyond 72 h. Hence, Vap CL 6 h samples were assessed for 120 h (Fig. 5(b)).
From Fig. 5(b) it was verified that Vap CL 6 h H2O2 levels dropped after 3 days, confirming GOx inactivation by day 4. This is due to the accumulation of H2O2 and reduction in pH rather than the depletion of glucose, which is in excess. Furthermore, a contributing factor is the gradual loss of mesh integrity by this timepoint. Moreover, the comparison between washed and unwashed samples made it evident that acetone wash significantly affected the gel's activity, with an approximate 85% reduction in maximum H2O2 concentration. As confirmed by FTIR spectra (Fig. 1), the wash removes a substantial amount of the encapsulated RO-101 gel, which results in the limited H2O2 production.
3.6 Antimicrobial susceptibility assays
The antimicrobial efficacy of ROS-producing products has been demonstrated both in vitro and in vivo against Gram-negative and Gram-positive bacteria, including antibiotic resistant species.36–38 Although further optimisation is required to remove unreacted GA and improve the encapsulation of RO-101 for a better control of the H2O2 release profile, the inhibiting activity of crosslinked PVA RO40 meshes was studied using disk diffusion and time-kill assays against S. Aureus Newman strain. PVA meshes without RO-101 were included as negative control.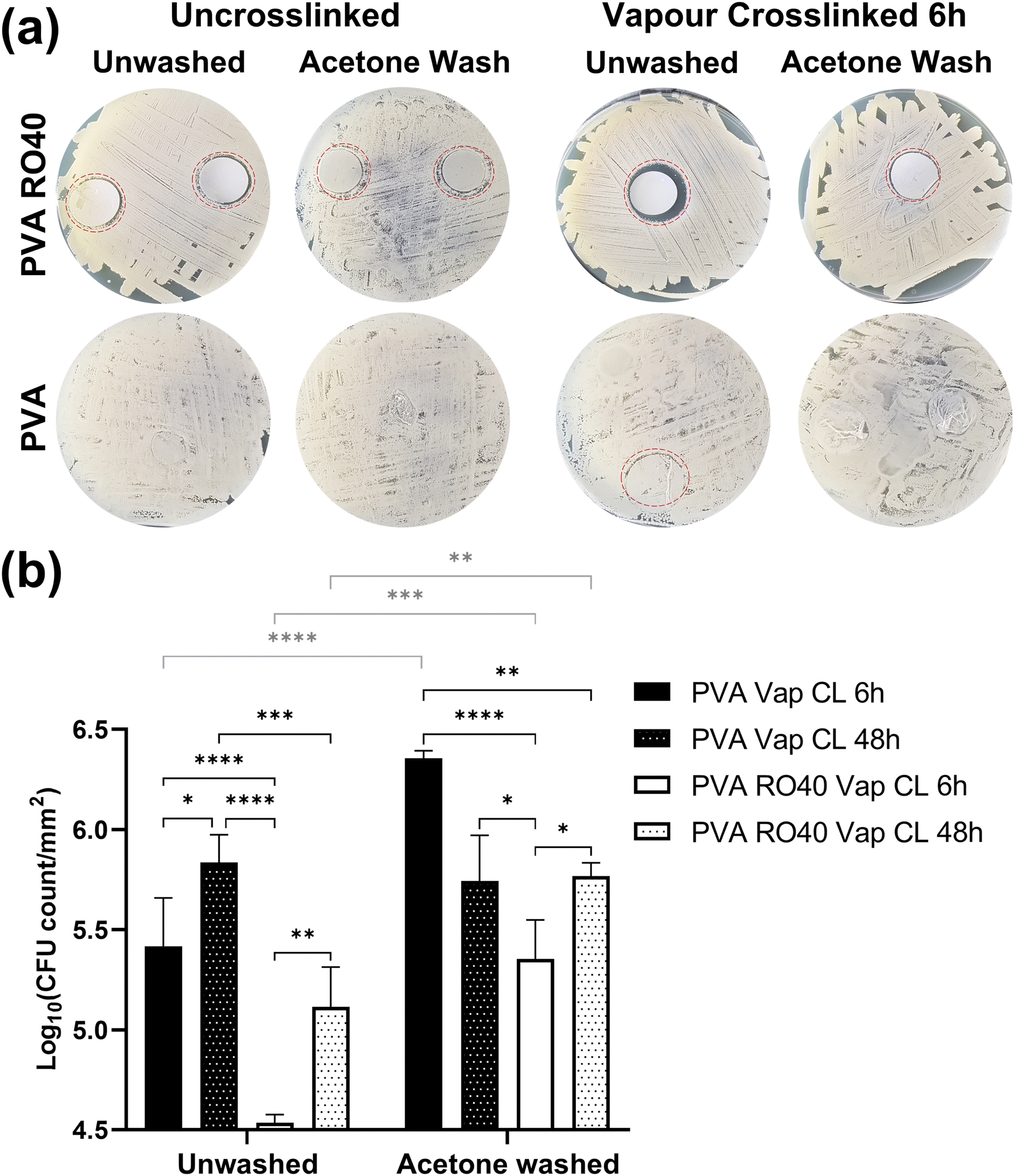 | ||
| Fig. 6 Antibiotic activity of vapour-crosslinked PVA RO40 meshes against S. aureus Newman strain illustrated by (a) representative agar plates showing zones of inhibitions, and (b) reductions in viable count after 24 h incubation expressed as Log10 CFU of living bacteria attached to mesh per mm2. Pure PVA meshes were included as negative controls. | ||
As expected, uncrosslinked PVA meshes showed no inhibition of bacteria growth. In Fig. 6(a) it is difficult to locate PVA samples since bacteria grew completely over them. The only visible PVA mesh was the unwashed Vap CL 6 h one (circled in red Fig. 6(a)), where a small indication of inhibition was identified. This implies that unreacted GA is contributing to Vap CL meshes’ antibiotic effect, and it emphasises the importance to establish an adequate process to remove all GA residuals without affecting RO-101's enzymatic activity. When PVA meshes were acetone-washed, S. Aureus colonies were able to grow like in the uncrosslinked cases, confirming that a considerable part of the cytotoxic GA residuals were removed with the wash. A similar phenomenon was observed with the PVA RO40 Vap CL 6 h washed sample, where a smaller inhibition was recorded when compared to the unwashed case. Still, as FTIR spectra and H2O2 release profiles show, RO-101 was still present in the washed mesh and it was able to support inhibition around the mesh, and bacteria was not able to grow on it either. All zones of inhibitions of samples containing RO-101 are larger than pure PVA, suggesting that a substantial portion of antimicrobial activity is attributed to the enzymatic production of H2O2 by the gel encapsulated in the fibres.
From this experiment it was qualitatively concluded that there is a cytotoxic effect caused by GA residuals, but this could be removed or reduced with an appropriate post-processing method. In this case, an acetone wash was able to reduce the cytotoxic effect whilst still preserving the antimicrobial activity of the RO-101 gel to some degree.
Fig. 6(b) highlights the significant reductions in viable count achieved by PVA RO40 Vap CL 6 h samples when compared to the mesh without RO-101 (PVA Vap CL 6 h). Average reductions were 0.88 ± 0.24 log units for the unwashed case, and 1.00 ± 0.20 for the acetone-washed samples. For samples vapour crosslinked for 48 h, RO-101 containing meshes reduced the CFU count by 0.72 ± 0.24 log units for the unwashed case but were not able to achieve any reduction when acetone-washed.
The samples with the highest viable bacteria count attached is acetone-washed PVA Vap CL 6 h, supporting an average log10 CFU count per mm2 of 6.36 ± 0.04. Overall, acetone-washed samples had significantly more viable bacteria attached when compared to their unwashed counterparts (statistical comparisons plotted in grey on Fig. 6(b)). This aligns to what was found in the agar diffusion assay, confirming that acetone effectively reduces the majority of the cytotoxic effect by unreacted GA.
The only pairwise comparison before and after washing that resulted in no significant difference was the case of PVA Vap CL 48 h. Furthermore, a smaller inhibitory effect was recorded in all samples when increasing the GA exposure from 6 h to 48 h, seemingly due to a lower amount of unreacted GA present in the fibres as they had more time to crosslink when the exposure was 48 h (as supported by SEM and FTIR analysis). This explains why in PVA Vap CL 48 h samples the wash did not affect the results. On the other hand, the wash prevented PVA RO40 Vap CL 48 h meshes to achieve a CFU count reduction since it removed the encapsulated gel from the mesh. Moreover, the shorter exposure to GA (6 h) may lead to reduced GOx immobilisation, allowing for a higher H2O2 production when compared to 48 h exposure time.
These assays demonstrated that even after significantly reducing the cytotoxic effect of unreacted GA with acetone washes, RO-101 containing meshes crosslinked for 6 h still demonstrated antibiotic activity against a Gram-positive bacterial strain commonly found in wounds. However, the 1 log reduction observed in the PVA RO40 samples are not clinically sufficient and significant further optimisation of the H2O2 production and removal of GA residues is required. Furthermore, determining the optimal antimicrobial H2O2 concentration required to be produced by the electrospun meshes is necessary. For example, Wang et al.39 demonstrated a synergistic photothermal and photocatalytic antimicrobial effect using a silicon nanowire array modified with gold–silver alloy nanoparticles. An antimicrobial effect was observed from 0.05 mg mL−1 H2O2 (∼20% killing efficiency) with it reaching a peak at 0.75–1.0 mg mL−1 (∼100% killing efficiency). However, comparison with this study and other H2O2 producing antimicrobial platforms is a challenge due to the differences in materials and method of H2O2 production.15,38–40
This preliminary study was designed as a proof of concept, with the aim of demonstrating the feasibility of incorporating the gel within the fibres and preserving its antimicrobial properties following crosslinking with GA. In future work, a broader spectrum of bacteria will be evaluated, including Gram-positive, Gram-negative, and antimicrobial resistant strains, to better assess its efficacy and potential for wider application.
3.7 Cell metabolic activity
Preliminary cytocompatibility of the PVA RO40 vapour-crosslinked electrospun meshes was assessed through evaluating cell metabolic activity of hADSCs for up to 14 days (Fig. 7). All samples are unwashed to verify the cytotoxicity of unreacted GA, and to discover if the produced H2O2 has any negative effect on cells.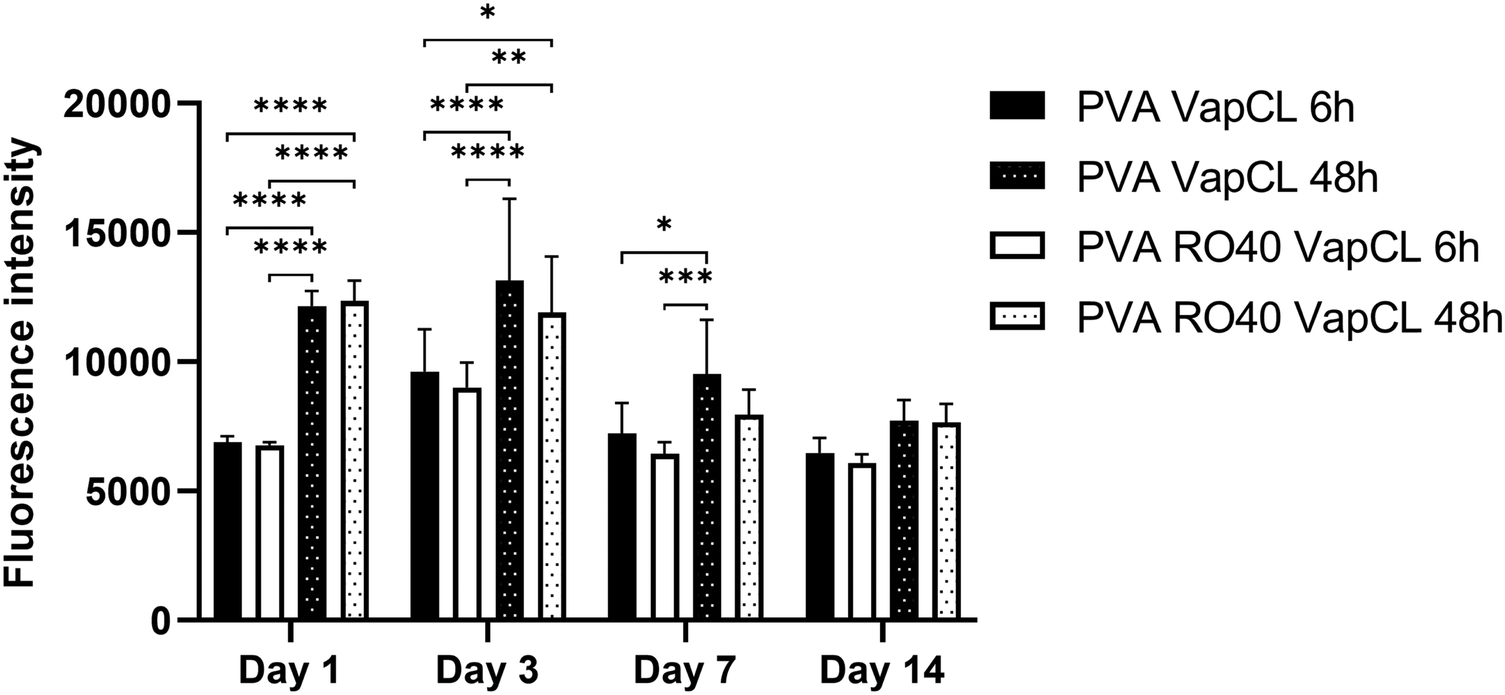 | ||
| Fig. 7 Metabolic activity of hADSCs at days 1, 3, 7, and 14 on PVA RO40 meshes crosslinked via GA vapour exposure for 6 h and 48 h. Pure PVA meshes were included as controls. | ||
The results showed no significant differences in any day between PVA RO40 samples when compared to PVA. Fluorescence intensity was only slightly lower from day 3 in PVA RO40 Vap CL samples for both 6 and 48 h crosslinking exposure time. The time point of day 3 corresponds to these sample's peak H2O2 release. Still, even the highest levels of enzymatically-produced H2O2 had no adverse effect on metabolic activity of hADSCs when compared to PVA samples, which suggests biocompatibility of samples containing RO-101 for mammalian cells.
The metabolic activity for all samples uniformly decreased after day 3, which confirms the cytotoxicity of unreacted GA residuals that were not removed from the fibres, since they were unwashed. Moreover, when samples were exposed to GA for 48 h, the recorded proliferation was significantly higher than samples crosslinked for 6 h. At 48 h exposure time, there should be less free unreacted aldehyde groups,21 and a more stable morphology in aqueous environments (ESI,† Fig. S3). In addition, these meshes displayed enhanced mechanical properties (Fig. 4), which may favour cell proliferation and differentiation.34
Vapour-crosslinked PVA RO40 electrospun meshes presented potential for tissue regeneration applications, since its H2O2 production is non-cytotoxic for mammalian cells. However, unreacted GA residuals could have a cytotoxic effect on cells which could negatively impact proliferation, and thus the healing process.32,33 Hence, a better method for GA removal without interfering with GOx enzymatic activity needs to be developed to improve the meshes biocompatibility and viability for a wound setting. Due to the enzymatic sensitivity of the PVA RO-101 meshes this is a significant challenge, as previously demonstrated (Fig. 5(b)). Repeated washing the samples with water would be inappropriate due to the premature activation of GOx, reducing the subsequent ROS production at later stages, and would begin to degrade the meshes (Fig. 3(c)). Alternatively, supercritical carbon dioxide has been shown to remove GA residues in biomaterials.41,42 This warrants further investigation as the process appears to be gentle enough to not change the biomaterial physicochemical properties. Thus, potentially could be suitable for the removal of GA residues in the developed PVA RO-101 meshes. Furthermore, different natural and synthetic polymers (e.g., alginate, gelatin, and polyethylene glycol diacrylate) which rely on different crosslinking modalities, avoiding the use of GA, can be explored as a carrier.
Additionally, comprehensive in vitro studies are required with skin specific cells and other cell types, for potential application in other tissues and organs, to ascertain the PVA RO-101 mesh biocompatibility. However, key aspects of the mesh need to be optimised prior to this including improving mesh stability and H2O2 production, and removal of GA residues.
4. Conclusions
In this study, RO-101 antimicrobial gel was successfully incorporated into PVA polymeric nanofibres for controlled delivery of H2O2 to a wound site, along with crosslinking of the mesh for improved aqueous stability. The encapsulation of the gel and crosslinking of the polymeric chains were confirmed through FTIR results, but unreacted GA residuals were also found. Acetone washes partially removed unreacted GA, but also resulted in loss of encapsulated gel.In situ crosslinking was found to be an inconsistent fabrication process, while vapour crosslinking with 48 h exposure time improved mesh mechanical stability in aqueous environments without affecting the interaction of the encapsulated gel with water molecules. The enzymatic activity of vapour crosslinked PVA RO40 meshes was not significantly affected compared to uncrosslinked meshes. The release profiles indicated that the enzyme immobilization caused by GA vapours allowed a more gradual H2O2 production. These findings suggest that the vapour crosslinking could be used to control the enzymatic activity and H2O2 release of PVA RO40 meshes, potentially improving their effectiveness for wound healing applications. Increasing the crosslinking exposure time significantly increased the strength and flexibility of PVA RO40 meshes. The trade-off between properties should be considered when designing the desired wound dressing. The meshes demonstrated antibiotic activity against a Gram-positive bacterial strain commonly found in wounds.
This proof-of-concept study suggests that vapour-crosslinked PVA RO40 electrospun mesh presents potential for tissue regeneration applications, as its H2O2 production is non-cytotoxic for mammalian cells. However, unreacted GA residuals could have a cytotoxic effect on cells, which could negatively impact proliferation and thus the healing process. Future work should establish a better method for GA removal without interfering with GOx enzymatic activity to improve the mesh's biocompatibility and viability for a wound setting. Overall, the developed mesh has potential for wound healing applications, providing a barrier against infection and promoting tissue regeneration.
Data availability
Data available on request from the authors due to commercial restrictions.Conflicts of interest
Joel Yupanqui Mieles received financial support from Matoke Holdings. Cian Vyas, Carl Diver, and Paulo Bartolo have patent pending to Matoke Holdings.Acknowledgements
Funding was provided by Matoke Holdings and the United Kingdom Engineering and Physical Sciences Research Council (EPSRC) Doctoral Prize Fellowship (EP/R513131/1) and partially supported by the Henry Royce Institute for Advanced Materials, funded through EPSRC grants (EP/R00661X/1, EP/S019367/1, EP/P025021/1, and EP/P025498/1).References
- G. Han and R. Ceilley, Chronic Wound Healing: A Review of Current Management and Treatments, Adv. Ther., 2017, 34(3), 599–610 CrossRef PubMed.
- H. Liu, C. Wang, C. Li, Y. Qin, Z. Wang and F. Yang, et al., A functional chitosan-based hydrogel as a wound dressing and drug delivery system in the treatment of wound healing, RSC Adv., 2018, 8(14), 7533–7549 RSC.
- Y. Hussein, E. M. El-Fakharany, E. A. Kamoun, S. A. Loutfy, R. Amin and T. H. Taha, et al., Electrospun PVA/hyaluronic acid/L-arginine nanofibers for wound healing applications: Nanofibers optimization and in vitro bioevaluation, Int. J. Biol. Macromol., 2020, 164, 667–676 CrossRef CAS PubMed.
- J. Qu, X. Zhao, Y. Liang, T. Zhang, P. X. Ma and B. Guo, Antibacterial adhesive injectable hydrogels with rapid self-healing, extensibility and compressibility as wound dressing for joints skin wound healing, Biomaterials, 2018, 183, 185–199 CrossRef CAS PubMed.
- M. Séon-Lutz, A.-C. Couffin, S. Vignoud, G. Schlatter and A. Hébraud, Electrospinning in water and in situ crosslinking of hyaluronic acid/cyclodextrin nanofibers: Towards wound dressing with controlled drug release, Carbohydr. Polym., 2019, 207, 276–287 CrossRef PubMed.
- J. Lev, M. Holba, M. Došek, L. Kalhotka, P. Mikula and D. Kimmer, A novel electrospun polyurethane nanofibre membrane – production parameters and suitability for wastewater (WW) treatment, Water Sci. Technol., 2014, 69(7), 1496–1501 CrossRef CAS PubMed.
- H. Maleki, A. A. Gharehaghaji and P. J. Dijkstra, A novel honey-based nanofibrous scaffold for wound dressing application, J. Appl. Polym. Sci., 2013, 127(5), 4086–4092 CrossRef CAS.
- J. Yupanqui Mieles, C. Vyas, E. Aslan, G. Humphreys, C. Diver and P. Bartolo, Honey: An Advanced Antimicrobial and Wound Healing Biomaterial for Tissue Engineering Applications, Pharmaceutics, 2022, 14(8), 1663 CrossRef CAS PubMed.
- Y. Tang, X. Lan, C. Liang, Z. Zhong, R. Xie and Y. Zhou, et al., Honey loaded alginate/PVA nanofibrous membrane as potential bioactive wound dressing, Carbohydr. Polym., 2019, 219, 113–120 CrossRef CAS PubMed.
- A. Naeimi, M. Payandeh, A. R. Ghara and F. E. Ghadi, In vivo evaluation of the wound healing properties of bio-nanofiber chitosan/polyvinyl alcohol incorporating honey and Nepeta dschuparensis, Carbohydr. Polym., 2020, 240, 116315 CrossRef CAS PubMed.
- S. Kanimozhi, G. Kathiresan, A. Kathalingam, H.-S. Kim and M. N. R. Doss, Organic nanocomposite Band-Aid for chronic wound healing: a novel honey-based nanofibrous scaffold, Appl. Nanosci., 2020, 10(5), 1639–1652 CrossRef CAS.
- S. Ghalei, J. Li, M. Douglass, M. Garren and H. Handa, Synergistic Approach to Develop Antibacterial Electrospun Scaffolds Using Honey and S-Nitroso-N-acetyl Penicillamine, ACS Biomater. Sci. Eng., 2021, 7(2), 517–526 CrossRef CAS PubMed.
- C. Dunnill, T. Patton, J. Brennan, J. Barrett, M. Dryden and J. Cooke, et al., Reactive oxygen species (ROS) and wound healing: the functional role of ROS and emerging ROS-modulating technologies for augmentation of the healing process, Int. Wound J., 2017, 14(1), 89–96 CrossRef PubMed.
- M. Dryden, Reactive oxygen species: a novel antimicrobial, Int. J. Antimicrob. Agents, 2018, 51(3), 299–303 CrossRef CAS PubMed.
- T. J. Hall, J. M. A. Blair, R. J. A. Moakes, E. G. Pelan, L. M. Grover and S. C. Cox, Antimicrobial emulsions: Formulation of a triggered release reactive oxygen delivery system, Mater. Sci. Eng., C, 2019, 103, 109735 CrossRef CAS PubMed.
- Q. Zhang, Y. Li, Z. Y. Lin, K. K. Y. Wong, M. Lin and L. Yildirimer, et al., Electrospun polymeric micro/nanofibrous scaffolds for long-term drug release and their biomedical applications, Drug Discovery Today, 2017, 22(9), 1351–1366 CrossRef CAS PubMed.
- F. Leonarta and C.-K. Lee, Nanofibrous Membrane with Encapsulated Glucose Oxidase for Self-Sustained Antimicrobial Applications, Membranes, 2021, 11(12), 997 CrossRef CAS PubMed.
- Q. Zhang, Z. Lin, W. Zhang, T. Huang, J. Jiang and Y. Ren, et al., Fabrication of green poly(vinyl alcohol) nanofibers using natural deep eutectic solvent for fast-dissolving drug delivery, RSC Adv., 2021, 11(2), 1012–1021 RSC.
- F. N. Parin, P. Terzioğlu, Y. Sicak, K. Yildirim and M. Öztürk, Pine honey–loaded electrospun poly(vinyl alcohol)/gelatin nanofibers with antioxidant properties, J. Text. Inst., 2021, 112(4), 628–635 CrossRef CAS.
- S. Bao, B. Chen, Y. Zhang and Y. Tang, Synthesis of coated solvent impregnated resins by PVA cross-linked with vapor-phase glutaraldehyde for adsorption of vanadium(IV), React. Funct. Polym., 2018, 128, 58–66 CrossRef CAS.
- A. G. Destaye, C.-K. Lin and C.-K. Lee, Glutaraldehyde Vapor Cross-linked Nanofibrous PVA Mat with in Situ Formed Silver Nanoparticles, ACS Appl. Mater. Interfaces, 2013, 5(11), 4745–4752 CrossRef CAS PubMed.
- R. P. Shaikh, P. Kumar, Y. E. Choonara, L. C. du Toit and V. Pillay, Crosslinked electrospun PVA nanofibrous membranes: elucidation of their physicochemical, physicomechanical and molecular disposition, Biofabrication, 2012, 4(2), 025002 CrossRef PubMed.
- [ISO] IOfS. ISO 11137-2:2013 Sterilization of health care products—Radiation—Part 2: Establishing the sterilization dose. 2013.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair and T. Pietzsch, et al., Fiji: an open-source platform for biological-image analysis, Nat. Methods, 2012, 9(7), 676–682 CrossRef CAS PubMed.
- H. S. Mansur, C. M. Sadahira, A. N. Souza and A. A. P. Mansur, FTIR spectroscopy characterization of poly(vinyl alcohol) hydrogel with different hydrolysis degree and chemically crosslinked with glutaraldehyde, Mater. Sci. Eng., C, 2008, 28(4), 539–548 CrossRef CAS.
- L. Betancor, F. López-Gallego, A. Hidalgo, N. Alonso-Morales, G. Dellamora-Ortiz and J. M. Guisán, et al., Preparation of a very stable immobilized biocatalyst of glucose oxidase from Aspergillus niger, J. Biotechnol., 2006, 121(2), 284–289 CrossRef CAS PubMed.
- J. L. House, E. M. Anderson and W. K. Ward, Immobilization techniques to avoid enzyme loss from oxidase-based biosensors: a one-year study, J. Diabetes Sci. Technol., 2007, 1(1), 18–27 CrossRef PubMed.
- R. V. Gadhave, P. A. Mahanwar and P. T. Gadekar, Effect of glutaraldehyde on thermal and mechanical properties of starch and polyvinyl alcohol blends, Des. Monomers Polym., 2019, 22(1), 164–170 CrossRef CAS PubMed.
- A. Luzio, E. V. Canesi, C. Bertarelli and M. Caironi, Electrospun Polymer Fibers for Electronic Applications, Materials, 2014, 7(2), 906–947 CrossRef CAS PubMed.
- W. Keun Son, J. Ho Youk, T. Seung Lee and W. H. Park, Effect of pH on electrospinning of poly(vinyl alcohol), Mater. Lett., 2005, 59(12), 1571–1575 CrossRef.
- C. E. Campiglio, S. Ponzini, P. De Stefano, G. Ortoleva, L. Vignati and L. Draghi, Cross-Linking Optimization for Electrospun Gelatin: Challenge of Preserving Fiber Topography, Polymers, 2020, 12(11), 2472 CrossRef CAS PubMed.
- Y. A. Nashchekina, O. A. Lukonina, D. M. Darvish, A. V. Nashchekin, V. Y. Elokhovskii and V. E. Yudin, et al., Biological and Rheological Properties of Collagen Cross-Linked with Glutaraldehyde, Tech. Phys., 2020, 65(9), 1535–1540 CrossRef CAS.
- J. E. Gough, C. A. Scotchford and S. Downes, Cytotoxicity of glutaraldehyde crosslinked collagen/poly(vinyl alcohol) films is by the mechanism of apoptosis, J. Biomed. Mater. Res., 2002, 61(1), 121–130 CrossRef CAS PubMed.
- Y. Wang, U. Armato and J. Wu, Targeting Tunable Physical Properties of Materials for Chronic Wound Care, Front. Bioeng. Biotechnol., 2020, 8, 584 CrossRef PubMed.
- P. F. Greenfield, J. R. Kittrell and R. L. Laurence, Inactivation of immobilized glucose oxidase by hydrogen peroxide, Anal. Biochem., 1975, 65(1), 109–124 CrossRef CAS PubMed.
- M. Dryden, G. Lockyer, K. Saeed and J. Cooke, Engineered honey: In vitro antimicrobial activity of a novel topical wound care treatment, J. Global Antimicrob. Resist., 2014, 2(3), 168–172 CrossRef PubMed.
- T. J. Hall, E. A. B. Hughes, H. Sajjad, S. A. Kuehne, M. M. Grant and L. M. Grover, et al., Formulation of a reactive oxygen producing calcium sulphate cement as an anti-bacterial hard tissue scaffold, Sci. Rep., 2021, 11(1), 4491 CrossRef CAS PubMed.
- T. J. Hall, I. Azoidis, I. A. Barroso, E. A. B. Hughes, L. M. Grover and S. C. Cox, Formulation of an antimicrobial superabsorbent powder that gels in situ to produce reactive oxygen, Mater. Sci. Eng., C, 2021, 118, 111479 CrossRef CAS PubMed.
- Z. Wang, X. Huang, S. Jin, H. Wang, L. Yuan and J. L. Brash, Rapid antibacterial effect of sunlight-exposed silicon nanowire arrays modified with Au/Ag alloy nanoparticles, J. Mater. Chem. B, 2019, 7(40), 6202–6209 RSC.
- C. O'Farrell, T. J. Hall, L. M. Grover and S. C. Cox, Formulation of an antibacterial topical cream containing bioengineered honey that generates reactive oxygen species, Biomater. Adv., 2022, 133, 112664 CrossRef PubMed.
- D. M. Casali, M. J. Yost and M. A. Matthews, Eliminating glutaraldehyde from crosslinked collagen films using supercritical CO2, J. Biomed. Mater. Res., Part A, 2018, 106(1), 86–94 CrossRef CAS PubMed.
- L. Baldino, S. Concilio, S. Cardea, I. De Marco and E. Reverchon, Complete glutaraldehyde elimination during chitosan hydrogel drying by SC-CO2 processing, J. Supercrit. Fluids, 2015, 103, 70–76 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma00395k |
| This journal is © The Royal Society of Chemistry 2024 |