
A survey of stapling methods to increase affinity, activity, and stability of ghrelin analogues†
Juan J.
Esteban
a,
Julia R.
Mason
a,
Jakob
Kaminski
a,
Rithwik
Ramachandran
b and
Leonard G.
Luyt
 *acd
*acd
aDepartment of Chemistry, University of Western Ontario, 1151 Richmond Street, London, Ontario N6A 3K7, Canada. E-mail: lluyt@uwo.ca
bDepartment of Physiology and Pharmacology, University of Western Ontario, 1151 Richmond Street, London, Ontario N6A 5C1, Canada
cDepartments of Medical Imaging and Oncology, University of Western Ontario, 1151 Richmond Street, London, Ontario N6A 3K7, Canada
dLondon Regional Cancer Program, Lawson Health Research Institute, 800 Commissioners Road East, London, Ontario N6A 4L6, Canada
First published on 30th November 2023
Abstract
The growth hormone secretagogue receptor (GHSR) is a G protein-coupled receptor which regulates various important physiological and pathophysiological processes in the body such as energy homeostasis, growth hormone secretion and regulation of appetite. As a result, it has been postulated as a potential therapeutic target for the treatment of cancer cachexia and other metabolic disorders, as well as a potential imaging agent target for cancers and cardiovascular diseases. Ghrelin is the primary high affinity endogenous ligand for GHSR and has limited secondary structure in solution, which makes it proteolytically unstable. This inherent instability in ghrelin can be overcome by incorporating helix-inducing staples that stabilize its structure and improve affinity and activity. We present an analysis of different stapling methods at positions 12 and 16 of ghrelin(1–20) analogues with the goal of increasing proteolytic stability and to retain or improve affinity and activity towards the GHSR. Ghrelin(1–20) analogues were modified with a wide range of chemical staples, including a lactam staple, triazole staple, hydrocarbon staple, Glaser staple, and xylene-thioether staple. Once synthesized, the receptor affinity and α-helicity were measured using competitive binding assays and circular dichroism spectroscopy, respectively. Generally, an increase in alpha-helicity using a flexible staple linker led to improved affinity towards GHSR. Ghrelin(1–20) analogues with a lactam, triazole, and hydrocarbon staple resulted in helical analogues with stronger affinity towards GHSR than unstapled ghrelin(1–20), a compound that lacks helical character. Compounds were also investigated for their agonist activity through β-arrestin 1 & 2 recruitment BRET assays and for their metabolic stability through serum stability analysis.
Introduction
The growth hormone secretagogue receptor (GHSR), also referred to as the ghrelin receptor, is a GPCR that has garnered attention for pharmaceutical development. The endogenous peptide ligand, ghrelin, is a 28 amino acid peptide hormone that possesses an octanoyl chain,1 which is post-translationally coupled onto the serine at the third position by ghrelin O-acyl transferase (GOAT).2,3 This unique modification is crucial for receptor recognition, as des-acyl ghrelin has no affinity (IC50 > 10 μM) towards the GHSR.4 When ghrelin binds to its native receptor, it exhibits a number of physiological functions such as energy homeostasis, growth hormone secretion and regulation of appetite.2,5 Ghrelin's orexigenic effects when bound to its receptor have resulted in the peptide being labelled as the “hunger hormone”.Due to this biological role that ghrelin plays, studies have probed its potential as a therapeutic agent for the treatment of cancer cachexia and other metabolic disorders.6 Cancer cachexia is reported to affect 50% of all cancer patients and 60–80% of advanced cancer patients. Its mortality rate is also described to be as high as 80% for cancer per year, while also being associated with poor-symptom status and poor quality of life for patients living with the complication.7,8 Treatment approaches for this disease range from anabolic steroids, anti-catabolic therapies, and nutritional interventions.9 However, to date there has not been an effective treatment developed for this illness. Ghrelin, which is the gastric-derived endogenous ligand of the GHSR, regulates energy homeostasis by transmitting peripheral nutritional information to the brain thereby exhibiting orexigenic effects.10 Activation of the central nervous system GHSRs by ghrelin has been demonstrated to control food anticipation and food-based learning in rodents.11 However, levels of acylated ghrelin have been found to be 50% higher in cancer patients with cachexia when compared to those without cachexia.12 This suggests that cachexia's anorexic effects overwhelm ghrelin's orexigenic properties and thus supraphysiologic doses must be used to yield positive results.13 As a result, it is theorized that modifying ghrelin's structure may serve as an effective structural template for the design of therapeutic agents for the treatment of this wasting disease.
Due to the ghrelin receptor's potential as a biomarker in a wide range of diseases and biological processes, there have also been a variety of imaging agents developed in the last decade to target this receptor.14 More specifically, this receptor has been shown to be differentially expressed in ovarian15 and prostate16 cancer, and in the hearts of dilated cardiomyopathy patients and patients with end-stage heart failure.14,17 Ghrelin's natural high affinity towards the receptor has made it an ideal template for the design of potential imaging agents targeting the ghrelin receptor. Thus, it has become a target of interest in the field of molecular imaging.18–20
Like many natural peptides, ghrelin's linear structure makes it susceptible to enzymatic degradation by proteases. As a result, ghrelin analogues that were developed as imaging agents have been noted as having poor in vitro stability in human serum, with structural modifications required in order to improve stability.21,22 One important modification is replacing the acyl connection on Ser3 with an amide linkage, to prevent ester hydrolysis.4 However, studies have demonstrated that when ghrelin binds to its receptor, part of its peptide structure folds into an α-helical conformation approximately from Pro7 to Gln14.23–25 This suggests that a section of ghrelin's sequence has helical character in certain environments and could be stabilized using a helix-inducing chemical staple, although in contrast, a NMR spectroscopy analysis describes considerable fluidity in the Glu8 to Ser18 region for truncated ghrelin(1–18).26
Chemical staples have become recognized as an effective molecular tool for modulating the pharmacodynamic and pharmacokinetic properties of peptides. The addition of a helix-inducing chemical staple into a peptide structure is a synthetic method for increasing affinity towards the target protein while also increasing peptide stability to enzymatic degradation and improving cell permeability. By locking the peptide into the desired biological conformation, staples can enhance a peptide's ability to form interactions with their native receptor, thereby increasing affinity towards their receptor target. Additionally, studies have demonstrated that the binding sites of many proteases found in humans are not large enough to permit an α-helical conformation to bind, thus resulting in improved serum stability.27 However, considering the wide range of chemical staples that have been developed, the question arises as to which staples are consistent at inducing helicity when incorporated into a peptide structure and also provide the preferential conformation for a particular binding site.
Strategies for inducing α-helicity in peptides have been around for decades, including metal ion clips,28 unnatural amino acids,29 and helix-nucleating templates.30 Staples have been notably effective at stabilizing the secondary structure of a peptide by reducing the entropic penalty associated with folding. The term ‘peptide staple’ was coined by Verdine and coworkers in their seminal paper published in 2000.31 In their work, they developed a hydrocarbon cross link using α,α-disubstituted amino acid residues bearing olefin tethers. This was the first example of a peptide where the use of a hydrocarbon tether was effective at enhancing its helicity and metabolic stability. In years to come, the hydrocarbon staple would be regarded as one of the most effective staples at increasing α-helicity in a range of peptide sequences and biological applications. Other peptide staples that are known for their ability to induce α-helicity are the lactam staple,32 triazole staple,33 xylene-thioether staple,34 and the more recently developed Glaser staple.35 Comparative studies have demonstrated that the lactam and triazole staples are as efficient as the hydrocarbon staple at increasing helical character in a peptide structure.36 The xylene-thioether staple is known for its efficient two-component cross-link which allows for bifunctionality of the linker unit, while the Glaser staple allows for increased rigidity in the linker structure.
In prior work, a lactam staple (Glu-Lys) was introduced into the peptide structure of [Dpr3(n-octanoyl)]ghrelin(1–20)-NH2 in what was referred to as a “staple scan” to discover the optimal location that yields a cyclic peptide with the highest affinity towards the ghrelin receptor. In the resulting publication, Lalonde and coworkers described how an i, i + 4 staple between the 12 and 16 position of [Dpr3]ghrelin(1–20) resulted in the highest affinity towards the ghrelin receptor with a reported IC50 of 7.9 nM.37 The resulting compound was then appended with a Cy5 fluorescent dye and was successful at imaging the ghrelin receptor in ovarian cancer cell lines using confocal microscopy.
Since the optimal position for an i, i + 4 lactam (Glu-Lys) staple in ghrelin(1–20) analogues was determined to be positions 12 and 16, the question arose whether different stapling techniques at this same location would yield compounds with greater affinity towards the ghrelin receptor, and if these techniques would have varied influence on the metabolic stability of ghrelin analogues. As a result, we present an investigation of five different helix inducing staples at positions 12 and 16 in truncated ghrelin(1–20) analogues. The five staples investigated are the triazole, lactam (Lys-Asp), hydrocarbon, xylene-thioether, and Glaser staples (Fig. 1). This selection of stapling techniques provides a range of conformations as it includes varying degrees of flexibility in the linker region. Once synthesized, these compounds were evaluated for their affinity towards the ghrelin receptor, for their ability to activate the ghrelin receptor, and for their metabolic stability in human serum. Selected compounds with desirable pharmacological properties have the potential to be developed into molecular imaging agents or used as therapeutic agents for cancer cachexia.
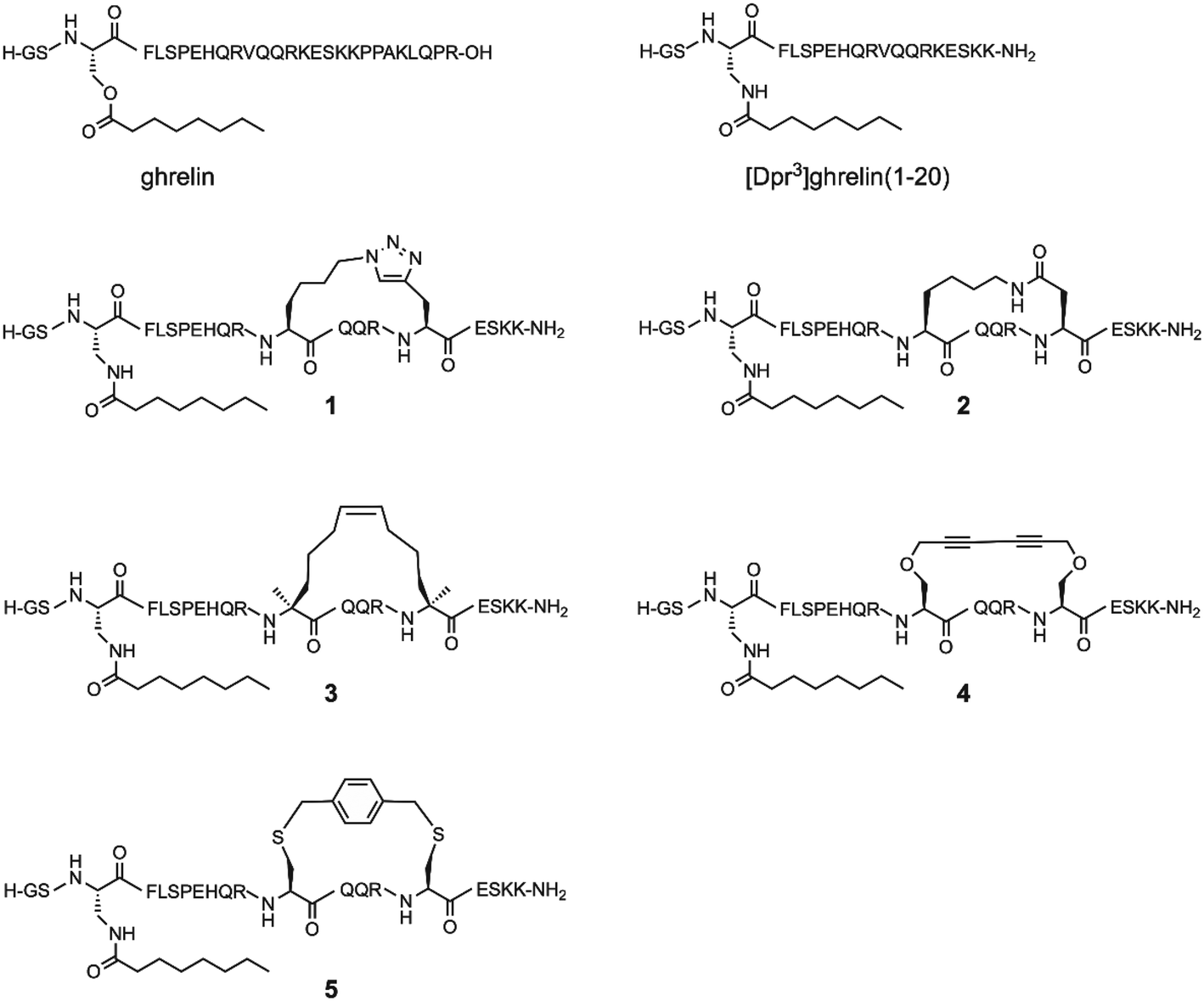 | ||
| Fig. 1 Structure of ghrelin, [Dpr3]ghrelin(1–20) and the five stapled ghrelin analogues. | ||
Results and discussion
To determine the effect of different stapling techniques on the bioactivity and stability of [Dpr3]ghrelin(1–20), five ghrelin analogues bearing helix-inducing staples were synthesized. For their synthesis, the natural ghrelin sequence had substitutions at positions 12 and 16 with residues bearing the functional groups required for the diverse stapling techniques with an i, i + 4 spacing (Scheme 1). Ghrelin's natural affinity towards the ghrelin receptor is largely due to its octanoyl chain present at Ser3. However, the instability of the ester bond requires that the Ser3 residue be substituted with an amine analogue, diaminopropionic acid (Dpr), in order to have the more stable amide bond connecting the octanoyl chain to the peptide. Studies have shown that the substitution of this ester bond with an amide bond showed no effect on ghrelin's affinity towards the ghrelin receptor.4 Due to the different cyclization methods used in this report, two different orthogonal protecting groups had to be used to protect the reactive amine of the Dpr3 residue to prevent any unwanted side reactions from occurring. For most peptides, an Alloc protecting group was used, which deprotects easily and effectively with a palladium catalyst and phenyl silane. For any suspected cyclizations that posed a problem with the Alloc protecting group, the protecting group Mtt, which deprotects with dilute acid (1% TFA), was used. Following the successful stapling and coupling of the octanoyl chain, peptides are cleaved from their solid support, purified by preparative HPLC and characterized by ESI+ mass spectrometry.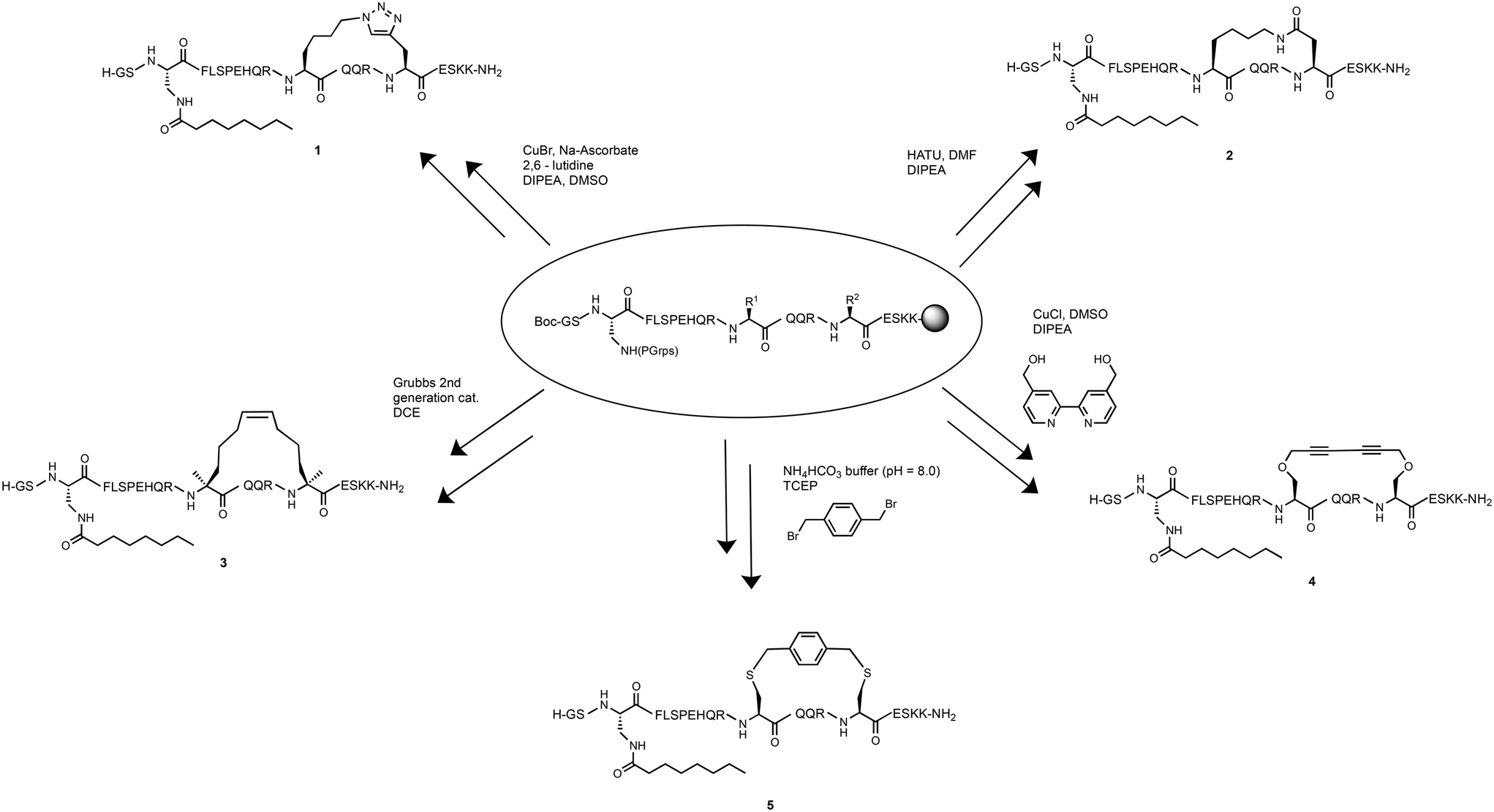 | ||
| Scheme 1 Synthetic scheme overview for the synthesis of ghrelin(1–20) analogues. | ||
Triazole stapled ghrelin(1–20) analogue
Triazole tethers have been documented to stabilize secondary peptide structures and produce helicity that is comparable to that of the lactam staples.38,39 Furthermore, the amino acid residues used for the formation of the triazole moiety are readily accessible and the optimal conditions of the copper assisted “click chemistry” reaction are established.40 As a result, compound 1, a triazole stapled ghrelin(1–20) analogue, was included as part of the study. To synthesize the 20 amino-acid long peptide, Fmoc-based automated SPPS was used with modifications of the truncated ghrelin amino acid sequence at positions 3, 12 and 16. Position 3 used an Alloc protecting group on the Dpr residue for the coupling of the octanoyl chain. Positions 12 and 16 were occupied with a lysine azide residue and a propargyl glycine residue, respectively. The side chains of these two residues were then cyclized in the copper-assisted azide–alkyne cycloaddition reaction using copper(I) bromide, sodium ascorbate, 2,6-lutidine and DIPEA. Once the macrocyclization was confirmed to have been successful using LCMS, the deprotection of the Alloc protecting group of Dpr3 and the subsequent coupling of the octanoyl chain was carried out to produce 1.Lactam (Lys-Asp) stapled ghrelin(1–20) analogue
While prior work demonstrated the efficiency of a lactam (Glu-Lys) staple at increasing the helicity and affinity for ghrelin(1–20) analogues,37 studies have suggested that a lactam (Lys-Asp) staple also provides an increase of helicity in peptides, and may perhaps stabilize the secondary structure of certain peptides due to the smaller stapling unit.32,41 Thus, a lactam (Lys-Asp) stapled ghrelin(1–20) analogue was synthesized as part of this project to further investigate the differences that the amide position of the lactam staple and ring size have on helicity and binding affinity towards the ghrelin receptor.The first attempt at synthesizing this compound involved using orthogonal protecting groups, with the Lys12 residue containing an Alloc group and the Asp16 containing an allyl ester group. However, analysis of a resin sample post-SPPS with LCMS revealed that a considerable amount of aspartimide formation had occurred during the automated synthesis. Aspartimide formation is a sequence dependent cyclization that occurs to aspartic acid residues in the presence of base, in this case due to the repetitive piperidine treatments required for Fmoc deprotection.42 Therefore, a new strategy had to be designed for the synthesis of this stapled analogue while considering ways to limit this unwanted cyclization.
The second attempt at the synthesis involved using alternative orthogonal protecting groups. The orthogonal phenylisopropyl ester (Pip) protecting group, which can be removed under mild acidic conditions, can provide sufficient steric bulk to prevent unwanted side reactions such as the removal of the amide hydrogen by piperidine seen in aspartimide formation.32 Thus, Asp16 was protected with the Pip group and the Lys12 residue was protected with an Mtt group, another acid labile group. Since both protecting groups are acid labile, the Dpr3 residue contained the orthogonal Alloc group as this protecting group is inert to acidic conditions. After the SPPS of the peptide, a small sample confirmed through LCMS that aspartimide formation was successfully kept to a minimum. Next, the lactamisation reaction using the coupling agent HATU and the base DIPEA was able to yield the desired resin-bound peptide macrocycle and subsequently compound 2.
Hydrocarbon stapled ghrelin(1–20) analogue
Another stapling technique that was investigated was the hydrocarbon staple. This stapling technique is formed from a ring closing metathesis (RCM) reaction between two non-natural amino acids bearing olefin tethers. Once the olefin tethers undergo the ring closing metathesis and the peptide macrocycle is formed, it provides a stabilization of the secondary structure of the peptide and has been observed to significantly increase protease resistance.43,44 The synthesis of the hydrocarbon stapled ghrelin(1–20) analogue, was attempted using the disubstituted unnatural amino acid residue (4′-pentenyl) alanine at positions 12 and 16 and with the Dpr3 being protected with the orthogonal Mtt group. Following SPPS however, it was discovered that a noticeable amount of incomplete coupling of the (4′-pentenyl) alanine residue had occurred. It was theorized that this was caused due to the increased steric bulk present near the amine caused by the disubstituted character of the amino acid. As a result, a manual coupling of the unnatural amino acid was completed with longer reaction times as well as the more efficient coupling agent, HATU. Following the successful synthesis with complete coupling of the unnatural amino acid, the deprotection of Mtt and coupling of octanoyl acid chain was carried out. The ring closing metathesis reaction was then attempted using 20 mol% of Grubbs 2nd generation catalyst in DCE in an open-air system. An open-air system was used to allow for the ethylene by product of the RCM reaction to escape, thereby shifting the equilibrium of the reaction to the products. This open-air system yielded the successfully cyclized peptide product 3.Glaser stapled ghrelin(1–20) analogue
A more recent development in helix-inducing stapling techniques is the Glaser staple. This rigid staple is formed from a Glaser–Hay coupling reaction involving two 1,3-diyne motifs. When incorporated into a peptide structure in both i, i + 4 and i, i + 7 residue spacing, a significant increase in helicity was observed, likely due to the added rigidity that is produced from the cyclization reaction.45,46 This stapling technique also offers increased solubility in water when compared to cyclic regions consisting of entirely hydrophobic surfaces.35 To synthesize this stapled peptide analogue, Dpr3 was protected with Mtt so that unwanted side reactions could not occur during the cyclization reaction. Following the deprotection of the Mtt group and coupling of the octanoyl chain, the cyclization reaction using a Glaser–Hay coupling, was attempted. This reaction involves the formation of a macrocycle between two propargyl serine residues, and was achieved by adding copper chloride, a 4,4′-bis(hydroxymethyl)-2,2′-bipyridine ligand and DIPEA to a vessel containing the resin-anchored peptide and allowing it to react for three days. The resulting stapled peptide product was the Glaser stapled ghrelin(1–20) analogue 4.Xylene-thioether stapled ghrelin(1–20) analogue
The inclusion of semi-rigid cross-linkers, through the formation of thioethers, has been a popular technique for stabilizing alpha-helical secondary structures. This is an example of a two-component stapling technique which is formed from the bisalkylation of sulfide moieties most commonly in an i, i + 4 residue spacing. This stapling technique has the advantage of allowing for more flexibility in design, as the cross-linking reactions are selective and compatible with unprotected peptides.34 While many cross-linkers can be used to react with Cys residues, a recent study showed that more rigid systems like bis(bromomethyl)benzene can provide increased helicity due to their ability for low-strain intramolecular cyclizations.47 While studies report that the different constitutional isomers of α,α′-dibromo-xylene (meta, ortho and para) are all able to provide significant increases in helicity, the α,α′-dibromo-p-xylene isomer was chosen for evaluation due to availability.34 Thus, a synthesis of a bis-thioether stapled ghrelin(1–20) analogue using the α,α′-dibromo-p-xylene linker was planned. The synthesis of ghrelin analogue first involved SPPS with two trityl protected cysteine residues at positions 12 and 16. Since the trityl protecting group is acid labile, the Dpr3 was orthogonally protected with an Alloc protecting group. Following the deprotection of the Alloc group and the coupling of the octanoyl chain, the peptide was deprotected using a cleavage cocktail that included ethane-1,2-dithiol (EDT) in order to prevent oxidation of the free thiols that are present after deprotection. For the cross-linker coupling reaction, the deprotected peptide product was then mixed in a NH4HCO3 buffer solution with tris(2-carboxyethyl)phosphine and α,α′-dibromo-p-xylene producing 5.Circular dichroism
The helical character of the stapled ghrelin(1–20) analogues was determined by obtaining the respective circular dichroism (CD) spectra of each peptide. CD measurements were performed at a concentration of 0.1 mM in sodium phosphate buffer. The distinctive spectra for an α-helical peptide contains a large positive band at 192 nm and two negative bands at 208 nm and 222 nm. It is common practice to report the mean residue ellipticity (MRE) of the negative band at 222 nm to compare the relative helicity of peptides, as seen in Table 1. Perhaps a more useful method for comparison, however, is to report the percent helicity of the peptide. Using the MRE value at 222 nm, a percent helicity for an individual peptide can be determined using a formula that considers the temperature, the number of peptide units, and the theoretical MRE values for a fully helical peptide and a random coil at 222 nm.41 The calculated value approximates what fraction of the peptide is in a helical conformation. The results from the CD experiments showed that the peptides with the highest increase in helicity, in comparison to the linear [Dpr3]ghrelin(1–20), were the triazole, lactam and hydrocarbon stapled analogues (1–3) (Fig. 2). These staples were able to produce peptides that were more helical than unstapled [Dpr3]ghrelin(1–20), which shows 0% helical character in water,37 and full-length human ghrelin. However, it should be noted that human ghrelin's CD was measured in water, and reports indicate that this peptide becomes more helical once it has interacted with the ghrelin receptor or in a lipid bilayer/water system.24 The Glaser and bisthioether staples (4 and 5) were also able to increase helicity in comparison to unstapled [Dpr3]ghrelin(1–20) but were significantly less helical than 1–3. This could indicate that a more rigid peptide staple is not compatible with the peptide sequence of [Dpr3]ghrelin(1–20) for increasing helicity in contrast to more flexible staples.| Compound | [θ]222 (water) | % helicity |
|---|---|---|
| Human ghrelin | −2884 | 12% |
| [Dpr3]ghrelin(1–20) | 2460 | 0% |
| 1 | −7996 | 26% |
| 2 | −11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 436 436 |
35% |
| 3 | −12332 |
39% |
| 4 | −2497 | 10% |
| 5 | −2840 | 12% |
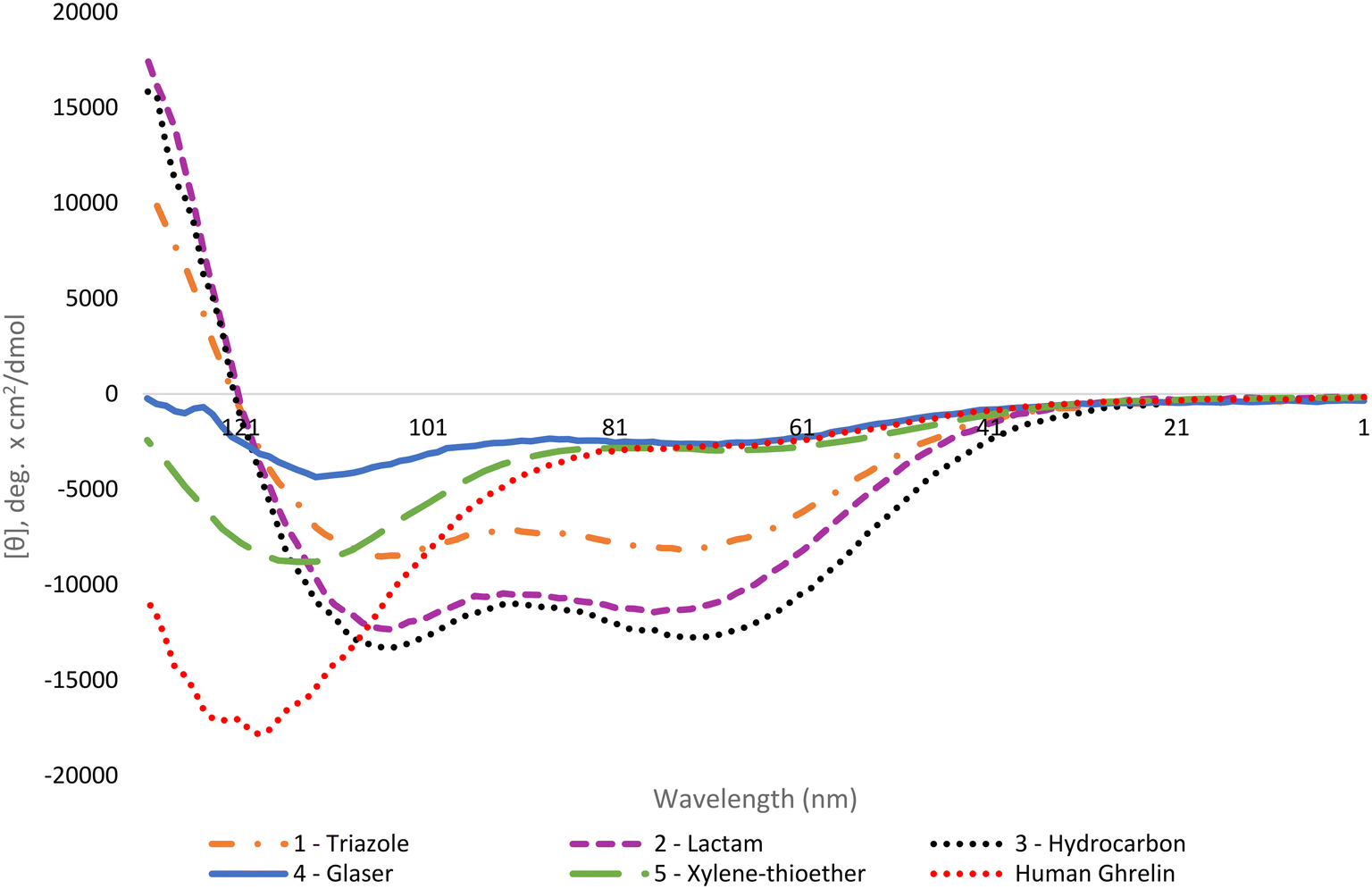 | ||
| Fig. 2 Circular dichroism spectra for the ghrelin(1–20) analogues and human ghrelin. | ||
Consistent with the literature, the results from the circular dichroism experiments indicate that the lactam, triazole and hydrocarbon staple are the most efficient at inducing α-helicity in a peptide structure.40 Interestingly, the lactam (Lys-Asp) staple saw a modest increase in percent helicity (35%) when compared to the previously reported lactam (Glu-Lys) staple which had a calculated percent helicity value of 27% under identical experimental conditions.37 This difference in helicity could be explained by the compatibility of ghrelin(1–20)'s sequence with the shorter stapling unit of the lactam (Lys-Asp) staple. Not surprisingly, the hydrocarbon stapled ghrelin analogues saw the greatest induction of α-helicity in ghrelin(1–20), as was predicted from the literature. It is postulated that part of the reason this staple is so effective at inducing helicity is due to the disubstituted nature of its unnatural amino acid residue from which the linker unit is formed. In fact, peptides have been reported to have achieved α-helical induction even when the unnatural amino acids have not been cyclized.48 A logical progression of this work would be to investigate if using disubstituted amino acid with the other staples investigated would yield similar percent helicity values as the hydrocarbon staple. Probing for the effect that this unique feature of the unnatural amino acid building blocks has on helicity would give insight for the design of more helical peptide compounds.
GHSR binding assays
To evaluate the binding affinity of the synthesized stapled ghrelin(1–20) analogues towards the GHSR, radioligand inhibition binding assays using [125I]-ghrelin on HEK-293 cells transiently transfected with the ghrelin receptor were performed. These cells were specifically chosen for their rapid proliferation and their compatibility with X-tremeGENE 9 DNA transfection. The averaged results of these assays, measured by calculating the concentration of the different stapled ghrelin analogues at 50% radioligand inhibition (IC50) can be seen in Table 2 along with their respective peptide sequences. In order to compare the efficiency of the different staples at increasing the affinity of ghrelin(1–20) analogues towards the ghrelin receptor, values obtained through a binding assay for both human ghrelin (7.57 nM) and [Dpr3]ghrelin(1–20) (2.28 nM) were also included in the table. The difference between these two values indicates that truncation of the native human ghrelin sequence to the first 20 amino acids leads to an increase binding affinity towards the receptor, which is consistent with the data found in the work by Bednarek and coworkers.4 In their paper, they used radioligand binding assays with a potent ghrelin receptor binder [35S]MK-0677 to compare the binding affinity of full-length human ghrelin (IC50 = 0.25 nM) and truncated ghrelin(1–23) (IC50 = 0.16 nM) and noticed an almost two-fold difference between the two. The results from the binding assays demonstrate that the triazole, lactam (Lys-Asp) and hydrocarbon staples (1–3) were able to increase the binding affinity of ghrelin(1–20) analogues. The three staples saw a varied increase in binding affinity to the ghrelin receptor in comparison to [Dpr3]ghrelin(1–20), with the triazole having almost a two-fold increase (0.92 nM) (Fig. 3). In contrast, the bulkier and more rigid stapling units, the Glaser and xylene-thioether staples (4 and 5), saw a decrease in binding affinity in comparison to [Dpr3]ghrelin(1–20).| Compound | Sequence | IC50 (nM) |
|---|---|---|
| Human ghrelin | H-GSS(octanoyl)FLSPEHQRVQQRKESKKPPAKLQPR-OH | 7.57 ± 1.51 |
| [Dpr3]ghrelin(1–20) | H-GS-Dpr(octanoyl)-FLSPEHQRVQQRKESKK-NH2 | 2.28 ± 0.48 |
| 1 | H-GS-Dpr(octanoyl)-FLSPEHQR[XQQRY]ESKK-NH2 | 0.92 ± 0.31 |
| 2 | H-GS-Dpr(octanoyl)-FLSPEHQR[KQQRD]ESKK-NH2 | 1.19 ± 0.40 |
| 3 | H-GS-Dpr(octanoyl)-FLSPEHQR[OQQRO]ESKK-NH2 | 1.44 ± 0.16 |
| 4 | H-GS-Dpr(octanoyl)-FLSPEHQR[ZQQRZ]ESKK-NH2 | 4.03 ± 0.65 |
| 5 | H-GS-Dpr(octanoyl)-FLSPEHQR[CQQRC]ESKK-NH2 | 4.93 ± 1.08 |
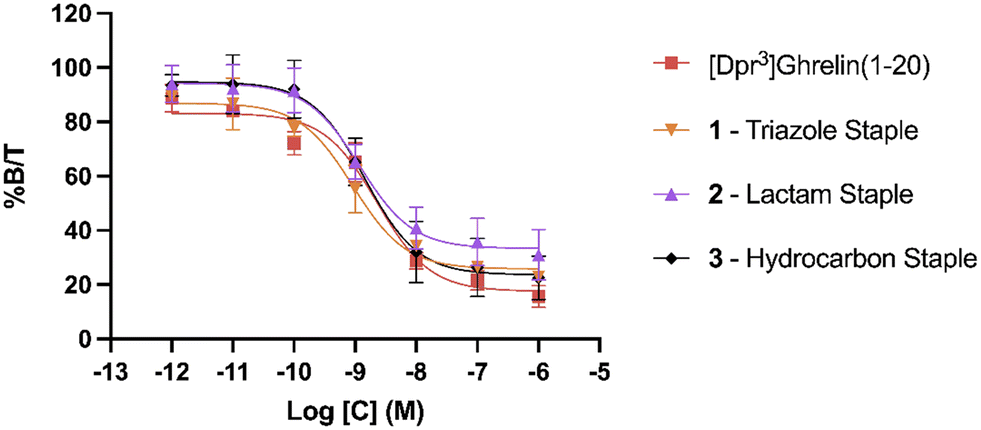 | ||
| Fig. 3 Radioligand binding assay IC50 curves for lactam stapled, triazole stapled and hydrocarbon stapled ghrelin(1–20) analogues. % B/T denotes the percentage of bound [125I]-ghrelin to the ghrelin receptor. Data are mean ± SEM. | ||
Clear differences are present in the degrees of freedom of the staple linker region, for peptides that saw an increase in binding affinity towards the ghrelin receptor (triazole, lactam, and hydrocarbon) in comparison to those that saw a decrease (Glaser, xylene-thioether). As a result, it is hypothesized that the rigidity of the stapling unit may influence the conformation of the peptide when bound to the ghrelin receptor, which may affect the interactions that take place in the binding pocket. Furthermore, a cryo-EM study published by Qin and coworkers described how the ECL2 of the ghrelin receptor sits directly over the ligand-binding pocket, thereby suggesting that interactions with the C-terminal region of a ghrelin analogue could occur.25 This indicates that the different stapling moieties may interact with these regions and could partly explain differences found in binding affinities for stapled analogues with similar degrees of freedom. Comparing the CD results to the binding affinity data towards the ghrelin receptor, there does not appear to be a clear correlation between increased helicity and increased binding affinity towards the ghrelin receptor. While the most helical peptides (1–3) were able to significantly increase binding affinity towards the receptor, the other two staples (4 and 5) that also saw an increase in helicity were weaker binders to the ghrelin receptor in comparison to the linear [Dpr3]ghrelin (1–20).
β-Arrestin recruitment BRET assays
As previously described, ghrelin has been proposed as a potential molecular template for the design of therapeutics targeting cancer cachexia due to ghrelin's orexigenic properties when bound to its receptor. The ghrelin receptor is expressed in the hippocampus and hypothalamic arcuate nucleus where it is said to play a role in regulating feeding behavior.49 Ligands activate GPCRs by binding to the active site and causing a conformational change in the receptor that activates heterotrimeric G proteins and dissociation of the α and βγ subunits. These then go on to activate intracellular second messenger signaling cascades. For the ghrelin receptor specifically, intracellular signaling occurs primarily via Gαq/11 and Gαi/o activation.50 Following activation of G protein mediated signaling, activated GPCR C-terminal serine and threonine residues are phosphorylated by GPCR kinases (GRKs) to facilitate β-arrestin recruitment, termination of G protein mediated signaling and receptor internalization through endocytosis. Though β-arrestins were first described as being part of the desensitization pathway for the ghrelin receptor, recent studies demonstrated that they can also act as signaling scaffolds. For example, β-arrestins have been linked to the adipogenic functions of the ghrelin/GHSR system.40 Therefore we used β-arrestin recruitment to the GHSR as a surrogate for the ability of the stapled ghrelin analogues to activate the ghrelin receptor. BRET assays were done in HEK293 cells transfected with a GHSR-1a-eYFP construct and either β-arrestin-1 or -2-RLuc construct. BRET signals are calculated as the ratio of the acceptor (eYFP) over the donor (RLuc) signal and agonist stimulated BRET signals (net BRET) were obtained by subtracting the basal BRET signal from signals obtained after agonist stimulation. The concentration effect curves for different compounds were calculated by plotting the net BRET signal against the agonist concentrations. Curve fitting was done using three-parameter, nonlinear regression models and EC50 and Emax values were calculated for each stapled ghrelin analogue.The results from the BRET assays can be found in Table 3. Notably, these assays revealed that the triazole and lactam (Lys-Asp) stapled analogues (1 and 2) activated the receptor with greater potency than unstapled [Dpr3]ghrelin(1–20) with EC50 values for β-arrestin 1 of 2.4 nM and 0.6 nM, and values for β-arrestin 2 of 32.0 nM and 12.6 nM, respectively (Fig. 4A and C). However, both analogues were partial agonists, with Emax values that were lower than unstapled [Dpr3]ghrelin(1–20). In contrast, the more rigid xylene-thioether and Glaser stapled analogues (4, 5) appear to be super-agonists that activated the receptor with greater efficacy than unstapled [Dpr3]ghrelin(1–20), with Emax values for β-arrestin 1 of 0.44 and 0.46 compared to 0.33 for the unstapled control (Fig. 4B). The lactam, hydrocarbon and xylene-thioether compounds (3–5) activated the receptor with slightly lower potency (EC50) than the unstapled ghrelin analogue.
| Compound | β-arr 1Emax (eYFP/RLuc) | β-arr 1 EC50 (nM) | β-arr 2Emax (eYFP/RLuc) | β-arr 2 EC50 (nM) |
|---|---|---|---|---|
| [Dpr3]ghrelin(1–20) | 0.331 ± 0.027 | 33.3 ± 13.6 | 0.550 ± 0.028 | 54.0 ± 5.12 |
| 1 – triazole staple | 0.259 ± 0.016* | 2.43 ± 0.29 | 0.394 ± 0.022*** | 32.0 ± 3.17 |
| 2 – lactam staple | 0.220 ± 0.016** | 0.57 ± 0.08 | 0.388 ± 0.020**** | 12.6 ± 3.55 |
| 3 – hydrocarbon staple | 0.383 ± 0.030 | 77.2 ± 15.9 | 0.547 ± 0.044 | 114 ± 42.5 |
| 4 – Glaser staple | 0.435 ± 0.028* | 40.5 ± 4.78 | 0.544 ± 0.042 | 62.8 ± 13.8 |
| 5 – xylene-thioether staple | 0.455 ± 0.029** | 91.7 ± 10.7 | 0.609 ± 0.028 | 71.6 ± 7.02 |
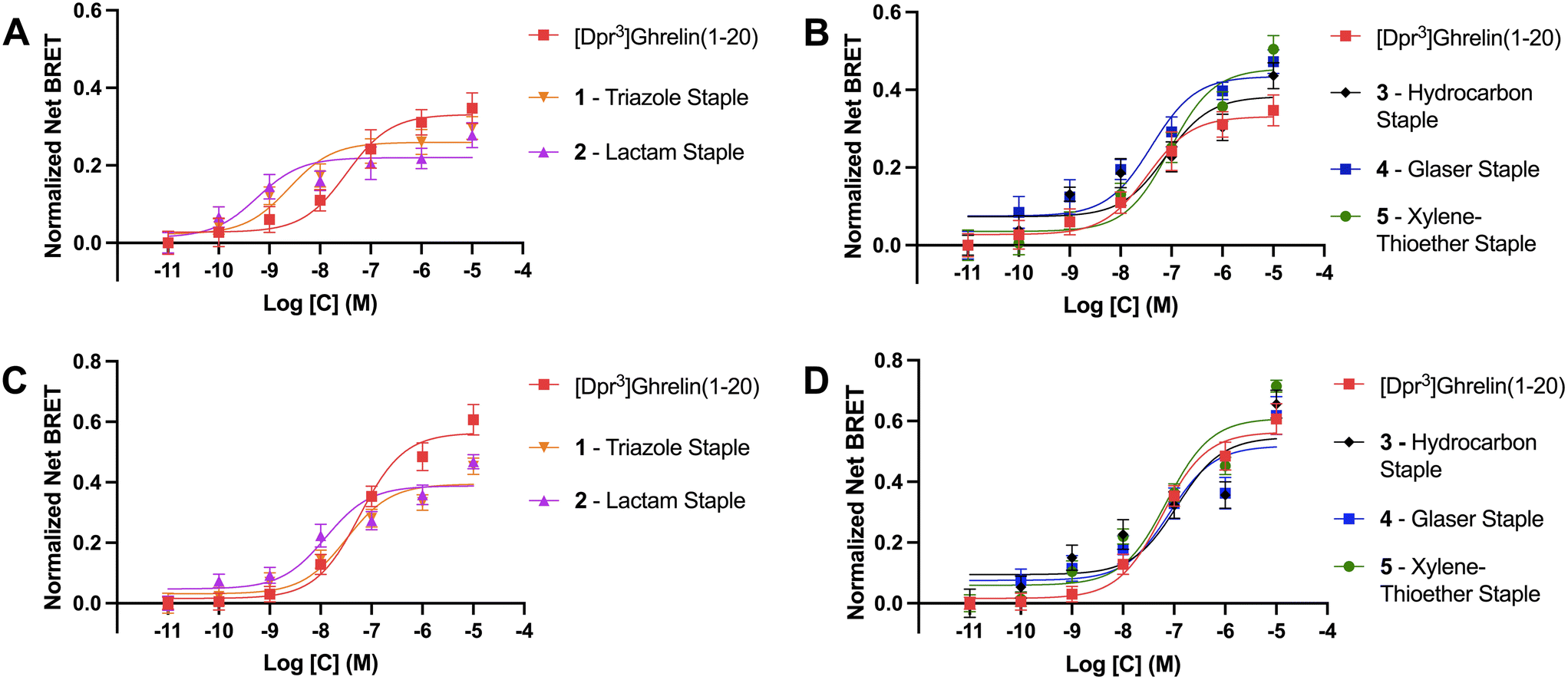 | ||
| Fig. 4 β-Arrestin recruitment BRET assay curves for the outlined compounds measured as a net BRET ratio (eYFP/RLuc). (A) and (B) are for β-arrestin 1, (C) and (D) are for β-arrestin 2. Data are mean ± SEM. | ||
Differences between the bioactivity of the stapled ghrelin analogues could be explained by variations in the flexibility of the staple units. The stapled compounds with the more flexible staples (1 and 2) appeared to activate the ghrelin receptor as potent partial agonists for both β-arrestin 1 & 2 recruitment. Partial agonists are ligands that are unable to induce maximal activation of a receptor population, suggesting that the more flexible staples allow for conformations of [Dpr3]ghrelin(1–20) that only stabilize the active conformation of the receptor partially but do so with great affinity. This is consistent with the data from the radioligand binding assays performed, where compounds 1 and 2 bound to the ghrelin receptor with higher affinity than unstapled [Dpr3]ghrelin(1–20). Conversely, the more rigid peptide staples (4 and 5) acted as super-agonists of the GHSR, with respect to β-arrestin 1 recruitment. Super-agonists are ligands that display higher efficacy than the receptor's endogenous agonist, and as such display an activation that surpasses the maximal activation of a receptor population induced by its endogenous ligand. This suggest that increased rigidity of the peptide staple unit allows for interactions of [Dpr3]ghrelin(1–20) that effectively stabilize the active conformation of the ghrelin receptor. Though an outlier with this trend does exist, where the hydrocarbon stapled ghrelin analogue (3) which has a flexible stapling unit like compounds 1 and 2, exhibited full agonist activity at a slightly reduced potency when compared to [Dpr3]ghrelin(1–20). While it is not known why this stapled compound demonstrates this unique bioactivity, it is postulated that its disubstituted amino acid residues may play a role in the ligand conformations that lead to the stabilization of the active conformation of the ghrelin receptor.
Serum stability assays
Like many natural peptides, ghrelin suffers from having poor in vivo stability due to the efficiency of proteases at hydrolysing amide bonds and this limits the usefulness of ghrelin for clinical translation.39 To test our hypothesis that increasing the α-helical character of ghrelin analogues would increase their metabolic stability from protease degradation, serum stability assays were conducted. For these assays, the most helical compound (3) and unstapled [Dpr3]ghrelin(1–20) were evaluated to see if there would be a significant difference in metabolic stability. Peptides were incubated in a solution of 25% human serum in PBS and aliquots would be periodically removed for quantification by HPLC/MS.As expected, compound 3 was stable from proteolysis by serum proteins for over 24 hours. This is in stark contrast with the unstapled [Dpr3]ghrelin(1–20) compound which had a calculated t1/2 < 5 hours (Fig. 5A and B). Therefore, it can be stated that inducing significant helicity into the ghrelin structure using chemical staples does yield increased metabolic stability from human serum proteins. As mentioned previously, studies have demonstrated that the active sites of human proteases do not permit an α-helical conformation to bind, which is consistent with the results seen from these assays.51 The proteolytic resistance exhibited by the hydrocarbon staple has also been well documented in the literature. In the work by Bird and coworkers, they demonstrated how the degree of α-helix stabilization and the number of inserted staples correlated with increased proteolytic resistance.38
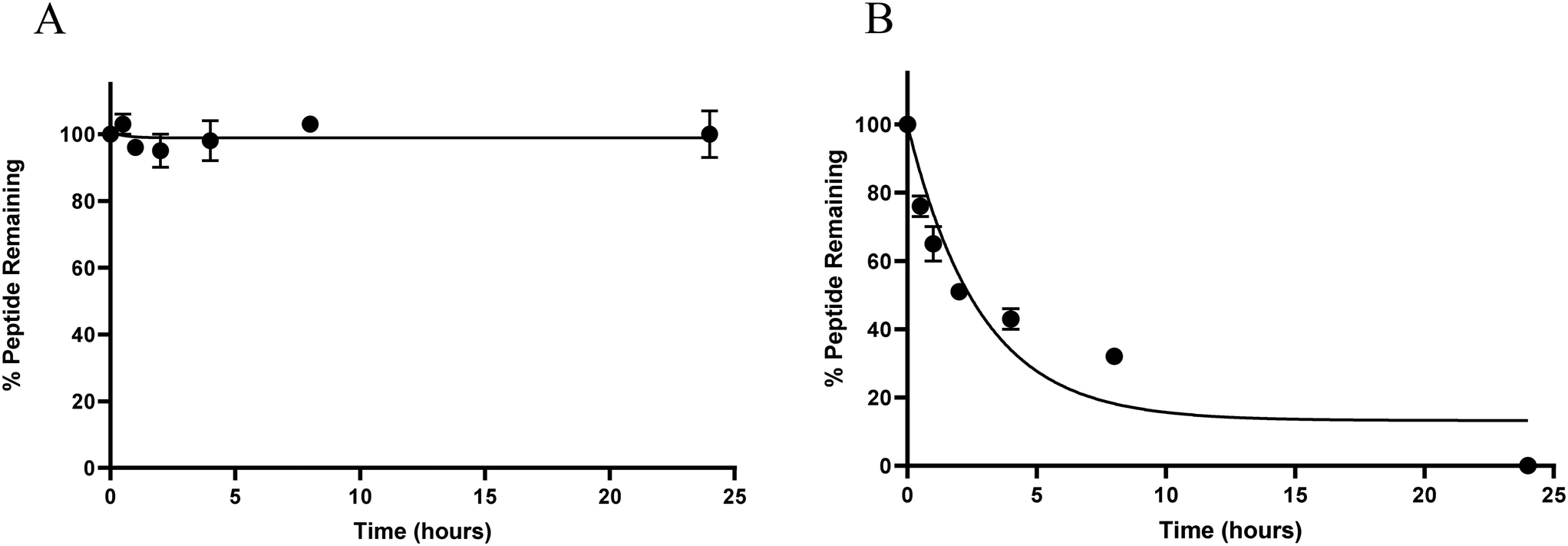 | ||
| Fig. 5 Human serum stability assay results for (A) 3 and (B) [Dpr3]ghrelin(1–20). | ||
Conclusion
The five ghrelin(1–20) analogues showcased in this work all contained distinct helix-inducing staples, providing cyclic peptides that have varying flexibility, lipophilicity, and potential for functionality. The stapled peptides were evaluated for their affinity towards and their ability to activate the ghrelin receptor. The results from the bioassays suggested that the flexibility of the stapling unit influenced both affinity and activity towards the ghrelin receptor. The more flexible peptide staples (1–3) were determined to have the greatest increase in binding affinity towards the ghrelin receptor in comparison to unstapled [Dpr3]ghrelin(1–20), whereas the more rigid staples (4 and 5) saw a decrease. Interestingly, some of the flexible linkers (1 and 2) activated the ghrelin receptor as potent partial agonists when compared to their unstapled counterpart. This contrasts with the more rigid linkers (4 and 5) which acted as superagonists for the ghrelin receptor. An outlier in the activity assays was discovered where compound 3, which has a flexible linker unit, also acted as a superagonist for the ghrelin receptor. The stapled peptides were also evaluated for their helical content and for their stability in human serum. It was discovered that the introduction of all peptide staples into the [Dpr3]ghrelin(1–20) structure resulted in an increase in α-helicity compared to their linear counterpart, albeit to a different extent. A trend was seen where the more flexible linker units (1–3) saw the greatest increase in percent helicity. Moreover, compound 5 was found to yield the greatest increase in helicity out of all the stapling techniques investigated with a 39% helical character. Compound 3 was then further evaluated in serum stability assays and was determined to be metabolically stable for over 24 hours. The work presented demonstrates that the incorporation of a peptide staple into ghrelin(1–20) analogues could be used as a template for the design of imaging agents targeting diseases such as ovarian and prostate cancer. The retained agonist properties of the stapled peptides suggests promise for their use as therapeutic agents for diseases such as cancer cachexia.Experimental
Peptide synthesis and characterization
All protected amino acids, coupling reagents, and resins were purchased from Novabiochem, Peptides International, and Chem-Impex. All reagents were obtained from Sigma-Aldrich. Peptides were synthesized using standard fluorenylmethyloxycarbonyl (Fmoc) based solid-phase peptide synthesis (SPPS) conditions on a Biotage (Uppsala, Sweden) SyroWave automated peptide synthesizer. Syntheses were done on a 0.1 mmol scale using Fmoc-protected Rink amide MBHA resin, which was allowed to swell before each synthesis with 2 mL of dichloromethane for 10 minutes. Fmoc deprotection was completed by adding 20% piperidine in dimethylformamide (DMF) for 12 minutes with 30 second vortexing, washing 3 times with DMF, and repeating the 20% piperidine in DMF for another 12 minutes with 30 second vortexing. Coupling of the desired amino acid was accomplished by adding the Fmoc-protected amino acid (4 eq.) in DMF, HCTU (4 eq.) in DMF and DIPEA in N-methyl pyrrolidinone for 1 hour in vortex. This amino acid coupling and deprotection cycle was continued until the desired sequence of the peptide was completed. In the case of (5), the coupling reaction for the disubstituted pentenyl alanine residues was performed twice with the coupling agent HATU due to incomplete coupling seen with standard coupling conditions (see below). After each synthesis, and in between modifications of the peptides, small samples of the resin-bound peptides were obtained and “micro-cleaved” by adding a 300 μL cleavage cocktail consisting of 95% trifluoroacetic acid, 2.5% triisopropylsilane, and 2.5% water for 1 hour in vortex. The resulting liquid was then precipitated with 1 mL of cold tert-butyl methyl ether (TBME), centrifuged for 10 minutes at 3000 rpm, and the mother liquor decanted. The resulting product was characterized using analytical reverse-phase high-performance liquid chromatography tandem mass spectrometry (LCMS) over gradients of acetonitrile (0.1% TFA) and water (0.1% TFA). The same procedure was used when fully cleaving the peptides from the resin with 5 mL cleavage cocktails and 20 mL of TBME for precipitating the peptides. Analytical and preparative reverse-phase LC-MS was performed on a system consisting of a Waters 600 controller, Waters prep degasser, and Waters MassLynx software. The UV absorbance was detected using a Waters 2998 Photodiode array detector. A preparative (Agilent Zorbax PrepHT SB-C18 column 21.2 × 150 mm, 5 μm) or analytical column (Agilent Zorbax SB-C18 column 4.6 × 150 mm, 5 μm) was used. The solvent system ran gradients of solvent A, 0.1% trifluoroacetic acid (TFA) in acetonitrile (ACN), and solvent B, 0.1% TFA in MilliQ (18.2 mΩ cm conductivity) water, over 10 minutes with a 5-minute wash. A flow rate of 20 mL min−1 or 1.50 mL min−1 was used for preparative and analytical runs, respectively. HRMS was run on an Agilent 6550 QToF mass spectrometer in ESI+ mode.:H2O solution containing 100 mM of NH4CO3 buffer. To this mixture 1.5 eq. of TCEP was added and allowed to stir for 1 hour. Then, 3 eq. of the α,α′-dibromo-p-xylene linker and allowed to stir for 2 hours. The reaction was the quenched using 5% HCl and purified using preparative LCMS to effectively yield compound 5. MS (ESI+) [M + 3H]3+m/z calculated: 854.4342, [M + 3H]3+m/z observed: 854.4370.
Circular dichroism and percent helicity
Circular dichroism (CD) spectra was obtained using a Jasco J-810 spectropolarimeter and recorded in the range of 185–260 nm. Peptide solutions were prepared with sodium phosphate buffer at a pH of 7.2 and a concentration of 0.1 mM. Measurements were performed using a cuvette with a path length of 1 mm and a scanning speed of 10–50 nm min−1 for each peptide. Five individual scans were averaged by the instrument to obtain the CD spectra. All spectra were collected at a temperature of 20 °C. The percent helicity for each peptide was calculate using eqn (1).40 In the equation, [θ]222 corresponds to the mean residue ellipticity of the peptide at 222 nm and [θ]0 corresponds to the mean residue ellipticity of a random coil conformation. [θ]max corresponds to the maximum theoretical mean residue ellipticity for a helix of n residues and is described in eqn (2), where x is an empirical constant equal to 3. [θ]∞ corresponds to the infinite molar ellipticity for an α-helix at a specific wavelength.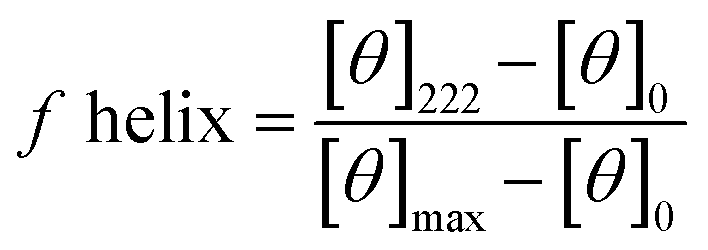 | (1) |
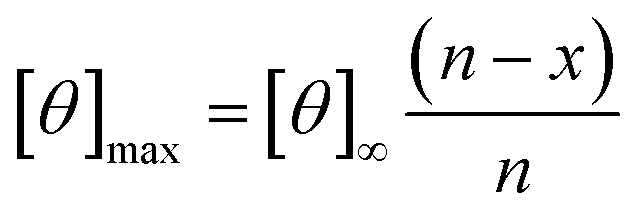 | (2) |
Radioligand-displacement binding assay
GHSR transfection of HEK293 cells
HEK293 cells (ATCC, Manassas VA) were maintained in T175 flask containing RPMI medium supplemented with 10% fetal bovine serum and were passaged 2–3 times per week. On the day of transfection, a fully confluent T175 flask of cells was seeded into four 10 cm3 plates. A solution was made for each plate containing 480 μL of opti-MEM and 15 μL of X-tremeGENE 9 DNA Transfection Reagent in a glass vial and set aside for 15 min to allow for complex to form. 5 μg of DNA of GHSR-1a-eYFP construct was added to each vial and allowed to sit for 20 minutes. The resulting 500 μL DNA complex solution was added dropwise to each 10 cm3 plate and allowed to incubate for 24 hours. The following day the cells were harvested and allowed to freeze to a temperature of −150 °C and stored until needed.Binding assay
Binding assays were performed using human [His[125I]]-ghrelin radioligand (PerkinElmer, Waltham MA) and GHSR-1a transfected HEK293 cells. A suspension of 50000 cells assay tube were incubated with the outlined ghrelin(1–20) analogues at concentrations of 10−5 to 10−11 M and 15 pM of human [His[125I]]-ghrelin radioligand in binding buffer. The binding buffer consisted of 25 mM HEPES (5 mM magnesium chloride, 1 mM calcium chloride, 2.5 mM EDTA, and 0.4% BSA, pH 7.4). Cell suspension was incubated for 20 min at 37.5 °C with stirring at 550 rpm. Immediately following incubation, the resulting cell suspension was centrifuged to produce a cell pellet which was then washed with a tris-HCl solution. The assay tubes were then place in a γ counter where the unbound radioligand was measured. The materials used were purchased from PerkinElmer and Sigma-Aldrich.
Beta-arrestin BRET assays
GHSR transfection of HEK293 cells
HEK293 cells (ATCC, Manassas VA) were maintained in T175 flask containing RPMI medium supplemented with 10% fetal bovine serum and were passaged 2–3 times per week. On the day of the transfection, two vials with 300 μL of opti-MEM and 9 μL of X-tremeGENE 9 DNA Transfection Reagent were set aside for 20 min to allow for a complex to form. 7.9 μg of DNA of GHSR-1a-eYFP construct and 300 μg of beta-arrestin 1 or 2 tagged with RLuc were added to each vial and allowed to sit for 40 minutes. 2 mL were then taken from a fully confluent HEK293 T175 flask, diluted to 12 mL and split evenly in a 6 well plate. The resulting DNA complex solution was then added dropwise to each well and allowed to incubate for 24 hours. The following day the cells were harvested and transferred onto a 96 well plate with each well containing 100 μL of the transfected HEK293 cell solution. 24 hours after transferring to the 96 well plate, the cells were ready for the BRET assay. The materials used were purchased from PerkinElmer and Sigma-Aldrich.BRET assay
Beta-arrestin BRET assays were performed as follows. A 96 well plate with the transfected HEK293 cells (see above) was plated for 24 hours. A drug plate containing a serial dilution of the outlined stapled analogues with a 10−4 to 10−10 concentration range was prepared using Hank's balanced salt solution (HBSS). The cell media from the 96 well plate was then discarded and to each well 90 μL of HBSS and 10 μL of drug was added for a final concentration range of 10−5 to 10−11. The plate was then allowed to sit at room temperature for 20 minutes. At the 15-minute mark, 10 μL of a 50 μM H-coelenterazine solution was added to each well for a final concentration of 5 μM. The plate is then allowed to sit for the remaining 5 minutes and then placed in a Mithras LB940 plate reader to measure the corresponding BRET signal. The resulting data was analyzed using Prism 8 by GraphPad, responses are expressed as net emission of eYFP/RLuc (calculated by subtracting baseline eYFP/RLuc ratio from treated eYFP/RLuc ratio) and normalized to a positive control ([Dpr3]ghrelin(1–20) at 10−5 M). The concentration-effect curves were fitted and analyzed using a non-linear regression dose–response simulation three-parameter model to calculate logEC50.| net BRET = BRET stimulus − BRET baseline | (3) |
 | (4) |
Serum stability assays
The stability of the stapled ghrelin analogues in human serum was assessed by incubating each peptide (1 mM) at 37 °C in a 25% serum PBS solution at pH 7.4. Sample aliquots were collected after 10 minutes of incubation in serum to allow for equilibrium between the peptide and serum. 15 μL aliquots of the peptide solution were removed in triplicate at 0, 30 min, 1 h, 2 h, 4 h, 8 h, and 24 h. 60 μL of 4% NH4OH(aq) was used to quench the reaction and residual salts and proteins were removed using a Waters Oasis HLB microextraction plate. The cartridge was activated using 200 μL of MeOH followed by 200 μL of water. The sample solution was loaded onto the cartridge and the column was washed with 200 μL of 5% MeOH(aq). The peptide was then eluted using 30 μL (×2) of 2% formic acid in MeOH. The resulting 60 μL aliquot of the eluted peptide was then analyzed by LCMS. The selected ion chromatogram corresponding to the m/z value of the peptide was obtained and the resulting MS peak was integrated to quantify the amount of intact peptide remaining at each time-point.Conflicts of interest
There is no conflict of interest to declare.Acknowledgements
We acknowledge financial support from the Natural Sciences and Engineering Research Council of Canada (NSERC) (RGPIN-2018-05477) and the Canadian Institutes of Health Research (CIHR) (469411). We also acknowledge the educational support of the Molecular Imaging Collaborative Program at Western University.References
- M. Kojima, H. Hosoda, Y. Date, M. Nakazato, H. Matsuo and K. Kangawa, Nature, 1999, 402, 656–660 CrossRef CAS PubMed
.
- J. Yang, M. S. Brown, G. Liang, N. V. Grishin and J. L. Goldstein, Cell, 2008, 132, 387–396 CrossRef CAS PubMed
- J. L. Hougland, Biochem. Soc. Trans., 2019, 47, 169–178 CrossRef CAS PubMed
- M. A. Bednarek, S. D. Feighner, S.-S. Pong, K. K. McKee, D. L. Hreniuk, M. V. Silva, V. A. Warren, A. D. Howard, A. Lex, H. Y. Van der Ploeg and J. V. Heck, J. Med. Chem., 2000, 43, 4370–4376 CrossRef CAS PubMed
- L. Mihalache, A. Gherasim, O. Niţă, M. C. Ungureanu, S. S. Pădureanu, R. S. Gavril and L. I. Arhire, Hormones, 2016, 15, 186–196 CrossRef
- A. Abizaid and J. L. Hougland, Trends Endocrinol. Metab., 2020, 2, 107–117 CrossRef
- J. M. Garcia, M. Garcia-Touza, R. A. Hijazi, G. Taffet, D. Epner, D. Mann, R. G. Smith, G. R. Cunningham and M. Marcelli, J. Clin. Endocrinol. Metab., 2005, 90, 2920–2926 CrossRef CAS
- F. Strasser, T. A. Lutz, M. T. Maeder, B. Thuerlimann, D. Bueche, M. Tschöp, K. Kaufmann, B. Holst, M. Brändle and R. Von Moos,
et al.
, Br. J. Cancer, 2008, 98, 300–308 CrossRef CAS
- S. von Haehling, M. Lainscak, J. Springer and S. D. Anker, Pharmacol. Ther., 2009, 121, 227–252 CrossRef CAS PubMed
- S. Yanagi, T. Sato, K. Kangawa and M. Nakazato, Cell Metab., 2018, 27, 786–804 CrossRef CAS
- J. F. Davis, D. L. Choi, D. J. Clegg and S. C. Benoit, Physiol. Behav., 2011, 103, 39–43 CrossRef CAS PubMed
- J. M. Garcia, M. Garcia-Touza, R. A. Hijazi, G. Taffet, D. Epner, D. Mann, R. G. Smith, G. R. Cunningham and M. Marcelli, J. Clin. Endocrinol. Metab., 2005, 90, 2920–2926 CrossRef CAS
- M. D. DeBoer, X. Zhu, P. R. Levasseur, A. Inui, Z. Hu, G. Han, W. E. Mitch, J. E. Taylor, H. A. Halem and J. Z. Dong,
et al.
, Endocrinology, 2008, 149, 827–835 CrossRef CAS PubMed
- A. Aleksova, A. P. Beltrami, E. Bevilacqua, L. Padoan, D. Santon, F. Biondi, G. Barbati, E. Stenner, G. G. Cappellari and R. Barazzoni,
et al.
, J. Clin. Med., 2019, 8, 1152 CrossRef CAS
- Y. Xu, X. Pang, M. Dong, F. Wen and Y. Zhang, Oncol. Rep., 2013, 30, 2063–2070 CrossRef CAS PubMed
- C. Lu, M. S. McFarland, R.-L. Nesbitt, A. K. Williams, S. Chan, J. Gomez-Lemus, A. M. Autran-Gomez, A. Al-Zahrani, J. L. Chin and J. I. Izawa,
et al.
, Prostate, 2012, 72, 825–833 CrossRef CAS PubMed
- R. Sullivan, V. K. Randhawa, A. Stokes, D. Wu, T. Lalonde, B. Kiaii, L. Luyt, G. Wisenberg and S. Dhanvantari, J. Endocr. Soc., 2019, 3, 748–762 CrossRef CAS PubMed
- M. D. Childs and L. G. Luyt, Mol. Imaging, 2020, 19, 1536012120952623 CrossRef PubMed
- C. L. Charron, J. Hou, M. S. McFarland, S. Dhanvantari, M. S. Kovacs and L. G. Luyt, J. Med. Chem., 2017, 60, 7256–7266 CrossRef CAS PubMed
- C. L. Charron, M. S. McFarland, S. Dhanvantari and L. G. Luyt, MedChemComm, 2018, 9, 1761–1767 RSC
- C. Chollet, R. Bergmann, J. Pietzsch and A. G. Beck-Sickinger, Bioconjugate Chem., 2012, 23, 771–784 CrossRef CAS
- M. D. Childs, A. Chandrabalan, D. Hodgson, R. Ramachandran and L. G. Luyt, ACS Pharmacol. Transl. Sci., 2023, 6, 1075–1086 CrossRef CAS PubMed
- M. Martín-Pastor, A. De Capua, C. J. P. Álvarez, M. D. Díaz-Hernández, J. Jiménez-Barbero, F. F. Casanueva and Y. Pazos, Bioorg. Med. Chem., 2010, 18, 1583–1590 CrossRef
- A. J. Beevers and A. Kukol, J. Biomol. Struct. Dyn., 2006, 23, 357–363 CrossRef CAS PubMed
- J. Qin, Y. Cai, Z. Xu, Q. Ming, S. Y. Ji, C. Wu, H. Zhang, C. Mao, D. D. Shen and K. Hirata,
et al.
, Nat. Commun., 2022, 13, 1–11 Search PubMed
- G. Ferré, M. Louet, O. Saurel and J. Banères,
et al.
, Proc. Natl. Acad. Sci. U. S. A., 2019, 116(35), 17525–17530 CrossRef
- J. D. A. Tyndall, T. Nall and D. P. Fairlie, Chem. Rev., 2005, 105, 973–999 CrossRef CAS PubMed
- M. Siedlecka, G. Goch, A. Ejchart, H. Sticht and A. Bierzyński, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 903–908 CrossRef CAS
- S. D. Bryant, R. Guerrini, S. Salvadori, C. Bianchi, R. Tomatis, M. Attila and L. H. Lazarus, J. Med. Chem., 1997, 40, 2579–2587 CrossRef CAS PubMed
- D. S. Kemp, T. J. Allen and S. L. Oslick, J. Am. Chem. Soc., 1995, 117, 6641–6657 CrossRef CAS
- C. E. Schafmeister, A. Julia Po and G. L. Verdine, J. Am. Chem. Soc., 2000, 122, 5891–5892 CrossRef CAS
- N. E. Shepherd, H. N. Hoang, V. S. Desai, E. Letouze, P. R. Young and D. P. Fairlie, J. Am. Chem. Soc., 2006, 128, 13284–13289 CrossRef CAS
- S. Ingale and P. E. Dawson, Org. Lett., 2011, 13, 2822–2825 CrossRef CAS
- N. S. A. Crone, A. Kros and A. L. Boyle, Bioconjugate Chem., 2020, 31, 834–843 CrossRef CAS PubMed
- P. A. Cistrone, A. P. Silvestri, J. C. J. Hintzen and P. E. Dawson, ChemBioChem, 2018, 19, 1031–1035 CrossRef CAS
- Y. H. Lau, P. de Andrade, Y. Wu and D. R. Spring, Chem. Soc. Rev., 2014, 44, 91–102 RSC
- T. Lalonde, T. G. Shepherd, S. Dhanvantari and L. G. Luyt, Pept. Sci., 2019, 111, e24055 CrossRef
- G. H. Bird, N. Madani, A. F. Perry, A. M. Princiotto, J. G. Supko, X. He, E. Gavathiotis, J. G. Sodroski and L. D. Walensky, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14093–14098 CrossRef CAS
- J. Tong, N. Dave, G. M. Mugundu, H. W. Davis, B. D. Gaylinn, M. O. Thorner, M. H. Tschöp, D. D'Alessio and P. B. Desai, Eur. J. Endocrinol., 2013, 168, 821–828 CAS
- M. Bouzo-Lorenzo, I. Santo-Zas, M. Lodeiro, R. Nogueiras, F. F. Casanueva, M. Castro, Y. Pazos, A. B. Tobin, A. J. Butcher and J. P. Camiña, Sci. Rep., 2016, 6, 22495 CrossRef CAS PubMed
- A. D. de Araujo, H. N. Hoang, W. M. Kok, F. Diness, P. Gupta, T. A. Hill, R. W. Driver, D. A. Price, S. Liras and D. P. Fairlie, Angew. Chem., Int. Ed., 2014, 53, 6965–6969 CrossRef CAS
- M. Mergler, F. Dick, B. Sax, P. Weiler and T. Vorherr, J. Pept. Sci., 2003, 9, 36–46 CrossRef CAS PubMed
- L. D. Walensky and G. H. Bird, J. Med. Chem., 2014, 57, 6275–6288 CrossRef CAS
- P. Barthe, C. Roumestand, S. Rochette and C. Vita, Protein Sci., 2000, 9, 942–955 CrossRef CAS PubMed
- C. Toniolo and E. Benedetti, Macromolecules, 1991, 24, 4004–4009 CrossRef CAS
- S. Verlinden, N. Geudens, K. Van Holsbeeck, M. Mannes, J. C. Martins, G. Verniest and S. Ballet, J. Pept. Sci., 2019, 25, e3194 CrossRef
- H. Jo, N. Meinhardt, Y. Wu, S. Kulkarni, X. Hu, K. E. Low, P. L. Davies, W. F. DeGrado and D. C. Greenbaum, J. Am. Chem. Soc., 2012, 134, 17704–17713 CrossRef CAS
- G. H. Bird, W. C. Crannell and L. D. Walensky, Curr. Protoc. Chem. Biol., 2011, 3, 99–117 CrossRef PubMed
- A. M. Wren, L. J. Seal, M. A. Cohen, A. E. Brynes, G. S. Frost, K. G. Murphy, W. S. Dhillo, M. A. Ghatei and S. R. Bloom, J. Clin. Endocrinol. Metab., 2001, 86, 5992–5992 CrossRef CAS
- J. P. Camiña, J. Neuroendocrinol., 2006, 18, 65–76 CrossRef
- L. K. Henchey, A. L. Jochim and P. S. Arora, Curr. Opin. Chem. Biol., 2008, 12, 692–697 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3md00441d |
| This journal is © The Royal Society of Chemistry 2024 |