
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective placement of functionalised DNA origami via thermal scanning probe lithography patterning†
Tingting
Zheng
a,
Caoimhe
O’Neill
b,
John F.
Marshall
c,
Thomas
Iskratsch
*b and
Matteo
Palma
 *a
*a
aDepartment of Chemistry, Queen Mary University of London, Mile End Road, London E1 4NS, UK. E-mail: m.palma@qmul.ac.uk
bSchool of Engineering and Materials Science, Queen Mary University of London, Mile End Road, London E1 4NS, UK. E-mail: t.iskratsch@qmul.ac.uk
cBarts Cancer Institute, Cancer Research UK Centre of Excellence, Queen Mary University of London, Charterhouse Square, London EC1M 6BQ, UK
First published on 4th November 2024
Abstract
Here we present a nanopatterning strategy utilising thermal scanning probe lithography (t-SPL) for the precise organisation of DNA origami into nanoarrays. The aim of this approach is to demonstrate control in the fabrication of nanoarray platforms exhibiting single-molecule accuracy. Combining the inherent programmability of DNA origami structures with t-SPL nanopatterning, we demonstrated the controlled immobilisation on surfaces of functionalised DNA origami – as proof of concept we employed gold nanoparticles (AuNPs) and quantum dots (QDs) – at predefined positions and in nanoarray configurations. This method holds great potential for the construction of hetero-functionalised biomolecular nanoarrays with single-molecule control, with applications in bionanotechnology and (nano)materials science.
1. Introduction
The ability to control the patterning of single molecules/moieties with nanoscale spatial resolution, is of great interest in a variety of fields, from optoelectronics to biomedicine.1–3 In this context, the integration of DNA nanotechnology approaches and lithographic patterning methodologies has allowed the development of large-scale nanoarrays of single molecules, enabling high-throughput designs and assays.4 Notable examples include the construction of nanoparticles arrays,5–9 the linking of molecular emitters to photonic crystal cavities as an ideal technique for the fabrication of hybrid nanophotonic devices,10 and the fabrication of biomimetic nanoarrays for multivalent investigations of ligand–receptor interactions in cancer cell spreading.11,12Due to its high programmability and remarkable chemical flexibility, DNA has indeed become a highly desirable material for the organisation of individual molecules and nanostructures with nanoscale spatial resolution and single-molecule control.13,14 DNA origami, a bottom-up construction approach, utilises a long viral DNA strand as a scaffold that can be folded into a predetermined shape by incorporating specifically designed short staple strands.15,16 Modular DNA origami nanostructures, comprising various moieties, such as biomolecules (proteins, aptamers, or RNA),17,18 nanoparticles (metal particles, quantum dots)19–21 and organic dyes,22 can be assembled with a resolution of 3–5 nm.14 This precision is achieved by attaching distinct molecules to the staple strands through site-specific modifications.
Large-scale organization of modular DNA origami can then be achieved through electrostatic or covalent immobilization onto activated binding sites, which have been lithographically defined on substrates.6,23–28 Top-down approaches for manufacturing and immobilising nanostructures have received great attention in chemical and biological research as they can attain nanoscale resolution and allow the hierarchical organisation of molecules and nanoscale moieties. Various techniques have been employed to this end, including electron-beam (e-beam) lithography,25 focused ion beam (FIB) lithography,11,29 nanoimprint lithography,23,30 colloidal lithography,7,31 scanning probe lithography,32,33 biotemplated lithography,34,35 and dip-pen nanolithography.36
In previous studies, we employed e-beam lithography and FIB patterning, to selectively fabricate and organize functionalised DNA origami scaffolds on surfaces in array configurations, in order to fabricate single-molecule nanoarrays of quantum dots,37 proteins,38 and cell-binding ligands for the investigation of cancer cells spreading11 and cardiomyocytes mechanosignaling.12,39 Although e-beam and FIB lithography techniques are reliable and commercially available, they are costly, time-consuming and have several drawbacks such as the requirement of ultrahigh vacuum, limiting their scale-up and application.40 Here, we employed thermal scanning probe lithography (t-SPL)41 as an alternative nanopatterning strategy. In this technique thermal energy from a heated tip is used to induce local material modifications, typically a removable thermolabile resist.41 t-SPL distinct qualities, including high-resolution nanolithography, closed-loop operation enabling simultaneous reading and writing for characterisation without prior development step, and non-invasiveness (avoiding proximity effects), have allowed its use for a variety of applications, from sub-10 nm resolution patterning,42 to nanoscale precise 3D fabrication,41–43 nanoelectrode patterning for 2D electronics,44 high-throughput protein nanopatterning,45 and the nanoscale tuneable reduction of graphene oxide.46
In the study presented here, we demonstrate the organisation of heterogenous functionalised DNA origami, incorporating AuNPs and QDs, in predefined positions within a nanoarray pattern created using t-SPL. This strategy allows for the construction of versatile and tuneable DNA-templated surfaces, that hold considerable promise for the fabrication of hetero-functionalised single-molecule nanoarrays, with broad applications in bionanotechnology and materials science.
2. Materials and methods
2.1. Preparation of DNA origami
DNA origami assembly via combining 10 nM single-stranded M13mp18 scaffold DNA and 100 nM staple strands in a 50 μL volume of 1× assembly buffer (1× TAE with 12.5 mM MgCl2) and annealed from 95 °C to 20 °C in a thermal cycler at a cooling rate of 1 °C per minute and keep in 4 °C after annealing. The mixture solution of self-assembled DNA origami was purified with Millipore Amicon Ultra 100 kDa spin columns in a centrifuge at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 4 min, three times, to remove unassembled staple strands. A UV-Vis spectrophotometer was used to detect the rough concentration of DNA origami products.
000 rpm for 4 min, three times, to remove unassembled staple strands. A UV-Vis spectrophotometer was used to detect the rough concentration of DNA origami products.
2.2. Preparation of AuNPs-ssDNA
Following mixing 1 mL of AuNPs with 4 μL Tween 80 (10% v/v, in water), the mixture was allowed to react for 30 min at room temperature. Next, 0.1 M PBS was added to the mixture. Thiol-ssDNAs were subsequently introduced into the AuNPs solution, resulting in a final DNA concentration of 10 μM. The mixture was then incubated for 2.5 hours at 50 °C. Any unbound ssDNAs were separated by centrifugation at 16000g for 15 minutes and washed three times with 1× assembly buffer. Finally, the resulting conjugates were re-dispersed in 1× assembly buffer.
2.3. DNA origami functionalisation
Amino anchors were assembled onto DNA origami for surface covalent immobilisation. The modification of the amino group on DNA origami is achieved by modified of an amino group at the 3′end of 15 chosen staple strands (A05, B05, C05, A13, B13, C13, A33, B33, C33, A42, B42, C42, A50, B50, C05, and C50) which are all positioned in the inner edges of triangular DNA origami.To modify DNA origami with AuNPs, AuNPs were conjugated with thiol single-stranded DNA (ssDNA-SH) through thiol-Au interactions. The resulting AuNPs-ssDNA complexes were designed to be complementary to the sticky ends predefined on the staple strands A48, A49, and A56 of DNA origami. AuNPs-ssDNA were then mixed with DNA origami in a 5:1 ratio in 1× assembly buffer. The mixture was incubated in the fridge overnight to allow for complete complementary binding with the sticky ends.
To modify DNA origami with streptavidin-QDs, commercially available streptavidin-modified QDs were directly attached to the biotinylated sticky ends on the DNA origami (located at B32 in a corner of the triangle). Streptavidin-QDs were mixed with biotinylated DNA origami in a 10:1 ratio in a 1× assembly buffer. The mixture was then incubated at room temperature for 30 minutes. Subsequently, the functional DNA origami was cast onto the surface of a freshly clean mica and characterised using atomic force microscopy (AFM).
2.4. AFM characterization of DNA origami
Dimension icon AFM (Bruker) was used to characterize the DNA origami structures. The DNA origami solution is diluted by 1× assembly buffer to around 1 nM for getting a good separation of the DNA nanostructures. Five micro litters of diluted DNA origami solution were cast on freshly cleaned mica and left to adsorb on the surface for 5 min. Subsequently, the substrate was washed with distilled water to remove non-absorbed origami and then blown dry by compressed air. ScanAsyst-Air tips with 0.4 N m−1 spring constant were used to scan the sample by AFM under ScanAsyst™ Mode. A resolution of 512 pixels per line with a 1 Hz scan rate was chosen for appropriate imaging of the DNA origami nanostructures.2.5. t-SPL nanopattern fabrication
Silicon chips are soaked and sonicated in acetone for 5 minutes, then rinsed with isopropanol. The silicon chips were dried with compressed air and cleaned using 2 minutes of Harrick plasma treatment. The resist films were prepared by spin coating polyhthalaldehyde (PPA, 5% in anisole). To achieve 80 nm thick films, PPA was spun at 3000 rpm for 35 s at ambient conditions, followed by incubation at 110 °C for 2 minutes to remove residual solvent. The substrate, patterned by t-SPL, was designed to match the size of the DNA origami. Nanoapertures for DNA origami placement was created by selectively removing the PPA resist. The PPA resist can be removed if needed via the use of N-methyl-2-pyrrolidone. All t-SPL processes were conducted using the NanoFrazor Explore system, Heidelberg Instruments, following the manufacturer's instructions. The generated patterns were packed in a clean 24-well plate and prepared for DNA origami immobilization.2.6. DNA origami covalent immobilisation
After oxygen plasma treatment, a 0.1% solution of carboxyethylsilane (CTES) in 5 mM Tris buffer (pH 8) was incubated on a shaker for 30 minutes to form carboxyl groups on the activated binding sites. The substrates were then washed three times with 1× assembly buffer to remove any free CTES. To covalently binding DNA origami to carboxyl sites, fifteen amino anchors along the inner edges of the triangular DNA origami were functionalised. One hundred microliters of the DNA origami solution (300 pM) were deposited onto the substrate and incubated on a shaker for 1 hour in a 24-well plate with a moist Kimwipe. After placement, the substrates were washed three times with 10 mM MOPS buffer (pH 8.1) containing 125 mM MgCl2. An equal volume of 50 mM EDC and 100 mM NHS in MOPS buffer was added to the substrates and incubated on a shaker for 10 min for covalent conjugation. The DNA origami covalently conjugated substrates were washed with 10 mM MOPS buffer (pH 8.1) containing 150 mM NaCl, then rinsed with PBS containing 125 mM NaCl to remove any noncovalently bound DNA origami, and subsequently rinsed with water and dry with compressed air. The samples were checked under AFM.3. Results and discussion
Triangular DNA origamis were chosen and synthesized in this project due to their inherent advantages such as stiffness and low tendency to folding and aggregation during assembly from solution to surfaces.15 To verify the construction of these DNA nanostructures, synthesized DNA origamis were cast on clean mica and imaged using atomic force microscopy (AFM). AFM imaging confirmed that triangle DNA origami contain three 120 nm edges and have a height of roughly 1.5–2 nm, which is consistent with the height of dsDNA (Fig. 1A). Different nanoparticles were selected in this proof-of-concept project, namely commercially available streptavidin modified quantum dots (QDs) (∼9.4 ± 0.8 nm) and ssDNA-conjugated gold nanoparticles (AuNPs) of different sizes (AuNPs 1-ssDNA, ∼8.7 ± 0.5 nm and AuNPs 2-ssDNA, ∼18.7 ± 4.8 nm) [Fig. S1 in the ESI†]. The DNA origami scaffold was modified with biotin to the end of a staple located in a corner of its triangular structure, which can bind to streptavidin modified QDs (Fig. 1B). For the binding of AuNPs to the DNA origami, the metal nanoparticles were modified with a thiol single-strand DNA (ssDNA-SH) with the aid of tween 80 (ref. 47) (Fig. S2, ESI†), to obtain AuNPs-ssDNA hybrids complimentary to predefined sticky ends on the DNA origami (Fig. 1C). Fig. 1D–H show the schematic design of DNA origami modified with QDs/AuNPs and the corresponding AFM topographic images confirming DNA origami's heterogenous functionalisation. AFM imaging allowed us to determine the yield of mono- and multi-valent functionalisation (Fig. S3, ESI†): 79.28% (88/111) with a QD (Fig. 1D), 83.77% (294/351) with AuNP 1 (Fig. 1E), 88.89% (176/198) with AuNP 2 (Fig. 1F), 65% (78/120) with QD and AuNP 1 (Fig. 1G), and 70.53% (67/95) for DNA origami with QD and AuNP 2 (Fig. 1H).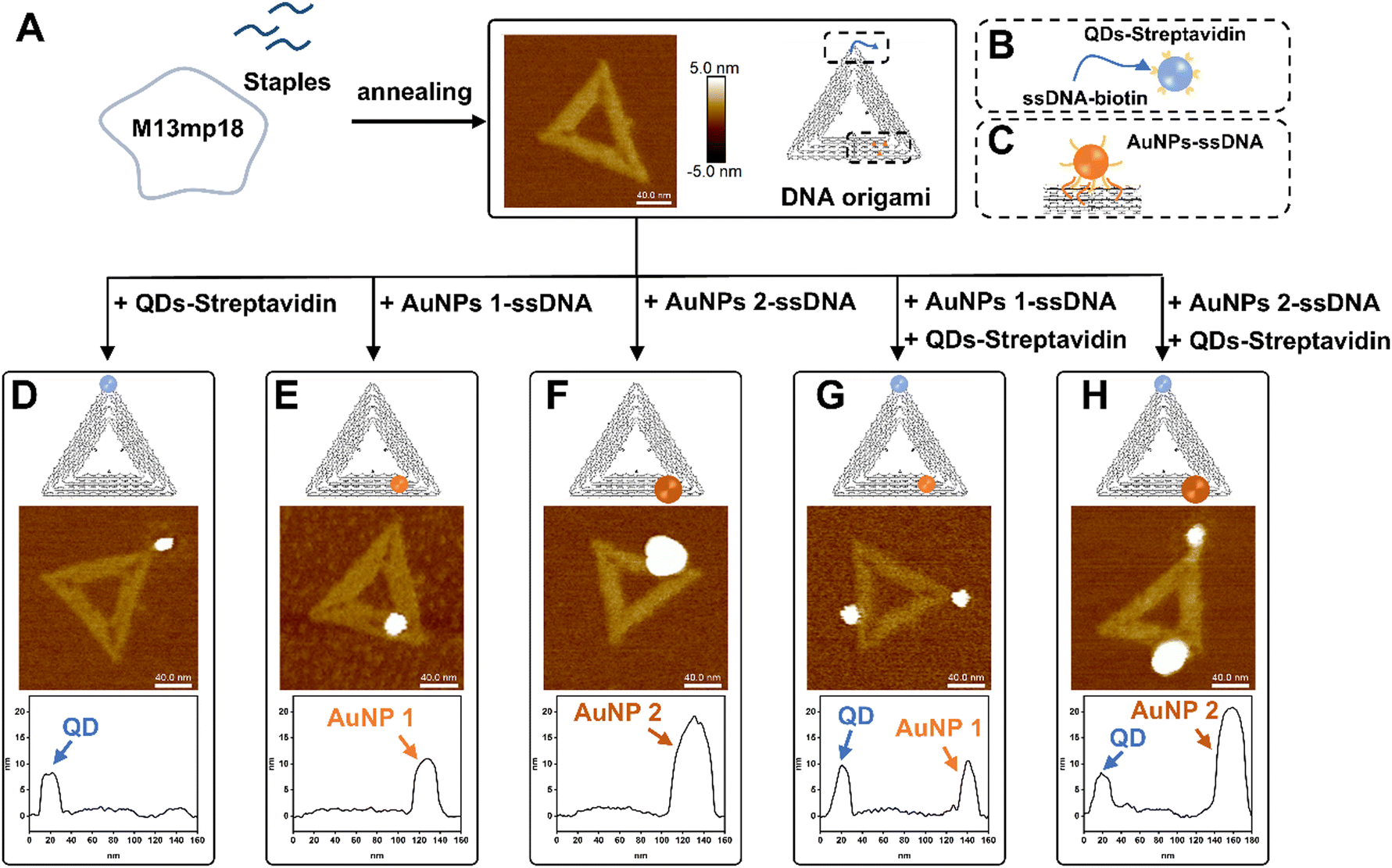 | ||
| Fig. 1 Synthesis and functionalisation of DNA origami with QDs and AuNPs. (A) Schematic of DNA origami synthesis. (B) Cartoon of DNA origami functionalised with QDs via biotin–streptavidin coupling, and (C) of ssDNA functionalised AuNPs complementary hybridisation on predefined sticky ends of DNA origami. Schematic, topographic AFM image and corresponding height profile of DNA origami modified with (D) one QD, (E) AuNP 1, (F) AuNP 2, (G) AuNP 1 and QD, and (H) AuNP 2 and QD. | ||
In order to organise the functionalised DNA origami in nanoarray configuration on surfaces, we employed t-SPL to nanopattern on silicon chips and create chemically patterned areas capable of selectively binding DNA nanostructures. The silicon substrates were coated with polyphthalaldehyde (PPA), as illustrated in Fig. 2A; subsequently t-SPL was employed to pattern areas matching the size and shape of the DNA origami we employed. Following the thermal scan probe etching process, the PPA decomposes from polymers into monomers, exhibiting topographic features as designed (Fig. 2B). Subsequently, selective removal of the PPA resist remaining in the nanoapertures was achieved by oxygen plasma etching, creating hydrophilic regions for the subsequent DNA origami placement (Fig. 2C). The initial thickness of PPA was ca. 80 nm, as measured by AFM, and was decreased by oxygen plasma treatment at a rate of ca.1.2 nm per second (Fig. S4, ESI†). The plasma activated silicon surfaces exhibiting hydroxyl groups were silanised using carboxyethylsilanetriol (CTES) to create carboxylic groups for subsequent immobilisation of amino-terminated DNA Origami, via amidation. Fifteen amino groups were introduced onto the inner edges of the triangular DNA origami as binding anchors; this allowed the amino DNA origami to bind covalently to the carboxylic substrates via EDC/NHS coupling (see the ESI† for details, and Fig. 2D and Fig. S5A, ESI†). This covalent binding strategy can be successfully achieved on various substrates, such as silicon, silicon dioxide and ITO glass as we show in Fig. S5B (ESI†).
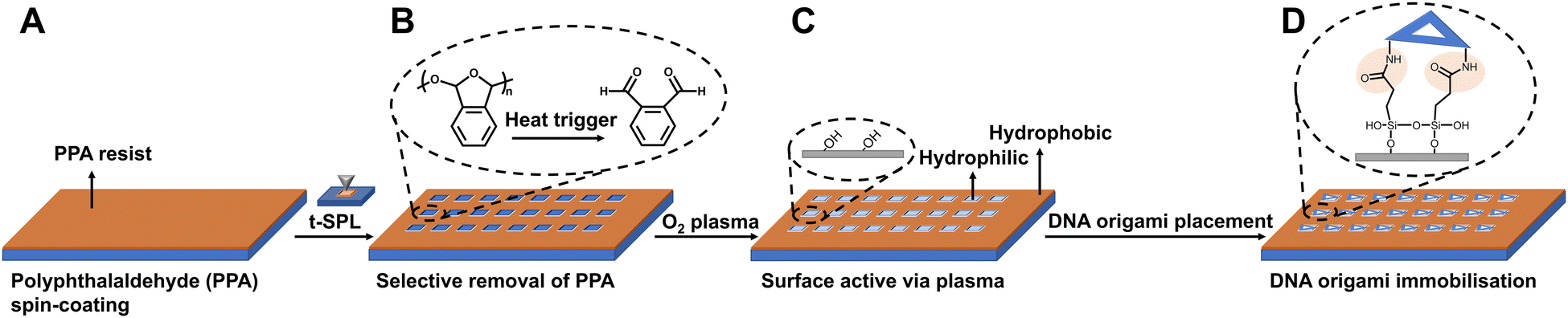 | ||
| Fig. 2 Schematic of the t-SPL patterning and DNA origami placement process. (A) Polyphthalaldehyde (PPA) spin coating on the silicon substrate; (B) selective removal of PPA via t-SPL; (C) oxygen plasma for removal of PPA in nanoapertures and activation of the surface; (D) activated surface functionalised with carboxyl group via silanisation, and convent binding with amino-modified DNA origami with the aid of EDC and NHS. | ||
The relationship between the size of the nanoapertures before and after plasma treatment was investigated, as nanoapertures could be activated and enlarged through oxygen plasma etching. To optimise the ideal nanoaperture size for single DNA origami placement, we set the resolution of t-SPL to 50 nm per pixel and patterned each nanoaperture in a size equivalent to one, 2- and 3-pixels. Fig. 3A–C show the patterning results, as imaged and analysed via AFM: highly uniform nanoarray apertures were obtained, with diameters of 50 nm, 100 nm, and 150 nm, for the aforementioned one, 2- and 3-pixels experiments, respectively. After oxygen plasma, the size of the nanoapertures increased to approximately 180 nm, 250 nm, and 300 nm, (Fig. 3D–I).
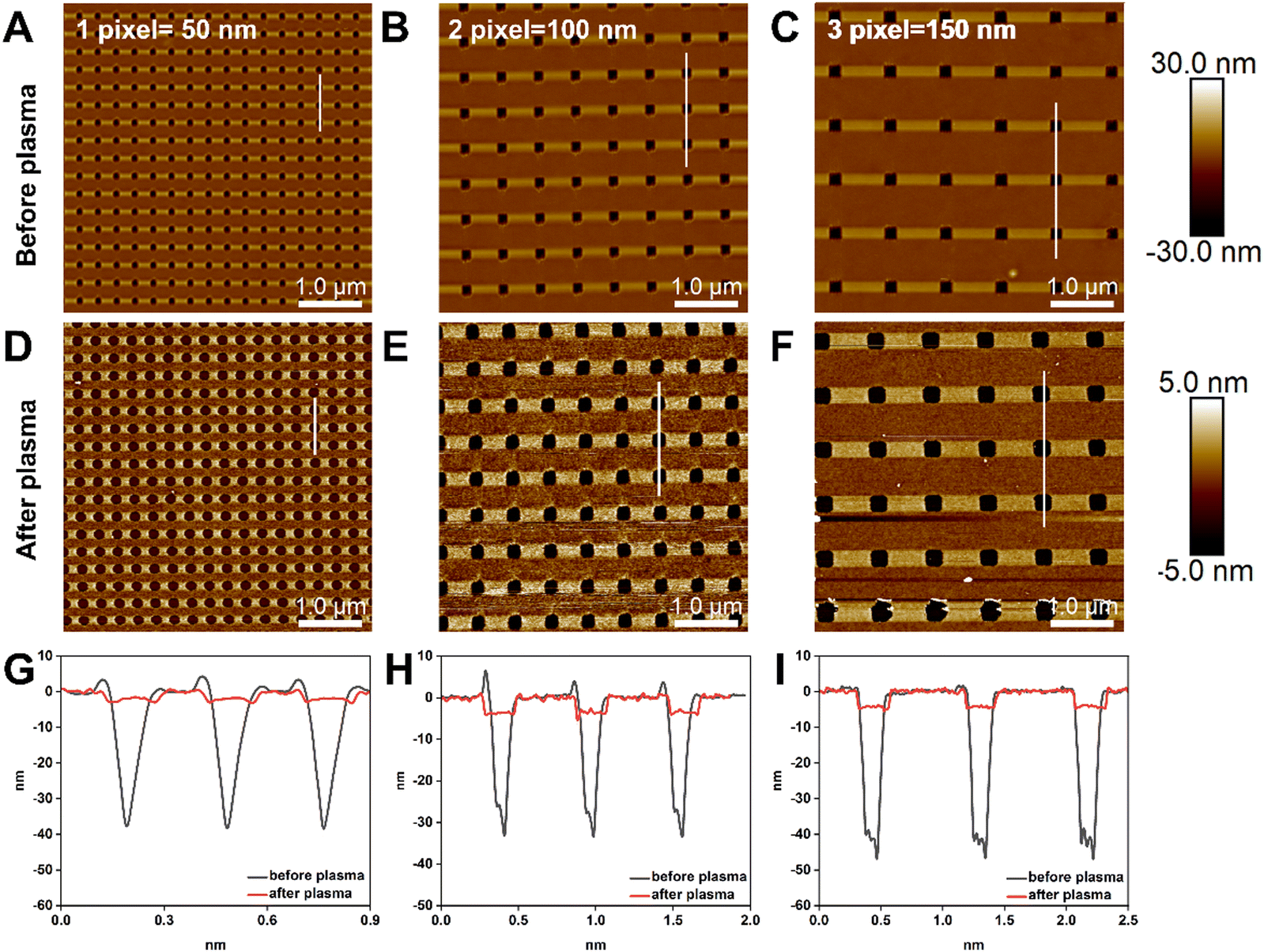 | ||
| Fig. 3 AFM topographical images and height profiles of nanoapertures activation and enlargement via oxygen plasma etching. (A)–(C) t-SPL patterning nanoaperture nanoarrays with 1-pixel, 2-pixel and 3-pixel, respectively. The resolution of per pixel is 50 nm. (D)–(F) Show each pattern after 60 seconds oxygen plasma leading to apertures of approximately 180 nm, 250 nm, and 300 nm. (G)–(I) AFM height profiles showing the depth and width change of the nanoapertures in each pattern (white lines in A–F), before (black line) and after (red line) oxygen plasma. | ||
We further proceeded to covalently tether DNA origami onto each activated nanoapertures of varying sizes, in order to confirm the optimal size for single DNA origami placement. DNA origami solutions were dropping cast on the nanopatterned surfaces and imaged via AFM in air. Fig. 4A shows -via AFM imaging- that the 180 nm nanoapertures were the optimal ones for the binding of a single DNA origami per hole (the size of DNA origami is around 120 nm, as shown in Fig. 1A). Differently, the 250 nm and 300 nm nanoapertures lead to the immobilisation of multiple DNA origami per nanoaperture (Fig. 4B and C). The yield of at least one DNA origami immobilisation per nanoaperture was found to be of 68 ± 17% (based on 152 nanoapertures), with 58.8 ± 13.6% single occupancy, for the 180 nm nanoapertures. Differently, the 250 nm nanoapertures exhibited an occupancy of at least one DNA origami with a yield of 96 ± 4% (59 nanoapertures), but only 13.6 ± 3.8% showed single occupancy. The 300 nm nanoapertures displayed 93.3 ± 11.5% (71 nanoapertures) immobilisation of at least one DNA origami, with only 11.3 ± 17.9% achieving single occupancy. These results are shown in Fig. S6 (ESI†).
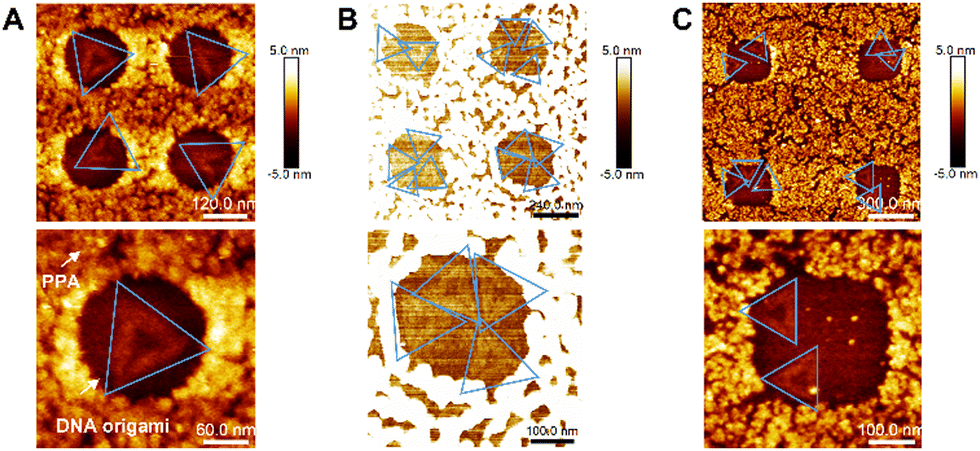 | ||
| Fig. 4 AFM topographical images of DNA origami placement on nanoapertures. (A) Single DNA origami binding in 180 nm nanoapertures; (B) multiple DNA origamis binding in 250 nm nanoapertures; (C) multiple DNA origamis binding in 300 nm nanoapertures. | ||
We further extended the nanopatterning strategy employed here, to the direct placement of DNA origami into nanoarrays. Fig. 5 demonstrates the successful assembly of DNA origami with a single AuNP1 (Fig. 5A and B) and multiple nanoparticles (AuNP1 and QD, Fig. 5C and D) on different sizes of nanoapertures (180 nm and 250 nm) in arrays. Analysis of the immobilisation yields of DNA origami functionalised with individual (AuNP 1) and multiple (AuNP 1 and QD) nanoparticles (NPs) is shown in Fig. 5E. Compared to unmodified DNA origami, the immobilisation yields of at least one NP(s)-functionalised DNA origami on both 180 nm and 250 nm nanoapertures decreased in all cases (Fig. 5E). For the 180 nm nanoapertures, the yield showed no significant change, remaining at 68 ± 6.4% (61 nanoapertures) for DNA origami functionalized with AuNP 1 only, and 75.3 ± 13.5% (111 nanoapertures) for multiple NPs-DNA origami architectures (i.e. DNA origami with both AuNP 1 and QD). On 250 nm nanoapertures the yield of immobilisation of at least one NP(s)-functionalised DNA origami decreased from 96 ± 4% to 69.3 ± 17.24% (48 nanoapertures) for DNA origami functionalised with a AuNP 1 only, and down to 61.2 ± 12% (104 nanoapertures) for multiple NPs-DNA origami constructs. This observed decrease in immobilisation efficiency is likely due to the multiple binding sites on each nanoparticle, which may lead to aggregation of DNA origami/nanoparticles in solution, reducing the concentration of individual DNA origami.
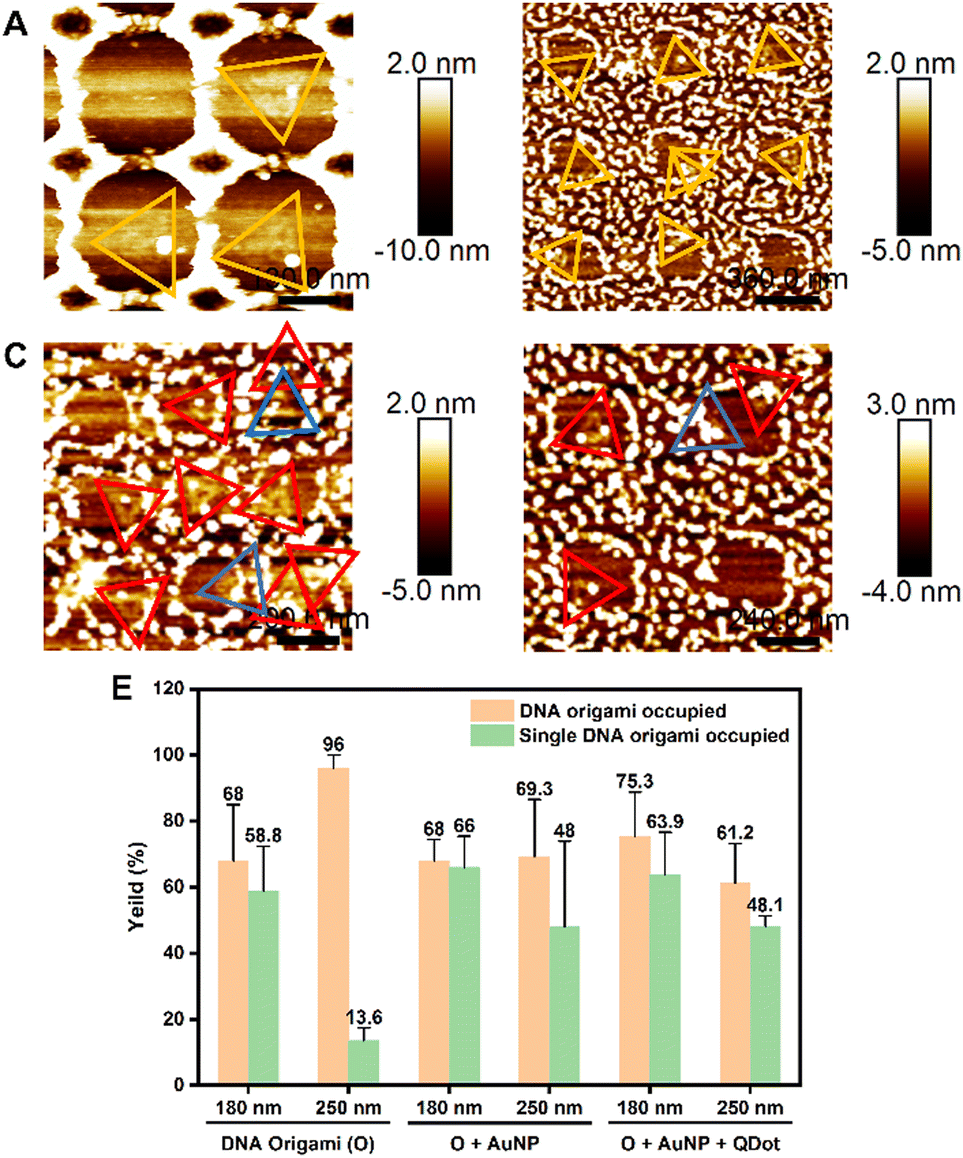 | ||
| Fig. 5 AFM images of nanoparticle-functionalised DNA origami immobilised on nanoapertures. (A) DNA origami with AuNP 1, immobilised in 180 nm nanoapertures; (B) DNA origami with AuNP1, immobilised in 250 nm nanoapertures; yellow triangle frames indicate DNA origami with AuNP 1; (C) DNA origami with AuNP1 and QD, immobilised in 180 nm nanoapertures; (D) DNA origami with AuNP1 and QD immobilised in 250 nm nanoapertures. Blue triangle frames indicate incomplete functionalised DNA origami (only one or no nanoparticle), while red triangle frames indicate DNA origami with multiple nanoparticles. (E) Analysis of DNA origami immobilization yields for 180 nm and 250 nm nanoapertures. | ||
Nevertheless, single DNA origami assembly efficiency did not decrease significantly on 180 nm nanoapertures: from 58.8 ± 13.6% to 66 ± 9.4% in DNA origami-AuNP 1, while multiple NPs-functionalised DNA origami exhibited a yield of single DNA origami occupancy of 63.9 ± 12.73%. Notably, on 250 nm nanoapertures the yield of single DNA origami occupancy improved, going from 13.6 ± 3.8% to 48 ± 25.9% for DNA origami-AuNP 1, and 48.1 ± 3.2%, multiple NPs-functionalised DNA origami (Fig. 5E). The increase in single occupancy yields in 250 nm nanoapertures may be due the increase in size for DNA origami-nanoparticle/s hybrids, making them more suitable for the 250 nm nanoapertures, preventing another DNA origami from entering the same nanoaperture. Another reason could be the decrease in concentration of individual DNA origami in the overall solution, caused by the aggregation of DNA origami/nanoparticle complexes. This aggregation reduces the likelihood of multiple DNA origami appearing in the same nanoaperture. Fig. S7 (ESI†) show additional representative AFM images of AuNP 1-functionalised DNA origami (Fig. S7A and B, ESI†) and multiple nanoparticle-functionalised DNA origami with AuNP 1 and QD immobilised in 180 nm and 250 nm nanoapertures (Fig. S7C and D, ESI†).
4. Conclusions
In this work we report a strategy for the fabrication of versatile platforms capable of assembling individual nanoscale moieties with precise single-molecule control in array configurations. As a proof of concept, we employed DNA origami functionalised with QDs and AuNPs as modular components, while t-SPL was utilised to pattern nanoapertures arrays for individual DNA origami placement. Following covalent binding aided by carboxyl silanisation on silicon substrates, we systematically investigated the yield of individual DNA origami occupancy per nanoaperture as a function of the parameters employed in the nanopatterning that directly relate to the size of the apertures -from 180 nm to 300 nm- to the yield of placement of 120 nm DNA origami functionalised with QDs and AuNPs. In this way, we demonstrated a hight degree of control in the directed assembly in nanoarray configuration of DNA origami functionalised with distinct nanoparticles. Future investigations will likely explore how to better control the number of DNA nanostructures per aperture and their orientation. The methodology presented here, and the findings reported, are of interest for the high-throughput and high-yield construction of single-molecule nanoarrays; these in turn have applications in a variety of fields from information technology to photonics and biomedicine.Data availability
Data for this article, including DNA sequences, experimental nanopatterning and functionalisation procedures, and materials are available in the ESI,† of the articleConflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge funding from the British Heart Foundation under award SP/F/23/150045 and the BBSRC under award BB/S001123/1. Moreover, we thank Eliott Clerc at Heidelberg NanoAG for patterned substrates. T. Z. was supported by the China Scholarship Council.Notes and references
- A. Nassereddine, A. Abdelrahman, E. Benard, F. Bedu, I. Ozerov, L. Limozin and K. Sengupta, Nano Lett., 2021, 21, 5606–5613 CrossRef CAS PubMed
.
- M. Palma, J. J. Abramson, A. A. Gorodetsky, E. Penzo, R. L. Gonzalez, M. P. Sheetz, C. Nuckolls, J. Hone and S. J. Wind, J. Am. Chem. Soc., 2011, 133, 7656–7659 CrossRef CAS PubMed
- I. Mayer, T. Karimian, K. Gordiyenko, A. Angelin, R. Kumar, M. Hirtz, R. Mikut, M. Reischl, J. Stegmaier, L. Zhou, R. Ma, G. U. Nienhaus, K. S. Rabe, P. Lanzerstorfer, C. M. Domínguez and C. M. Niemeyer, Nano Lett., 2024, 24, 1611–1619 CrossRef CAS PubMed
- R. J. Kershner, L. D. Bozano, C. M. Micheel, A. M. Hung, A. R. Fornof, J. N. Cha, C. T. Rettner, M. Bersani, J. Frommer, P. W. K. Rothemund and G. M. Wallraff, Nat. Nanotechnol., 2009, 4, 557–561 CrossRef CAS PubMed
- C. Sikeler, F. Haslinger, I. V. Martynenko and T. Liedl, Adv. Funct. Mater., 2024, 34, 2404766 CrossRef CAS
- A. M. Hung, C. M. Micheel, L. D. Bozano, L. W. Osterbur, G. M. Wallraff and J. N. Cha, Nat. Nanotechnol., 2010, 5, 121–126 CrossRef CAS PubMed
- R. Guan, Z. Hu, Q. Wang, H. Li, M. Khan, H. Hu, W. Chen, N. Liu and X. Lan, ACS Photonics, 2024, 11, 3351–3358 CrossRef CAS
- I. V. Martynenko, E. Erber, V. Ruider, M. Dass, G. Posnjak, X. Yin, P. Altpeter and T. Liedl, Nat. Nanotechnol., 2023, 18, 1456–1462 CrossRef CAS PubMed
- B. Teschome, S. Facsko, K. V. Gothelf and A. Keller, Langmuir, 2015, 31, 12823–12829 CrossRef CAS PubMed
- A. Gopinath, E. Miyazono, A. Faraon and P. W. K. Rothemund, Nature, 2016, 535, 401–405 CrossRef CAS PubMed
- D. Huang, K. Patel, S. Perez-Garrido, J. F. Marshall and M. Palma, ACS Nano, 2019, 13, 728–736 CrossRef CAS PubMed
- W. Hawkes, E. Marhuenda, P. Reynolds, C. O’Neill, P. Pandey, D. G. Samuel Wilson, M. Freeley, D. Huang, J. Hu, S. Gondarenko, J. Hone, N. Gadegaard, M. Palma and T. Iskratsch, Philos. Trans. R. Soc. B Biol. Sci., 2022, 377, 20220021 CrossRef CAS PubMed
- N. C. Seeman, Nano Lett., 2020, 20, 1477–1478 CrossRef CAS PubMed
- P. Zhan, A. Peil, Q. Jiang, D. Wang, S. Mousavi, Q. Xiong, Q. Shen, Y. Shang, B. Ding, C. Lin, Y. Ke and N. Liu, Chem. Rev., 2023, 123, 3976–4050 CrossRef CAS PubMed
- P. W. K. Rothemund, Nature, 2006, 440, 297–302 CrossRef CAS PubMed
- F. Hong, F. Zhang, Y. Liu and H. Yan, Chem. Rev., 2017, 117, 12584–12640 CrossRef CAS PubMed
- J. Hellmeier, R. Platzer, V. Mühlgrabner, M. C. Schneider, E. Kurz, G. J. Schütz, J. B. Huppa and E. Sevcsik, ACS Nano, 2021, 15, 15057–15068 CrossRef CAS PubMed
- T. Zheng, Q. Tang, L. Wan, Y. Zhao, R. Xu, X. Xu, H. Li and D. Han, Nano Lett., 2023, 23, 2081–2086 CrossRef CAS PubMed
- W. Fang, S. Jia, J. Chao, L. Wang, X. Duan, H. Liu, Q. Li, X. Zuo, L. Wang, L. Wang, N. Liu and C. Fan, Sci. Adv., 2019, 5, eaau4506 CrossRef PubMed
- D. Huang, L. Haddad, F. Rahman, M. Palma and A. Sapelkin, RSC Adv., 2022, 12, 23778–23785 RSC
- R. Wang, C. Nuckolls and S. J. Wind, Angew. Chem. Int. Ed., 2012, 51, 11325–11327 CrossRef CAS PubMed
- J. Schnitzbauer, M. T. Strauss, T. Schlichthaerle, F. Schueder and R. Jungmann, Nat. Protoc., 2017, 12, 1198–1228 CrossRef CAS PubMed
- E. Penzo, R. Wang, M. Palma and S. J. Wind, J. Vac. Sci. Technol. B Nanotechnol. Microelectron. Mater. Process. Meas. Phenom., 2011, 29, 06F205 Search PubMed
- R. M. Shetty, S. R. Brady, P. W. K. Rothemund, R. F. Hariadi and A. Gopinath, ACS Nano, 2021, 15, 11441–11450 CrossRef CAS PubMed
- A. Gopinath and P. W. K. Rothemund, ACS Nano, 2014, 8, 12030–12040 CrossRef CAS PubMed
- K. Brassat, S. Ramakrishnan, J. Bürger, M. Hanke, M. Doostdar, J. K. N. Lindner, G. Grundmeier and A. Keller, Langmuir, 2018, 34, 14757–14765 CrossRef CAS PubMed
- A. Gopinath, C. Thachuk, A. Mitskovets, H. A. Atwater, D. Kirkpatrick and P. W. K. Rothemund, Science, 2021, 371, eabd6179 CrossRef CAS PubMed
- K. Tapio, C. Kielar, J. M. Parikka, A. Keller, H. Järvinen, K. Fahmy and J. J. Toppari, Chem. Mater., 2023, 35, 1961–1971 CrossRef CAS
- A. A. Tseng, Small, 2005, 1, 924–939 CrossRef CAS PubMed
- M. Schvartzman, M. Palma, J. Sable, J. Abramson, X. Hu, M. P. Sheetz and S. J. Wind, Nano Lett., 2011, 11, 1306–1312 CrossRef CAS PubMed
- J. Huang, S. V. Gräter, F. Corbellini, S. Rinck, E. Bock, R. Kemkemer, H. Kessler, J. Ding and J. P. Spatz, Nano Lett., 2009, 9, 1111–1116 CrossRef CAS PubMed
- D. Wang, V. K. Kodali, W. D. Underwood II, J. E. Jarvholm, T. Okada, S. C. Jones, M. Rumi, Z. Dai, W. P. King, S. R. Marder, J. E. Curtis and E. Riedo, Adv. Funct. Mater., 2009, 19, 3696–3702 CrossRef CAS
- R. Garcia, A. W. Knoll and E. Riedo, Nat. Nanotechnol., 2014, 9, 577–587 CrossRef CAS PubMed
- B. Shen, V. Linko, K. Tapio, S. Pikker, T. Lemma, A. Gopinath, K. V. Gothelf, M. A. Kostiainen and J. J. Toppari, Sci. Adv., 2018, 4, eaap8978 CrossRef PubMed
- P. Piskunen, B. Shen, A. Keller, J. J. Toppari, M. A. Kostiainen and V. Linko, ACS Appl. Nano Mater., 2021, 4, 529–538 CrossRef CAS
- S. W. Lee, B.-K. Oh, R. G. Sanedrin, K. Salaita, T. Fujigaya and C. A. Mirkin, Adv. Mater., 2006, 18, 1133–1136 CrossRef CAS
- D. Huang, M. Freeley and M. Palma, Sci. Rep., 2017, 7, 45591 CrossRef CAS PubMed
- K. Cervantes-Salguero, M. Freeley, R. E. A. Gwyther, D. D. Jones, J. L. Chávez and M. Palma, Biophys. Rev., 2022, 3, 031401 CrossRef CAS PubMed
- W. Hawkes, D. Huang, P. Reynolds, L. Hammond, M. Ward, N. Gadegaard, J. F. Marshall, T. Iskratsch and M. Palma, Faraday Discuss., 2019, 219, 203–219 RSC
- D. K. Oh, H. Jeong, J. Kim, Y. Kim, I. Kim, J. G. Ok and J. Rho, J. Mech. Sci. Technol., 2021, 35, 837–859 CrossRef
- E. Albisetti, A. Calò, A. Zanut, X. Zheng, G. M. de Peppo and E. Riedo, Nat. Rev. Methods Primer, 2022, 2, 32 CrossRef CAS
- X. Liu, M. Kumar, A. Calo, E. Albisetti, X. Zheng, K. B. Manning, E. Elacqua, M. Weck, R. V. Ulijn and E. Riedo, ACS Appl. Mater. Interfaces, 2019, 11, 41780–41790 CrossRef CAS PubMed
- K. M. Carroll, A. J. Giordano, D. Wang, V. K. Kodali, J. Scrimgeour, W. P. King, S. R. Marder, E. Riedo and J. E. Curtis, Langmuir, 2013, 29, 8675–8682 CrossRef CAS PubMed
- X. Zheng, A. Calò, E. Albisetti, X. Liu, A. S. M. Alharbi, G. Arefe, X. Liu, M. Spieser, W. J. Yoo, T. Taniguchi, K. Watanabe, C. Aruta, A. Ciarrocchi, A. Kis, B. S. Lee, M. Lipson, J. Hone, D. Shahrjerdi and E. Riedo, Nat. Electron., 2019, 2, 17–25 CrossRef CAS
- X. Liu, M. Kumar, A. Calo’, E. Albisetti, X. Zheng, K. B. Manning, E. Elacqua, M. Weck, R. V. Ulijn and E. Riedo, Faraday Discuss., 2019, 219, 33–43 RSC
- Z. Wei, D. Wang, S. Kim, S.-Y. Kim, Y. Hu, M. K. Yakes, A. R. Laracuente, Z. Dai, S. R. Marder, C. Berger, W. P. King, W. A. de Heer, P. E. Sheehan and E. Riedo, Science, 2010, 328, 1373–1376 CrossRef CAS PubMed
- S. Xu, H. Yuan, A. Xu, J. Wang and L. Wu, Langmuir, 2011, 27, 13629–13634 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma00828f |
| This journal is © The Royal Society of Chemistry 2024 |