
Sirtuin 1-activating derivatives belonging to the anilinopyridine class displaying in vivo cardioprotective activities†
Giulia
Bononi‡
a,
Valentina
Citi‡
a,
Alma
Martelli
 ab,
Giulio
Poli
a,
Tiziano
Tuccinardi
ab,
Carlotta
Granchi
*ab,
Lara
Testai
*ab,
Vincenzo
Calderone
ab and
Filippo
Minutolo
ab
ab,
Giulio
Poli
a,
Tiziano
Tuccinardi
ab,
Carlotta
Granchi
*ab,
Lara
Testai
*ab,
Vincenzo
Calderone
ab and
Filippo
Minutolo
ab
aDepartment of Pharmacy, University of Pisa, Via Bonanno 6, 56126 Pisa, Italy. E-mail: giulia.bononi@farm.unipi.it; valentina.citi@unipi.it; alma.martelli@unipi.it; giulio.poli@unipi.it; tiziano.tuccinardi@unipi.it; carlotta.granchi@unipi.it; lara.testai@unipi.it; vincenzo.calderone@unipi.it; filippo.minutolo@unipi.it
bCenter for Instrument Sharing of the University of Pisa (CISUP), Lungarno Pacinotti 43, 56126 Pisa, Italy
First published on 5th December 2023
Abstract
Sirtuin 1 (SIRT1) is an enzyme that relies on NAD+ cofactor and functions as a deacetylase. It has been associated with various biological and pathological processes, including cancer, diabetes, and cardiovascular diseases. Recent studies have shown that compounds that activate SIRT1 exhibit protective effects on the heart. Consequently, targeting SIRT1 has emerged as a viable approach to treat cardiovascular diseases, leading to the identification of several SIRT1 activators derived from natural or synthetic sources. In this study, we developed anilinopyridine-based SIRT1 activators that displayed significantly greater potency in activating SIRT1 compared to the reference compound resveratrol, as demonstrated in enzymatic assays. In particular, compounds 8 and 10, representative 6-aryl-2-anilinopyridine derivatives from this series, were further investigated pharmacologically and found to reduce myocardial damage caused by occlusion and subsequent reperfusion in vivo, confirming their cardioprotective properties. Notably, the cardioprotective effects of 8 and 10 were significantly superior to that of resveratrol. Significantly, compound 10 emerged as the most potent among the tested compounds, demonstrating the ability to substantially decrease the size of the ischemic area at a dosage one hundred times lower (0.1 mg kg−1) than that of resveratrol/compound 1. These promising findings open avenues for expanding and optimizing this chemical class of potent SIRT1 activators as potential agents for cardioprotection.
1. Introduction
Sirtuins are a class of NAD+-dependent proteins present in both Eukaryotes and Prokaryotes, implicated in the regulation of metabolic/cellular homeostasis.1 They have received great attention since their discovery, when they were recognized to play an important role in the yeast Saccharomyces cerevisiae.2 The most studied sirtuin is SIRT1, a NAD+-dependent nicotinamide-adenine dinucleotide histone deacetylase, which is involved in the regulation of numerous biological functions. Thanks to its antioxidant properties and its role in regulating homeostasis, it is widely investigated as a possible therapeutic target for reducing aging-related disorders. Indeed, low sirtuin activity is implicated in physiological aging processes leading to the onset of a wide variety of cardiovascular and metabolic diseases, including acute cardiac syndromes, atherosclerosis, cardiomyopathy, hypertrophy and heart failure, arrhythmias, hypertension, metabolic syndrome, obesity and fatty liver, diabetes and dyslipidemia.3Several studies during the last years have explored the role of sirtuins and NAD+ in ischemia–reperfusion (IR) injury.4 IR injury arises clinically during myocardial infarction and the period of ischemia can cause tissue injury and cell death as a result of reduced ATP levels. This damage is exacerbated by rapid reperfusion, which results in the induction of oxidative stress and apoptosis.5,6 IR injury increases with age and periods of ischemia or hypoxia in cardiomyocytes are associated with a reduction in endogenous nicotinamide phosphoribosyl transferase (NAMPT), as well as downregulation of SIRT1.
Therefore, increasing SIRT1 activity can be viewed as a potential strategy for reducing IR injury. Indeed, SIRT1 knockout mice exhibit enhanced cardiac injury after exposure to IR, while mice overexpressing SIRT1 are protected from IR damage.7 Overexpression of SIRT1 promotes upregulation of antioxidant pathways mediated by FOXO1 and manganese superoxide dismutase (MnSOD) and downregulation of pro-apoptotic pathways mediated by caspase 3 and Bax.8 A similar role of SIRT1 is observed in rats exposed to IR injury. Other in vitro studies have shown the same anti-apoptotic role of SIRT1 by mediating FOXO1 expression, in the context of hypoxia.9
The activity of SIRT1 can be increased with molecules known as sirtuin activating compounds (STACs). One of the first STACs, resveratrol 1 (Fig. 1), was discovered in a screen for molecules that increased the activity of human SIRT1 and extended the lifespan of yeast.10 Since then, there have been several generations of STACs, with increasing potency and specificity, including SRT1720 2 and SRT2104 3 (Fig. 1).11 STACs act as allosteric activators of SIRT1, binding to the STAC-binding domain in its N-terminus, thus increasing the binding affinity of a substrate for SIRT1. In addition, other natural compounds such as naringenin 4 and quercetin 5 (Fig. 1) have been reported to activate SIRT1.12 Starting from computational studies of the structure of resveratrol 1 and naringenin 4, synthetic molecules were designed and tested as SIRT1 activators, such as compounds 6 and 7 (Fig. 1), which showed very promising results in IR models.13–15
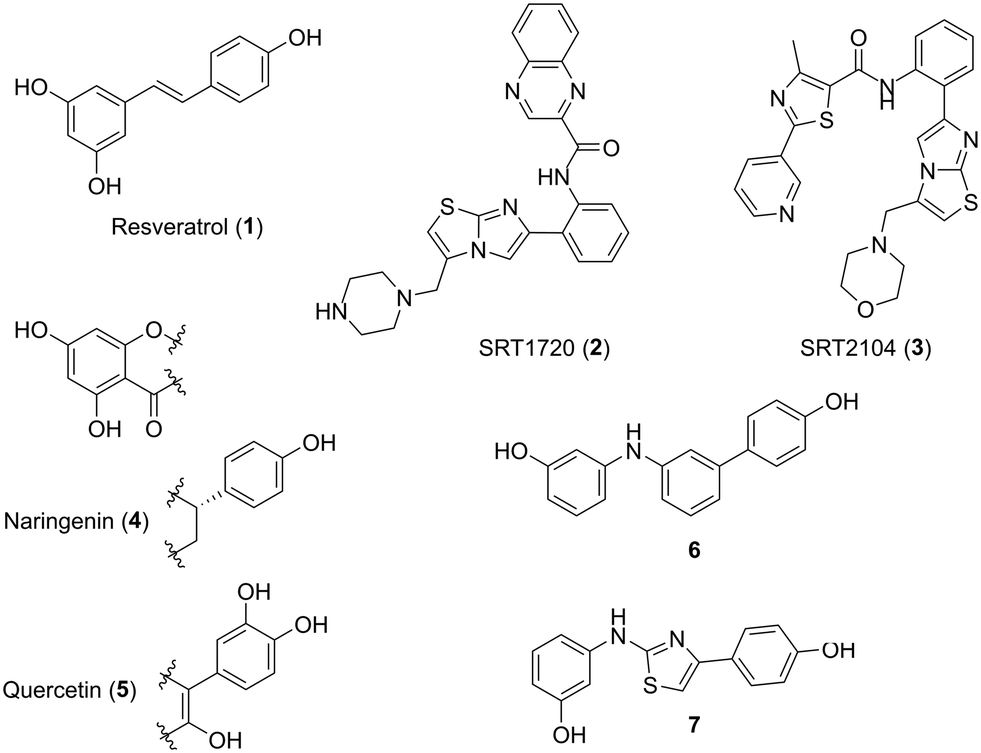 | ||
| Fig. 1 Main STACs reported in literature. | ||
Considering our previously published series of SIRT1 activators (compounds 6 and 7 are the most representative examples),14,15 we were intrigued by the possibility of substituting the central phenyl or thiazole portions with pyridine rings, varying the position of the nitrogen atom with respect to the double phenolic substitution, considering that pyridine moieties are often used in drug candidates thanks to their favourable features such as basicity, water solubility, and hydrogen bond-forming ability.16 In this context, we synthesized four new subclasses of compounds belonging to four different scaffolds (scaffolds 1–4, Fig. 2), maintaining the two peripheral phenolic rings A and B in which the hydroxyl groups are present in meta or para positions: N,6-di(hydroxyphenyl)pyridin-2-amine derivatives 8–11 (scaffold 1), N,2-di(hydroxyphenyl)pyridin-4-amine derivatives 12–15 (scaffold 2), N,5-di(hydroxyphenyl)pyridine-3-amine derivatives 16–19 (scaffold 3) and N,4-di(hydroxyphenyl)pyridin-2-amine derivatives 20–23 (scaffold 4).
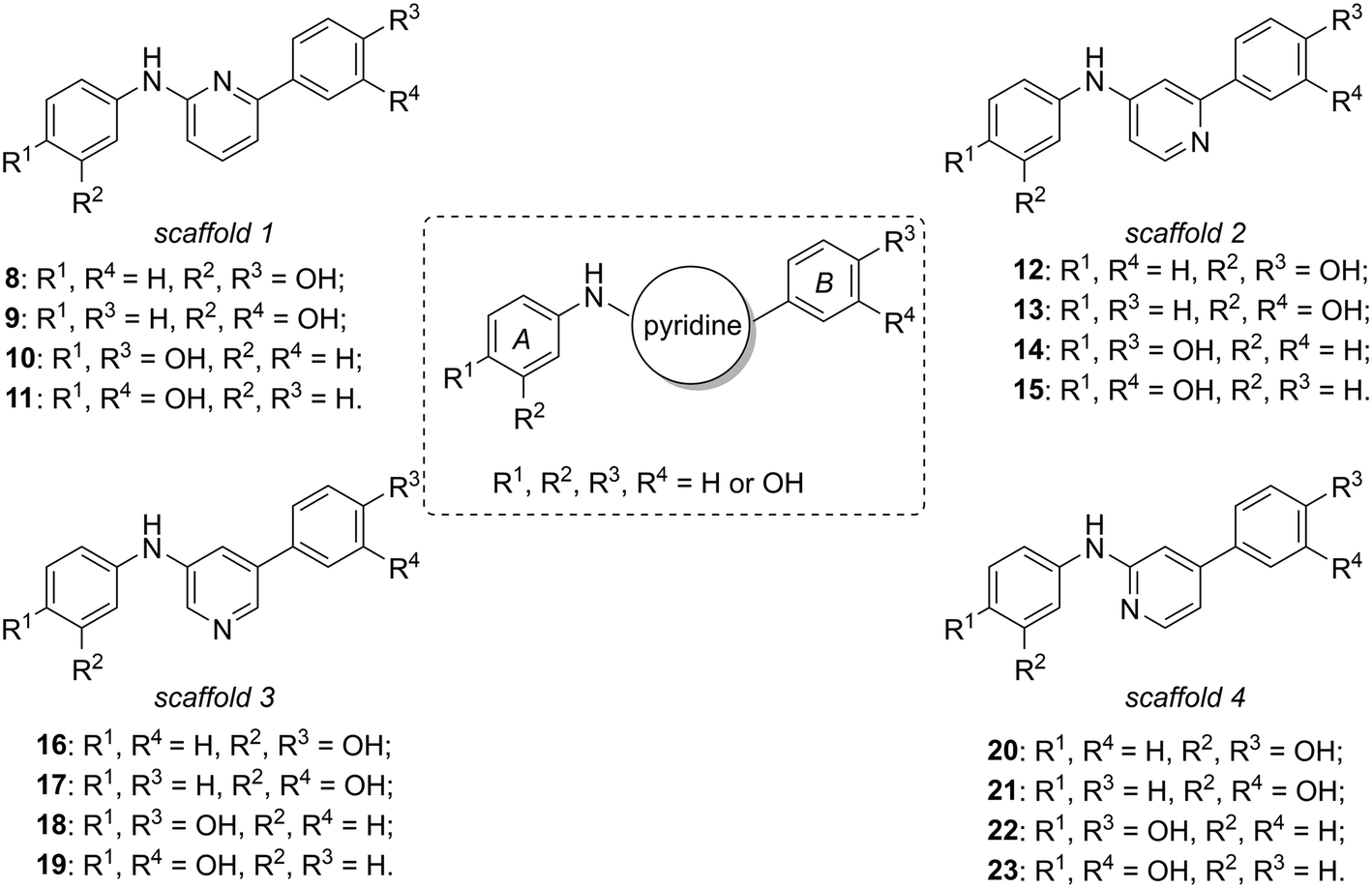 | ||
| Fig. 2 Anilinopyridine derivatives developed as STACs. The general scaffold is represented in the central dashed box and the four new groups of compounds belonging to different chemical classes are named as scaffold 1 (compounds 8–11), scaffold 2 (compounds 12–15), scaffold 3 (compounds 16–19) and scaffold 4 (compounds 20–23). | ||
The straightforward synthesis for the obtainment of compounds 8–11 and 16–23 is outlined in Scheme 1: commercially available 2-amino-6-bromopyridine 24, 3-amino-5-bromopyridine 25 or 2-amino-4-bromopyirdine 26 were reacted with 3- or 4-methoxybenzeneboronic acid in tetrakis(triphenylphosphine)palladium(0)-catalyzed Suzuki reactions17 to afford intermediates 27–32, then they were condensed with 3- or 4-bromoanisole by adopting previously reported conditions15,18–20 and, finally, dimethoxy-substituted derivatives 33–44 were deprotected by boron tribromide to the corresponding final phenolic compounds.
 | ||
| Scheme 1 Synthesis of derivatives 8–11 and 16–23. Reagents and conditions: (a) 3- or 4-methoxybenzeneboronic acid, Pd(OAc)2, PPh3, K2CO3, anhydrous toluene, 100 °C, 18 h [91–99%]; (b) 3- or 4-bromoanisole, Pd2dba3, XPhos, K3PO4, anhydrous toluene, 100 °C, 20–26 h [20–99%]; (c) 1 M BBr3 in CH2Cl2, anhydrous CH2Cl2, −15 to 0 °C, then RT, 2–3 h [20–99%]. | ||
Preparation of compounds 12–15 (scaffold 2, Fig. 2) started from 4-amino-2-bromopyridine 45 and followed the same synthetic scheme previously described when 45 was coupled with 4-methoxybenzeneboronic acid (intermediates 49, 50 and 52, Scheme 2). Differently, in the case of 3-methoxybenzeneboronic acid we preferred to perform a precautionary protection of the anilino group of compound 45 (to avoid the formation of side products we otherwise obtained in the Suzuki reaction of 45) and then react Boc-protected intermediate 46 in the Suzuki reaction to obtain compound 47. Trifluoroacetic acid-mediated deprotection of 47 led to the obtainment of aniline derivative 48, which was subjected to the Pd2dba3-catalyzed reaction to form the C–N bond (compounds 51 and 53). The last BBr3-promoted step (step e, Scheme 2) allowed the formation of the desired compounds 12–15.
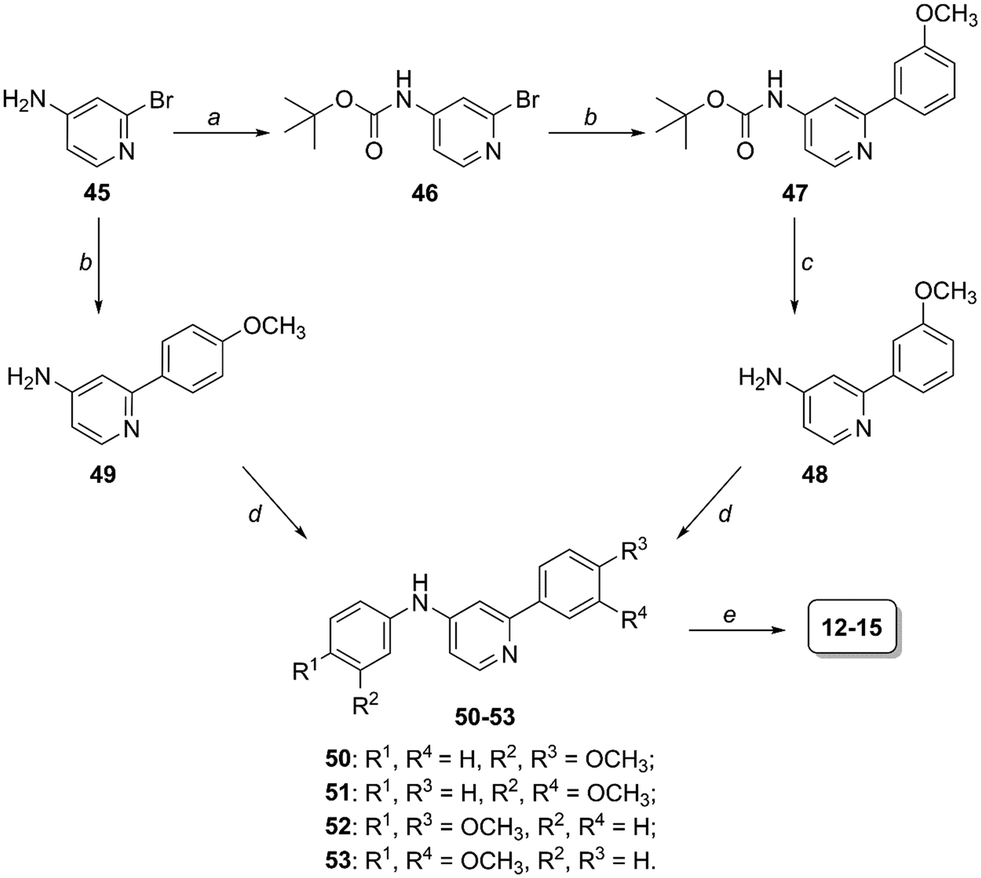 | ||
| Scheme 2 Synthesis of derivatives 12–15. Reagents and conditions: (a) (Boc)2O, Et3N, anhydrous THF, 0 °C to RT, overnight [72%]; (b) 3- or 4-methoxybenzeneboronic acid, Pd(OAc)2, PPh3, K2CO3, anhydrous toluene, 100 °C, 18 h [84–96%]; (c) TFA, anhydrous CH2Cl2, 0 °C to RT, 2 h [99%]; (d) 3- or 4-bromoanisole, Pd2dba3, XPhos, K3PO4, anhydrous toluene, 100 °C, 20–26 h [33–83%]; (e) 1 M BBr3 in CH2Cl2, anhydrous CH2Cl2, −15 to 0 °C, then RT, 2–3 h [45–78%]. | ||
2. Results
2.1 In vitro SIRT1 activation
The newly synthetized compounds were tested for their ability to activate SIRT1 enzyme. Compound 1 100 μM was used as reference compound and the activation obtained was set as 100%. SIRT1 activation obtained by the tested compounds was compared to that of compound 1 and reported as a percentage of activation.Compounds 9 (characterized by hydroxylic groups on both aromatic rings A and B in meta position) and 8 (characterized by hydroxylic groups on aromatic ring A in meta position and B in para position) led to SIRT1 activation comparable to compound 1. The other two derivatives were 11, carrying the phenolic OH of the A-ring in para, and that of the B-ring in meta, and 10, which has both the hydroxylic groups substituted in para position on ring A and ring B. 11 showed a significant enhancement in the activation of SIRT1, leading to an activation of the enzyme almost twice as high as compound 1-mediated activation. Finally, a marked improvement occurred using 10 which increased threefold compared with compound 1-promoted activation (Fig. 3, Table 1).
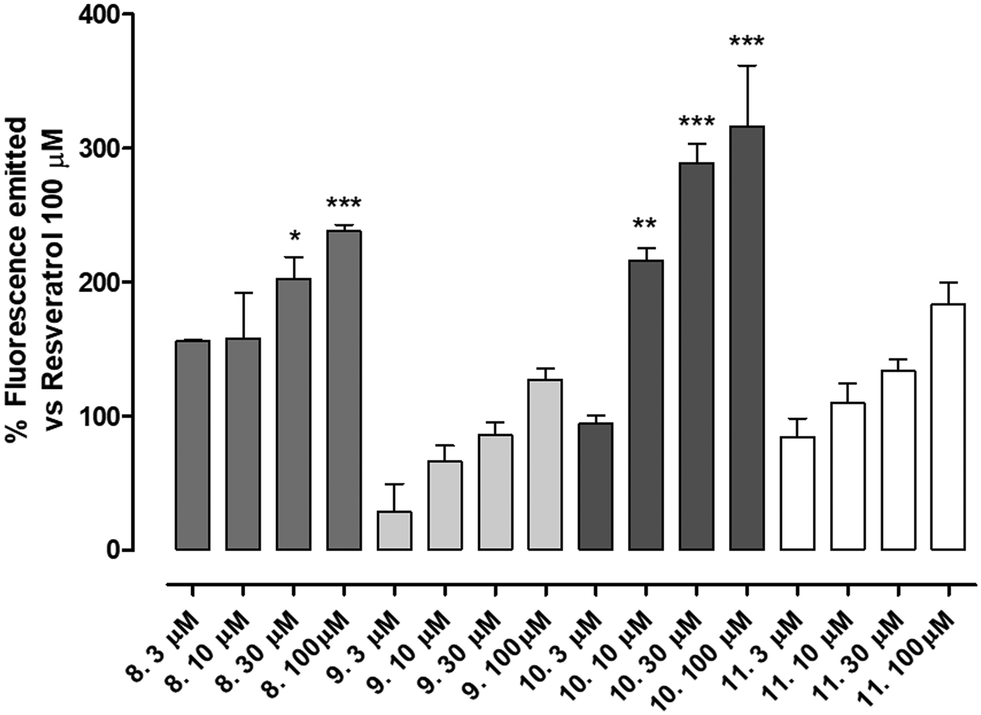 | ||
| Fig. 3 The figure shows the activation of SIRT1 expressed as percentage of fluorescence emitted in comparison to that of resveratrol 100 μM of increasing concentration of compounds 8, 9, 10 and 11. Data are expressed as mean ± SEM. Data are expressed as mean ± SEM. Statistical analysis (one way ANOVA followed by Bonferroni post test) was performed. * indicates significant difference vs. resveratrol 100 μM (*p < 0.05; **p < 0.01; ***p < 0.001). Number of replicates = 6. | ||
| Compound | Structure | Activity % |
|---|---|---|
| 8 |
|
130.9 ± 23.9 |
| 9 |
|
115.3 ± 10.7 |
| 10 |
|
316.3 ± 45.6 |
| 11 |
|
183.6 ± 15.9 |
| 12 |
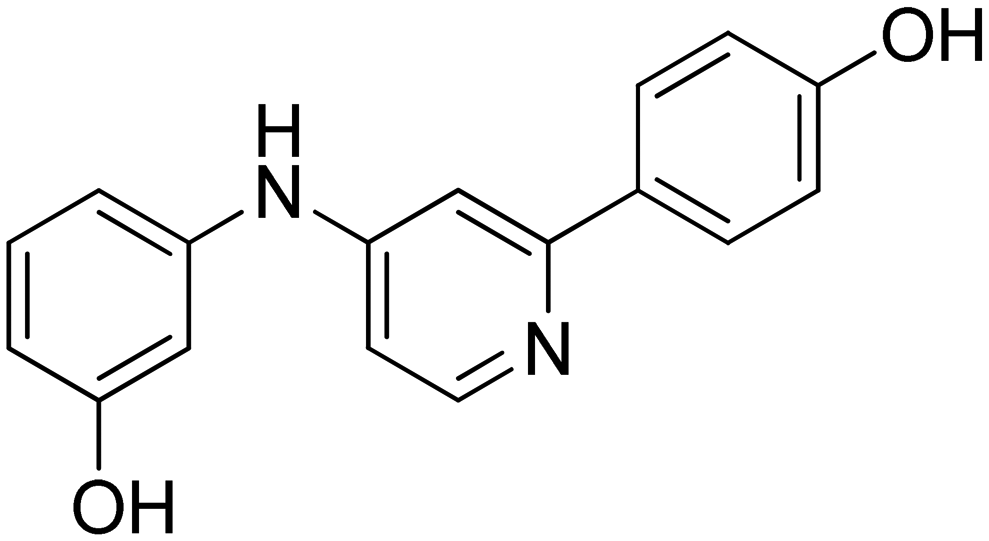
|
n.a. |
| 13 |
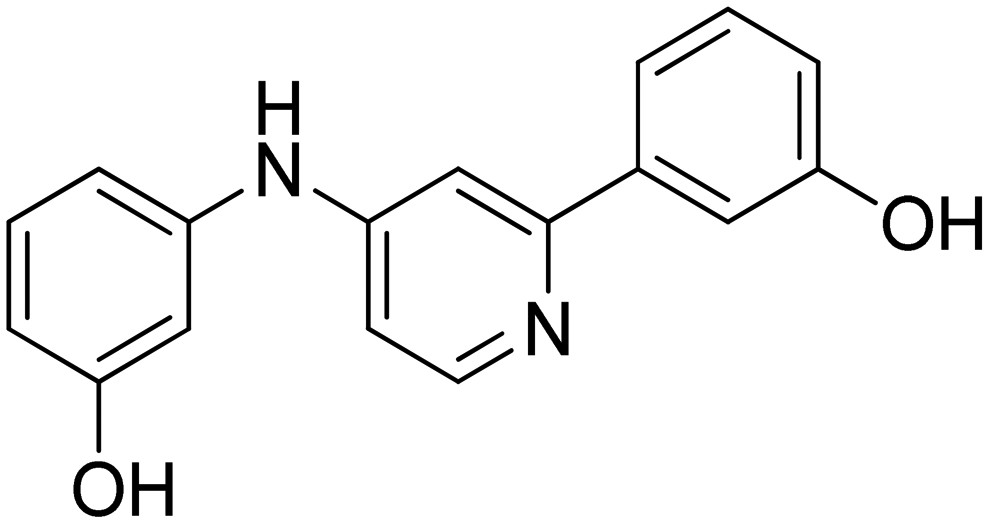
|
104.3 ± 10.1 |
| 14 |
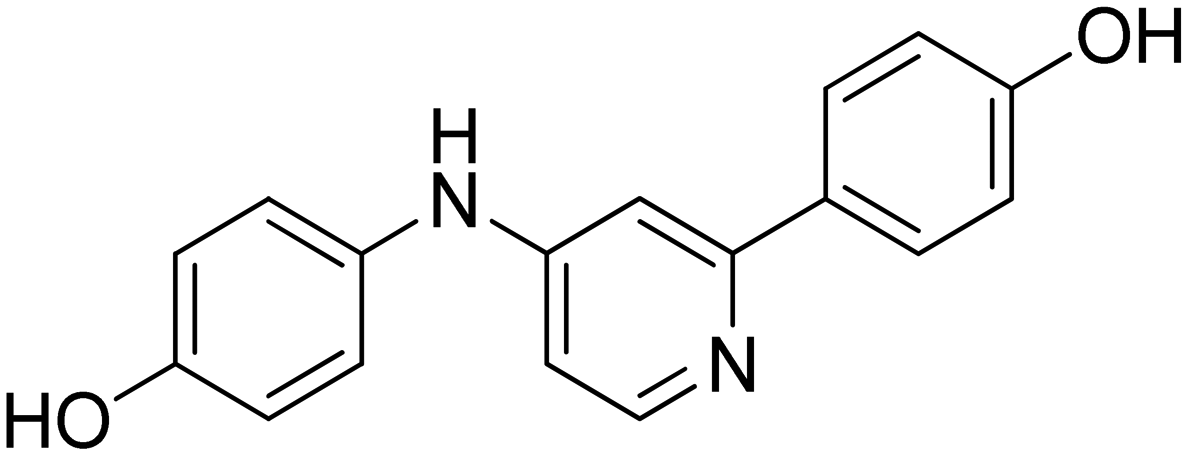
|
n.a. |
| 15 |
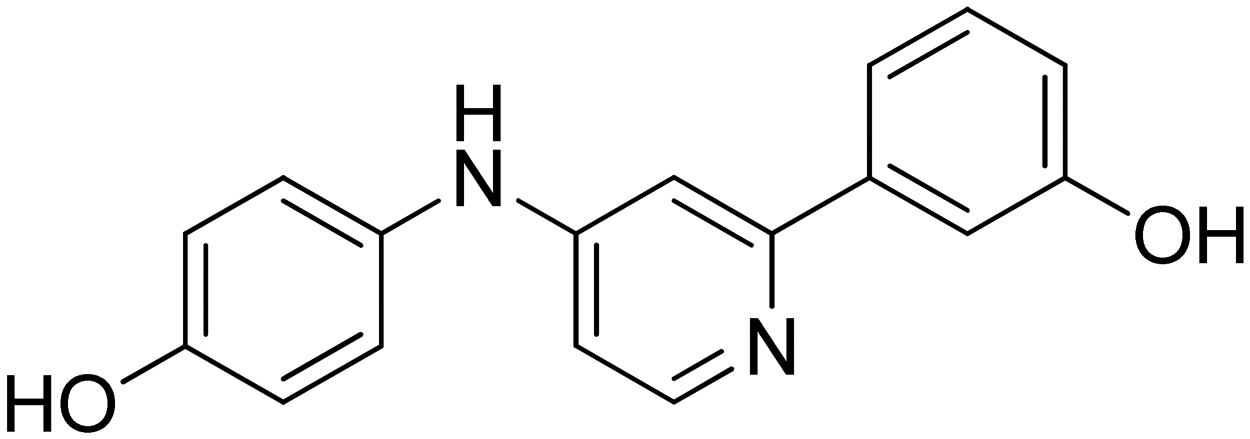
|
n.a. |
| 16 |
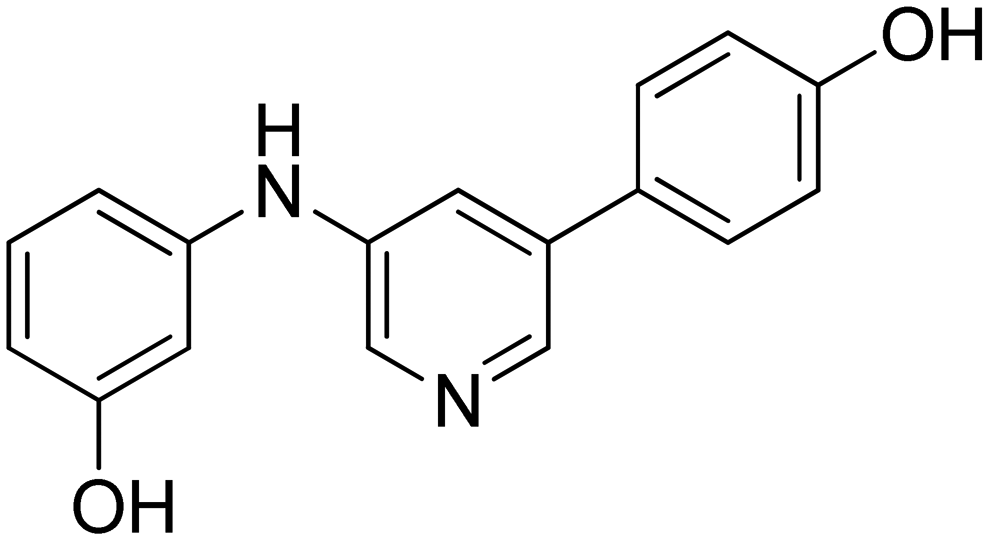
|
18.8 ± 7.2 |
| 17 |

|
28.8 ± 5.9 |
| 18 |
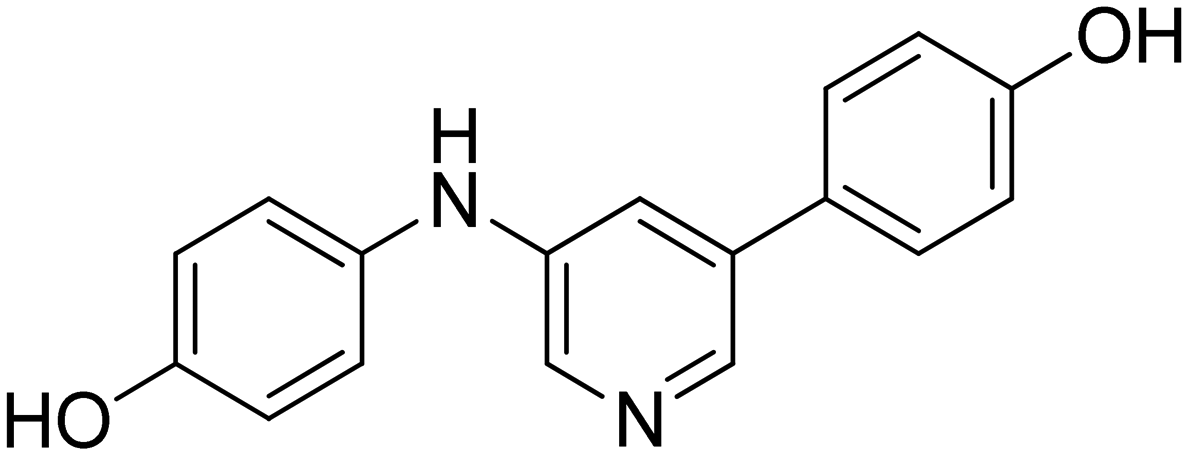
|
n.a. |
| 19 |
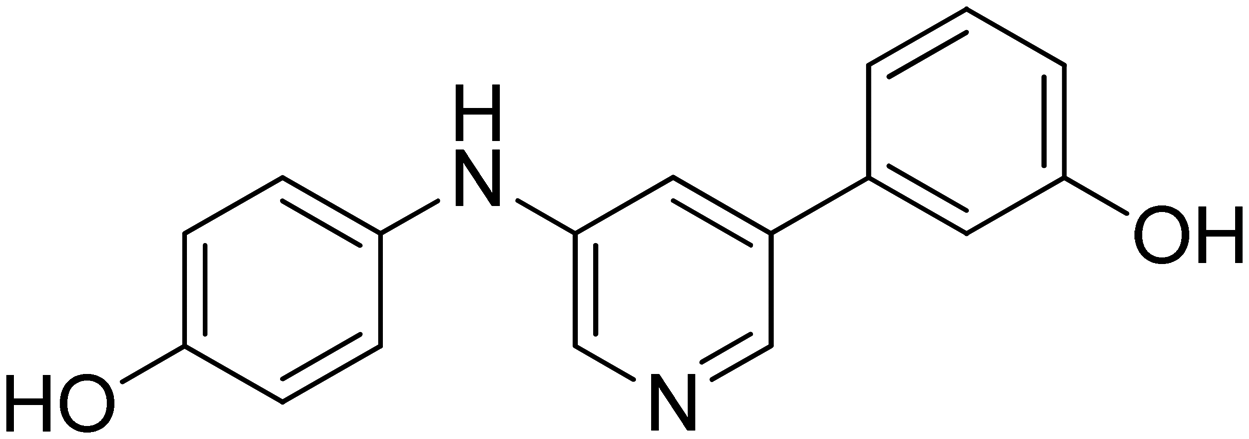
|
n.a. |
| 20 |
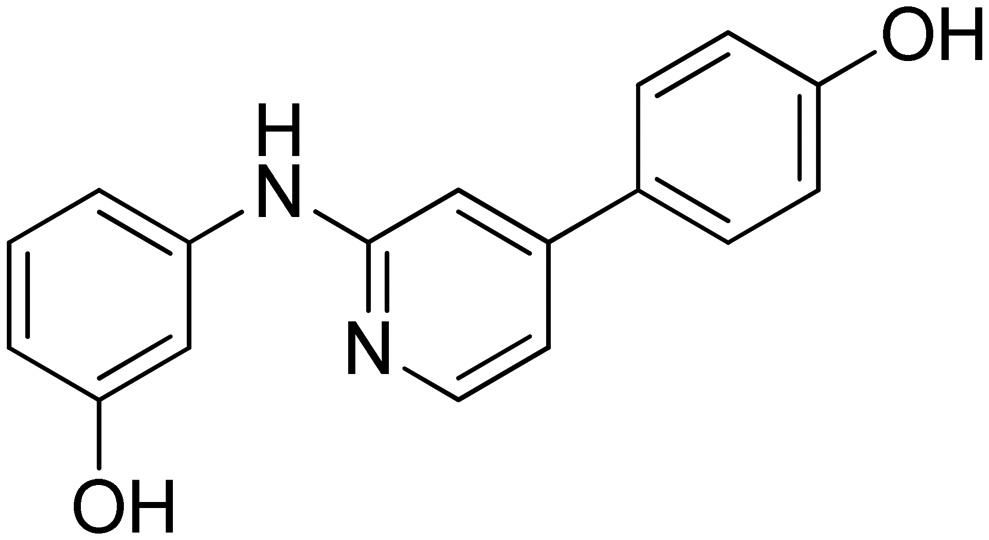
|
49.1 ± 4.2 |
| 21 |
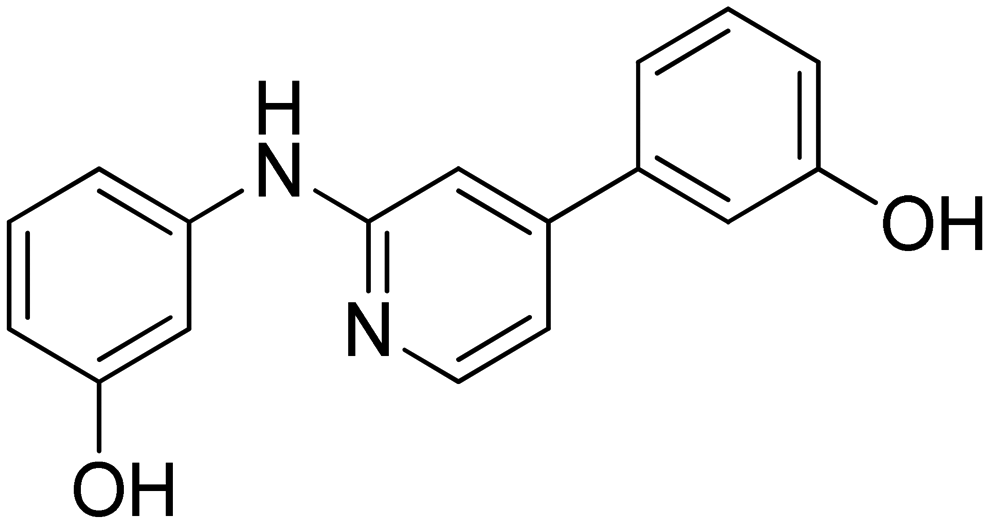
|
96.7 ± 5.0 |
| 22 |
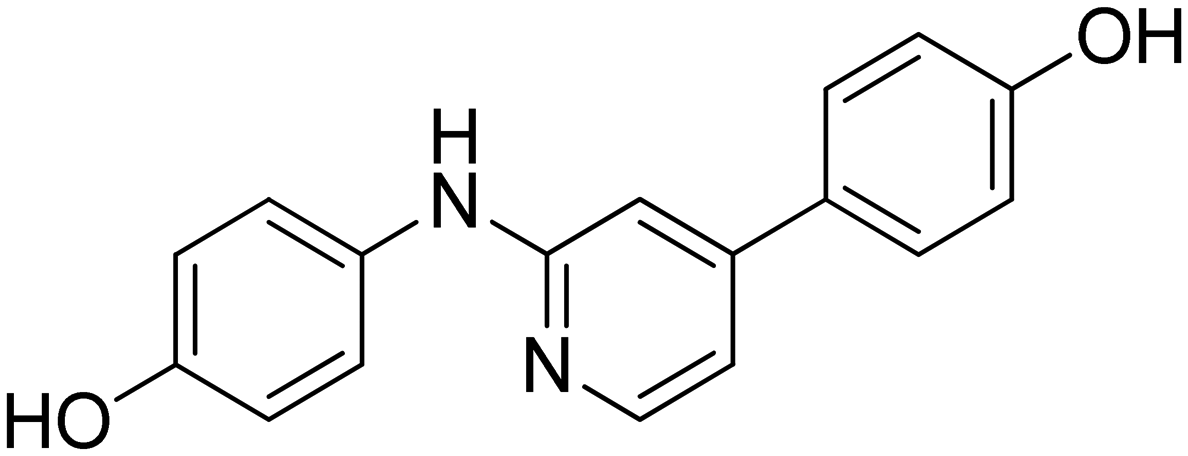
|
28.4 ± 10.8 |
| 23 |
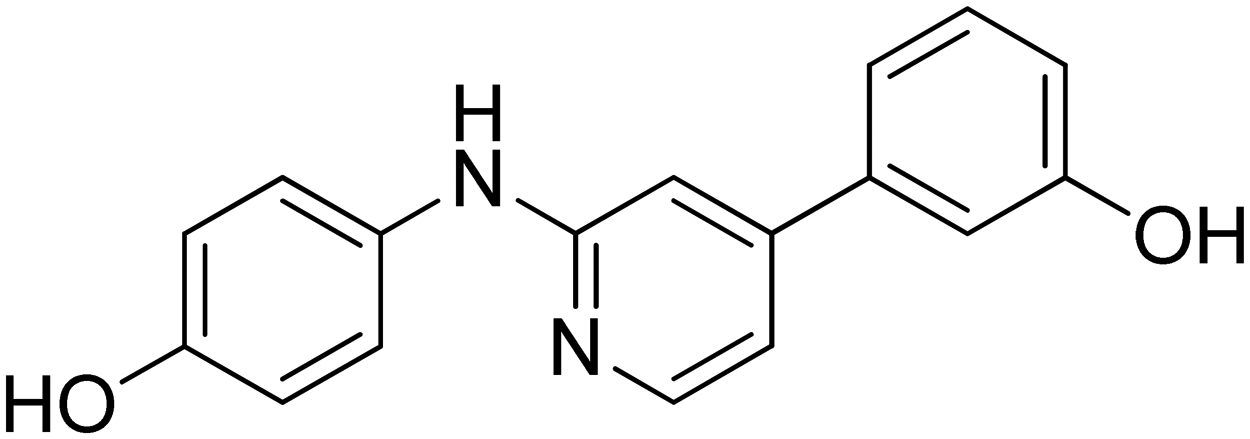
|
29.6 ± 4.2 |
All the compounds showed a clear concentration-dependent response. Compounds 8, 10 and 11 maintained SIRT-1 activation comparable to that of compound 1 even when incubated at the minimum concentration tested (Fig. 3).
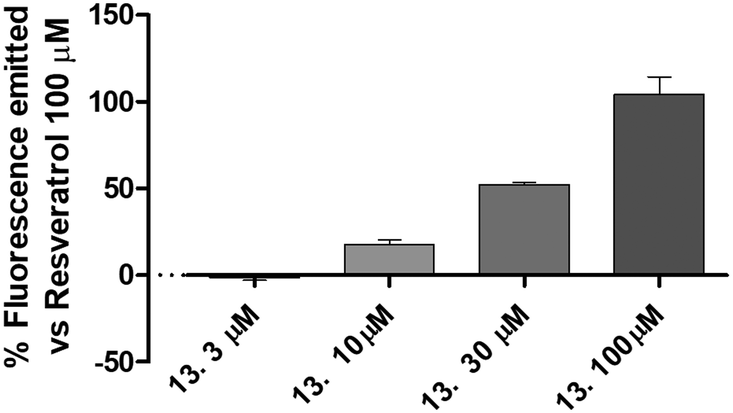 | ||
| Fig. 4 The figure shows the activation of SIRT1 expressed as percentage of fluorescence emitted in comparison to that of resveratrol 100 μM of increasing concentration of compound 13. Data are expressed as mean ± SEM. Data are expressed as mean ± SEM. Statistical analysis (one way ANOVA followed by Bonferroni post test) was performed. Number of replicates = 6. | ||
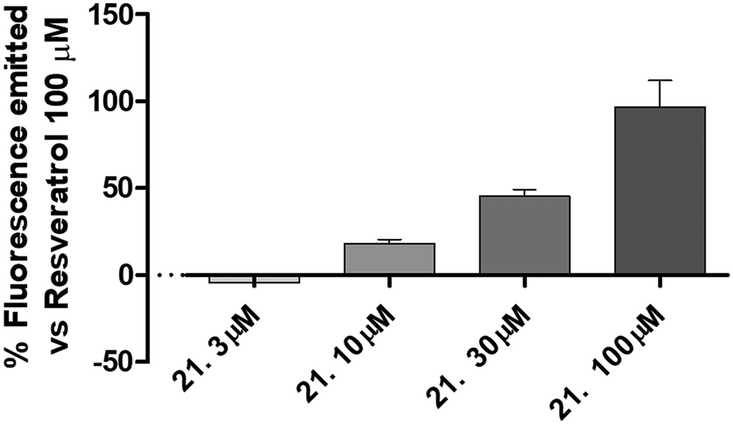 | ||
| Fig. 5 The figure shows the activation of SIRT1 expressed as percentage of fluorescence emitted in comparison to that of resveratrol 100 μM of increasing concentration of compound 21. Data are expressed as mean ± SEM. Statistical analysis (one way ANOVA followed by Bonferroni post test) was performed. Number of replicates = 6. | ||
The best compound of this series (21) was selected to evaluate the concentration-response curve on SIRT1 activation. As reported in Fig. 4, there is a clear concentration-dependent response, with a complete loss of activity at the concentration of 3 μM.
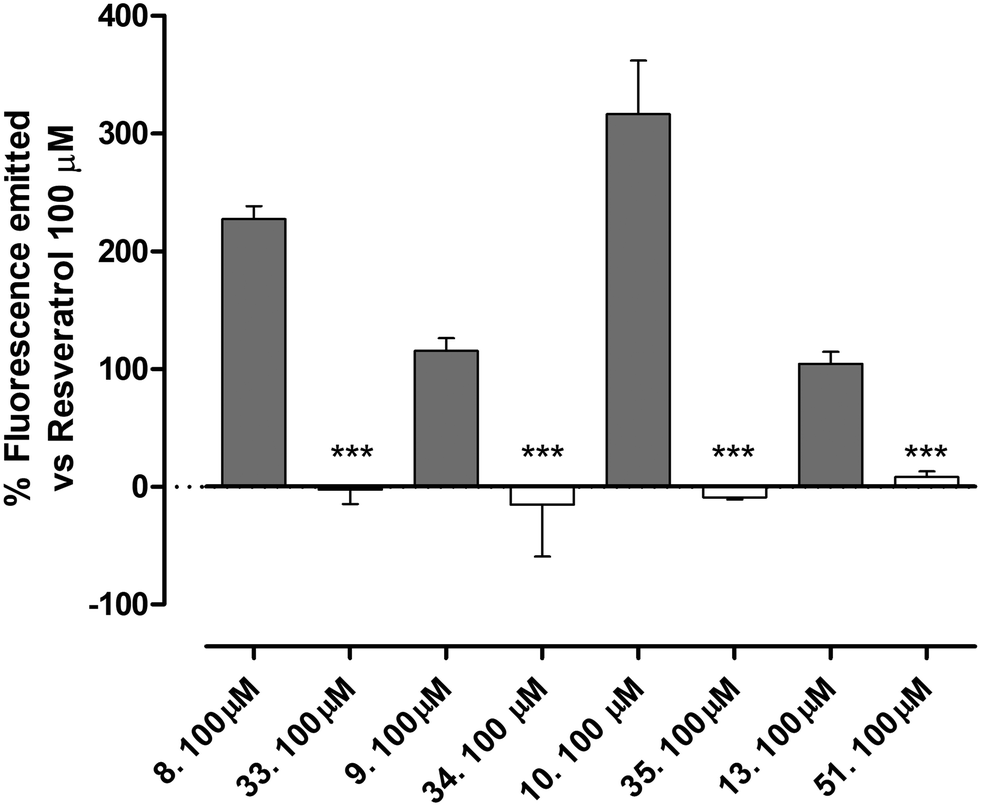 | ||
| Fig. 6 The figure shows the activation of SIRT1 expressed as percentage of fluorescence emitted in comparison to that of resveratrol 100 μM of increasing concentration of compound 8 and the methoxy derivative 33; of compound 9 and methoxy derivative 34; of compound 10 and methoxy derivative 35; of compound 13 and methoxy derivative 51. Data are expressed as mean ± SEM. Statistical analysis (t test student) was performed. * indicates significant difference vs. the relative O-methoxy derivative (***p < 0.001). Number of replicates = 6. | ||
2.2 Molecular modeling studies
With the aim of shedding light onto the potential binding mode of the new series of SIRT1 ligands, docking and molecular dynamics (MD) simulation studies were performed using compound 10, the most active compound of the series, as a reference. The compound was docked into the structure of SIRT1 in complex with p53-AMC peptide and the parent diarylamine ligand 6 (Fig. 1) predicted in our previous study.14 Since both compound 6 and its thiazole-based derivative 7 were predicted to occupy two different subpockets of the receptor14,15 one located above the coumarin moiety of the p53-AMC peptide, and a second one placed at its side (Fig. S49†), compound 10 was initially docked into each of the two subpockets. Surprisingly, the results of the two docking calculations suggested that one single molecule of the ligand could interact with the protein binding site occupying portions of both subpockets at the same time, since the results of the two docking studies converged in the same binding mode (see Materials and methods for details). The ternary SIRT1/p53-AMC/10 complex predicted by docking was then refined through a 100 ns molecular dynamics (MD) simulation study. As shown in Fig. 7, displaying the minimized average structure of the refined ternary complex obtained as a result of the MD simulation, the arylaminopyridine moiety of the ligand occupies the subpocket located above the coumarin moiety of the p53-AMC peptide. In particular, the pyridine ring of compound 10 is sandwiched between I223 from one side and the bicyclic core of the peptide from the other, forming lipophilic interactions with I223 and a π–π stacking with the peptide AMC moiety. Moreover, the NH group of the ligand forms an H-bond with the backbone carbonyl group of the peptide, whereas the adjacent phenolic moiety forms a stable water-bridged H-bond with R446 and shows hydrophobic interactions mainly with I227 and P447. The other phenolic moiety of the ligand occupies a portion of the second subpocket of SIRT1 binding site (Fig. S49†), forming hydrophobic interactions with P212, L215 and G415, a π-based interaction with the backbone amide moiety connecting F414 and G415, as well as a water-mediated H-bond with D298 and the backbone oxygen of F414. Finally, the central pyridine ring of compound 10 is involved in a complex H-bond network mediated by two different water molecules that connects the ligand pyridine nitrogen, the backbone nitrogen of the p53-AMC peptide, and the side chain of N226, which in turn forms a direct H-bond with the terminal backbone oxygen of the peptide (Fig. 7). This network of interactions, is believed to be crucial for the stability of the binding mode of compound 10, strongly contributing to firmly anchor the ligand to the backbone of the p53-AMC peptide. This is consistent with the SAR data derived from the biological evaluation of the series of compounds, demonstrating that the proper position of the pyridine nitrogen with respect to the NH group is essential for obtaining high potency ligands. At the same time, the predicted binding mode of compound 10 highlights the importance of both the presence of the terminal OH groups and their optimal position within the structure of the ligand for allowing it to form interactions with key anchoring residues placed at the two extremities of SIRT1 binding site, and to occupy two different receptor subpockets. This is in agreement with the detrimental effect on potency obtained in derivatives bearing either one or both OH groups in different positions with respect to compound 10, as well as with the lack of activity of the O-methylated analogues. | ||
| Fig. 7 Minimized average structure of SIRT1 in complex with p53-AMC peptide (green) and compound 10 (orange). Relevant H-bonds are highlighted as black dashed lines. For clarity, the side chains of R1, H2, and K3 of p53-AMC are not shown. | ||
2.3 Cardioprotective activity of selected SIRT1-activators in acute myocardial infarct model
Vehicle-treated animals submitted to in vivo LAD ligature showed a marked damage at myocardial level; indeed, the AI/ALV was 46 ± 2% (Fig. 8). In sham animals, submitted to the same experimental protocol, but without occlusion/reperfusion, the extension of AI/ALV was very negligible (11 ± 2%, Fig. 8). In addition, the IPC procedure induced a significant reduction of the ischemic injury if compared to vehicle group (AI/ALV = 21 ± 2%, Fig. 8). The pharmacological pre-treatment with reference compound 1 (10 mg kg−1), according to literature18 significantly protected the myocardium from the ischemic damage if compared to vehicle, resulting in an injured area of 30 ± 2% (Fig. 8). Finally, animals pre-treated with the best newly synthesized SIRT1 activators of these series (8 and 10) showed a remarkable protection against IR injury. 8 (at the same dose of compound 1), showed evident cardioprotection if compared to vehicle, which reached levels obtained in the sham group (AI/ALV = 6 ± 3%). Of note, 8 at 10 mg kg−1 was able to promote a much higher level of protection than that compound 1 (30 ± 2%), thus we tested a lower dose of 8 (1 mg kg−1) and, still, observed a significant protection (31 ± 1%), which was comparable to those of IPC procedure and compound 1 groups. Since 10 was endowed with a higher potency of activation on isolated SIRT1 enzyme than 8 (see the Fig. 3), it was administered to rats at lower doses. Interestingly, compound 10 at 1 mg kg−1 showed a marked reduction of the injury extension (AI/ALV = 12 ± 2%), like the highest dose (10 mg kg−1) of 8 and to sham. Finally, at the dose of 0.1 mg kg−110 was still active as anti-ischemic agent, inducing a protection level (AI/ALV = 28 ± 3%, Fig. 8), that is like that of compound 1 at 10 mg kg−1.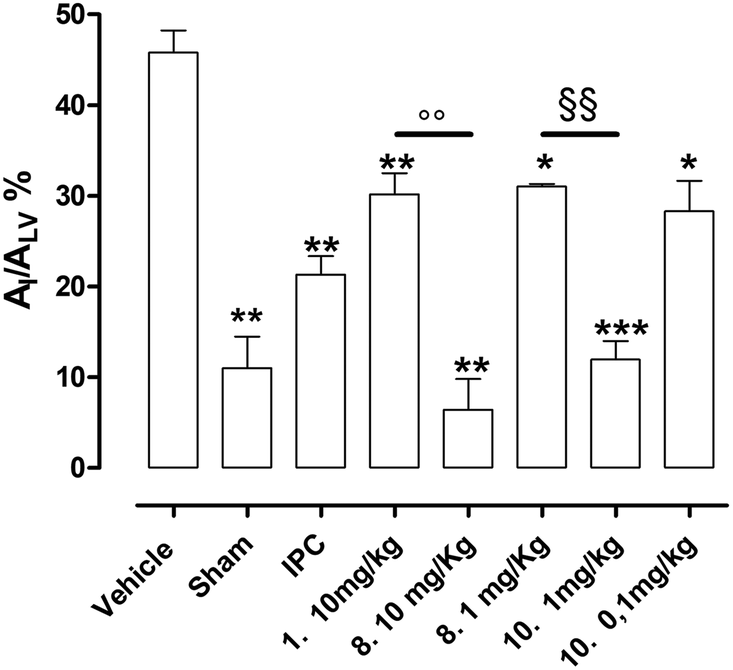 | ||
| Fig. 8 The histograms show the percentage of the ischemic area (AI) compared with the total area of the left ventricle (ALV) in the different groups. Data are expressed as mean ± SEM. * indicates significant difference vs. vehicle (*p < 0.05; **p < 0.01; ***p < 0.001). ○ indicates significant difference vs. compound 1 (resveratrol) group (○○p < 0.01). § indicates significant difference vs. compound 8 (1 mg kg−1) group (§§p < 0.01). (Number of animals used = 5). | ||
3. Discussion
Sirtuin family are NAD+-dependent deacetylases and, among them, SIRT1 is the most extensively studied sirtuin for its numerous implications on human health. Currently, it is well accepted that a positive modulation of SIRT1 may contribute to slow down the evolution of neurodegenerative diseases and of cardiovascular and metabolic disorders, as well as to prolong the life-span extension.21Among cardiovascular diseases, myocardial infarct is one of the main causes of death in the world.22 IR injury is a complex multi-factorial pathological process, that occurs when a reperfusion period (i.e. the restoring of blood supply) follows a prolonged ischemic event. Indeed, although the most effective strategy to reduce ischemic damage is an early reperfusion, on the other hand paradoxically the reperfusion in itself is responsible for an additional damage.23 Indeed, at the beginning of reperfusion a burst of ROS production, responsible for the downstream dangerous events such as peroxidation of protein, mitochondrial DNA damage and mitochondrial dysfunction, is widely documented;24 therefore, pharmaceutical strategies addressed to the containment of these unfavorable events are currently being considered as promising approaches. Resveratrol is often considered as the lead compound for SIRT1-activators and numerous papers confirm its anti-ischemic cardioprotection.25–29 However, the clinical use of this polyphenol is limited by its disadvantageous pharmacokinetic profile. Therefore, great efforts are currently being focused on the obtainment of novel agents able to stimulate SIRT1 enzyme with a more favorable bioavailability than that of resveratrol. In this direction, several SIRT1-activators have been developed by us and other groups, demonstrating interesting cardioprotective profiles against IR injury in in vivo and ex vivo models.14,15 In this regard, recently we published two papers in which, starting from the stilbenic structure of resveratrol, thiazole-derivatives were obtained and were demonstrated to have promising SIRT1-activating activities in occlusion/reperfusion models.14,15
The results obtained with the newly synthesized class of pyridine-based di-phenolic compounds herein reported suggest the crucial role of the positions of the pyridine endocyclic nitrogen atom in order to reach an efficient SIRT1-activation. In particular, the best structural arrangement is represented by the compounds comprised in scaffold 1 (8–11, Fig. 2), where both peripheral aryl rings A and B (Fig. 2) are placed in the two α positions with respect to the pyridine nitrogen atom.
Among the compounds belonging to the scaffold 1 class, the most active derivatives possess at least one para-hydroxy group in either ring A or B, with the maximum activating property reached when both hydroxy groups are in para position (compound 10). Our molecular modeling studies suggested that a single molecule of compound 10 is able to occupy two different subpockets of SIRT1 catalytic site, by interacting with the bound p53-AMC peptide and with residues located at the extremities of the protein binding site. In particular, the biding mode predicted for compound 10 suggested that the proper position of the pyridine nitrogen is crucial for firmly anchoring the ligand to SIRT1 catalytic site, and that both the presence and the para-position of the terminal hydroxyl groups of rings A and B are fundamental for enabling its interactions with the different subpockets, in agreement with the experimental data.
Both 8 and 10 proved to markedly reduce myocardial damage induced by the in vivo IR procedure at a lower dose than that of lead compound resveratrol. Of note, 10 resulted to be the most active compound, since it was able to significantly reduce the extension of ischemic area at a hundred times lower dose if compared to resveratrol/compound 1.
4. Conclusions
Taken together, these results demonstrate unequivocally the efficacy of synthetic strategy that led to identify the aryl-anilinopyridine structure as a new scaffold for SIRT1 activators to be employed as cardioprotective agents in ischemia. Future pharmacokinetic studies will be necessary to confirm the improvement of bioavailability than resveratrol and the toxicity profile, in order to have useful information about their efficacy and safety to translate to human use.5. Materials and methods
5.1 Synthesis. General procedures and materials
All solvents and chemicals were used as purchased without further purification. Chromatographic separations were performed on silica gel columns by flash chromatography (Kieselgel 40, 0.040–0.063 mm; Merck). Reactions were followed by thin layer chromatography (TLC) on Merck aluminum silica gel (60 F254) sheets that were visualized under a UV lamp. Evaporation was performed in vacuo (rotating evaporator). Sodium sulfate was always used as the drying agent. Proton (1H) and carbon (13C) NMR spectra were obtained with a Bruker Avance III 400 MHz spectrometer using the indicated deuterated solvents. Chemical shifts are given in parts per million (ppm) (δ relative to residual solvent peak for 1H and 13C). 1H-NMR spectra are reported in this order: multiplicity and number of protons. Standard abbreviations indicating the multiplicity were used as follows: s = singlet, d = doublet, dd = doublet of doublets, ddd = doublet of doublet of doublets, t = triplet, dt = doublet of triplets, m = multiplet and bs = broad singlet. HPLC analysis was used to determine purity: all target compounds (i.e., assessed in biological assays) were ≥95% pure by HPLC, as confirmed via UV detection (λ = 254 nm). Analytical reversed-phase HPLC was conducted using a Kinetex EVO C18 column (5 μm, 150 × 4.6 mm, Phenomenex, Inc.); eluent A, water + 0.1% TFA; eluent B, CH3CN for method A or MeOH for method B; after 5 min at 25% B, a gradient was formed from 25% to 75% of B in 5 min and held at 75% of B for 10 min; flow rate was 1 mL min−1. HPLC analyses were performed at 254 nm. The ESI-MS spectra were recorded by direct injection at a 5 μL min−1 flow rate in an Orbitrap Q Exactive Plus high-resolution mass spectrometer (Thermo Fischer Scientific Inc., Germany), equipped with an H-ESI source. The working conditions were as follows: positive polarity, spray voltage 3400 V, capillary temperature 290 °C, S-lens RF level 50, the sheath gas was set at 24, and the auxiliary gas was set at 5 (arbitrary units). For acquisition and analysis, Xcalibur 4.2 software (Thermo) was used. For spectra acquisition, a nominal resolution (at m/z 200) of 140![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 was used. Yields refer to isolated and purified products derived from non-optimized procedures.
000 was used. Yields refer to isolated and purified products derived from non-optimized procedures.
6-(4-Methoxyphenyl)pyridin-2-amine 27. 91% yield from 24 and 4-methoxybenzeneboronic acid. 1H-NMR (CDCl3) δ (ppm): 3.85 (s, 3H), 4.67 (bs, 2H), 6.42 (dd, 1H, J = 8.1, 0.7 Hz), 6.97 (AA′XX′, 2H, JAX = 9.0 Hz, JAA′/XX′ = 2.6 Hz), 7.03 (dd, 1H, J = 7.6, 0.7 Hz), 7.49 (dd, 1H, J = 8.1, 7.6 Hz), 7.90 (AA′XX′, 2H, JAX = 9.0 Hz, JAA′/XX′ = 2.6 Hz).
6-(3-Methoxyphenyl)pyridin-2-amine 28. 99% yield from 24 and 3-methoxybenzeneboronic acid. 1H-NMR (CDCl3) δ (ppm): 3.89 (s, 3H), 4.67 (bs, 2H), 6.48 (dd, 1H, J = 8.1, 0.6 Hz), 6.93 (ddd, 1H, J = 8.2, 2.6, 1.0 Hz), 7.08 (dd, 1H, J = 7.5, 0.7 Hz), 7.34 (t, 1H, J = 7.9 Hz), 7.46–7.55 (m, 3H).
5-(4-Methoxyphenyl)pyridin-3-amine 29. 94% yield from 25 and 4-methoxybenzeneboronic acid. 1H-NMR (CDCl3) δ (ppm): 3.50–3.90 (bs, 2H), 3.85 (s, 3H), 6.98 (AA′XX′, 2H, JAX = 8.8 Hz, JAA′/XX′ = 2.6 Hz), 7.12 (dd, 1H, J = 2.5, 2.0 Hz), 7.48 (AA′XX′, 2H, JAX = 8.8 Hz, JAA′/XX′ = 2.6 Hz), 8.04 (d, 1H, J = 2.6 Hz), 8.23 (d, 1H, J = 1.9 Hz).
5-(3-Methoxyphenyl)pyridin-3-amine 30. 99% yield from 25 and 3-methoxybenzeneboronic acid. 1H-NMR (CDCl3) δ (ppm): 3.86 (s, 3H), 6.93 (ddd, 1H, J = 8.2, 2.6, 0.8 Hz), 7.07 (t, 1H, J = 2.0 Hz), 7.10–7.15 (m, 1H), 7.16 (t, 1H, J = 2.3 Hz), 7.37 (t, 1H, J = 8.0 Hz), 8.09 (d, 1H, J = 2.5 Hz), 8.25 (d, 1H, J = 1.5 Hz).
4-(4-Methoxyphenyl)pyridin-2-amine 31. 98% yield from 26 and 4-methoxybenzeneboronic acid. 1H-NMR (CDCl3) δ (ppm): 3.86 (s, 3H), 4.45–4.55 (bs, 2H), 6.68 (dd, 1H, J = 1.5, 0.7 Hz), 6.86 (dd, 1H, J = 5.4, 1.6 Hz), 6.98 (AA′XX′, 2H, JAX = 8.9 Hz, JAA′/XX′ = 2.6 Hz), 7.54 (AA′XX′, 2H, JAX = 8.9 Hz, JAA′/XX′ = 2.6 Hz), 8.08 (dd, 1H, J = 5.4, 0.6 Hz).
4-(3-Methoxyphenyl)pyridin-2-amine 32. 97% yield from 26 and 3-methoxybenzeneboronic acid. 1H-NMR (CDCl3) δ (ppm): 3.86 (s, 3H), 4.64 (bs, 2H), 6.70 (dd, 1H, J = 1.5, 0.7 Hz), 6.88 (dd, 1H, J = 5.5, 1.6 Hz), 6.96 (ddd, 1H, J = 8.3, 2.6, 0.9 Hz), 7.10 (t, 1H, J = 2.1 Hz), 7.17 (ddd, 1H, J = 7.7, 1.6, 0.9 Hz), 7.37 (t, 1H, J = 7.9 Hz), 8.14 (dd, 1H, J = 5.4, 0.5 Hz).
Tert-butyl(2-(3-methoxyphenyl)pyridin-4-yl)carbamate 47. 96% yield from 46 and 3-methoxybenzeneboronic acid. 1H-NMR (CDCl3) δ (ppm): 1.55 (s, 9H), 3.89 (s, 3H), 6.77 (bs, 1H), 6.96 (ddd, 1H, J = 8.2, 2.6, 0.8 Hz), 7.24 (dd, 1H, J = 5.6, 2.1 Hz), 7.36 (t, 1H, J = 8.0 Hz), 7.49–7.54 (m, 1H), 7.57 (t, 1H, J = 2.0 Hz), 7.77 (d, 1H, J = 1.8 Hz), 8.51 (d, 1H, J = 5.5 Hz).
2-(4-Methoxyphenyl)pyridin-4-amine 49. 84% yield from 45 and 3-methoxybenzeneboronic acid. 1H-NMR (CDCl3) δ (ppm): 3.85 (s, 3H), 4.20 (bs, 2H), 6.46 (dd, 1H, J = 5.6, 2.3 Hz), 6.90 (d, 1H, J = 2.2 Hz), 6.96 (AA′XX′, 2H, JAX = 9.0 Hz, JAA′/XX′ = 2.6 Hz), 7.87 (AA′XX′, 2H, JAX = 9.0 Hz, JAA′/XX′ = 2.6 Hz), 8.28 (d, 1H, J = 5.6 Hz).
N-(3-Methoxyphenyl)-6-(4-methoxyphenyl)pyridin-2-amine 33. 99% yield from 27 and 3-bromoanisole. 1H-NMR (CDCl3) δ (ppm): 3.83 (s, 3H), 3.87 (s, 3H), 6.60 (ddd, 1H, J = 8.1, 2.5, 0.8 Hz), 6.60–6.67 (bs, 1H), 6.78 (d, 1H, J = 8.2 Hz), 6.92 (ddd, 1H, J = 8.0, 2.0, 0.7 Hz), 7.98 (AA′XX′, 2H, JAX = 9.0 Hz, JAA′/XX′ = 2.6 Hz), 7.13–7.18 (m, 2H), 7.24 (t, 1H, J = 8.1 Hz), 7.55 (t, 1H, J = 7.9 Hz), 7.97 (AA′XX′, 2H, JAX = 9.0 Hz, JAA′/XX′ = 2.5 Hz).
N,6-Bis(3-methoxyphenyl)pyridin-2-amine 34. 64% yield from 28 and 3-bromoanisole. 1H-NMR (CDCl3) δ (ppm): 3.83 (s, 3H), 3.91 (s, 3H), 6.62 (ddd, 1H, J = 8.3, 2.5, 0.7 Hz), 6.83 (d, 1H, J = 7.8 Hz), 6.91–6.98 (m, 2H), 7.18–7.25 (m, 3H), 7.37 (t, 1H, J = 7.9 Hz), 7.53–7.62 (m, 2H), 7.64 (t, 1H, J = 2.1 Hz).
N,6-Bis(4-methoxyphenyl)pyridin-2-amine 35. 83% yield from 27 and 4-bromoanisole. 1H-NMR (acetone-d6) δ (ppm): 3.78 (s, 3H), 3.86 (s, 3H), 6.65 (dd, 1H, J = 8.3, 0.6 Hz), 6.91 (AA′XX′, 2H, JAX = 9.0 Hz, JAA′/XX′ = 2.9 Hz), 7.01 (AA′XX′, 2H, JAX = 9.0 Hz, JAA′/XX′ = 2.6 Hz), 7.19 (dd, 1H, J = 7.5, 0.6 Hz), 7.54 (dd, 1H, J = 8.2, 7.6 Hz), 7.66 (AA′XX′, 2H, JAX = 9.0 Hz, JAA′/XX′ = 2.9 Hz), 8.01 (bs, 1H), 8.05 (AA′XX′, 2H, JAX = 9.0 Hz, JAA′/XX′ = 2.6 Hz).
6-(3-Methoxyphenyl)-N-(4-methoxyphenyl)pyridin-2-amine 36. 48% yield from 28 and 4-bromoanisole. 1H-NMR (CDCl3) δ (ppm): 3.82 (s, 3H), 3.90 (s, 3H), 6.64 (dd, 1H, J = 8.3, 0.6 Hz), 6.91 (AA′XX′, 2H, JAX = 8.9 Hz, JAA′/XX′ = 2.9 Hz), 6.95 (ddd, 1H, J = 8.2, 2.6, 0.9 Hz), 7.13 (dd, 1H, J = 7.5, 0.7 Hz), 7.32 (AA′XX′, 2H, JAX = 8.8 Hz, JAA′/XX′ = 2.7 Hz), 7.36 (t, 1H, J = 8.0 Hz), 7.49–7.56 (m, 2H), 7.58–7.61 (m, 1H).
N-(3-Methoxyphenyl)-5-(4-methoxyphenyl)pyridin-3-amine 37. 64% yield from 29 and 3-bromoanisole. 1H-NMR (CDCl3) δ (ppm): 3.80 (s, 3H), 3.86 (s, 3H), 5.91 (bs, 1H), 6.57 (dd, 1H, J = 8.3, 2.4 Hz), 6.68 (t, 1H, J = 2.2 Hz), 6.72 (dd, 1H, J = 8.0, 2.1 Hz), 6.99 (AA′XX′, 2H, JAX = 8.8 Hz, JAA′/XX′ = 2.5 Hz), 7.22 (t, 1H, J = 8.1 Hz), 7.49 (AA′XX′, 2H, JAX = 8.8 Hz, JAA′/XX′ = 2.6 Hz), 7.59 (t, 1H, J = 2.2 Hz), 8.34 (d, 1H, J = 2.6 Hz), 8.37 (d, 1H, J = 1.9 Hz).
N,5-Bis(3-methoxyphenyl)pyridin-3-amine 38. 63% yield from 30 and 3-bromoanisole. 1H-NMR (CDCl3) δ (ppm): 3.80 (s, 3H), 3.86 (s, 3H), 5.88 (bs, 1H), 6.57 (dd, 1H, J = 8.0, 2.1 Hz), 6.67 (t, 1H, J = 2.2 Hz), 6.72 (dd, 1H, J = 8.0, 1.5 Hz), 6.94 (ddd, 1H, J = 8.2, 2.5, 0.7 Hz), 7.08 (t, 1H, J = 2.1 Hz), 7.10–7.16 (m, 1H), 7.22 (t, 1H, J = 8.1 Hz), 7.38 (t, 1H, J = 8.0 Hz), 7.60 (t, 1H, J = 2.3 Hz), 8.37 (d, 1H, J = 2.5 Hz), 8.39 (d, 1H, J = 1.8 Hz).
N,5-Bis(4-methoxyphenyl)pyridin-3-amine 39. 20% yield from 29 and 4-bromoanisole. 1H-NMR (acetone-d6) δ (ppm): 3.79 (s, 3H), 3.84 (s, 3H), 6.91–6.96 (m, 2H), 7.00–7.06 (m, 2H), 7.16–7.22 (m, 2H), 7.34 (s, 1H), 7.45 (s, 1H), 7.53–7.58 (m, 2H), 8.21 (bs, 1H).
5-(3-Methoxyphenyl)-N-(4-methoxyphenyl)pyridin-3-amine 40. 57% yield from 30 and 4-bromoanisole. 1H-NMR (CDCl3) δ (ppm): 3.82 (s, 3H), 3.85 (s, 3H), 5.69 (bs, 1H), 6.90 (AA′XX′, 2H, JAX = 9.0 Hz, JAA′/XX′ = 2.5 Hz), 6.93 (ddd, 1H, J = 8.2, 2.6, 0.9 Hz), 7.05 (t, 1H, J = 2.1 Hz), 7.09–7.16 (m, 3H), 7.33–7.39 (m, 2H), 8.23 (d, 1H, J = 2.6 Hz), 8.28 (d, 1H, J = 1.8 Hz).
N-(3-Methoxyphenyl)-4-(4-methoxyphenyl)pyridin-2-amine 41. 40% yield from 31 and 3-bromoanisole. 1H-NMR (CDCl3) δ (ppm): 3.82 (s, 3H), 3.85 (s, 3H), 6.62 (ddd, 1H, J = 8.2, 2.4, 0.8 Hz), 6.90–7.01 (m, 5H), 7.06–7.10 (m, 1H), 7.25 (t, 1H, J = 8.1 Hz), 7.54 (AA′XX′, 2H, JAX = 8.9 Hz, JAA′/XX′ = 2.6 Hz), 8.21 (dd, 1H, J = 5.4, 0.5 Hz).
N,4-Bis(3-methoxyphenyl)pyridin-2-amine 42. 42% yield from 32 and 3-bromoanisole. 1H-NMR (CDCl3) δ (ppm): 3.82 (s, 3H), 3.86 (s, 3H), 6.62 (ddd, 1H, J = 8.4, 2.4, 0.8 Hz), 6.73 (bs, 1H), 6.91–6.99 (m, 3H), 7.00 (t, 1H, J = 2.2 Hz), 7.07–7.12 (m, 2H), 7.16 (ddd, 1H, J = 7.6, 1.6, 1.0 Hz), 7.25 (t, 1H, J = 8.1 Hz), 7.37 (t, 1H, J = 7.9 Hz), 8.25 (d, 1H, J = 5.6 Hz).
N,4-Bis(4-methoxyphenyl)pyridin-2-amine 43. 22% yield from 31 and 4-bromoanisole. 1H-NMR (CDCl3) δ (ppm): 3.82 (s, 3H), 3.84 (s, 3H), 6.51 (bs, 1H), 6.83–6.85 (m, 1H), 6.88 (dd, 1H, J = 5.4, 1.6 Hz), 6.91 (AA′XX′, 2H, JAX = 8.9 Hz, JAA′/XX′ = 2.9 Hz), 6.95 (AA′XX′, 2H, JAX = 8.9 Hz, JAA′/XX′ = 2.5 Hz), 7.24–7.30 (m, 2H), 7.50 (AA′XX′, 2H, JAX = 8.9 Hz, JAA′/XX′ = 2.8 Hz), 8.16 (d, 1H, J = 5.3 Hz).
4-(3-Methoxyphenyl)-N-(4-methoxyphenyl)pyridin-2-amine 44. 26% yield from 32 and 4-bromoanisole. 1H-NMR (CDCl3) δ (ppm): 3.82 (s, 3H), 3.85 (s, 3H), 6.57 (bs, 1H), 6.84–6.96 (m, 5H), 7.07 (t, 1H, J = 2.0 Hz), 7.13 (ddd, 1H, J = 7.6, 1.7, 1.0 Hz), 7.24–7.30 (m, 2H), 7.35 (t, 1H, J = 7.9 Hz), 8.18 (dd, 1H, J = 5.3, 0.6 Hz).
N-(3-Methoxyphenyl)-2-(4-methoxyphenyl)pyridin-4-amine 50. 83% yield from 49 and 3-bromoanisole. 1H-NMR (CDCl3) δ (ppm): 3.82 (s, 3H), 3.85 (s, 3H), 6.24 (bs, 1H), 6.69 (dd, 1H, J = 7.8, 2.2 Hz), 6.75–6.85 (m, 3H), 6.97 (AA′XX′, 2H, JAX = 8.9 Hz, JAA′/XX′ = 2.5 Hz), 7.22 (d, 1H, J = 2.0 Hz), 7.29 (d, 1H, J = 8.0 Hz), 7.87 (AA′XX′, 2H, JAX = 8.8 Hz, JAA′/XX′ = 2.5 Hz), 8.35 (d, 1H, J = 5.7 Hz).
N,2-Bis(3-methoxyphenyl)pyridin-4-amine 51. 33% yield from 48 and 3-bromoanisole. 1H-NMR (CDCl3) δ (ppm): 3.82 (s, 3H), 3.88 (s, 3H), 6.21 (bs, 1H), 6.69 (dd, 1H, J = 8.1, 2.4 Hz), 6.77 (t, 1H, J = 2.2 Hz), 6.79–6.85 (m, 2H), 6.95 (ddd, 1H, J = 8.1, 2.6, 0.9 Hz), 7.25 (d, 1H, J = 2.2 Hz), 7.28 (t, 1H, J = 8.1 Hz), 7.34 (t, 1H, J = 7.9 Hz), 7.42–7.46 (m, 1H), 7.51 (t, 1H, J = 2.1 Hz), 8.38 (d, 1H, J = 5.7 Hz).
N,2-Bis(4-methoxyphenyl)pyridin-4-amine 52. 57% yield from 49 and 4-bromoanisole. 1H-NMR (CDCl3) δ (ppm): 3.83 (s, 3H), 3.84 (s, 3H), 6.09 (bs, 1H), 6.59 (dd, 1H, J = 5.8, 2.2 Hz), 6.89–6.99 (m, 4H), 7.03 (d, 1H, J = 2.0 Hz), 7.17 (AA′XX′, 2H, JAX = 8.8 Hz, JAA′/XX′ = 2.7 Hz), 7.84 (AA′XX′, 2H, JAX = 9.0 Hz, JAA′/XX′ = 2.5 Hz), 8.28 (d, 1H, J = 5.8 Hz).
2-(3-Methoxyphenyl)-N-(4-methoxyphenyl)pyridin-4-amine 53. 51% yield from 48 and 4-bromoanisole. 1H-NMR (CDCl3) δ (ppm): 3.84 (s, 3H), 3.87 (s, 3H), 5.99 (bs, 1H), 6.63 (dd, 1H, J = 5.7, 2.3 Hz), 6.90–6.97 (m, 3H), 7.07 (d, 1H, J = 2.2 Hz), 7.17 (AA′XX′, 2H, JAX = 8.8 Hz, JAA′/XX′ = 2.8 Hz), 7.33 (t, 1H, J = 7.9 Hz), 7.42 (dt, 1H, J = 7.7, 1.3 Hz), 7.49 (t, 1H, J = 2.0 Hz), 8.32 (d, 1H, J = 5.7 Hz).
:2 as eluent to afford intermediate 46 (72% yield). 1H-NMR (CDCl3) δ (ppm): 1.52 (s, 9H), 6.68 (bs, 1H), 7.18 (dd, 1H, J = 5.6, 2.0 Hz), 7.64 (d, 1H, J = 1.9 Hz), 8.17 (d, 1H, J = 5.7 Hz).
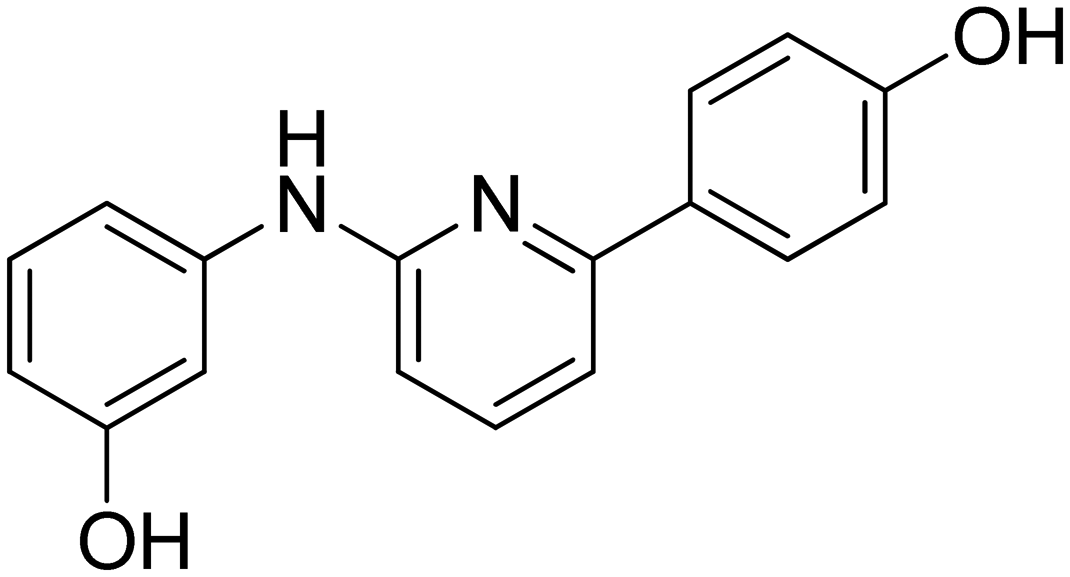
3-((6-(4-Hydroxyphenyl)pyridin-2-yl)amino)phenol 8. Yellow solid. 92% yield from 33. 1H-NMR (acetone-d6) δ (ppm): 6.44 (ddd, 1H, J = 7.9, 2.4, 1.0 Hz), 6.73 (d, 1H, J = 8.1 Hz), 6.93 (AA′XX′, 2H, JAX = 8.7 Hz, JAA′/XX′ = 2.4 Hz), 7.10 (t, 1H, J = 8.0 Hz), 7.15–7.19 (m, 1H), 7.21 (d, 1H, J = 7.5 Hz), 7.46 (t, 1H, J = 2.2 Hz), 7.56 (t, 1H, J = 7.9 Hz), 8.00 (AA′XX′, 2H, JAX = 8.8 Hz, JAA′/XX′ = 2.5 Hz), 8.15 (bs, 1H), 8.18 (s, 1H), 8.55 (s, 1H). 13C-NMR (acetone-d6) δ (ppm): 106.49, 108.82, 108.89, 110.46, 110.83, 116.17 (2C), 128.90 (2C), 130.19, 132.15, 138.75, 144.09, 155.99, 156.72, 158.78, 159.12. HPLC analysis (method A): retention time = 10.758; peak area 99% (254 nm). HRMS: m/z for C17H15N2O2 [M + H]+ calculated: 279.11335, found: 279.11246.
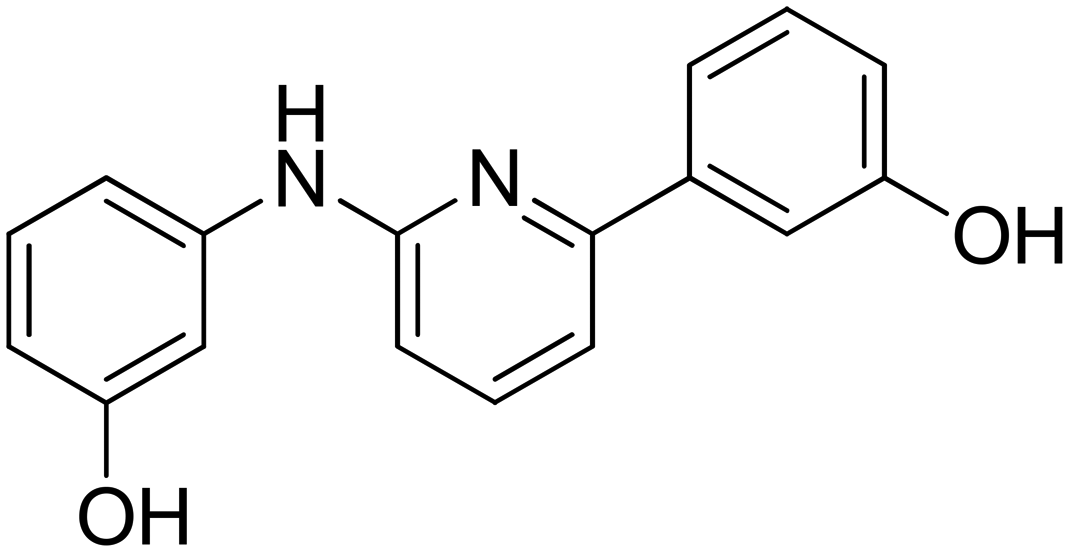
3-(6-((3-Hydroxyphenyl)amino)pyridin-2-yl)phenol 9. Yellow solid. 99% yield from 34. 1H-NMR (acetone-d6) δ (ppm): 6.44 (ddd, 1H, J = 8.0, 2.4, 1.0 Hz), 6.81 (dd, 1H, J = 8.3, 0.6 Hz), 6.88 (ddd, 1H, J = 8.0, 2.5, 0.9 Hz), 7.10 (t, 1H, J = 8.1 Hz), 7.21–7.31 (m, 3H), 7.40 (t, 1H, J = 2.2 Hz), 7.54–7.64 (m, 3H), 8.18 (s, 1H), 8.20 (bs, 1H), 8.41 (s, 1H). 13C-NMR (acetone-d6) δ (ppm): 106.46, 108.99, 109.96, 110.85, 111.60, 114.47, 116.49, 118.75, 130.25, 130.36, 138.80, 142.19, 143.98, 155.93, 156.80, 158.61, 158.77. HPLC analysis (method A): retention time = 11.006; peak area 98% (254 nm). HRMS: m/z for C17H15N2O2 [M + H]+ calculated: 279.11335, found: 279.11255.
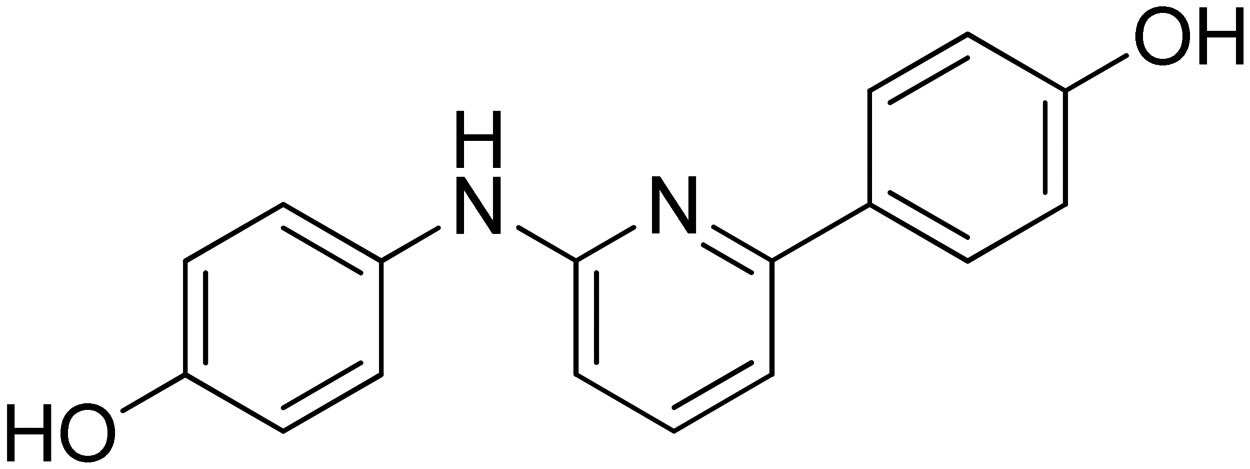
4-(6-((4-Hydroxyphenyl)amino)pyridin-2-yl)phenol 10. Yellow-orange solid. 78% yield from 35. 1H-NMR (acetone-d6) δ (ppm): 6.60 (dd, 1H, J = 8.2, 0.5 Hz), 6.82 (AA′XX′, 2H, JAX = 8.8 Hz, JAA′/XX′ = 2.8 Hz), 6.91 (AA′XX′, 2H, JAX = 8.8 Hz, JAA′/XX′ = 2.5 Hz), 7.14 (dd, 1H, J = 7.5, 0.5 Hz), 7.46–7.56 (m, 3H), 7.86 (bs, 1H), 7.93–8.01 (m, 3H), 8.53 (s, 1H). 13C-NMR (acetone-d6) δ (ppm): 107.42, 109.60, 116.10 (2C), 116.15 (2C), 122.48 (2C), 128.77 (2C), 132.25, 134.80, 138.64, 153.27, 155.88, 157.54, 159.03. HPLC analysis (method A): retention time = 10.535; peak area 97% (254 nm). HRMS: m/z for C17H15N2O2 [M + H]+ calculated: 279.11335, found: 279.11249.
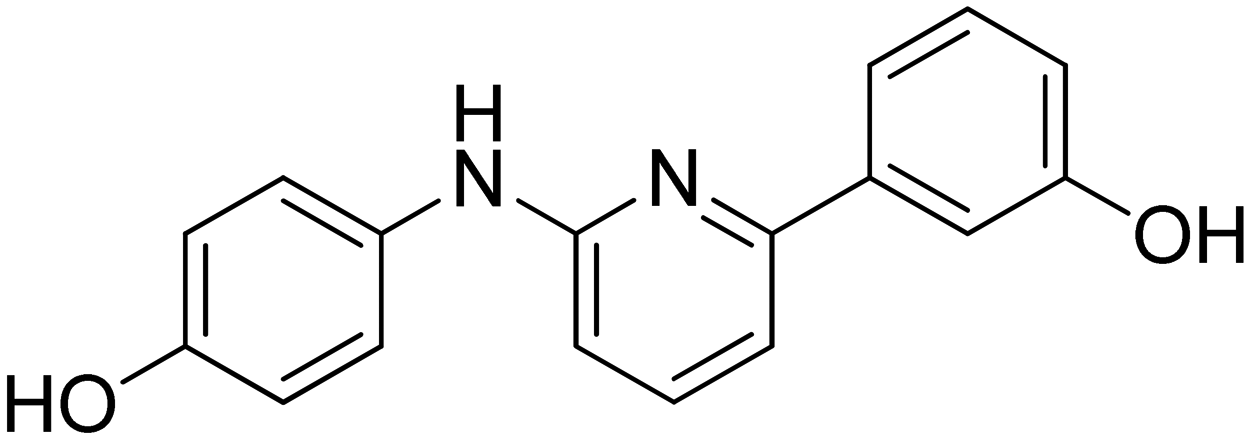
3-(6-((4-Hydroxyphenyl)amino)pyridin-2-yl)phenol 11. Yellow solid. 81% yield from 36. 1H-NMR (acetone-d6) δ (ppm): 6.68 (dd, 1H, J = 8.4, 0.6 Hz), 6.82 (AA′XX′, 2H, JAX = 8.9 Hz, JAA′/XX′ = 2.8 Hz), 6.86 (ddd, 1H, J = 8.0, 2.5, 0.9 Hz), 7.18 (dd, 1H, J = 7.5, 0.6 Hz), 7.27 (t, 1H, J = 7.9 Hz), 7.50–7.58 (m, 4H), 7.62 (t, 1H, J = 2.0 Hz), 7.94 (bs, 1H), 7.99 (s, 1H), 8.40 (s, 1H). 13C-NMR (acetone-d6) δ (ppm): 108.64, 110.70, 114.38, 116.18 (2C), 116.38, 118.64, 122.39 (2C), 130.28, 134.71, 138.68, 142.27, 153.27, 155.77, 157.55, 158.54. HPLC analysis (method A): retention time = 10.721; peak area 99% (254 nm). HRMS: m/z for C17H15N2O2 [M + H]+ calculated: 279.11335, found: 279.11258.
3-((2-(4-Hydroxyphenyl)pyridin-4-yl)amino)phenol 12. Yellow solid. 62% yield from 50. 1H-NMR (DMSO-d6) δ (ppm): 6.43 (ddd, 1H, J = 8.1, 2.1, 1.0 Hz), 6.62–6.68 (m, 2H), 6.80 (dd, 1H, J = 5.6, 2.2 Hz), 6.83 (AA′XX′, 2H, JAX = 8.8 Hz, JAA′/XX′ = 2.5 Hz), 7.13 (t, 1H, J = 8.0 Hz), 7.26 (d, 1H, J = 2.1 Hz), 7.77 (AA′XX′, 2H, JAX = 8.7 Hz, JAA′/XX′ = 2.5 Hz), 8.21 (d, 1H, J = 5.7 Hz), 8.72 (s, 1H), 9.43 (s, 1H), 9.65 (s, 1H). 13C-NMR (DMSO-d6) δ (ppm): 104.53, 106.55, 107.52, 109.57, 110.59, 115.30 (2C), 127.65 (2C), 129.37, 130.06, 130.31, 141.77, 149.78, 150.82, 156.86, 158.19. HPLC analysis (method B): retention time = 11.111; peak area 99% (254 nm). HRMS: m/z for C17H15N2O2 [M + H]+ calculated: 279.11335, found: 279.11261.
3-(4-((3-Hydroxyphenyl)amino)pyridin-2-yl)phenol 13. Orange solid. 76% yield from 51. 1H-NMR (DMSO-d6) δ (ppm): 6.42–6.48 (m, 1H), 6.62–6.69 (m, 2H), 6.77–6.82 (m, 1H), 6.86 (dd, 1H, J = 5.6, 2.2 Hz), 7.14 (t, 1H, J = 8.3 Hz), 7.25 (t, 1H, J = 7.8 Hz), 7.29–7.35 (m, 2H), 7.37 (t, 1H, J = 1.9 Hz), 8.26 (d, 1H, J = 5.6 Hz), 8.76 (s, 1H), 9.46 (s, 1H), 9.50 (s, 1H). 13C-NMR (DMSO-d6) δ (ppm): 105.62, 106.72, 108.22, 109.76, 110.73, 113.23, 115.71, 117.03, 129.57, 130.12, 140.83, 141.65, 149.98, 150.94, 156.81, 157.59, 158.25. HPLC analysis (method B): retention time = 11.136; peak area 99% (254 nm). HRMS: m/z for C17H15N2O2 [M + H]+ calculated: 279.11335, found: 279.11258.
4-(4-((4-Hydroxyphenyl)amino)pyridin-2-yl)phenol 14. Yellow solid. 45% yield from 52. 1H-NMR (DMSO-d6) δ (ppm): 6.58 (dd, 1H, J = 5.7, 2.2 Hz), 6.74–6.84 (m, 4H), 7.01–7.07 (m, 3H), 7.72 (AA′XX′, 2H, JAX = 8.8 Hz, JAA′/XX′ = 2.4 Hz), 8.12 (d, 1H, J = 5.7 Hz), 8.39 (s, 1H), 9.31 (bs, 1H), 9.58 (bs, 1H). 13C-NMR (DMSO-d6) δ (ppm): 103.15, 106.21, 115.27 (2C), 115.91 (2C), 124.01 (2C), 127.60 (2C), 130.43, 131.45, 149.53, 152.64, 153.84, 156.62, 158.10. HPLC analysis (method B): retention time = 10.811; peak area 99% (254 nm). HRMS: m/z for C17H15N2O2 [M + H]+ calculated: 279.11335, found: 279.11255.
3-(4-((4-Hydroxyphenyl)amino)pyridin-2-yl)phenol 15. Orange solid. 78% yield from 53. 1H-NMR (DMSO-d6) δ (ppm): 6.64 (dd, 1H, J = 5.7, 2.2 Hz), 6.74–6.82 (m, 3H), 7.05 (AA′XX′, 2H, JAX = 8.7 Hz, JAA′/XX′ = 2.7 Hz), 7.09 (d, 1H, J = 2.0 Hz), 7.19 (t, 1H, J = 7.8 Hz), 7.29 (dt, 1H, J = 7.8, 1.4 Hz), 7.33 (t, 1H, J = 2.0 Hz), 8.16 (d, 1H, J = 5.6 Hz), 8.46 (s, 1H), 9.32 (s, 1H), 9.46 (s, 1H). 13C-NMR (DMSO-d6) δ (ppm): 104.23, 106.91, 113.17, 115.57, 115.93 (2C), 116.98, 124.15 (2C), 129.50, 131.33, 140.99, 149.76, 152.71, 153.95, 156.60, 157.53. HPLC analysis (method B): retention time = 10.800; peak area 97% (254 nm). HRMS: m/z for C17H15N2O2 [M + H]+ calculated: 279.11335, found: 279.11124.
3-((5-(4-Hydroxyphenyl)pyridin-3-yl)amino)phenol 16. Yellow solid. 43% yield from 37. 1H-NMR (DMSO-d6) δ (ppm): 6.30 (ddd, 1H, J = 8.0, 2.2, 0.8 Hz), 6.55 (ddd, 1H, J = 8.0, 2.0, 0.8 Hz), 6.59 (t, 1H, J = 2.1 Hz), 6.87 (AA′XX′, 2H, JAX = 8.7 Hz, JAA′/XX′ = 2.5 Hz), 7.05 (t, 1H, J = 8.0 Hz), 7.48 (AA′XX′, 2H, JAX = 8.7 Hz, JAA′/XX′ = 2.5 Hz), 7.55 (t, 1H, J = 2.3 Hz), 8.20–8.25 (m, 2H), 8.31 (s, 1H), 9.30 (bs, 1H), 9.65 (bs, 1H). 13C-NMR (DMSO-d6) δ (ppm): 103.76, 107.87, 108.19, 115.90 (2C), 119.60, 127.96 (2C), 128.04, 130.07, 135.80, 137.28, 138.13, 139.99, 143.63, 157.65, 158.27. HPLC analysis (method B): retention time = 12.489; peak area 95% (254 nm). HRMS: m/z for C17H15N2O2 [M + H]+ calculated: 279.11335, found: 279.11194.
3-(5-((3-Hydroxyphenyl)amino)pyridin-3-yl)phenol 17. Light-yellow solid. 32% yield from 38. 1H-NMR (DMSO-d6) δ (ppm): 6.51 (ddd, 1H, J = 8.0, 2.2, 0.8 Hz), 6.73–6.81 (m, 2H), 7.00 (ddd, 1H, J = 8.1, 2.4, 0.9 Hz), 7.19 (t, 1H, J = 3.0 Hz), 7.22–7.29 (m, 2H), 7.48 (t, 1H, J = 7.9 Hz), 7.76 (t, 1H, J = 2.2 Hz), 8.43 (d, 1H, J = 1.9 Hz), 8.49 (d, 1H, J = 2.5 Hz), 8.56 (s, 1H), 9.52 (s, 1H), 9.81 (s, 1H). 13C-NMR (DMSO-d6) δ (ppm): 104.03, 108.11, 108.38, 113.53, 115.09, 117.55, 119.94, 130.12, 130.17, 135.93, 138.06, 138.35, 138.82, 140.16, 143.45, 157.92, 158.31. HPLC analysis (method B): retention time = 12.669; peak area 98% (254 nm). HRMS: m/z for C17H15N2O2 [M + H]+ calculated: 279.11335, found: 279.11169.
4-(5-((4-Hydroxyphenyl)amino)pyridin-3-yl)phenol 18. Orange-yellow solid. 20% yield from 39. 1H-NMR (acetone-d6) δ (ppm): 6.95 (AA′XX′, 2H, JAX = 8.8 Hz, JAA′/XX′ = 2.8 Hz), 7.03 (AA′XX′, 2H, JAX = 8.8 Hz, JAA′/XX′ = 2.6 Hz), 7.26 (AA′XX′, 2H, JAX = 8.9 Hz, JAA′/XX′ = 2.8 Hz), 7.65 (AA′XX′, 2H, JAX = 8.9 Hz, JAA′/XX′ = 2.6 Hz), 8.13 (dd, 1H, J = 2.4, 1.7 Hz), 8.18 (d, 1H, J = 2.4 Hz), 8.30 (s, 1H). 13C-NMR (acetone-d6) δ (ppm): 117.13, 117.26, 117.30, 123.44, 124.94, 125.86 (2C), 126.80, 129.19, 129.33, 129.47, 129.59, 131.39, 147.44, 154.19, 156.19, 160.10. HPLC analysis (method A): retention time = 12.022; peak area 99% (254 nm). HRMS: m/z for C17H15N2O2 [M + H]+ calculated: 279.11335, found: 279.11249.
3-(5-((4-Hydroxyphenyl)amino)pyridin-3-yl)phenol 19. Light-yellow solid. 31% yield from 40. 1H-NMR (DMSO-d6) δ (ppm): 6.75 (AA′XX′, 2H, JAX = 8.8 Hz, JAA′/XX′ = 2.9 Hz), 6.78 (ddd, 1H, J = 8.1, 2.4, 0.8 Hz), 6.93 (t, 1H, J = 2.0 Hz), 6.98–7.03 (m, 3H), 7.25 (t, 1H, J = 7.9 Hz), 7.29 (t, 1H, J = 2.2 Hz), 8.01 (s, 1H), 8.09 (d, 1H, J = 1.2 Hz), 8.14 (d, 1H, J = 2.3 Hz), 9.16 (s, 1H), 9.57 (s, 1H). 13C-NMR (DMSO-d6) δ (ppm): 113.42, 114.96, 115.64, 115.96 (2C), 117.01, 117.45, 122.35 (2C), 130.12, 132.91, 135.95, 136.68, 139.09, 142.45, 152.95, 157.85. HPLC analysis (method B): retention time = 12.378; peak area 97% (254 nm). HRMS: m/z for C17H15N2O2 [M + H]+ calculated: 279.11335, found: 279.11276.
3-((4-(4-Hydroxyphenyl)pyridin-2-yl)amino)phenol 20. Yellow solid. 44% yield from 41. 1H-NMR (DMSO-d6) δ (ppm): 6.25–6.33 (m, 1H), 6.84–6.92 (m, 2H), 6.93–7.05 (m, 4H), 7.28 (s, 1H), 7.49–7.57 (m, 2H), 8.13 (d, 1H, J = 5.4 Hz), 8.90 (s, 1H), 9.19 (bs, 1H), 9.79 (bs, 1H). 13C-NMR (DMSO-d6) δ (ppm): 105.11, 106.78, 107.67, 109.17, 111.75, 115.93 (2C), 127.74 (2C), 128.47, 129.21, 142.92, 147.82, 148.12, 156.64, 157.68, 158.45. HPLC analysis (method B): retention time = 12.767; peak area 97% (254 nm). HRMS: m/z for C17H15N2O2 [M + H]+ calculated: 279.11335, found: 279.11252.
3-(2-((3-Hydroxyphenyl)amino)pyridin-4-yl)phenol 21. Light-yellow solid. 63% yield from 42. 1H-NMR (DMSO-d6) δ (ppm): 6.27–6.34 (m, 1H), 6.85 (ddd, 1H, J = 8.0, 2.4, 0.9 Hz), 6.95 (dd, 1H, J = 5.4, 1.5 Hz), 6.99–7.06 (m, 4H), 7.10 (ddd, 1H, J = 7.7, 1.7, 1.0 Hz), 7.27–7.33 (m, 2H), 8.18 (d, 1H, J = 5.3 Hz), 8.96 (s, 1H), 9.20 (s, 1H), 9.66 (s, 1H). 13C-NMR (DMSO-d6) δ (ppm): 105.18, 107.70, 107.79, 109.21, 112.20, 113.22, 115.94, 117.22, 129.21, 130.18, 139.43, 142.77, 147.94, 148.40, 156.62, 157.68, 157.95. HPLC analysis (method B): retention time = 12.880; peak area 97% (254 nm). HRMS: m/z for C17H15N2O2 [M + H]+ calculated: 279.11335, found: 279.11252.
4-(2-((4-Hydroxyphenyl)amino)pyridin-4-yl)phenol 22. Yellow solid. 74% yield from 43. 1H-NMR (DMSO-d6) δ (ppm): 6.69 (AA′XX′, 2H, JAX = 8.9 Hz, JAA′/XX′ = 2.8 Hz), 6.84–6.90 (m, 4H), 7.40 (AA′XX′, 2H, JAX = 8.9 Hz, JAA′/XX′ = 2.7 Hz), 7.50 (AA′XX′, 2H, JAX = 8.7′ Hz, JAA′/XX′ = 2.5 Hz), 8.03–8.07 (m, 1H), 8.62 (s, 1H), 8.96 (s, 1H), 9.75 (bs, 1H). 13C-NMR (DMSO-d6) δ (ppm): 105.43, 110.89, 115.17 (2C), 115.86 (2C), 120.85 (2C), 127.66 (2C), 128.64, 133.35, 147.89, 147.98, 151.84, 157.28, 158.33. HPLC analysis (method B): retention time = 12.262; peak area 97% (254 nm). HRMS: m/z for C17H15N2O2 [M + H]+ calculated: 279.11335, found: 279.11108.
3-(2-((4-Hydroxyphenyl)amino)pyridin-4-yl)phenol 23. Light-yellow solid. 63% yield from 44. 1H-NMR (DMSO-d6) δ (ppm): 6.70 (AA′XX′, 2H, JAX = 8.8 Hz, JAA′/XX′ = 2.7 Hz), 6.80–6.90 (m, 3H), 7.01 (t, 1H, J = 2.0 Hz), 7.03–7.09 (m, 1H), 7.28 (t, 1H, J = 7.9 Hz), 7.40 (AA′XX′, 2H, JAX = 8.9 Hz, JAA′/XX′ = 2.7 Hz), 8.09 (d, 1H, J = 5.5 Hz), 8.68 (s, 1H), 8.98 (bs, 1H), 9.63 (bs, 1H). 13C-NMR (DMSO-d6) δ (ppm): 106.31, 111.34, 113.15, 115.20 (2C), 115.82, 117.17, 121.03 (2C), 130.13, 133.17, 139.61, 148.03, 148.29, 151.98, 157.29, 157.90. HPLC analysis (method B): retention time = 12.607; peak area 98% (254 nm). HRMS: m/z for C17H15N2O2 [M + H]+ calculated: 279.11335, found: 279.11240.
5.2 In vitro screening on isolated SIRT1 enzyme
The effects of the new compounds on SIRT1 activity were evaluated by a direct enzymatic assay using a SIRT1 Direct Fluorescent Screening Assay Kit (Cayman Chemical, Ann Arbor, MI, USA), following the protocol user guide. Before performing the test, the possible interferences of the compounds under examination with the fluorophore and/or developer were evaluated according to manufacturer's protocol. Briefly, the fluorescence was analyzed with the EnSpire spectrofluorometer (PerkinElmer, Waltham, MA, USA) at a wavelength of 350–360 nm in excitation and 450–465 nm in emission. The increase in recorded fluorescence was directly proportional to the activation of SIRT1. Data obtained were analyzed by removing baseline and normalizing to the fluorescence value vs. vehicle (DMSO 0.1%; corresponding to 0%) and the SIRT1 reference activator resveratrol 100 μM (named compound 1) (Sigma-Aldrich, St. Louis, MO, USA) at which was assigned the value of 100%.5.3 Docking calculations
The structure of compound 10 was generated using the latest version of MolBook UNIPI.30 The compound was docked into the structure of SIRT1 in complex with p53-AMC peptide and the parent diarylamine ligand 6 (Fig. 1) predicted in our previous study.14 The docking calculations were performed with GOLD software, using the GoldScore fitness function. The ligand was subjected to two different docking studies related to the two different receptor sub-sites (Fig. S49†) occupied by our previously published SIRT1 inhibitors.14,15 In each calculation, the docking site included all of the residues that stayed within a 10 Å shell from the specific bound ligand in the reference complex. The compound was subjected to 100 genetic algorithm runs, in which the “allow early termination” option was deactivated, while the possibility for the ligand to flip ring corners was activated. All other settings were left as their defaults. The root mean-squared deviation (RMSD) threshold for pose clustering was set to 2.0 Å. Only clusters of poses including more than 5% of solutions (more than 5 different poses) were considered. The best docked conformation belonging to the best cluster of solutions (top-scored pose) was considered for each docking study, whose results converged in the same binding mode.5.4 Molecular dynamics simulations
MD simulations were carried out using AMBER, version 20, using the ff14SB force field. General AMBER force field (GAFF) parameters were used for the ligand, whose partial charges were assigned using the Antechamber suite of AMBER 20, based on the AM1-BCC method. The predicted SIRT1–peptide-ligand complex was placed at the centre of a rectangular parallelepiped box and solvated with a 15 Å water cap, generated using TIP3P explicit solvent model. Sodium ions were added to neutralize the solvated system, which was then energy minimized using a two-step protocol. In the first step, only the minimization of the solvent was performed by applying a harmonic restraint of 100 kcal mol−1 Å−2 on the solute atoms. In the second step, 5000 cycles of steepest descent followed by conjugate gradient (CG) were used to minimize the whole system, until a reaching a convergence of 0.05 kcal Å−1 mol−1. The minimized complex was used as input for the MD simulations, which were run using particle mesh Ewald (PME) electrostatics, with a cut-off of 10 Å for the non-bonded interactions and periodic boundary conditions. The SHAKE algorithm was used to constrain all of the bonds involving hydrogen atoms and a time step of 2.0 fs was thus used for the simulations, following a protocol optimized from previous studies.31 Initially, a heating stage of 0.5 ns, in which the temperature of the system was raised from 0 to 300 K, was performed using constant-volume periodic boundary conditions. An equilibration stage of constant-pressure periodic boundary MD was run for 3 ns, keeping the temperature of the system at a constant value of 300 K using the Langevin thermostat. A second constant-pressure equilibration stage of 3 ns was then performed at 300 K for reaching an optimal relaxation of the ligand–protein binding conformation and full equilibration. This second equilibration stage was performed using the hydrogen mass repartition (HMR) scheme32 and a time step of 4.0 fs, since this technique proved to be useful for reducing the simulation time while maintaining an unbiased MD protocol.33,34 Finally, a 100 ns production step was performed, maintaining the same constant pressure and temperature conditions, using the HMR scheme and a time step of 4.0 fs. All the protein α carbons were subjected to a harmonic potential of 10 kcal mol−1 Å−2 during all MD stages. The final structure of the ternary SIRT1/p53-AMC/10 complex corresponded to the average of the last 50 ns of MD simulation minimized by the CG method until a convergence of 0.05 kcal mol−1 Å−2. The average structure was obtained using the Cpptraj program implemented in AMBER 20.5.5 In vivo acute myocardial infarction
Adult male Wistar rats (350–450 g) were housed in humidity and temperature controlled conditions (22 °C and 50%, respectively) with food and water ad libitum and exposed to 12 h:12 h light/dark cycles. The experimental protocol for coronary occlusion/reperfusion has been performed, according to European (EEC Directive 2010/63) and Italian (D.L. 4 March 2014 no. 26) legislation (protocol number 909/2016/PR, 29 June 2016), as previously published,14,19 with minor modifications. All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Pisa University and approved by the Animal Ethics Committee of Pisa. Briefly, two hours before the experimental procedures, rats received an i.p. injection of compounds 8 (at the dose 1 or 10 mg kg−1), 10 (at the dose 0.1 or 1 mg kg−1), compound 1 (as reference drug, at the dose 10 mg kg−1) or their vehicle (DMSO, 1 ml kg−1). Then, rats were anaesthetized with thiopental sodium (70 mg kg−1, i.p.). The trachea was intubated and connected to a rodent ventilator (mod. 7025 Ugo Basile, Comerio, Italy) for artificial ventilation with room air (stroke volume, 1 ml/100 g body weight; 70 strokes per min). Electrocardiogram (ECG) was continuously measured by lead II (Mindray, PM5000, 2 Biological Instruments, Varese-Italy). The chest was opened by a left thoracotomy. A 6–0 surgical needle was passed around the left anterior descending coronary artery (LAD), located between the base of the pulmonary artery and left atrium. The occlusion (LAD ligature) was induced by clamping a polypropylene tube (PE50) in which were inserted the ends of the suture. The successful occlusion was confirmed by observing regional cyanosis downstream of the ligature, and by ST elevation in the ECG recording. When the snare was released, the reperfusion was initiated. The acute infarct protocol consisted of 30 minutes of occlusion/120 minutes of reperfusion (IR). A group of vehicle-pretreated animals was submitted to an ischemic preconditioning (IPC) procedure, achieved by 3 cycles of 5 minutes of occlusion/10 minutes of reperfusion, followed by 30 minutes of coronary occlusion and 120 minutes of reperfusion. Moreover, another group of animals was assigned to sham procedure, consisting in the submitting them to all surgical procedures without occlusion/reperfusion. Each experimental group was composed of 5 animals. At the end of reperfusion, rats were euthanized by an overdose of thiopental sodium, then hearts were quickly excised, mounted on a Langendorff apparatus (Radnoti, California, USA) and perfused for about 10 minutes with Krebs solution at 37 °C to wash out the coronary blood vessels. Then, the atria and right ventricle were removed from the hearts, and the left ventricle was dried, frozen at −20 °C, and cut into 4–5 transverse slices from apex to base of equal thickness (about 2 mm). The slices were treated with 2,3,5-triphenyltetrazolium chloride (TTC) solution (1% w/v in phosphate buffer, pH 7.4) at 37 °C for 20 minutes. In the presence of dehydrogenase enzymes a reaction between TTC and the cofactor NADH occurs, forming an oxidized formazan derivative, which stains the viable cells with intense red color; while the dead cells remain of a light pink color. Then, the slices were fixed overnight in 10% formaldehyde and finally photographed, to planimetrically evaluate the extension of infarct area (AI/ALV %) (The GIMP 2). t-student test was selected as statistical analysis, and the difference between groups was considered statistically different when p < 0.05.
Author contributions
C. G. and F. M. designed the compounds, G. B. synthesized the compounds, L. T. and Vi. Ca. designed the biological experiments, Va. Ci. executed the biological experiments, L. T., Va. Ci., G. P. and T. T. designed the computational studies, G. P. executed the computational studies, C. G., G. B. and G. P. prepared the manuscript, A. M., F. M., T. T. and Vi. Ca. revised the manuscript, Vi. Ca. and F. M. supervised the work.Conflicts of interest
There is no conflict of interest to declare.Acknowledgements
This work was supported by the University of Pisa Grant “Epidrugs: una strategia innovativa nelle patologie cardiovascolari e neurodegenerative età-correlate”—grant number PRA_2022_60 (Va. Ci.).Notes and references
- A. A. Sauve and D. Y. Youn, Sirtuins: NAD(+)-dependent deacetylase mechanism and regulation, Curr. Opin. Chem. Biol., 2012, 16, 535–543, DOI:10.1016/j.cbpa.2012.10.003.
- L. Guarente, Sirtuins in aging and disease, Cold Spring Harbor Symp. Quant. Biol., 2007, 72, 483–488, DOI:10.1101/sqb.2007.72.024.
- N. Poulose and R. Raju, Sirtuin regulation in aging and injury, Biochim. Biophys. Acta, 2015, 1852, 2442–2455, DOI:10.1016/j.bbadis.2015.08.017.
- E. Pantazi, M. A. Zaouali, M. Bejaoui, E. Folch-Puy, H. Ben Abdennebi and J. Rosello-Catafau, Role of sirtuins in ischemia-reperfusion injury, World J. Gastroenterol., 2013, 19, 7594–7602, DOI:10.3748/wjg.v19.i43.7594.
- C. K. Singh, G. Chhabra, M. A. Ndiaye, L. M. Garcia-Peterson, N. J. Mack and N. Ahmad, The Role of Sirtuins in Antioxidant and Redox Signaling, Antioxid. Redox Signaling, 2018, 28, 643–661, DOI:10.1089/ars.2017.7290.
- M. Neri, I. Riezzo, N. Pascale, C. Pomara and E. Turillazzi, Ischemia/Reperfusion Injury following Acute Myocardial Infarction: A Critical Issue for Clinicians and Forensic Pathologists, Mediators Inflammation, 2017, 2017, 7018393, DOI:10.1155/2017/7018393.
- M. N. Sanz, L. Grimbert, M. Moulin, M. Gressette, C. Rucker-Martin, C. Lemaire, M. Mericskay, V. Veksler, R. Ventura-Clapier and A. Garnier, Inducible Cardiac-Specific Deletion of Sirt1 in Male Mice Reveals Progressive Cardiac Dysfunction and Sensitization of the Heart to Pressure Overload, Int. J. Mol. Sci., 2019, 20, 5005, DOI:10.3390/ijms20205005.
- Y. Kobayashi, Y. Furukawa-Hibi, C. Chen, Y. Horio, K. Isobe, K. Ikeda and N. Motoyama, SIRT1 is critical regulator of FOXO-mediated transcription in response to oxidative stress, Int. J. Mol. Med., 2005, 16, 237–243 CAS.
- G. Luo, Z. Jian, Y. Zhu, Y. Zhu, B. Chen, R. Ma, F. Tang and Y. Xiao, Sirt1 promotes autophagy and inhibits apoptosis to protect cardiomyocytes from hypoxic stress, Int. J. Mol. Med., 2019, 43, 2033–2043, DOI:10.3892/ijmm.2019.4125.
- M. T. Borra, B. C. Smith and J. M. Denu, Mechanism of human SIRT1 activation by resveratrol, J. Biol. Chem., 2005, 280, 17187–17195, DOI:10.1074/jbc.M501250200.
- M. Pacholec, J. E. Bleasdale, B. Chrunyk, D. Cunningham, D. Flynn, R. S. Garofalo, D. Griffith, M. Griffor, P. Loulakis and B. Pabst, SRT1720, SRT2183, SRT1460, and resveratrol are not direct activators of SIRT1, J. Biol. Chem., 2010, 285, 8340–8351, DOI:10.1074/jbc.M109.088682.
- L. Testai, E. Piragine, I. Piano, L. Flori, E. Da Pozzo, V. Miragliotta, A. Pirone, V. Citi, L. Di Cesare Mannelli, S. Brogi, C. Gheladini and V. Calderone, The Citrus Flavonoid Naringenin Protects the Myocardium from Ageing-Dependent Dysfunction: Potential Role of SIRT1, Oxid. Med. Cell. Longevity, 2020, 2020, 4650207, DOI:10.1155/2020/4650207.
- Z. Cui, X. Zhao, F. K. Amevor, X. Du, Y. Wang, D. Li, G. Shu, Y. Tian and X. Zhao, Therapeutic application of quercetin in aging-related diseases: SIRT1 as a potential mechanism, Front. Immunol., 2022, 13, 943321, DOI:10.3389/fimmu.2022.943321.
- G. Bononi, V. Citi, M. Lapillo, A. Martelli, G. Poli, T. Tuccinardi, C. Granchi, L. Testai, V. Calderone and F. Minutolo, Sirtuin 1-Activating Compounds: Discovery of a Class of Thiazole-Based Derivatives, Molecules, 2022, 27, 6535, DOI:10.3390/molecules27196535.
- G. Bononi, L. Flori, V. Citi, C. Acciai, V. Nocilla, A. Martelli, G. Poli, T. Tuccinardi, C. Granchi, L. Testai, V. Calderone and F. Minutolo, New Synthetic Analogues of Natural Polyphenols as Sirtuin 1-Activating Compounds, Pharmaceuticals, 2022, 15, 339, DOI:10.3390/ph15030339.
- S. De, S. K. A. Kumar, S. K. Shah, S. Kazi, N. Sarkar, S. Banerjee and S. Dey, Pyridine: the scaffolds with significant clinical diversity, RSC Adv., 2022, 12, 15385–15406, 10.1039/d2ra01571d.
- F. Perez and A. Minatti, Palladium-catalyzed C,N-cross coupling reactions of 3-halo-2-aminopyridines, Org. Lett., 2011, 13, 1984–1987, DOI:10.1021/ol200371n.
- M. D. Charles, P. Schultz and S. L. Buchwald, Efficient pd-catalyzed amination of heteroaryl halides, Org. Lett., 2005, 7, 3965–3968, DOI:10.1021/ol0514754.
- L. Testai, A. Martelli, A. Marino, V. D'Antongiovanni, F. Ciregia, L. Giusti, A. Lucacchini, S. Chericoni, M. C. Breschi and V. Calderone, The activation of mitochondrial BK potassium channels contributes to the protective effects of naringenin against myocardial ischemia/reperfusion injury, Biochem. Pharmacol., 2013, 85, 1634–1643, DOI:10.1016/j.bcp.2013.03.018.
- V. Calderone, L. Testai, A. Martelli, S. Rapposelli, M. Digiacomo, A. Balsamo and M. C. Breschi, Anti-ischemic properties of a new spiro-cyclic benzopyran activator of the cardiac mito-KATP channel, Biochem. Pharmacol., 2010, 79, 39–47, DOI:10.1016/j.bcp.2009.07.017.
- H. Li, F. Zheng, Y. Zhang, J. Sun, F. Gao and G. Shi, Resveratrol, novel application by preconditioning to attenuate myocardial ischemia/reperfusion injury in mice through regulate AMPK pathway and autophagy level, J. Cell. Mol. Med., 2022, 26, 4216–4229, DOI:10.1111/jcmm.17431.
- C. Granchi and F. Minutolo, Activators of Sirtuin-1 and their Involvement in Cardioprotection, Curr. Med. Chem., 2018, 25, 4432–4456, DOI:10.2174/0929867325666180214115438.
- N. Salari, F. Morddarvanjoghi, A. Abdolmaleki, S. Rasoulpoor, A. A. Khaleghi, L. A. Hezarkhani, S. Shohaimi and M. Mohammadi, The global prevalence of myocardial infarction: a systematic review and meta-analysis, BMC Cardiovasc. Disord., 2023, 23, 206, DOI:10.1186/s12872-023-03231-w.
- L. Testai, S. Rapposelli, A. Martelli, M. C. Breschi and V. Calderone, Mitochondrial potassium channels as pharmacological target for cardioprotective drugs, Med. Res. Rev., 2015, 35, 520–553, DOI:10.1002/med.21332.
- S. Cano-Carrillo, E. Lozano-Velasco, J. M. Castillo-Casas, C. Sanchez-Fernandez and D. Franco, The Role of ncRNAs in Cardiac Infarction and Regeneration, J. Cardiovasc. Dev. Dis., 2023, 10, 123, DOI:10.3390/jcdd10030123.
- H. Li, F. Zheng, Y. Zhang, J. Sun, F. Gao and G. Shi, Resveratrol, novel application by preconditioning to attenuate myocardial ischemia/reperfusion injury in mice through regulate AMPK pathway and autophagy level, J. Cell. Mol. Med., 2022, 26, 4216–4229, DOI:10.1111/jcmm.17431.
- T. Li, Y. Tan, S. Ouyang, J. He and L. Liu, Resveratrol protects against myocardial ischemia-reperfusion injury via attenuating ferroptosis, Gene, 2022, 808, 145968, DOI:10.1016/j.gene.2021.145968.
- G. Xu, X. Zhao, J. Fu and X. Wang, Resveratrol increase myocardial Nrf2 expression in type 2 diabetic rats and alleviate myocardial ischemia/reperfusion injury (MIRI), Ann. Palliat. Med., 2019, 8, 565–575, DOI:10.21037/apm.2019.11.25.
- N. Fourny, C. Lan, E. Seree, M. Bernard and M. Desrois, Protective Effect of Resveratrol against Ischemia-Reperfusion Injury via Enhanced High Energy Compounds and eNOS-SIRT1 Expression in Type 2 Diabetic Female Rat Heart, Nutrients, 2019, 11, 105, DOI:10.3390/nu11010105.
- S. Galati, M. Di Stefano, M. Macchia, G. Poli and T. Tuccinardi, MolBook UNIPI—Create, Manage, Analyze, and Share Your Chemical Data for Free, J. Chem. Inf. Model., 2023, 63, 3977, DOI:10.1021/acs.jcim.3c00278.
- C. Granchi, G. Bononi, R. Ferrisi, E. Gori, G. Mantini, S. Glasmacher, G. Poli, S. Palazzolo, I. Caligiuri, F. Rizzolio, V. Canzonieri, T. Perin, J. Gertsch, A. Sodi, E. Giovannetti, M. Macchia, F. Minutolo, T. Tuccinardi and A. Chicca, Design, synthesis and biological evaluation of second-generation benzoylpiperidine derivatives as reversible monoacylglycerol lipase (MAGL) inhibitors, Eur. J. Med. Chem., 2021, 209, 112857, DOI:10.1016/j.ejmech.2020.112857.
- C. W. Hopkins, S. Le Grand, R. C. Walker and A. E. Roitberg, Long-Time-Step Molecular Dynamics through Hydrogen Mass Repartitioning, J. Chem. Theory Comput., 2015, 11, 1864–1874, DOI:10.1021/ct5010406.
- G. Poli, I. Barravecchia, G. C. Demontis, A. Sodi, A. Saba, S. Rizzo, M. Macchia and T. Tuccinardi, Predicting potentially pathogenic effects of hRPE65 missense mutations: a computational strategy based on molecular dynamics simulations, J. Enzyme Inhib. Med. Chem., 2022, 37, 1765, DOI:10.1080/14756366.2022.2090547.
- G. Poli, G. C. Demontis, A. Sodi, A. Saba, S. Rizzo, M. Macchia and T. Tuccinardi, An in silico toolbox for the prediction of the potential pathogenic effects of missense mutations in the dimeric region of hRPE65, J. Enzyme Inhib. Med. Chem., 2023, 38, 2162047, DOI:10.1080/14756366.2022.2162047.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3md00489a |
| ‡ These authors equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2024 |