
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMinimalist columnar liquid crystals: influence of fluorination on the mesogenic behavior of tetramethoxytriphenylene derivatives†
Parikshit
Guragain
 a,
Mitchell
Powers
*b,
Brett
Ellman
c and
Robert J
Twieg
a
a,
Mitchell
Powers
*b,
Brett
Ellman
c and
Robert J
Twieg
a
aDepartment of Chemistry and Biochemistry, Kent State University, Kent, Ohio 44242, USA
bDepartment of Physics, Gettysburg College, Gettysburg, Pennsylvania 17325, USA. E-mail: mpowers@gettysburg.edu
cDepartment of Physics, Kent State University, Kent, Ohio 44242, USA
First published on 22nd October 2024
Abstract
Conventional columnar liquid crystals are almost invariably functionalized with multiple long aliphatic tails. In contrast, the molecule 1,2,3,4-tetrafluoro-6,7,10,11-tetramethoxytriphenylene is unusual as it has a broad columnar mesophase despite the fact that it has only four minimal-length methoxy tails distributed equally in two rings. Here, the full set of analogous tetramethoxytriphenylene molecules with zero to four fluorine atoms in the remaining ring are synthesized using photocyclodehydrofluorination (PCDHF) or a modified Scholl process for the final ring closure reaction, and their mesogenic properties are examined. Six out of the ten target molecules were found to be mesogenic. We found that a minimum of two fluorine substituents are required to form the mesophase, and one of them must be at the 1-position. These extremely short-tailed columnar liquid crystals possess a relatively simple molecular structure and can serve as model systems for understanding the interactions that are fundamental to the formation of a columnar mesophase, its properties and potential applications.
Introduction
Discotic liquid crystals (DLCs) are of special interest due in part to their ability to self-assemble into long columns which can act as organic semiconductors, and have potentially useful technological applications.1 Since their first discovery in 1977,2 the importance of DLCs has been evident in their applications in a variety of organic electronics including organic photovoltaics (OPVs), organic field effect transistors (OFETs), organic light emitting diodes (OLEDs), and other devices.3A wide variety of molecular motifs have been identified as suitable building blocks for forming the columnar structures, the vast majority of which rely on surrounding the molecular cores with long, flexible, aliphatic tails in order to form a buffer between columns and sustain a liquid crystal mesophase. Recently we have discovered a new set of materials which are based on triphenylene, one of the most widely studied molecular cores used in the field of discotic liquid crystals research. These compounds are unusual as they lack the long tails that are more commonly found on similar liquid crystals.4–6 In fact, in a small number of cases, there are no tails at all!7–10 Our efforts have focused on the integration of fluorine atoms throughout the available substitution sites of the triphenylene core. The change in the charge distribution caused by the presence of highly electronegative fluorine atoms leads to significant changes to the physical, chemical and thermal properties of the compounds. In this study, we have varied the number and location of the fluorine atoms substituted around the triphenylene core, which has previously been shown to have a significant impact on the intra- and intermolecular interactions within these materials and accordingly plays a crucial role in determining their mesogenic (liquid crystalline) properties.11,12
In ref. 7, we reported the synthesis and mesogenic properties of a family of tetrafluorinated tetraalkoxy triphenylenes which were mesogenic despite featuring unusually short alkoxy tails. This set of compounds included 1,2,3,4-tetrafluoro-6,7,10,11-tetramethoxytriphenylene, which features a columnar mesophase over a broad temperature range (K 198 Colh 310 I 307 Colh 182 K). To further explore the role of fluorination in creating a columnar mesophase, we have synthesized a set of compounds based on 6,7,10,11-tetramethoxytriphenylene which vary in the extent of fluorination of the non-methoxy bearing ring. This set includes all ten potential compounds with between zero and four fluorine substituents, of which six feature a columnar mesophase. This mesogenic behavior can be induced by selective fluorination, with as few as two fluorines being able to support a columnar mesophase in these compounds.7,8
Results and discussion
The set of compounds 1–10 described in this work are shown in Fig. 1. From the onset three of these compounds, 4, 8, and 10, were already known in the literature (curiously, they are three out of the four nonmesogenic substances discussed here). The compounds are formally derived from 10, the nonfluorinated 2,3,6,7-tetramethoxytriphenylene, which is not mesogenic and has a reported melting point at 214 °C.13 The other compounds previously described in the literature are 2,3-difluoro-6,7,10,11-tetramethoxytriphenylene, 4, and 2-fluoro-6,7,10,11-tetramethoxytriphenylene, 8, with reported melting points near 212 °C and 195 °C, respectively.14 The isotropic melting points for our compounds 8 and 10 are in agreement with the reported melting points, whereas in our study we found compound 4 to have a melting point about twenty degrees higher than previously reported.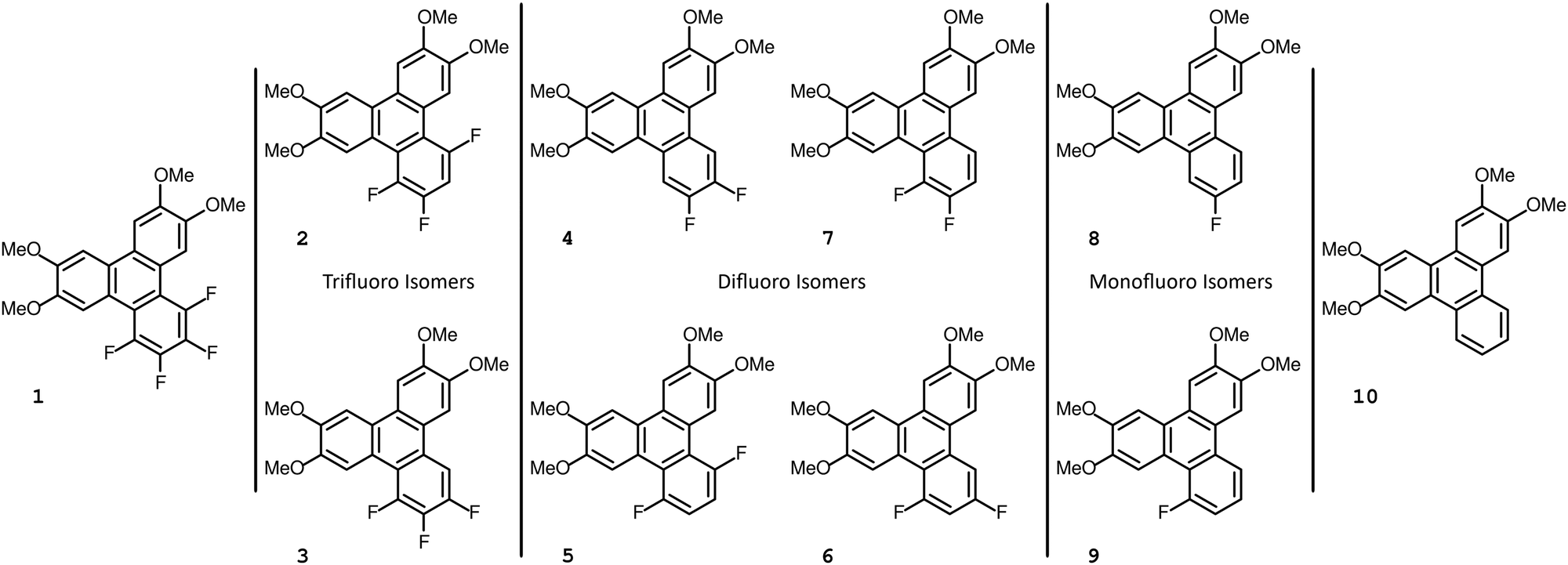 | ||
| Fig. 1 The structures of the targeted tetramethoxy triphenylene compounds, with the different isomers grouped together. Compounds 4, 8, 9 and 10 are non-mesogenic, with the rest all featuring a columnar mesophase. | ||
Synthetic procedures
The synthesis of compounds 1 and 2 is shown in Scheme 1. The synthesis of compound 2 uses the potassium salt of 2,3,5,6-tetrafluorobenzoic acid which decarboxylates more slowly than the potassium salt of pentafluorobenzoic acid (a copper(I) intermediate is actually involved in the decarboxylation)15 and this reaction also produces a diarylated by-product through the direct arylation of the acidic C–H bond of polyfluoroarene.15,16 Nonetheless, this route is still useful and convenient for the preparation of 2. The 2,3,5,6-tetrafluorobenzoic acid itself is readily obtained by the selective reduction at the para fluorine site in perfluorobenzoic acid with zinc metal,17 which when treated with t-BuOK gives the potassium salt of 2,3,5,6-tetrafluorobenzoic acid.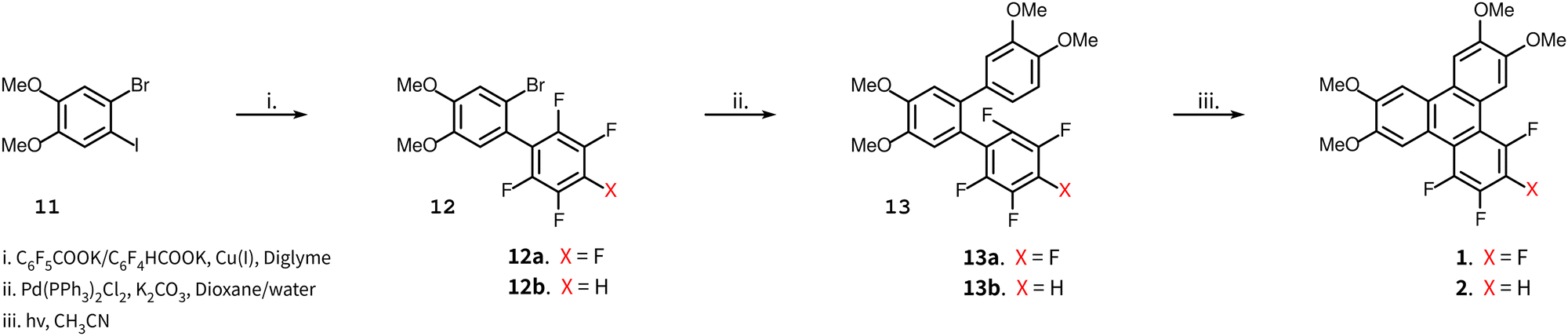 | ||
| Scheme 1 Synthesis of target compounds 1 and 2. | ||
Compound 3 was obtained as shown in Scheme 2 which has a modified step compared to Scheme 1. The decarboxylative coupling reaction with the potassium salt of 2,3,4,5-tetrafluorobenzoic acid is even slower than the 2,3,5,6-tetrafluorinated salt where the best yield obtained was around 50% (and is much less efficient than the perfluorinated salt, where the yield was around 90%). Some process development was performed (discussed later in some detail) before we undertook the first reaction of this route. Starting with 1,2-diiodo-4,5-dimethoxyiodobenzene 14, a Suzuki coupling reaction with 3,4-dimethoxyphenylboronic acid was carried out to obtain the iodobiphenyl 15. In the next step, copper(I) mediated decarboxylative coupling with the potassium salt of 2,3,4,5-tetrafluorobenzoic acid under modified conditions (dimethylacetamide, DMAc, as a solvent and Tris 2(2-methoxyethoxy)ethyl amine, TDA-1, as an additive) was used to obtain terphenyl 16 in yields up to 73%. DMAc has been suggested to work better in fluoroarene systems containing fewer fluorine atoms.15 The use of standard conditions (diglyme) gave poor yields typically in the range of 0–5%. For the preparation of the terphenyl 16, a Suzuki coupling reaction was followed by a decarboxylative coupling reaction under modified conditions. The sequence of these reactions (first and second) might not have made a difference if the decarboxylative cross-coupling between 2-bromo-4,5-dimethoxyiodobenzene and the potassium salt of 2,3,4,5-tetrafluorobenzoic acid had a good yield, though this was not the case in our experience. Thus, a Suzuki reaction followed by a decarboxylative coupling reaction proved most expedient. Finally, on irradiation, the o-terphenyl 16 loses HF in the PCDHF reaction to produce the target compound 3.
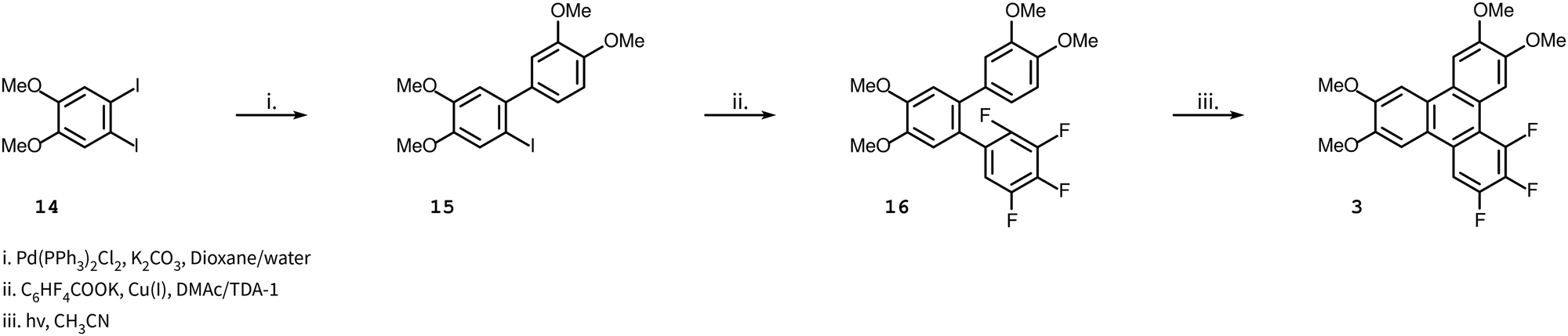 | ||
| Scheme 2 Synthesis of target molecule 3. | ||
The synthesis of the remaining target compounds 4–10 all follow the same route up to the penultimate step. Starting with 1-bromo-2-iodo-4,5-dimethoxybenzene 11, a Suzuki coupling reaction with 3,4-dimethoxyphenylboronic acid was carried out to obtain biphenyl 18 with high selectivity at the iodine site. In the subsequent step, a second Suzuki reaction was carried out with a set of partially fluorinated phenylboronic acids to obtain terphenyls 19a–g. These terphenyls were converted to the final triphenylene by two different routes. An intermolecular Scholl reaction facilitated by the presence of methoxy groups produced target compounds 4–6, and A PCDHF reaction was used to produce the target compounds 7–10 (Scheme 3). In Scholl-type ring closures, the reaction takes place with the loss of an H2 molecule from the ortho positions of two terminal rings in the o-terphenyl 19e–g whereas, in PCDHF-type ring closure, the reaction takes place with the loss of an HF molecule from ortho positions of two terminal rings in o-terphenyl 19a–d. So, in cases with a suitable terphenyl 19 that can close a ring via the loss of an HF molecule, we have utilized a PCDHF reaction. Terphenyls 19a–d all had an ortho fluorine and an ortho hydrogen atom suitable for a PCDHF reaction to give the desired product.
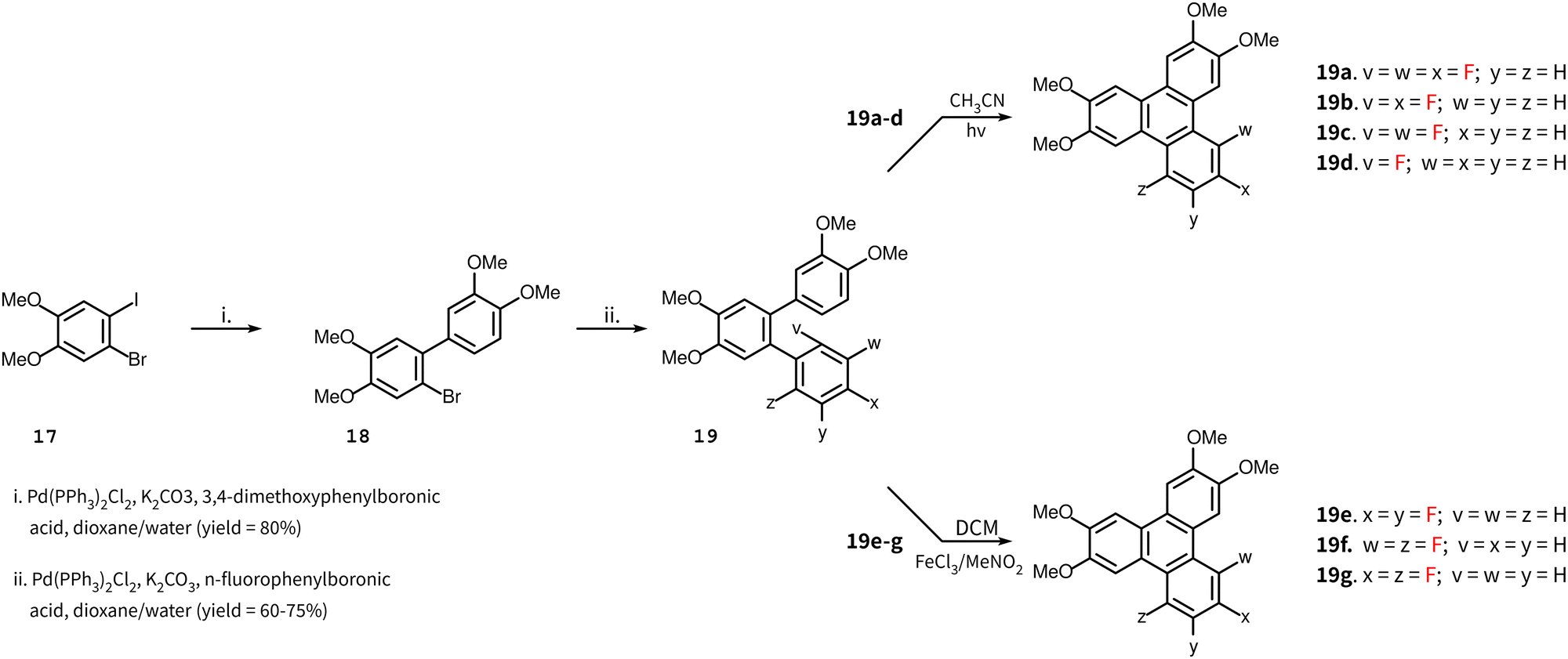 | ||
| Scheme 3 Synthesis of target molecules 4–10. Four of them (7–10) via PCDHF and three of them (4–6) via Scholl reaction. | ||
Phase behaviour
With all the target compounds 1–10 in hand, their phase behavior was studied, as summarized in Table 1. A selection of their microscopic textures are shown in Fig. 2. Phase identification was made on the basis of polarized microscopy observations except for compound 1, which was confirmed to have a hexagonal columnar mesophase (Colh) based on previously reported small angle X-ray diffraction studies,7 for other compounds we are unable to unambiguously identify the mesophase structure and therefore report their mesophase as a generic columnar mesophase (Col). Several interesting trends are apparent that give us an insight into the effect of fluorination on these materials and the interactions that are present in the short tailed mesogens (compounds 1–3 and 5–7).| Entry | Number of Fluorines | Location of Fluorination | Phase behaviour (°C [kJ mol−1]) |
|---|---|---|---|
| 1 | Four | 1,2,3,4-Tetrafluoro | K 198 [19.5] Colh 310 [18.4] I 307 [7.7] Colh 182 [9.5] K |
| 2 | Three | 1,2,4-Trifluoro | K 202 [20.04] Col 261 [12.93] I 259 [11.09] Col 175 [16.02] K |
| 3 | 1,2,3-Trifluoro | K 228 [21.65] Col 257 [5.68] I 254 [4.51] Col 210 [22.67] K | |
| 4 | Two | 2,3-Difluoro | K 237 [55.24] I 228 [42.78] K |
| 5 | 1,4-Difluoro | K 203 [22.04] Col 216 [14.11] I 214 [14.25] Col 193 [20.25] K | |
| 6 | 1,3-Difluoro | K 199 [19.10] Col 221 [11.27] I 218 [11.52] Col 182 [14.42] K | |
| 7 | 1,2-Difluoro | K 197 Col 198 [24.35]* I 195 [7.69] Col 175 [13.80] K | |
| 8 | One | 2-Fluoro | K 198 [31.90] I 167 [31.88] K |
| 9 | 1-Fluoro | K 197 [34.12] I 188 [34.22] K | |
| 10 | Zero | Nonfluorinated | K 215 [43.52] I 165 [33.48] K |
 | ||
| Fig. 2 Polarized light photomicrographs of the columnar mesophase of compounds 2 (T = 246 °C, left), 6 (T = 216 °C, middle) and 7 (T = 185 °C right) all taken on cooling. | ||
One of the clearest trends is the increase in the isotropic transition temperature that occurs with increased fluorination (Fig. 3). This holds for all of the compounds, with the exception of the nonfluorinated compound 10, which has a transition temperature in excess of the monofluoro compounds and on par with the average isotropic transition temperature of the difluoro compounds. One potential explanation of this trend, in the fluorinated compounds, is that it may be the product of the additional fluorine substituents increasing the strength, or number, of favourable interactions in the crystal and liquid crystal phase, provided that a similar increase does not occur in the isotropic state. This would result in a lowering of the Gibbs free energy of the crystal or liquid crystal phases, and if no effect of similar magnitude were to occur in the isotropic phase, the transition temperature would be expected to increase.
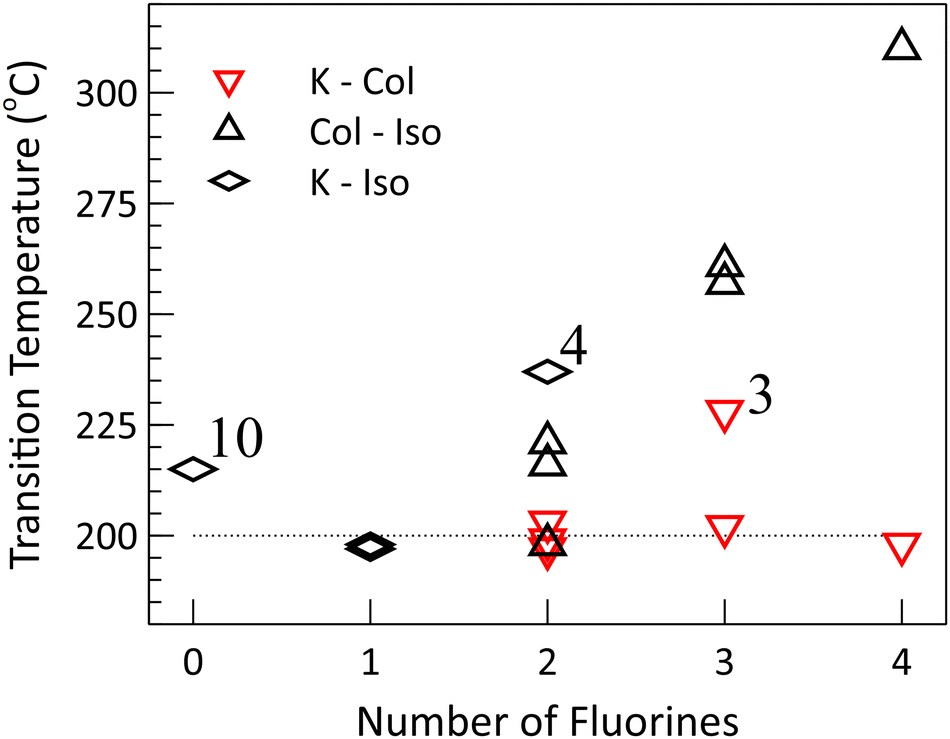 | ||
| Fig. 3 Phase transition temperatures for compounds 1–10, grouped based on the extent of fluorination. Plot markers show the transition temperatures on heating (see Table 1). The isotropic transition tends to increase in temperature with fluorination, while the crystal transition is nearly constant across the different compounds (the dotted line is a guide for the eye). | ||
A second, seemingly complementary, trend occurs for five of the six mesogenic compounds (excluding 3) which all possess nearly identical crystal to liquid crystal transition temperatures of approximately 200 °C (+/−3 °C). In this case, the opposite situation may be occurring in that the change in intermolecular interactions that follows from the fluorine substitution occurs in both the crystal and liquid crystal states approximately equally, effecting the free energy of both phases such that the transition temperature remains constant. Curiously, the melting point of the non-mesogenic monofluorinated compounds also fall within this range (which, of course, may be serendipitous).
There are few deviations from this trend. Among the non-mesogens, the non-fluorinated 10 and the difluorinated 4 have melting points of 215 °C and 237 °C, respectively. The sole outlier amongst the mesogens is the trifluoro compound 3, which becomes columnar at 228 °C. However, given the magnitude and small number of these deviations, the similarity between the transition temperatures across this set of compounds remains of significant interest.
If our interpretation of the mesophase behaviour in terms of a simplified view of the free energy is valid, we have to reconcile these observations. Specializing to the mesogenic case, the free energy of the crystal and liquid crystal phases must be lowered relative to the isotropic phase (in order to increase the isotropic transition temperature), without substantially lowering one relative to the other (in order to maintain a constant melting point near 200 °C). The effect of fluorination must also be sensitive to the position of the substituents, with the 1 and 4 positions behaving quite differently than the 2 and 3 positions. For example, all of the mesogenic compounds feature a fluorine in the 1 position (two of which are shown in Fig. 2), though a single fluorine at that site is insufficient to induce a mesophase, as demonstrated by compound 9.
There are several candidates for the source of this behaviour: (1) substituents may affect the symmetry of the molecule. For example, 1, 4, 5, and 10 all possess a mirror plane orthogonal to the plane of the molecule while the rest of the compounds do not. This may qualitatively change packing in both the crystal and columnar phases. (2) The three-dimensional conformation of the molecules may change due to steric effects, causing the fluorinated ring to twist as a result of crowding of the bay region near the 1 and 4 position (for example, in compounds 5, and 10). This will affect packing as well as the degree of conjugation of the triphenylene core, which may change intermolecular interaction energies. (3) The addition of highly electronegative fluorine atoms will allow for strong electrostatic interactions between molecules and influence the formation of hydrogen bond networks. To a lesser extent, it will also affect the van der Waals forces between molecules which, given that the short tails allow for short range intercolumn interactions, may be of increased significance. Of course, the true cause is likely a combination of these and other effects.
The symmetry of fluorine substitution, on its own, appears unlikely to be responsible for these trends. Even though compounds 1, 4, 5 and 10 are all symmetrically substituted, their properties vary widely. Some of these differences may be explained by deviations away from planarity resulting from the fluorination that causes the molecules to break symmetry, especially for 1 and 5 which each have fluorines in the 1 and 4 positions (as has been seen in similar compounds, including 1,2,3,4-tetrafluorotriphenylene18 and other fluorinated triphenylenes19–21). Compound 4, with its fluorines positioned far from the bays where they are less likely to warp the molecule, is potentially the compound to demonstrate the largest effect of symmetry.
In Fig. 4, we see that the symmetric compound 4 and non-symmetric compound 7 both undergo a substantial change in entropy through their phase transitions compared to the other compounds. This is an interesting pair, as their crystal states have both the highest (4 at 237 °C) and lowest (7 at 197 °C) melting points. So, while the total change in entropy would suggest that these compounds both possess highly ordered crystal structures, the difference in their melting points indicates an important difference in the nature of the cohesive forces that hold them together.
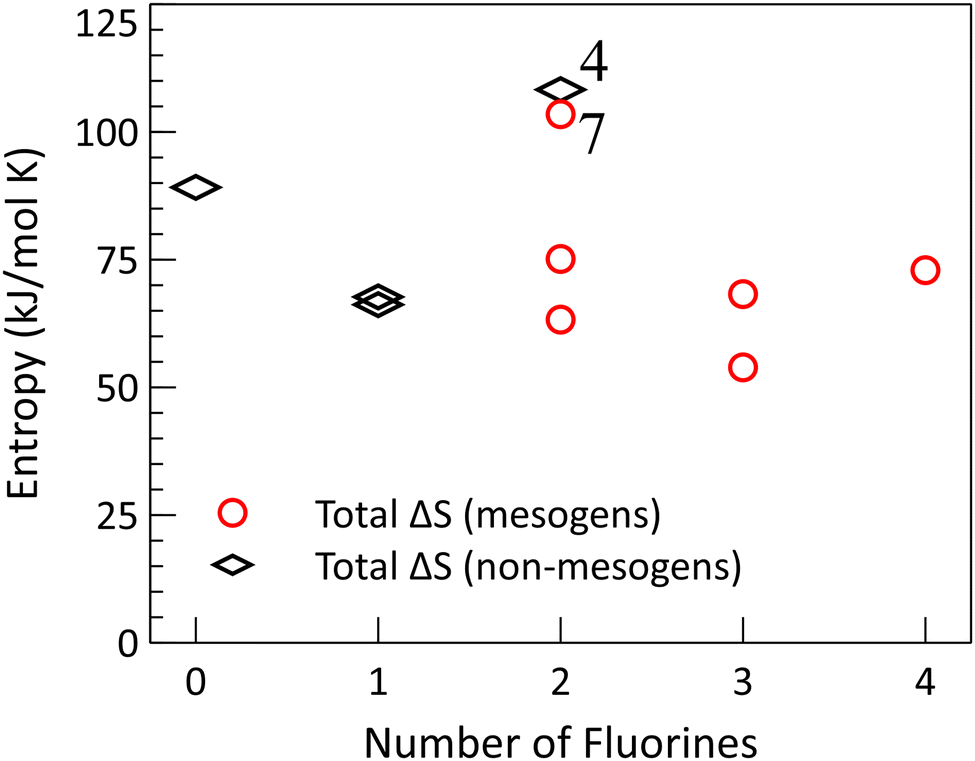 | ||
| Fig. 4 Transition entropy for compounds 1–10, arranged as in Fig. 3. Entropies are calculated as the transition enthalpies (taken on heating) divided by the transition temperatures. For the mesogens, the entropies of each transition have been added together for comparison with the non-mesogens. | ||
As for the effect fluorination has on the intermolecular interactions, we make a point to distinguish between directional and non-directional forces. This distinction is in response to the different effect that fluorination is observed to have on the crystal to liquid crystal transition compared to the liquid crystal to isotropic transition. Van der Waals interactions, for example, will behave similarly regardless of whether the system is in an ordered or disordered state. In contrast, electrostatic interactions between molecules (including the formation of hydrogen bond networks) are far more sensitive to molecular orientations. Substituents, especially electronegative ones like fluorine, are known to have a significant effect on these interactions.20–24 Altering these interactions provides a mechanism for influencing the two transition temperatures separately. While this could potentially explain some of the effects we have observed, more information, including crystal structures, are necessary to resolve this question with less ambiguity.
These observations aid our understanding of the connection between structure and liquid crystallinity. However, there is a caveat that we are yet to address: the extremely short methoxy tails can still exhibit complex behavior. This is illustrated by the long-standing problem of determining the conformers present near room temperature in o-dimethoxybenzene (which we take as a proxy for the methoxylated rings in our compounds)25,26 While the details seem unclear, both theory and experiment are consistent with multiple geometries of the methoxy groups, including both in-plane (e.g., anti–anti) as well as out-of-plane (anti–gauche) configurations. This will result in a distribution of multipole moments and molecular shapes. Therefore, even methoxy tails are a source of disorder and interactions that may vary appreciably between phases, particularly with regard to edge–edge intermolecular interactions (which are important in the columnar phase, especially when the methoxy tails allow the molecules to pack together closely). A truly tail-free mesogen,4,27,28 in which the tails are replaced by single atoms, would avoid this problem.
NMR characterization
The fluorine NMR spectra of our compounds (Fig. 5) can be understood quantitatively using the theory of statistical substituent chemical shift (SSCS),29 whereby 19F chemical shifts are estimated by summing contributions from each additional substituent (including additional fluorine atoms) on the fluorinated benzene ring:| Chemical shift F (ppm) = −113.9 + ∑Zortho + ∑Zmeta + ∑Zpara |
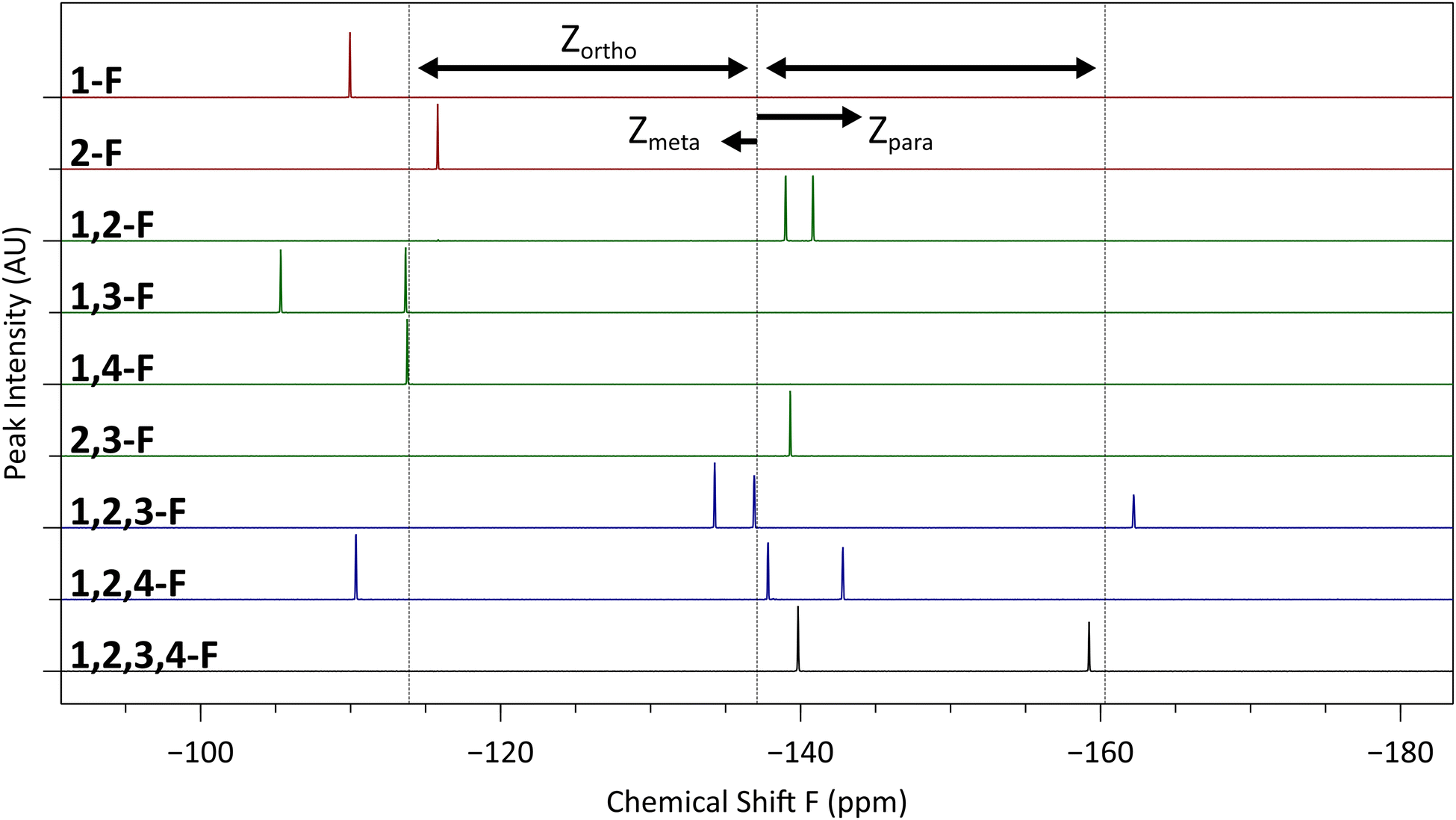 | ||
| Fig. 5 Stacked 19F NMRs of compounds 1 to 9, arranged with increasing number of fluorine atoms from top to bottom. The effect of substituents at various positions (ortho, meta and para) are shown, along with a reference line (dashed) showing the effect of one or two ortho substituents on the compounds. | ||
As an example, we consider compound 1, with its 1,2,3,4-tetrafluorinated ring. This substitution pattern has two sets of two equivalent fluorines. Those in the 1- or 4-positions each have an ortho, meta, and para fluorine substituents. We therefore compute a chemical shift of −141.7 ppm (−113.9 − 23.2 − 6.6 + 2 ppm), in excellent agreement with the observed resonance at about −140 ppm. The 2- and 4-positions, however, have two ortho and one meta fluoro substituents, giving a predicted resonance peak with a chemical shift of −158.3 ppm (−113.9 − 2(23.2) + 2 ppm), again in good agreement with the observed value of approximately −159 ppm. It is worth noting that Zortho is much larger in absolute value than the contributions of meta or para fluorine atoms, and so the gross features of an NMR spectrum may often be rationalized solely on their basis (e.g., in the 1,2-F compound 7, there are two closely spaced peaks with large negative shifts corresponding to the two fluorines with ortho neighbours).
Due to the strongly electron withdrawing nature of the fluorine substituents, we may expect the simple assumption of additivity of the shifts due to each substituent to break down as a result of multiple fluorine substituents exhausting the electron charge density in the fluorinated ring. The fact that the observed shifts continue to be in agreement with this additive model even in the tetrafluorinated compound is presumably due to the ability of the fluorine substituents to use the remainder of the large conjugated triphenylene system as a charge reservoir without depleting it.
Experimental
General information
The chemicals used in this research were purchased from distributors including Combi-Blocks, TCI America, Acros Organics, and Matrix scientific and they were used directly without further purification. Silica gel (Silicycle SiliaFlash F60, 230–400 mesh) was used for column chromatography, and silica gel plates (Scientific Adsorbents Inc.) were used for thin layer chromatography (TLC).A Rayonet photochemical reactor (16 × 254 nm lamps) was used for the photochemical reactions. An acetonitrile solution (0.5–1.5 mmol) of the terphenyls in a quartz tube (approximately 40 cm × 25 mm with 24/40 ST joint) was irradiated in the Rayonet.
A Bruker 400 NMR (400 MHz for 1H-NMR; 376 MHz for 19F-NMR and 101 MHz for 13C-NMR) and Agilent 500 NMR (500 MHz for 1H-NMR; 470 MHz for 19F-NMR and 126 MHz for 13C-NMR) were used for NMR data acquisition.
Differential scanning calorimetry (DSC) was run on a DSC from TA instruments (TA instruments Inc., Newcastle, DE, USA) for phase study and transition temperatures and enthalpies were obtained using Thermal Advantage Software (version 1.1A, TA Instruments Inc., Newcastle, DE, USA).
A polarizing optical microscope (POM) Nikon Eclipse E600 POL was employed to observe phase transitions and confirm melting points, along with a Mettler Toledo FP82HT Hot Stage and Mettler Toledo FP90 Central Processor temperature controller (Mettler Toledo, USA-Hightstown, NJ).
Process development summary for the synthesis of compound 3
The standard conditions (diglyme as a solvent with 20 mol% loading of copper(I) catalyst) give good to excellent yields for the decarboxylative coupling between an iodoaromatic (1.0 equiv.) and potassium pentafluorobenzoate (∼1.5 equiv.).15Decarboxylative coupling using potassium 2,3,5,6-tetrafluorobenzoate is slower and despite some side reactions still works almost as well. Generally, the best yields are obtained when there are at least two ortho fluorine atoms in the benzoic acid salt. If the same conditions are implemented with the potassium salt of 2,3,4,5-tetrafluorobenzoic acid the reaction is significantly less efficient. This motivated further development of a process to carry out the decarboxylative cross-coupling with the potassium salt of 2,3,4,5-tetrafluorobenzoic acid. We selected a model system (Scheme 4), using 2-iodobiphenyl as the standard aryl iodide material and tried different sets of conditions involving changing solvents, transition metal(s) catalysts, and amine co-catalysts.30
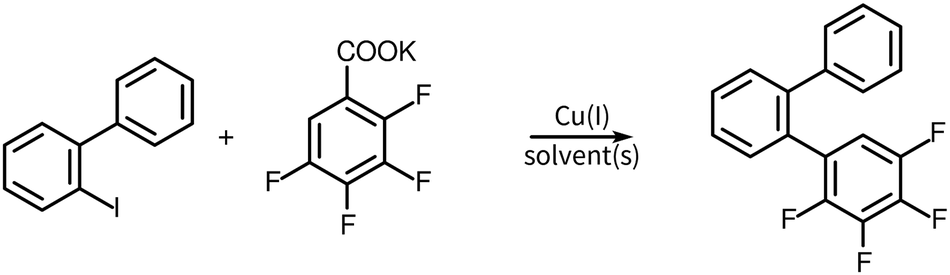 | ||
| Scheme 4 A model reaction designed for this process development study. | ||
The reaction mechanism of the copper-catalyzed decarboxylative reaction is not entirely clear. It is thought that after anion exchange between the copper(I) catalyst and the carboxylate group, the copper forms a complex with the C–C bond between the aromatic carbon and the carboxylate carbon. This is followed by a decarboxylation process to produce a phenylcopper(I) species which then reacts with the arylhalides.15,31 The process goes through oxidative addition and reductive elimination sequences, at a rate limited by the oxidative addition step. As a result, a sigma donor ligand like an aromatic or aliphatic amine will increase the electron density near the copper leading to an increase in the reaction rate. These chelating ligands also increase the rate of the decarboxylation step.32,33 Aliphatic amines like TDA-1 or TMEDA are more efficient compared to aromatic amines like 1,10-phenanthroline and quinoline.30 Moreover, TDA-1 is also a good phase transfer catalyst which can assist solubilization of a carboxylate in organic solvents.34 Thus, we have tried to enhance the rate of this decarboxylative cross coupling reaction using aliphatic amine solvents, and other cosolvents along with copper(I) catalysts in order to obtain the desired product with a high yield, which otherwise would not be accessible.
The results of the process study are summarized in Table 2. All the reactions were refluxed at the boiling point and whenever there was a use of TDA-1 temperature was maintained around 185 °C (the oil bath temperature).35 The best results in the model system involved the use of DMAc as a solvent, copper(I) iodide as a catalyst, and TDA-1 (1 equiv.) as an additive (entry 9). These conditions were then used to synthesize the terphenyl photoreaction precursor of the target material 3 and the yield was reproducible at 73%. This is in sharp contrast to the same reaction under standard conditions using diglyme and copper(I) iodide, which had a yield of 0%.
| Entry | Solvent/additives | Catalyst(s) | Yield (%) |
|---|---|---|---|
| 1 | Diglyme/TMEDA (20 mol%) | CuI (20 mol%) | 12 |
| 2 | Diglyme/TDA-1 (25 mol%) | CuI (20 mol%) | 22 |
| 3 | Diglyme/TDA-1 (1 equiv.) | CuI (20 mol%) | 28 |
| 4 | Diglyme/TMEDA | CuI (20 mol%) | 0 |
| 5 | Diglyme/TDA-1 (1 equiv.) | CuI (20 mol%) + Pd(OAc)2 (1.5 mol%) | 34 |
| 6 | Diglyme/TDA-1 (1 equiv.) | CuCl (20 mol%) + Pd(OAc)2 (1.5 mol%) | 17 |
| 7 | Diglyme/TDA-1 (1 equiv.) | Cu2O (20 mol%) + Pd(OAc)2 (1.5 mol%) | 0 |
| 8 | DMAc/TDA-1 (1 equiv.) | CuI (20 mol%) + Pd(OAc)2 (1.5 mol%) | 15 |
| 9 | DMAc/TDA-1 (1 equiv.) | CuI (20 mol%) | 52 |
| 10 | DMAc | CuI (20 mol%) | 37 |
| 11 | NMP/TDA-1 (1 equiv.) | CuI (20 mol%) | 30 |
| 12 | NMP | CuI (20 mol%) | 18 |
| 13 | DMI | CuI (20 mol%) | 0 |
| 14 | DMPU | CuI (20 mol%) | 0 |
| 15 | DMI/TDA-1 (1 equiv.) | CuI (20 mol%) | 0 |
| 16 | DMPU/TDA-1 (1 equiv.) | CuI (20 mol%) | 5 |
| 17 | Diglyme | CuI (20 mol%) | 0 |
Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2′,1′′-terphenyl (0.5–1.5 mmol) and acetonitrile (60.0 mL) were placed in a quartz tube (100 cm × 10 cm). The mixture was irradiated with 16 × 254 nm lamps in a Rayonet for 12–24 hours until TLC showed the completion of the reaction. The reaction mixture was allowed to cool down to room temperature (if the product precipitated out, it was filtered off and washed with acetonitrile) and transferred to a recovery flask. The reaction mixture was adsorbed on some silica gel. This loaded silica gel was placed at the top of a column and was eluted with a mixture of ethyl acetate and hexanes. Fractions containing pure product were combined and concentrated to afford a pale-yellow solid product which was recrystallized to obtain white crystals. (yield: 18–71%).
:2′,1′′-terphenyl (1 equiv.) was placed in a 100 mL round bottom flask and DCM (25.0 mL) was added under a nitrogen atmosphere. FeCl3 (3.0 equiv.) dissolved in nitromethane (50 mg FeCl3 per 1.0 mL) was added dropwise to the mixture while stirring and left for four to five hours at room temperature at which time the reaction was found to be complete by TLC. A mixture of methanol and water (1:1) was added and left stirring. The mixture was extracted with DCM and the organic portion was dried over anhydrous Na2SO4 and vacuum filtered. Silica gel was added to the filtrate and the solvent was evaporated under reduced pressure. The silica gel with adsorbed products was placed at the top of a column and eluted with a mixture of ethyl acetate and hexanes. Fractions containing the desired product were combined and concentrated to get a cream-colored solid which was recrystallized to give white crystals (yield: 68–85%).
:2′,1′′-terphenyl (0.5–1.5 mmol) and acetonitrile (60.0 mL) were placed in a quartz tube (100 cm × 10 cm). The mixture was irradiated with 16 × 254 nm lamps in a Rayonet for 12–24 hours until TLC showed the completion of the reaction. The reaction mixture was allowed to cool down to room temperature (if the product precipitated out, it was filtered off and washed with acetonitrile) and transferred to a recovery flask. The reaction mixture was adsorbed on some silica gel. This loaded silica gel was placed at the top of a column and was eluted with a mixture of ethyl acetate and hexanes. Fractions containing pure product were combined and concentrated to afford a pale-yellow solid product which was recrystallized to obtain white crystals. (yield: 18–71%).
:2′,1′′-terphenyl (1 equiv.) was placed in a 100 mL round bottom flask and DCM (25.0 mL) was added under a nitrogen atmosphere. FeCl3 (3.0 equiv.) dissolved in nitromethane (50 mg FeCl3 per 1.0 mL) was added dropwise to the mixture while stirring and left for four to five hours at room temperature at which time the reaction was found to be complete by TLC. A mixture of methanol and water (1:1) was added and left stirring. The mixture was extracted with DCM and the organic portion was dried over anhydrous Na2SO4 and vacuum filtered. Silica gel was added to the filtrate and the solvent was evaporated under reduced pressure. The silica gel with adsorbed products was placed at the top of a column and eluted with a mixture of ethyl acetate and hexanes. Fractions containing the desired product were combined and concentrated to get a cream-colored solid which was recrystallized to give white crystals (yield: 68–85%).
Additional synthetic details and full spectroscopic characterization for all of the intermediates and final products can be found in ESI.†
Conclusions
Fluorinated tetramethoxy columnar liquid crystals which differ in the number and location of fluorine atoms were synthesized and examined. Most of the synthesis was possible using the PCDHF method, and in those cases where the use of PCDHF was not effective a modified Scholl process was employed. Six out of the ten target molecules of interest were found to have a columnar mesophase. In fact, merely two fluorine atoms, strategically placed on a ring, were found to be sufficient to support a columnar mesophase in the 6,7,10,11-tetramethoxytriphenylene system. While the extremely short methoxy tails probably contribute a significant amount of disorder to the system, these compounds still prove to be useful as models for probing the reasons why some compounds form columnar mesophases and what the underlying mechanisms are. From an application standpoint, it is interesting to consider if the columnar mesophase of these short-tailed compounds behaves as a quasi-1D conductor, similar to other longer tailed columnar mesogens where the columns are widely separated by insulating tails, or do the shortened tails enable charge to move in a higher dimensional environment. Further work, including structural and transport studies, is warranted.Author contributions
P. Guragain and R. Twieg developed, and carried out, the synthesis of the compounds which were characterized by P. Guragain and M. Powers. R. Twieg and B. Ellman supervised the work, provided resources and acquired funding. P. Guragain wrote the original draft and M. Powers created the visualizations. All authors contributed the review and editing of the final draft.Data availability
The data supporting this article have been included as part of the ESI.† This includes additional synthesis information, NMR and HRMS data and DSC scans.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors gratefully acknowledge NSF grant 1809536 for funding this research. The authors also thank Prof. Dirk Friedrich for his assistance in obtaining HRMS data on several new compounds.References
- S. Sergeyev, W. Pisula and Y. H. Geerts, Chem. Soc. Rev., 2007, 36, 1902–1929 RSC.
- S. Chandrasekhar, B. K. Sadashiva and K. A. Suresh, Pramana, 1977, 9, 471–480 CrossRef CAS.
- F. M. Mulder, J. Stride, S. J. Picken, P. H. J. Kouwer, M. P. De Haas, L. D. A. Siebbeles and G. J. Kearley, J. Am. Chem. Soc., 2003, 125, 3860–3866 CrossRef CAS.
- J. Barbera, O. A. Rakitin, M. B. Ros and T. A. S. Torroba, Angew. Chem., Int. Ed., 1998, 37, 296–299 CrossRef CAS.
- D. Pucci, I. Aiello, A. Aprea, A. Bellusci, A. Crispini and M. Ghedini, Chem. Commun., 2009, 1550–1552 RSC.
- V. Percec, A. E. Dulcey, M. Peterca, M. Ilies, S. Nummelin, M. J. Sienkowska and P. A. Heiney, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 2518–2523 CrossRef CAS.
- Z. Li, M. Powers, K. Ivey, S. Adas, B. Ellman, S. D. Bunge and R. J. Twieg, Mater. Adv., 2022, 3, 534–546 RSC.
- P. Guragain, M. Powers, J. Portman, B. Ellman and R. J. Twieg, Mater. Adv., 2023, 4, 4129–4137 RSC.
- M. Powers, R. J. Twieg, J. Portman and B. Ellman, J. Chem. Phys., 2022, 157, 134901 CrossRef CAS PubMed.
- A. N. Cammidge, R. J. Turner, R. D. Beskeni and T. Almutairi, Liq. Cryst., 2017, 44, 2018–2028 CAS.
- M. Hird, Chem. Soc. Rev., 2007, 36, 2070–2095 RSC.
- C. Tschierske, Top. Curr. Chem., 2012, 318, 1–108 CrossRef CAS PubMed.
- M. Ichihara, H. Suzuki, B. Mohr and K. Ohta, Liq. Cryst., 2007, 34, 401–410 CrossRef CAS.
- Y. Wu, W. Zhang, Q. Peng, C. K. Ran, B. Q. Wang, P. Hu, K. Q. Zhao, C. Feng and S. K. Xiang, Org. Lett., 2018, 20, 2278–2281 CrossRef CAS.
- R. Shang, Y. Fu, Y. Wang, Q. Xu, H. Z. Yu and L. Liu, Angew. Chem., Int. Ed., 2009, 48, 9350–9354 CrossRef CAS PubMed.
- M. Lafrance, C. N. Rowley, T. K. Woo and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 8754–8756 CrossRef CAS PubMed.
- S. S. Laev and V. D. Shteingarts, Tetrahedron Lett., 1997, 38, 3765–3768 CrossRef CAS.
- D. M. Cho, S. R. Parkin and M. D. Watson, Org. Lett., 2005, 7, 1067–1068 CrossRef CAS.
- K. C. Rippy, N. J. DeWeerd, I. V. Kuvychko, Y. S. Chen, S. H. Strauss and O. V. Boltalina, ChemPlusChem, 2018, 83, 1067–1077 CrossRef CAS PubMed.
- J. He, Y. Chen, P. Hu, B. Wang, K. Zhao and B. Donnio, J. Mol. Liq., 2024, 414, 126218 CrossRef CAS.
- R. Yoshida, H. Sugiyama and Y. Segawa, Chem. Lett., 2024, 53, upae048 CrossRef.
- S. E. Wheeler and K. N. Houk, J. Chem. Theory Comput., 2009, 5, 2301–2312 CrossRef CAS PubMed.
- M. M. Zhou, J. He, H. M. Pan, Q. Zeng, H. Lin, K. Q. Zhao, P. Hu, B. Q. Wang and B. Donnio, Chem. – Eur. J., 2023, 29, e202301829 CrossRef CAS PubMed.
- S. Melandri, Phys. Chem. Chem. Phys., 2011, 13, 13901–13911 RSC.
- C. Vande Velde, E. Bultinck, K. Tersago, C. Van Alsenoy and F. Blockhuys, Int. J. Quantum Chem., 2007, 107, 670–679 CrossRef CAS.
- O. V. Dorofeeva, I. F. Shishkov, N. M. Karasev, L. V. Vilkov and H. Oberhammer, J. Mol. Struct., 2009, 933, 132–141 CrossRef CAS.
- M. D. Powers ( 2022). Tail-Free Discotic Liquid Crystals [Doctoral dissertation, Kent State University].
- P. Guragain ( 2023), Synthesis and properties of ferronematic and short tail columnar liquid crystals. [Doctoral dissertation, Kent State University].
- C. A. L. Mahaffy and J. R. Nanney, J. Fluor. Chem., 1994, 67, 67–74 CrossRef CAS.
- G. Cahiez, A. Moyeux, O. Gager and M. Poizat, Adv. Synth. Catal., 2013, 355, 790–796 CrossRef CAS.
- N. Rodríguez and L. J. Goossen, Chem. Soc. Rev., 2011, 40, 5030–5048 RSC.
- T. Cohen and R. A. Schambach, J. Am. Chem. Soc., 1970, 3189–3190 CrossRef CAS.
- T. Cohen, R. W. Berninger and J. T. Wood, J. Org. Chem., 1978, 43, 837–848 CrossRef CAS.
- A. McKillop and L. S. Mills, Synth. Commun., 1987, 17, 647–655 CrossRef CAS.
- D. Villemin and M. Letulle, Synth. Commun., 1989, 19, 2833–2839 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma00933a |
| This journal is © The Royal Society of Chemistry 2024 |