
Impact of dipeptide on ADC physicochemical properties and efficacy identifies Ala–Ala as the optimal dipeptide†
Lu
Wang
 *a,
Adrian D.
Hobson
a,
Julia
Fitzgibbons
a,
Axel
Hernandez
Jr.
a,
Ying
Jia
a,
Zhou
Xu
b,
Zhongyuan
Wang
b,
Yajie
Yu
b and
Xiang
Li
b
*a,
Adrian D.
Hobson
a,
Julia
Fitzgibbons
a,
Axel
Hernandez
Jr.
a,
Ying
Jia
a,
Zhou
Xu
b,
Zhongyuan
Wang
b,
Yajie
Yu
b and
Xiang
Li
b
aAbbVie Bioresearch Center, 381 Plantation Street, Worcester, Massachusetts 01605, USA. E-mail: lu.wang@abbvie.com
bWuXi AppTec, 168 Nanhai Road, Tianjin Economic-Technological Development Area TEDA, TJS 300457, China
First published on 27th November 2023
Abstract
Side chains of natural occurring amino acids vary greatly in terms of charge state, polarity, size and hydrophobicity. Using a linear synthetic route, two amino acids were sequentially coupled to a potent glucocorticoid receptor modulator (GRM) to afford a library of dipeptide-GRM linker payloads with a range of in silico properties. The linker payloads were conjugated to a mouse anti-TNF antibody through interchain disulfide Cys. Impact of various dipeptide linkers on ADC physical properties, including solubility, hydrophobicity, and aggregation were evaluated and the in silico properties pI, Log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P and tPSA of the linker drugs used to correlate with these properties. ADCs were screened in a GRE luciferase reporter assay to compare their in vitro efficacy. Data identified Ala–Ala as a superior dipeptide linker that allowed a maximum drug load of 10 while affording ADCs with low aggregation.
P and tPSA of the linker drugs used to correlate with these properties. ADCs were screened in a GRE luciferase reporter assay to compare their in vitro efficacy. Data identified Ala–Ala as a superior dipeptide linker that allowed a maximum drug load of 10 while affording ADCs with low aggregation.
Introduction
Antibody-drug conjugates (ADC), combining the benefits of the high specificity for disease-associated antigen from a monoclonal antibody and therapeutic effects from a drug (payload), seek to increase the therapeutic index of a small molecule drug by delivering an efficacious dose to targeted tissues while minimising systemic exposure of the free drug. The linker which connects the antibody with the therapeutic drug is a key component of an ADC. Linker choice determines ADC stability and intracellular payload release, which ultimately affects efficacy and therapeutic index of the ADC.1 An optimal ADC linker should be stable in the circulation and then effectively release the drug only at the intended site. Two types of linkers, non-cleavable and cleavable depending on their metabolic fate in vitro, are currently employed by ADCs in the clinic.2 ADCs that have a non-cleavable linker show superior stability in vivo. Their mechanism of action relies on proteolytic degradation of the antibodies to release the payload in a modified form (with linker and additional amino acid from the antibody still attached), which alters the properties of the payload. Of the 13 FDA-approved ADCs, trastuzumab emtansine and belantamab mafodotin, the latter being withdrawn in November 2022, are the only two ADCs utilising a non-cleavable linker. In contrast, ADCs with cleavable linkers can release the unmodified payload by responding to changes in the local environment following internalization to the cell. Hydrazone linkers are acid-labile, utilising the acidic environment found in the endosome (pH 5.5–6.5) and lysosome (pH 4.5) compartments to release the payload.3 Their application has been used with gemtuzumab ozogamicin and inotuzumab ozogamicin. Dipeptide linkers,4 designed to be selectively cleaved by lysosomal proteases, have gained significant attention in the development of ADCs due to their improved plasma stability compared to hydrazone linkers. For instance, an ADC with a Val–Cit dipeptide linker,5 cleaved by cathepsin B, was shown to be >100 times more stable than a hydrazone linked ADC in human plasma.6 Val–Cit, in conjunction with the self-immolative p-amino benzyl carbamate (PABC) group has been successfully implemented in brentuximab vedotin, polatuzumab vedotin, enfortumab vedotin and tisotumab vedotin using the microtubulin inhibitor payload monomethyl auristatin E (MMAE).7 Another dipeptide linker, Val–Ala, has been used in loncastuximab tesirine with the DNA crosslinking payload SG3199.8 Other types of cleavable linkers, such as glucuronide9–11 and disulfide,12,13 have also been reported in the literature.Both the linker and the payload can contribute to an increase in hydrophobicity observed for the ADC compared to the parental antibody.14,15 Decrease in thermal stability, aggregation formation and accelerated in vivo clearance have all been suggested to arise from the increased hydrophobicity of the ADC. While limiting the average DAR to a lower range of 2–4 can help to attenuate the negative impact on hydrophobicity, this approach is less feasible with moderately active payloads or targets with a low cell surface expression level. Alternatively, implementation of more hydrophilic linkers can be a viable strategy to reduce overall ADC hydrophobicity by offsetting the hydrophobicity associated with the payload. Work has been done to incorporate various hydrophilic moieties into the linker with different payloads, leading to ADCs with improved physicochemical properties,16 PK profiles,17–19 or therapeutic index.20,21 By introduction of a charged sulfonate group or a short polyethylene glycol (PEG) spacer in a linker it was demonstrated that maytansinoid ADCs with higher DAR could be prepared which showed an enhancement of in vitro potency without sacrificing their aggregation.22 Similarly, tethering a linear PEG24 in different locations of a glucuronide cleavable linker paired with an MMAE payload it was found that the PK of the resulting ADCs paralleled the trends in their apparent hydrophobicity as measured by hydrophobic interaction chromatography (HIC) retention time.23 These studies suggested that hydrophilic PEG24 appended to the side chain of the linker provided good shielding of the payload hydrophobicity.
Peptide linkers are protease cleavable and constructed from amino acids. Side chains of naturally occurring amino acids vary in terms of charge state, polarity, size, and hydrophobicity. Combination of different amino acids provides an efficient way to modulate the linker properties, and hence influence the properties of resulting ADCs as well. In this study, we synthesised a library of dipeptide-linked glucocorticoid receptor modulator (GRM) payloads and conjugated them to a mouse anti-TNF antibody. These ADCs were then used to investigate the impact of the various dipeptide linkers on ADC physical properties including solubility, hydrophobicity and aggregation. In silico properties of the linker drugs were used to correlate with these measured properties. ADCs were further screened in a GRE luciferase reporter assay to compare their in vitro efficacy. Knowledge gained from this work will contribute to the understanding of the impact from a specific amino acid to the overall ADC properties and help with future peptide linker design.
Results and discussion
Previous work in our group has identified compound C1 as a highly potent glucocorticoid receptor modulator.24 The aniline nitrogen in compound C1 provides a suitable site for linker attachment. To secure access to a diverse set of dipeptides, amino acids in P1 and P2 positions of the linker were selected from Gly, Ala, Phe, Val, Ser, Asn, Gln, His, Cit, Glu, Lys and Arg to cover changes in size, polarity, charge, and hydrophobicity (Fig. 1). The two amino acids were attached to compound C1 sequentially using standard amide-bond coupling procedures. Since the undesired oxidation of the hydroxy at C21 to the carboxylic acid (affording an inactive compound) and minor epimerization of the amino acids side chains were found to be associated with deprotection of Fmoc under basic conditions,25 the use of amino acids protected with the acid labile Boc group was preferred for synthesis. Pairing them with TFA deprotection minimised both the unwanted carboxylic acid formation and epimerization. When the amino acid carried a reactive side chain (OH, COOH, NH2, imidazole or guanidine) that could interfere with the amide coupling reaction, Fmoc-protected amino acids were used, while the side chain group was protected with a Boc group to afford an orthogonally acid labile protecting group. Subsequent removal of the Fmoc group with piperidine released the amine to facilitate further coupling reactions, while the Boc protecting group on the amino acid side chain remained intact. After sequential installation of two amino acids of choice, reaction with bromoacetic acid afforded the desired drug linker C4 (Scheme 1). One more deprotection step with TFA was executed on compound C4 if an amino acid side chain had a protecting group. The bromoacetamide portion of drug linker C4 provides a site for Cys conjugation and serves as a stable attachment between the antibody and dipeptide linker. All the synthesised linker drugs and their calculated properties are summarised in Table 1.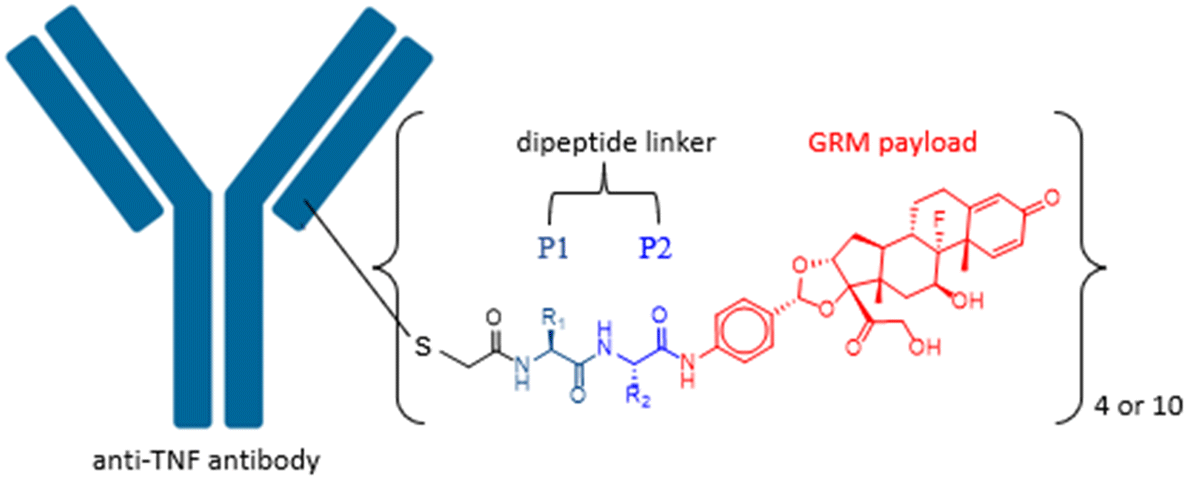 | ||
| Fig. 1 Anti-TNF GRM ADC with a dipeptide linker. | ||
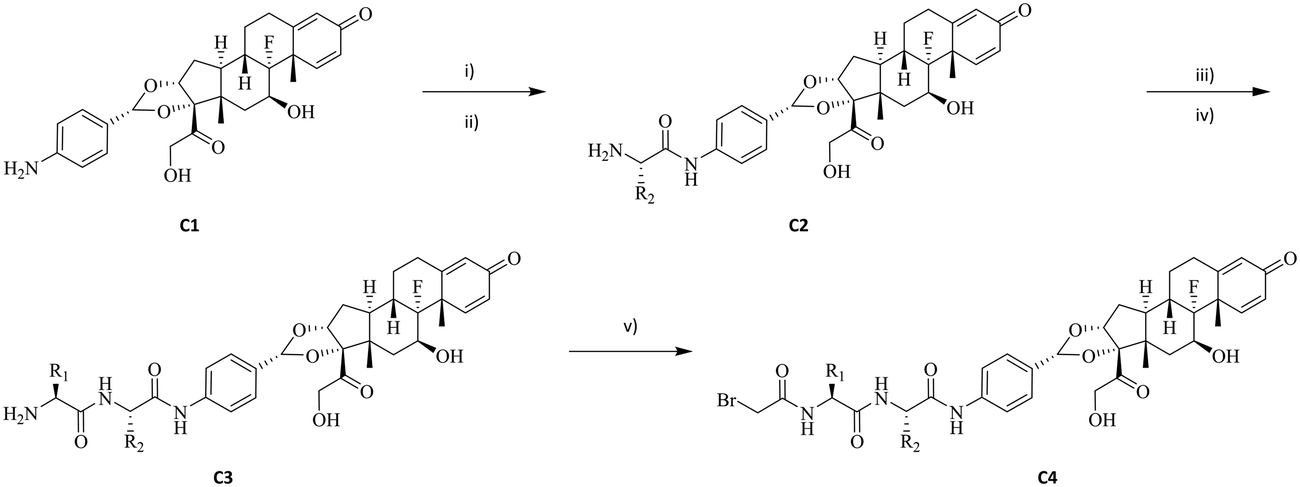 | ||
| Scheme 1 Synthetic route for the bromoacetamide–dipeptide-GRM. Regents and condition: (i) Boc-protected or Fmoc-protected amino acid in P2 position, BEP, DIEA, DMF; (ii) TFA (for Boc-protected amino acid) or piperidine (for Fmoc-protected amino acid), DCM; (iii) Boc-protected or Fmoc-protected amino acid in P1 position, BEP, DIEA, DMF; (iv) TFA (for Boc-protected amino acid) or piperidine (for Fmoc-protected amino acid), DCM; (v) 2-bromoacetic acid, BEP, DIEA, DMF. | ||
| Linker drug | P1 | P2 | cLogP |
cLogDa |
tPSA/Å2 |
|---|---|---|---|---|---|
| a Data calculated at pH 7.4. | |||||
| LD1 | Ala | Ala | 2.3 | 2.3 | 180 |
| LD2 | Ala | Arg | 0.8 | −1.0 | 242 |
| LD3 | Ala | Gln | 1.1 | 1.1 | 223 |
| LD4 | Ala | Glu | 1.9 | −1.2 | 218 |
| LD5 | Ala | Gly | 1.7 | 1.7 | 180 |
| LD6 | Ala | His | 1.9 | 1.8 | 209 |
| LD7 | Ala | Lys | 2.0 | −0.4 | 206 |
| LD8 | Ala | Ser | 1.2 | 1.2 | 201 |
| LD9 | Ala | Asn | 0.8 | 0.8 | 223 |
| LD10 | Ala | Val | 3.2 | 3.2 | 180 |
| LD11 | Ala | Phe | 4.0 | 4.0 | 180 |
| LD12 | Gly | Ala | 1.7 | 1.7 | 180 |
| LD13 | Gly | Gln | 0.6 | 0.6 | 223 |
| LD14 | Gly | Glu | 1.4 | −1.8 | 218 |
| LD15 | Gly | Gly | 1.2 | 1.2 | 180 |
| LD16 | Gly | Ser | 0.7 | 0.7 | 201 |
| LD17 | Phe | Lys | 3.6 | 1.2 | 206 |
| LD18 | Ser | Ala | 1.2 | 1.2 | 201 |
| LD19 | Ser | Asn | −0.2 | −0.2 | 244 |
| LD20 | Ser | Gln | 0.1 | 0.1 | 244 |
| LD21 | Ser | Glu | 0.9 | −2.4 | 238 |
| LD22 | Ser | Gly | 0.7 | 0.7 | 201 |
| LD23 | Val | Ala | 3.2 | 3.2 | 180 |
| LD24 | Val | Arg | 1.6 | −0.1 | 242 |
| LD25 | Val | Cit | 2.1 | 2.1 | 235 |
| LD26 | Val | Asn | 1.7 | 1.7 | 223 |
| LD27 | Val | Gln | 2.0 | 2.0 | 223 |
| LD28 | Val | Glu | 2.8 | −0.3 | 218 |
| LD29 | Val | Gly | 2.6 | 2.6 | 181 |
| LD30 | Val | Ser | 2.1 | 2.1 | 201 |
A total of 30 linker drugs were conjugated to a monoclonal mouse anti-TNF antibody to generate two sets of ADCs with different drug-to-antibody ratios (DARs) by using an appropriate reducing reagent as shown in Scheme 2. The mouse anti-TNF IgG2a antibody,26 has five interchain disulfides. Three of the interchain disulfides are between the heavy chains in the hinge region and one between light chain and heavy chain in each Fab region. A partial selective reduction of the antibody with a mild reducing reagent, DPhPEA, followed by conjugation with LD1–30 at pH ∼8.5 afforded the first set of ADCs (ADC1–ADC27) with an average DAR of around 4. Even though this afforded ADCs with a heterogeneous DAR, the DAR4 species was highly enriched in which conjugation occurred on Cys that previously formed the interchain disulfides between light chain and heavy chain. Alternatively, a maximal drug load of 10 was achieved followed by full reduction of all five interchain disulfides in the mouse anti-TNF antibody with TCEP (ADC28–ADC56).
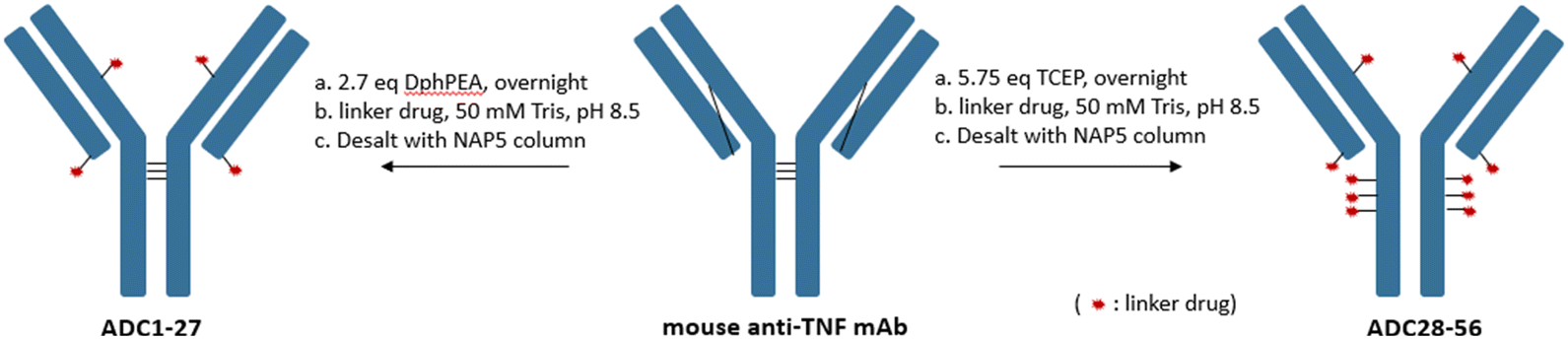 | ||
| Scheme 2 Generation of DAR4 (DPhPEA) and DAR10 (TCEP) ADCs. | ||
Effects of the linker-payloads on the ADC properties, including retention time by HIC, aggregation by size exclusion chromatography (SEC) and sample recovery after NAP column desalting are summarised in Tables 2 and 3. With the same linker drug, longer retention time on HIC, higher aggregation and lower sample recovery were consistently observed for ADCs with a DAR value of 10 versus 4. This is not surprising as the linker drugs are hydrophobic by nature and so the more linker drugs conjugated to an antibody, the greater the changes incurred to the ADCs.
| ADC | Linker | DAR | DAR4 peak HIC RT/min | Agg/% |
|---|---|---|---|---|
| Ab | — | 0 | 3.37 | 1.6 |
| ADC1 | Ala–Ala | 3.8 | 4.90 | 1.0 |
| ADC2 | Ala–Arg | 3.6 | 4.52 | 1.1 |
| ADC3 | Ala–Gln | 3.7 | 4.83 | 1.8 |
| ADC4 | Ala–Glu | 3.3 | 4.65 | 1.3 |
| ADC5 | Ala–Gly | 3.7 | 4.77 | 2.1 |
| ADC6 | Ala–His | 3.5 | 4.90 | 1.7 |
| ADC7 | Ala–Lys | 2.4 | 4.18 | 1.9 |
| ADC8 | Ala–Ser | 4.2 | 4.82 | 1.4 |
| ADC9 | Gly–Ala | 3.8 | 4.80 | 1.7 |
| ADC10 | Gly–Gln | 3.7 | 4.71 | 1.3 |
| ADC11 | Gly–Glu | 3.2 | 4.57 | 1.9 |
| ADC12 | Gly–Gly | 3.6 | 4.74 | 1.9 |
| ADC13 | Gly–Ser | 3.7 | 4.69 | 1.3 |
| ADC14 | Phe–Lys | 3.7 | 4.68 | 3.1 |
| ADC15 | Ser–Ala | 3.8 | 4.87 | 1.8 |
| ADC16 | Ser–Asn | 3.5 | 4.72 | 1.5 |
| ADC17 | Ser–Gln | 3.6 | 4.79 | 1.1 |
| ADC18 | Ser–Glu | 3.5 | 4.63 | 1.7 |
| ADC19 | Ser–Gly | 3.7 | 4.77 | 1.8 |
| ADC20 | Val–Ala | 3.8 | 5.10 | 1.3 |
| ADC21 | Val–Arg | 3.4 | 4.73 | 1.7 |
| ADC22 | Val–Cit | 3.6 | 4.98 | 1.2 |
| ADC23 | Val–Asn | 3.5 | 4.90 | 1.1 |
| ADC24 | Val–Gln | 4.1 | 5.02 | 1.3 |
| ADC25 | Val–Glu | 3.4 | 4.79 | 2.0 |
| ADC26 | Val–Gly | 3.6 | 4.99 | 1.3 |
| ADC27 | Val–Ser | 3.7 | 4.98 | 1.5 |
| ADC | Dipeptide | DAR | DAR10 peak HIC RT/min | Agg/% | ADC yield/% |
|---|---|---|---|---|---|
| a Not measured due to low sample recovery. | |||||
| Ab | — | 0.0 | 3.37 | 1.6 | — |
| ADC28 | Ala–Ala | 10 | 6.52 | 2.8 | 55.3 |
| ADC29 | Ala–Arg | 9.9 | 5.45 | 20.5 | 80.8 |
| ADC30 | Ala–Gln | 9.9 | 6.38 | 3.1 | 9.9 |
| ADC31 | Ala–Glu | NAa | NAa | NAa | 2.5 |
| ADC32 | Ala–Gly | 10 | 6.49 | 7.9 | 60.9 |
| ADC33 | Ala–His | 9.6 | 6.33 | 13.5 | 70.8 |
| ADC34 | Ala–Ser | 10 | 6.42 | 5.0 | 53.1 |
| ADC35 | Ala–Asn | 10 | 6.45 | 6.3 | 29.9 |
| ADC36 | Ala–Val | 10 | 6.92 | 13.6 | 49.7 |
| ADC37 | Ala–Phe | 9.8 | 7.42 | 44.7 | 43.4 |
| ADC38 | Gly–Ala | 10 | 6.29 | 7.7 | 51.0 |
| ADC39 | Gly–Gln | 10 | 6.12 | 3.5 | 18.2 |
| ADC40 | Gly–Glu | NAa | NAa | NAa | 1.9 |
| ADC41 | Gly–Gly | 10 | 6.26 | 8.2 | 54.1 |
| ADC42 | Gly–Ser | 10 | 6.14 | 6.2 | 62.1 |
| ADC43 | Phe–Lys | 9.8 | 5.72 | 50.9 | 70.5 |
| ADC44 | Ser–Ala | 10 | 6.48 | 4.9 | 47.6 |
| ADC45 | Ser–Asn | 9.6 | 6.19 | 12.2 | 38.3 |
| ADC46 | Ser–Gln | 9.6 | 6.29 | 8.4 | 16.1 |
| ADC47 | Ser–Glu | NAa | NAa | NAa | 0.6 |
| ADC48 | Ser–Gly | 10 | 6.33 | 6.1 | 43.2 |
| ADC49 | Val–Ala | 10 | 6.74 | 12.9 | 45.7 |
| ADC50 | Val–Arg | 9.6 | 5.77 | 36.4 | 68.9 |
| ADC51 | Val–Cit | 9.9 | 6.54 | 21.5 | 54.4 |
| ADC52 | Val–Asn | 9.8 | 6.46 | 13.9 | 40.8 |
| ADC53 | Val–Gln | 10 | 6.66 | 5.8 | 10.2 |
| ADC54 | Val–Glu | NAa | NAa | NAa | 1.2 |
| ADC55 | Val–Gly | 10 | 6.45 | 7.6 | 44.5 |
| ADC56 | Val–Ser | 10 | 6.38 | 9.1 | 41.4 |
ADC recovery data in Table 3 helped to identify any significant changes in ADC solubility that occurred either in the conjugation media or formulation buffer. ADCs in Table 2 with a DAR value of ∼4 had a consistent recovery of around 80%. In comparison, the set of DAR10 ADCs in Table 3 had much lower recovery in general (30–50%). This could be explained by the cumulative hydrophobicity from the increased number of linker drugs negatively impacting the aqueous solubility of the resulting ADC. However, 4 ADCs (ADC31, ADC40, ADC47, ADC54) were found to have extremely low recovery (<2%), even though the conjugation mixture remained clear before desalting with a NAP column. Interestingly, dipeptide linkers for these ADCs were not the most hydrophobic. Instead, Ser–Glu, linker for ADC47, was the most hydrophilic dipeptide synthesised in the study and showed the lowest recovery (0.5%). On the other hand, ADC49 with the most hydrophobic dipeptide linker, Val–Ala, had a recovery of 45.7% which was on par with the rest of the group. Clearly, linker hydrophobicity cannot rationalise these results. The four ADCs with essentially no sample recovery had Glu in the P2 position, an amino acid carrying a negative charge on its side chain. We reasoned that the additional charge from Glu caused a change in isoelectric point (pI) for corresponding ADCs. pI is an important property of a protein and affects its solubility at a given pH. Proteins tend to be the least soluble and precipitate near their pI. To approximate the pI value for a DAR4 or a DAR10 ADC, we performed the calculation by appending 4 or 10 such amino acids to the sequence of parental mouse anti-TNF antibody (Table 4). Addition of 10 Glu to the antibody resulted in a 1.6 drop in the calculated pI, from 8.72 to 7.12. During the desalting step with NAP columns, mixing conjugation buffer (pH 8.5) with storage buffer (pH 6.0) most likely dropped the pH close to the pI of the ADC and the resulting ADC precipitation was most likely the cause for the extremely low recovery seen on these ADCs. With the lower DAR4 ADCs, the pI value for the corresponding ADCs (ADC4, ADC14, ADC21, ADC28) was only lowered by ∼0.8 and solubilities were not impacted by this smaller change in pI.
| Modification | Calculated pI | Change in pI relative to parental antibody |
|---|---|---|
| Anti TNF | 8.72 | — |
| Anti TNF + 4 Gln | 8.72 | 0 |
| Anti TNF + 10 Gln | 8.72 | 0 |
| Anti TNF + 4 Glu | 7.96 | −0.8 |
| Anti TNF + 10 Glu | 7.12 | −1.6 |
| Anti TNF + 4 Lys | 9.10 | 0.4 |
| Anti TNF + 10 Lys | 9.40 | 0.7 |
| Anti TNF + 4 Arg | 9.12 | 0.4 |
| Anti TNF + 10 Arg | 9.42 | 0.7 |
Peptide linkers containing positively charged amino acids (Lys, Arg) also impacted the pI value of the ADCs by increasing it. However, they did not change the pI as significantly as the negatively charged amino acids did. For example, the pI change for 10 Arg was calculated to be only 0.7 higher than the parental antibody compared to the drop of 1.6 in pI for 10 Glu, while the calculated pIs for ADCs (ADC2, ADC7, ADC14 and ADC21 of DAR4 and ADC43, ADC50 of DAR10) were all above 9. Recovery for DAR10 ADCs (ADC29, ADC43, ADC50) containing Lys or Arg in the linker were between 68–80%, which was higher than the rest of ADCs and additionally, the extra positive charge did help improve the solubility of the ADCs.
Sample recoveries for another set of ADCs (ADC31, ADC40, ADC47, ADC54) containing Gln in P2 were on the lower end (<20%). While Gln being a hydrophilic and neutral amino acid, the observed low sample recovery could not be explained by either hydrophobicity or pI change of the ADC. On the other hand, recoveries for ADC35, ADC45 and ADC52 that had Asn in P2 were comparable to other ADCs in the group ADCs. We did not have a reasonable explanation for these results. Interestingly, the contribution of amino acids has on protein solubility in ammonium sulphate by single mutation of a surface exposed amino acid from a ribonuclease has been studied and it was shown that mutants with hydrophilic amino acids such as Asn and Gln showed even lower solubility than the more hydrophobic Ala mutant27 suggesting that amide groups from the amino acid side chains contributed unfavourably to protein solubility. This result is somewhat in line with our observation, even though our ADCs were formulated in histidine buffer rather than in ammonium sulphate. In addition, our Asn-containing ADCs did not seem to negatively impact solubility.
ADC hydrophobicity by HIC
Being a very important physical property, hydrophobicity often correlates well with ADC aggregation propensity and stability. HIC separates biomolecules based on their hydrophobicity28 with more hydrophobic ADCs being identified by a longer retention time by HIC. Hence, we used HIC retention time to rank order hydrophobicity for our ADCs. The lipophilicity of small molecules is commonly quantified as LogP (or LogD for ionizable compounds). Calculated LogP (cLogP) or calculated LogD (cLogD) of linker drugs was used to build a relationship with ADC hydrophobicity. Upon plotting ADC HIC retention time (Tables 2 and 3) against cLogP (or cLogD7.4) of its linker drug (Table 1) in Fig. 2, some correlation was observed with a R2 value of ∼0.6 for line fitting for both the DAR4 and DAR10 species. In general, linker drugs with a higher cLogP (or cLogD) resulted in higher hydrophobicity of the resulting ADCs. LD11, with the highest cLogP of 3.95 afforded ADC37, which had the longest retention time by HIC (7.42 min). Since all the ADCs we made had the same payload, we could assign any hydrophobicity change in the ADCs to the structural modifications in the dipeptide linkers. This shows that regardless of their small size relative to the larger payload the dipeptide linkers play a significant role in affecting the ADC hydrophobicity. In addition, the slope of the fitted line showed a 2.4-fold increase from DAR4 to DAR10, matching a 2.5-fold change in DAR value clearly demonstrating that the linker impact on ADC hydrophobicity was additive.
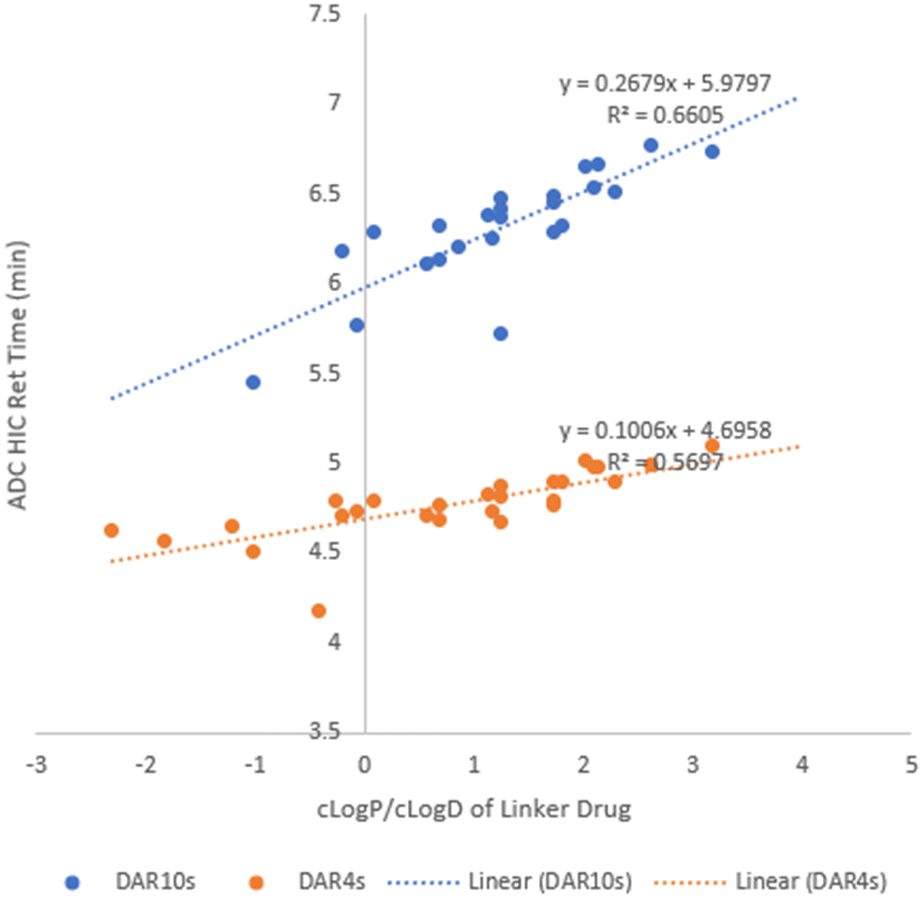 | ||
| Fig. 2 ADC HIC retention time vs. cLogP/cLogD of linker-payloads. | ||
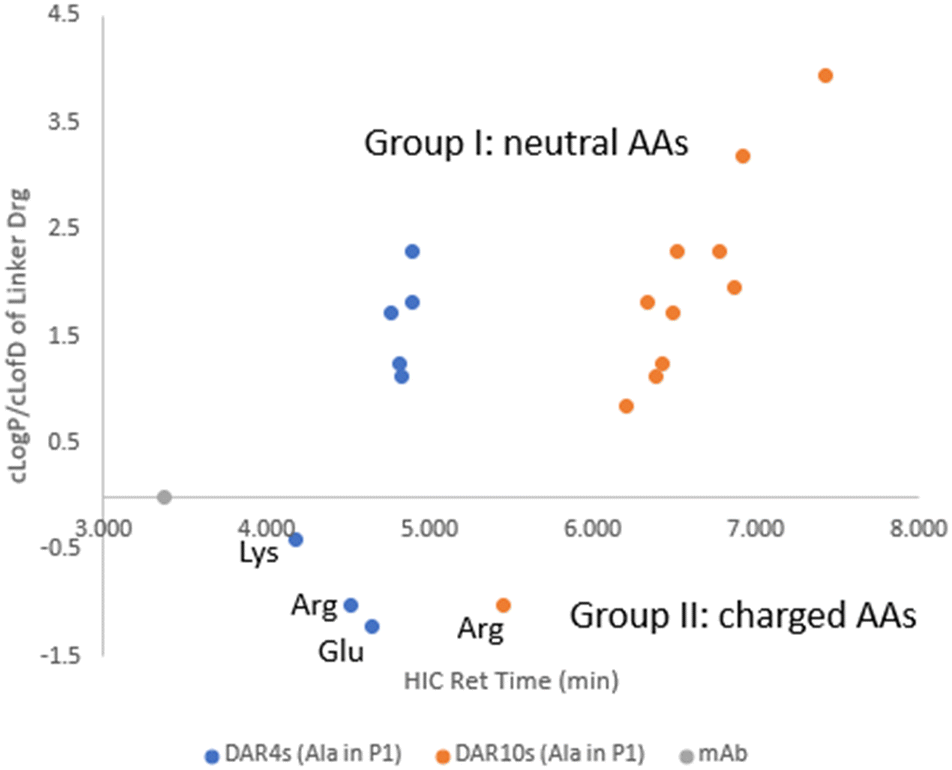
Two subsets of ADCs (ADC1–ADC8 with DAR4 and ADC28–ADC37 with DAR10) had an Ala–X linker, where X in P2 position of the dipeptide covered a variety of amino acids. A separate plot of cLogP/cLogD7.4 of linker drug against ADC HIC retention time allowed the evaluation of the contribution of the individual amino acids at position P2 had to the ADC hydrophobicity (Fig. 3). Compared to parental antibody, retention time of both sets of ADCs shifted to the right side on HIC, indicating an increase in hydrophobicity. These ADCs were grouped into two based on whether the side chain of amino acid X was charged under neutral pH. For group I X had a neutral side chain (Asn, Gln, Ser, Gly, His, Ala, Val and Phe) and for group II X had an ionizable side chain (Glu, Arg and Lys).
 | ||
| Fig. 3 cLogP/cLogD of linker-payload vs. ADC HIC retention time for ADCs with an Ala–X dipeptide linker. | ||
Within group I, the general trend existed that ADC HIC retention time correlated well with the cLogP of the linker drug. Within this group when X had a neutral polar side chain (Ser, Asn, Gln), more hydrophilic ADCs correlated with the lower cLogP of the linker drugs. However, contributions from these neutral polar groups were relatively small, especially for the DAR4 ADCs. For example, a decrease of 1.16 in cLogP from LD1 to LD3 only resulted in a decrease in HIC retention time of 0.07 min from ADC1 to ADC3 which are both DAR4, and a decrease of 0.14 min from ADC28 to ADC30 which are both DAR10. On the other hand, amino acids with large hydrophobic side chains (Val and Phe) had a much greater impact on increasing ADC hydrophobicity. HIC retention time was shifted to the more hydrophobic side by 0.4 min from ADC28 to ADC36 (both DAR10), resulting from a cLogP increase of 0.9 from LD1 to LD10.
Linker drugs in group II incorporated either a positive (Lys, Arg) or negative charge (Asp, Glu). These charged groups significantly lowered the cLogD of the linker drugs. Corresponding ADCs were indeed the most hydrophilic and separation of ADC hydrophobicity was more noticeable than with the ADCs in group I. A small increase of 0.2 in cLogD from LD4 to LD2 shifted the ADC to the more hydrophilic side by 0.13 min from ADC4 to ADC2 for DAR4. However, ADC HIC retention time did not correlate with cLogD of the linker drugs in group II with an ionizable side chain but was found to be related to the charge state of the side chains. Order of observed ADC hydrophobicity in this group followed Lys ≪ Arg < Glu for X in P2, which suggested that positively charged amino acids, in particularly Lys, reduced the ADC hydrophobicity more effectively than negatively charged amino acids.
ADC aggregation by SEC
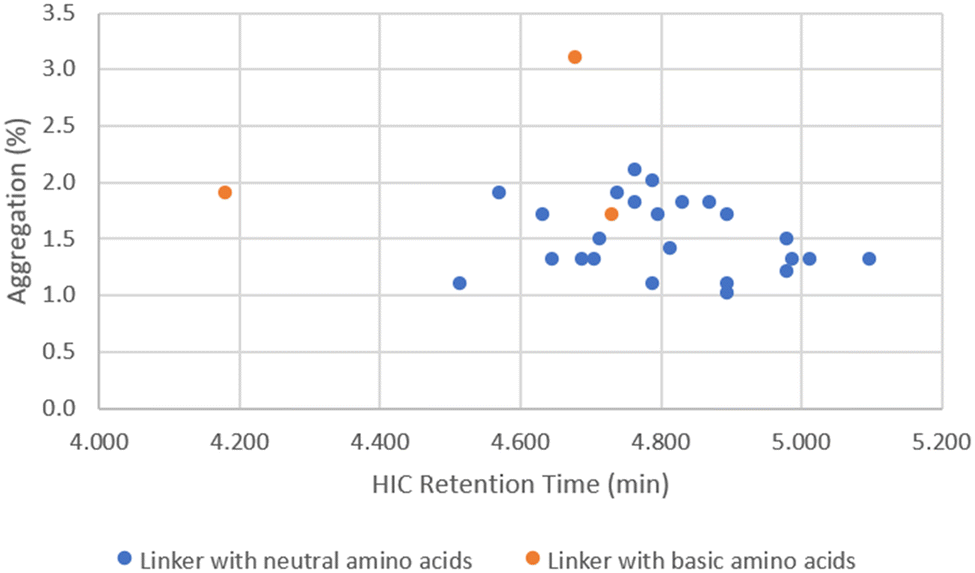
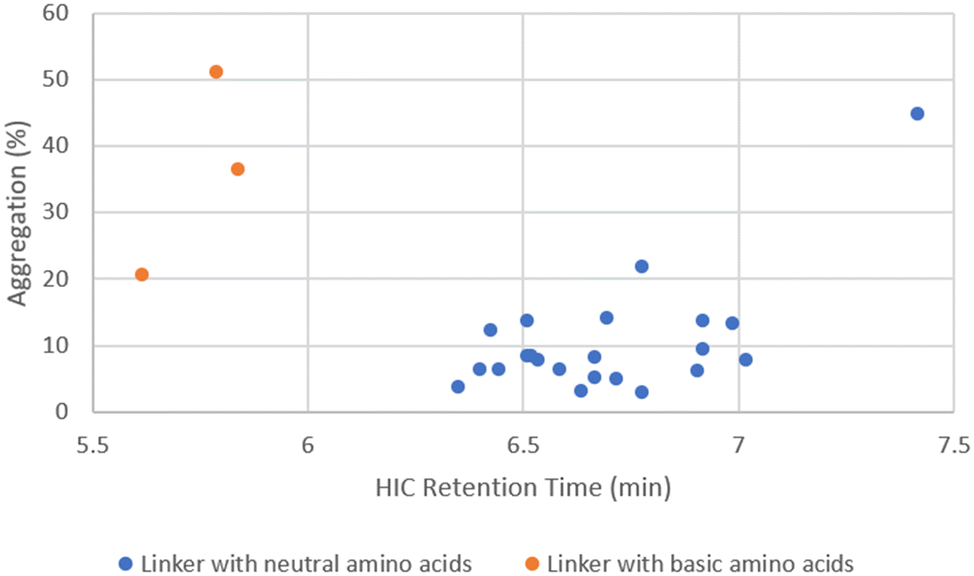
When DAR values were ∼4, ADCs showed little variation in aggregation level (1–3%), even though the cLogP/cLogD values of the linker drugs spanned over 5 units (Fig. 4). While the unconjugated antibody had 1.6% aggregation, the highest aggregation was only 3.1% with ADC14 which had a Phe–Lys linker. As the drug load was increased to 10, a larger separation in aggregation occurred. Upon plotting ADC aggregation against HIC retention time (an indication of ADC hydrophobicity), ADCs clustered in to two distinct groups (Fig. 5). One cluster of ADCs containing 3 ADCs (ADC29, ADC43 and ADC50) were to the left side of the graph, which represents lower cLogP and typically indicates the more hydrophilic ADCs. They were separated from the other cluster of ADCs by at least 0.5 min and were highly aggregated (>20%). This was counterintuitive as aggregation was often associated with increased hydrophobicity. Dipeptide linkers for ADC29, ADC43 and ADC50 were Phe–Lys, Ala–Arg and Val–Arg, respectively. A common feature they all share is a basic amino acid in the P2 position. This data suggested that ADC apparent hydrophobicity and aggregation did not always correlate. While linkers with positively charged amino acids afforded ADCs with lower hydrophobicity, they also resulted in higher levels of aggregation. Unfortunately, ADC31, ADC40, ADC47 and ADC50 with a linker containing Glu, another amino acid identified earlier to reduce hydrophobicity, had no recovery during preparation and so we were unable to evaluate the impact of acidic amino acid residues on ADC aggregation.
 | ||
| Fig. 4 ADC aggregation vs. HIC retention time for DAR4 ADCs. | ||
 | ||
| Fig. 5 ADC aggregation vs. HIC retention time for DAR10 ADCs. | ||
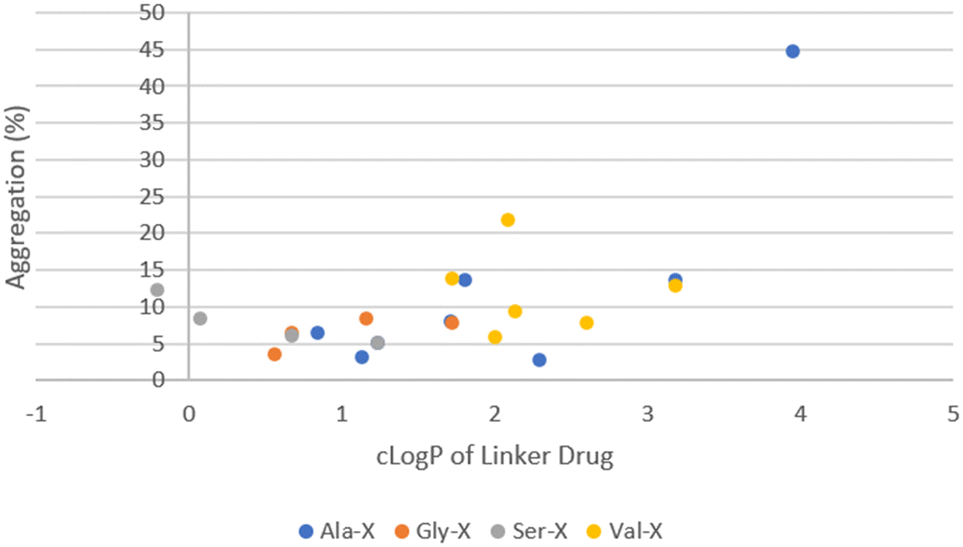
The remaining ADCs formed the second cluster located in the lower right portion of the graph in Fig. 5. These ADCs only contained neutral amino acids in the linker. Although they were more hydrophobic (higher cLogP), their aggregation levels were all below 20% with only two exceptions, ADC51 with a Val–Cit linker and ADC37 with an Ala–Phe linker. Overall, the level of aggregation in this group was random. Since all the ADCs in this group had Ala, Gly, Ser or Val in P1, the aggregation was plotted against cLogP of linker drugs after grouping them based on the amino acid at P1 position (Fig. 6). Linker hydrophobicity now rationalised some of the observed data. Linkers containing Val, the most hydrophobic amino acid among the four in this analysis, in general afforded ADCs with higher aggregation than the rest of the group. Using an arbitrarily assigned aggregation cutoff of 5%, there was no ADC in the Val group that fell below the threshold. Reducing the cLogP of the linker drugs as seen in LD29, LD30 and LD27 with a Val–Gly, Val–Ser and Val–Gln linker compared to ADC23 with a Val–Ala linker lowered aggregation for the corresponding ADC from 12.9% to 7.6%, 9.1%, 5.8%, respectively. For LD1, LD10 and LD11 the cLogP value increased from 2.3, 3.2 to 4.0 respectively. The same trend of increase in aggregation was also observed for the corresponding ADC28, ADC36 and ADC37 (2.8%, 13.6% and 44.7%, respectively). However, aggregation for some ADCs could not be explained by hydrophobicity of the linker drugs. For example, LD25 with Val–Cit was less hydrophobic than LD23 with Val–Ala (cLogP 2.1 vs. 3.2). This was reflected by an earlier elution of the corresponding ADC51 as compared to ADC49 (HIC retention time 6.54 min vs. 6.74 min). Surprisingly, aggregation was higher for ADC51 than ADC49 (21.5% vs. 13.9%). Similarly, in a separate study it was reported that a Val–Cit based MMAE ADC was found to have a shorter HIC retention time but higher aggregation than a Val–Ala based MMAE ADC.29 Citrulline (Cit) is structurally very similar to arginine and involves by replacing the guanidine group of Arg with a urea group. While Arg was positively charged at a neural pH, which we had previously shown to unfavourably promote aggregation, Cit had no net charge. We hypothesised that high polar surface area (PSA), a parameter used to quantify polarity of molecules, might cause repulsion between linker with high polarity and hydrophobic payload, leading to higher aggregation. For LD23 to LD25, the PSA increased from 180 Å2 to 235 Å2. Aggregation also rose from 13.9% in the corresponding ADC49 to 21.5% in ADC51. Other neutral amino acids contributing to PSA increase include Ser, Asn and Gln (Table 5). However, ADCs that had only one of these amino acids in the linker did not seem to result in higher aggregation. We further reasoned that contributions to PSA increase from any single amino acid was relatively small compared to Cit. It would require the presence of two low to medium polarity amino acids to result an increase in the aggregation. This hypothesis was supported by comparing ADC45, ADC46 (Ser–Asn and Ser–Gln) with ADC44, ADC48 (Ser–Ala, Ser–Gly). Both ADC45 and ADC46 were more hydrophilic and also more highly aggregated than ADC44 and ADC48 (HIC retention time 6.19 min, 6.29 min vs. 6.48 min, 6.33 min; aggregation. 12.7%, 8.8% vs. 6.1%, 4.9%, respectively). The observed SEC data correlated to a corresponding higher PSA of 244 Å2 for LD19 and LD20 and 200 Å2 for LD18 and LD22. Another interesting trend was that ADCs with a X–Asn linker were consistently higher in aggregation than ADCs with a X–Gln linker, even though they were slightly more hydrophilic by either HIC retention time or cLogP (Table 6). The difference between X–Asn and X–Gln linked ADCs was most noticeable when X was Val. Differences in aggregation have also reported in a study of Asn versus Gln repeat peptides.30 Asn and Gln are very similar in structure and differ only by the length of their side chain. As conjugation processes involve breaking up the interchain disulfides in antibodies, this could potentially cause some minor conformational changes. We reasoned that with the extra methylene group in Gln extending out the hydrophilic amide group further, it allows more flexibility to accommodate conformational changes.
 | ||
| Fig. 6 ADC aggregation vs. cLogP of linker-payload for DAR10 ADCs containing only neutral amino acids in the linker. | ||
| Amino acid | Contribution to PSA/Å2 |
|---|---|
| Ser | 21 |
| Asn/Gln | 43 |
| Cit | 55 |
| ADC | Dipeptide | HIC Ret time/min | Agg/% |
|---|---|---|---|
| ADC35 | Ala–Asn | 6.45 | 6.3 |
| ADC30 | Ala–Gln | 6.38 | 3.1 |
| ADC45 | Ser–Asn | 6.19 | 12.7 |
| ADC46 | Ser–Gln | 6.29 | 8.8 |
| ADC52 | Val–Asn | 6.46 | 14.2 |
| ADC53 | Val–Gln | 6.66 | 5.8 |
As demonstrated earlier, multiple factors potentially contribute to ADC aggregation. However, as a general rule, linkers containing amino acids with low (Ser) to medium (Gln) hydrophobicity and low polarity result in ADC with low aggregation. Of all the ADCs in this study, only 5 ADCs with a DAR value of 10 had 5% aggregation or less. These were ADC28, ADC30, ADC34, ADC39 and ADC44 with an Ala–Ala, Ala–Gln, Ala–Ser, Gly–Gln and Ser–Ala linker, respectively. It is worth noting that 3 of them had Ala in P1 position, indicating Ala in P1 was more favoured. Switching the order of amino acids in P1 and P2 position led to different linkers to investigate how the order of P1 and P2 would affect ADC properties.
To enable this comparison four pairs of ADCs were made (Table 7). Although their retention time on HIC differed to a certain extent, their SEC data was quite comparable. This suggested that the order of amino acids in the linker had minimal impact on the level of aggregation. Based on this data, we predicted that Ala, which was favoured in P1 position, should also be favoured in P2 position. To our delight, LD1 with an Ala–Ala dipeptide indeed afforded ADC28 a DAR10 ADC with the lowest aggregation in the entire study of only 2.8%, which had significantly lower aggregation than DAR10 ADCs with Val–Cit and Val–Ala linkers, these being the two linkers that have been most widely used in the ADC field.
| ADC | Dipeptide | HIC Ret time/min | Agg/% |
|---|---|---|---|
| ADC44 | Ser–Ala | 6.48 | 4.90 |
| ADC34 | Ala–Ser | 6.42 | 5.00 |
| ADC38 | Gly–Ala | 6.29 | 7.70 |
| ADC32 | Ala–Gly | 6.49 | 7.90 |
| ADC42 | Gly–Ser | 6.14 | 6.20 |
| ADC48 | Ser–Gly | 6.33 | 6.10 |
| ADC36 | Ala–Val | 6.92 | 13.6 |
| ADC49 | Val–Ala | 6.74 | 12.9 |
ADC in vitro activity
ADC1–ADC27 with a DAR value of ∼4 were screened in a GRE luciferase reporter assay in K562 cells over expressing mTNF and wild-type K562 cells to assess potency resulting from targeted delivery vs. nonspecific uptake. All the ADCs were inactive in the wild-type cells, suggesting the activity seen in mTNF over expressed cells was through targeted ADC uptake followed by intracellular release of compound C1. As with other bioassays, a 2 to 3-fold variation in value was deemed to be normal with this GRE reporter assay. Based on the EC50 of the DAR4 ADCs ranging from 0.5 to 1.5 μg mL−1 (3.3 to 10 nM) (Table 8), no significant difference in GRE reporter activity was observed. Although there might be a difference in payload release rate with different dipeptide linkers, comparable EC50s suggested that the accumulated intracellular drug level over the time of assay was similar. In addition, the EC50 value for compound C1 in the GRE reporter assay was 18 nM while the calculated EC50 for ADC with a DAR of 4 was 0.68 μg mL−1 (4.25 nM), assuming a 100% drug release. Since the observed EC50s were on par with the theoretical one, this suggested that compound C1 was efficiently released over time regardless of the different peptide linkers.| ADC | LD | Dipeptide | DAR | K562 EC50/ug mL−1 | |
|---|---|---|---|---|---|
| mTNF GREa | WT GREa | ||||
| a Data is single point. | |||||
| ADC1 | LD1 | Ala–Ala | 3.8 | 0.92 | >50 |
| ADC2 | LD2 | Ala–Arg | 3.6 | 0.74 | >50 |
| ADC3 | LD3 | Ala–Gln | 3.7 | 0.95 | >15 |
| ADC4 | LD4 | Ala–Glu | 3.3 | 0.84 | >50 |
| ADC5 | LD5 | Ala–Gly | 3.7 | 0.72 | >23 |
| ADC6 | LD6 | Ala–His | 3.5 | 0.82 | >50 |
| ADC7 | LD7 | Ala–Lys | 2.4 | 0.51 | >50 |
| ADC8 | LD8 | Ala–Ser | 4.2 | 0.44 | >34 |
| ADC9 | LD12 | Gly–Ala | 3.8 | 1.49 | >44 |
| ADC10 | LD13 | Gly–Gln | 3.7 | 0.60 | >50 |
| ADC11 | LD14 | Gly–Glu | 3.2 | 1.40 | >50 |
| ADC12 | LD15 | Gly–Gly | 3.6 | 0.54 | >38 |
| ADC13 | LD16 | Gly–Ser | 3.7 | 0.81 | >50 |
| ADC14 | LD17 | Phe–Lys | 3.7 | 0.78 | >50 |
| ADC15 | LD18 | Ser–Ala | 3.8 | 1.05 | >29 |
| ADC16 | LD19 | Ser–Asn | 3.5 | 0.80 | >50 |
| ADC17 | LD20 | Ser–Gln | 3.6 | 1.08 | >50 |
| ADC18 | LD21 | Ser–Glu | 3.5 | 1.69 | >20 |
| ADC19 | LD22 | Ser–Gly | 3.7 | 1.03 | >13 |
| ADC20 | LD23 | Val–Ala | 3.8 | 0.48 | >26 |
| ADC21 | LD24 | Val–Arg | 3.4 | 0.86 | >50 |
| ADC22 | LD25 | Val–Cit | 3.6 | 0.72 | >50 |
| ADC23 | LD26 | Val–Asn | 3.5 | 0.53 | >36 |
| ADC24 | LD27 | Val–Gln | 4.1 | 1.26 | >19 |
| ADC25 | LD28 | Val–Glu | 3.4 | 0.79 | >50 |
| ADC26 | LD29 | Val–Gly | 3.6 | 0.55 | >19 |
| ADC27 | LD30 | Val–Ser | 3.7 | 0.55 | >42 |
Conclusion
A library of 30 dipeptide-GRM linker drugs was prepared using a linear synthetic route. The dipeptides included the clinically validated Val–Cit and Val–Ala along with many dipeptide combinations that had not previously been published. By incorporating the same payload to all 30 linker-drugs any variation in properties of the resulting ADCs were attributed to the dipeptide linker. For property assessment the linker drugs were conjugated to a mouse anti-TNF antibody through interchain disulfide Cys to afford ADCs with both DAR4 and DAR10. The high DAR of 10 was selected to provide an extreme test to identify the dipeptides that facilitated ADCs with the most optimal drug-like properties. Sample recovery, hydrophobic interaction chromatography (HIC) and size exclusion chromatography (SEC) were all used to evaluate the impact of the dipeptide linkers on various ADC properties.While cLogP and PSA are routinely used to guide small molecule drug discovery, they were found to have little predictive benefit for dipeptide selection. Comparing Ala–Ala, Ala–Arg and Ala–Ser the aggregation of the DAR10 ADCs was counterintuitively inversely related to the PSA. The cLogP was more predictive for the retention by HIC with the both the retention time and cLogP for three dipeptides being in the order Arg < Ser < Ala. However, there is then no predictive correlation between the HIC retention time and aggregation. Based on these observations the use of cLogP and PSA to predict the properties of resulting ADCs is not recommended. Interestingly Val–Ala and Val–Cit afforded ADCs that were amongst the most aggregated, 12.9% and 21.5% respectively.
The impact from charged amino acids, such as Glu, Lys and Arg were found to be twofold. First, these amino acids add an extra carboxylic or amino group to the antibody, leading to a pI change in the ADC. Depending on the pI of the antibody and the targeted DAR value, precipitation can occur when pH of the conjugation media or formulation buffer approaches pI value of the ADC. It is therefore important to consider both your target DAR and pI of the parental antibody to help guide selection of the dipeptide linker. Second, while charged amino acids were very effective in lowering an ADC's apparent hydrophobicity, positively charged amino acids significantly increased the level of aggregation (negatively charged amino acids were not evaluated in this study). Neutral amino acids play a minor role in impacting ADC properties compared to charged amino acids. In general, there is a correlation between ADC apparent hydrophobicity with cLogP of dipeptide-GRM linker drugs. However, aggregation is found to be related to multiple factors, such as polarity and length of the side chain, rather than just hydrophobicity. Dipeptide linkers containing one amino acid with high hydrophobicity (Val, Phe) or high polarity (Cit), or both amino acids with low to medium polarity (Ser, Asn, Gln) typically resulted in higher levels of aggregation. Dipeptide linkers containing amino acids with low hydrophobicity (Gly, Ala) and single amino acid of low or medium polarity (Ser, Asn, Gln) were preferred for maintaining low aggregation.
Five dipeptide linkers, Ala–Ala, Ala–Gln, Ala–Ser, Gly–Gln and Ser–Ala were found to afford DAR10 ADCs with 5% aggregation or less. Among these, Ala–Ala was identified as the best dipeptide linker for ADCs with high DARs. When assessed for their in vitro potency in a GRE luciferase reporter assay there was little difference in the 27 dipeptide linkers that were tested demonstrating that they are all efficiently cleaved and released comparable levels of payload.
In conclusion, based on synthetic accessibility, physicochemical properties of high DAR10 ADCs and in vitro potency in a cellular assay, Ala–Ala was identified as the optimal dipeptide for linker-drugs with GRM payload C1. Additional studies are planned to investigate the impact of other payloads, lower DAR and cysteine mutants.
Abbreviations
| ADC | Antibody-drug conjugate |
| Agg | Aggregation |
| BEP | 2-Bromo-1-ethylpyridin-1-ium tetrafluoroborate |
| cLogP | Calculated LogP |
| cLogD | Calculated LogD |
| DAR | Drug antibody ratio |
| DCM | Dichloromethane |
| DIEA | N-Ethyl-N-isopropylpropan-2-amine |
| DMF | Dimethylformamide |
| DMSO | Dimethyl sulfoxide |
| DPhPEA | 2-(Diphenylphosphino)ethylamine |
| GRE | Glucocorticoid response element |
| EDTA | Ethylenediaminetetraacetic acid |
| HIC | Hydrophobic interaction chromatography |
| HPLC | High performance liquid chromatography |
| LCMS | Liquid chromatography mass spectrometry |
| LD | Linker-drug |
| mAb | Monoclonal antibody |
| NMR | Nuclear magnetic resonance; pI isoelectric point |
| PSA | Polar surface area |
| RT | Retention time |
| SEC | Size exclusion chromatography |
| TCEP | Tris(2-carboxyethyl)phosphine |
| TFA | Trifluoroacetic acid |
| TNF | Tumor necrosis factor |
Author contributions
Investigation, Lu Wang, Adrian D. Hobson, Julia Fitzgibbons, Axel Hernandez Jr, Ying Jia, Zhou Xu, Zhongyuan Wang, Yajie Yu, and Xiang Li; writing – original draft, Lu Wang; writing – review and editing, Adrian D. Hobson. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors LW, ADH, JF, AH and YJ are employees of AbbVie. ZX, ZW, YY and XL are employees of WuXi AppTec and have no funding to disclose. The design, study conduct, and financial support for this research were provided by AbbVie. AbbVie participated in the interpretation of data, review, and approval of the publication. The authors would like to sincerely thank AbbVie employee Kristine Frank for reading the manuscript and providing helpful comments.References
- Z. Su, D. Xiao, F. Xie, L. Lu, Y. Wang, S. Fan, X. Zhou and S. Li, Antibody-drug conjugates: Recent advances in linker chemistry, Acta Pharm. Sin. B, 2021, 11, 3889–3907 CrossRef CAS PubMed.
- D. Su and D. Zhang, Linker Design Impacts Antibody-Drug Conjugate Pharmacokinetics and Efficacy via modulating the stability and payload release efficiency, Front. Pharmacol., 2021, 12, 687926 CrossRef CAS PubMed.
- A. G. Polson, J. Calemine-Fenaux, P. Chan, W. Chang, E. Christensen, S. Clark, F. J. Sauvage, D. Eaton, K. Elkins, J. M. Elliott, G. Frantz, R. N. Fuji, A. Gray, K. Harden, G. S. Ingle, N. M. Kljavin, H. Koeppen, C. Nelson, S. Prabhu, H. Raab, S. Ross, D. S. Slaga, J. Stephan, S. J. Scales, S. D. Spencer, R. Vandlen, B. Wranik, S. Yu, B. Zheng and A. Ebens, Antibody-drug conjugates for the treatment of non-Hodgkin's lymphoma: target and linker-drug selection, Cancer Res., 2009, 69, 2358–2364 CrossRef CAS PubMed.
- F. M. de Groot, L. W. van Berkom and H. W. Scheeren, Synthesis and biological evaluation of 2′-carbamate-linked and 2′-carbonate-linked prodrugs of paclitaxel: selective activation by the tumor-associated protease plasmin, J. Med. Chem., 2000, 43, 3093–3102 CrossRef CAS PubMed.
- G. M. Dubowchik, R. A. Firestone, L. Padilla, D. Willner, S. J. Hofstead, K. Mosure, J. O. Knipe, S. J. Lasch and P. A. Trail, Cathepsin B-labile dipeptide linkers for lysosomal release of doxorubicin from internalizing immunoconjugates: model studies of enzymatic drug release and antigen-specific in vitro anticancer activity, Bioconjugate Chem., 2002, 13, 855–869 CrossRef CAS PubMed.
- S. O. Doronina, B. E. Toki, M. Y. Torgov, B. A. Mendelsohn, C. G. Cerveny, D. F. Chace, R. L. DeBlanc, R. P. Gearing, T. D. Bovee, C. B. Siegall, J. A. Francisco, A. F. Wahl, D. L. Meyer and P. D. Senter, Development of potent monoclonal antibody auristatin conjugates for cancer therapy, Nat. Biotechnol., 2003, 21, 778–784 CrossRef CAS PubMed.
- P. D. Senter and E. L. Sievers, The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma, Nat. Biotechnol., 2012, 30, 631–637 CrossRef CAS PubMed.
- J. A. Hartley, Antibody-drug conjugates (ADCs) delivering pyrrolobenzodiazepine (PBD) dimers for cancer therapy, Expert Opin. Biol. Ther., 2020, 21, 931–943 CrossRef PubMed.
- P. J. Burke, J. Z. Hamilton, T. A. Pires, J. R. Setter, J. H. Hunter, J. H. Cochran, A. B. Waight, K. A. Gordon, B. E. Toki, K. K. Emmerton, W. Zeng, I. J. Stone, P. D. Senter, R. P. Lyon and S. C. Jeffrey, Development of Novel Quaternary Ammonium Linkers for Antibody–Drug Conjugates, Mol. Cancer Ther., 2016, 15, 938–945 CrossRef CAS PubMed.
- P. J. Burke, J. Z. Hamilton, T. A. Pires, H. W. H. Lai, C. I. Leiske, K. K. Emmerton, A. B. Waight, P. D. Senter, R. P. Lyon and S. C. Jeffrey, Glucuronide-Linked Antibody–Tubulysin Conjugates Display Activity in MDR+ and Heterogeneous Tumor Models, Mol. Cancer Ther., 2018, 17, 1752–1760 CrossRef CAS PubMed.
- F. Li, Y. Li, L. Zhang, C. K. Zhang, H. A. Hu, J. Zhang, Y. Pan, J. Jung, S. H. Lee, H. Ryu, Y. Park, H. Yu and A. N. Tse, Abstract LBA008: CS5001, a novel ROR1-targeting antibody drug conjugate (ADC) armed with tumor-cleavable β-glucuronide linkers and pyrrolobenzodiazepine (PBD) prodrugs for hematological and solid malignancies, Mol. Cancer Ther., 2021, 20, LBA008 CrossRef.
- M. H. Lee, J. L. Sessler and J. S. Kim, Disulfide-Based Multifunctional Conjugates for Targeted Theranostic Drug Delivery, Acc. Chem. Res., 2015, 48, 2935–2946 CrossRef CAS PubMed.
- D. Zhang, T. H. Pillow, Y. Ma, J. Cruz-Chuh, K. R. Kozak, J. D. Sadowsky, G. D. L. Phillips, J. Guo, M. Darwish, P. Fan, J. Chen, C. He, T. Wang, H. Yao, Z. Xu, J. Chen, J. Wai, Z. Pei, C. E. C. A. Hop, S. C. Khojasteh and P. S. Dragovich, Linker Immolation Determines Cell Killing Activity of Disulfide-Linked Pyrrolobenzodiazepine Antibody–Drug Conjugates, ACS Med. Chem. Lett., 2016, 7, 988–993 CrossRef CAS PubMed.
- A. D. Hobson, J. Xu, D. Welch, C. C. Marvin, M. J. McPherson, B. Gates, X. Liao, M. Hollmann, M. Gattner, K. Dzeyk, H. Sarvaiya, M. M. Fettis, A. K. Bichoff, L. Wang, L. Wang, J. Fitzgibbons, P. Salomon, A. Hernandez Jr, Y. Jia, C. A. Goess, S. L. Mathieu and L. C. Santora, Optimization of Drug-Linker to Enable Long Term Storage of Antibody Dug Conjugate for Subcutaneous Dosing, J. Med. Chem., 2023, 66(13), 9161–9173 CrossRef CAS PubMed.
- A. D. Hobson, J. Xu, C. M. Marvin, D. S. Welch, M. J. McPherson, B. Gates, X. Liao, M. Hollmann, M. Gattner, K. Dzeyk, H. Sarvaiya, V. M. Shenoy, M. M. Fettis, A. K. Bischoff, L. Wang, L. C. Santora, L. Wang, J. Fitzgibbons, P. Salomon, A. Hernandez Jr., Y. Jia, C. A. Goess, S. L. Mathieu, S. H. Bryant, M. E. Larsen, B. Cui and Y. Tian, Discovery of ABBV-154 an anti-TNF Glucocorticoid Receptor Modulator Immunology Antibody Drug Conjugate (iADC), J. Med. Chem., 2023, 66(17), 12544–12558 CrossRef CAS PubMed.
- T. Shao, T. Chen, Y. Chen, X. Liu, Y. L. Chen, Q. Wang, T. Zhu, M. Guo, H. Li, D. Ju and C. Wang, Construction of Paclitaxel-Based Antibody-Drug Conjugates with a PEGylated Linker to Achieve superior Therapeutic index, Signal Transduction Targeted Ther., 2020, 5, 132 CrossRef CAS PubMed (3 pages).
- R. P. Lyon, T. D. Bovee, S. O. Doronina, P. J. Burke, J. H. Hunter, H. D. Neff-LaFord, M. Jonas, M. E. Anderson, J. R. Setter and P. D. Senter, Reducing hydrophobicity of homogeneous antibody-drug conjugates improves pharmacokinetics and therapeutic index, Nat. Biotechnol., 2015, 33, 733–735 CrossRef CAS PubMed.
- P. J. Burke, J. Z. Hamilton, S. C. Jeffrey, J. H. Hunter, S. O. Doronina, N. M. Okeley, J. B. Miyamoto, M. E. Anderson, I. J. Stone, M. L. Ulrich, J. K. Simmons, E. E. McKinney, P. D. Senter and R. P. Lyon, Optimization of a PEGylated glucuronide-monomethylauristatin E linker for antibody–drug conjugates, Mol. Cancer Ther., 2017, 16, 116–123 CrossRef CAS PubMed.
- T. Tedeschinia, B. Campara, A. Grigoletto, M. Bellinia, M. Salvalaioa, Y. A. Matsunob, H. Suzukib Yoshiokab and G. Pasut, Polyethylene glycol-based linkers as hydrophilicity reservoir for antibody-drug conjugates, J. Controlled Release, 2021, 337, 431–447 CrossRef PubMed.
- Y. Y. Ha, Y. Anami, C. M. Yamazaki, W. Xiong, C. M. Haase, S. D. Olson, J. Lee, N. T. Ueno, N. Zhang, Z. An and K. Tsuchikama, An Enzymatically Cleavable Tripeptide Linker for Maximizing the Therapeutic Index of Antibody–Drug Conjugates, Mol. Cancer Ther., 2022, 21, 1449–1461 CrossRef PubMed.
- T. Satomaa, H. Pynnönen, A. Vilkman, T. Kotiranta, V. Pitkänen, A. Heiskanen, B. Herpers, L. S. Price, J. Helin and J. Saarinen, Hydrophilic auristatin glycoside payload enables improved antibody-drug conjugate efficacy and biocompatibility, Antibodies, 2018, 7, 15–28 CrossRef CAS PubMed.
- R. Y. Zhao, S. D. Wilhelm, C. Audette, G. Jones, B. A. Leece, A. C. Lazar, V. S. Goldmacher, R. Singh, Y. Kovtun, W. C. Widdison, J. M. Lambert and R. V. J. Chari, Synthesis and evaluation of hydrophilic linkers for antibody-maytansinoid conjugates, J. Med. Chem., 2011, 54(10), 3606–3623 CrossRef CAS PubMed.
- R. P. Lyon, T. D. Bovee, S. O. Doronina, P. J. Burke, J. H. Hunter, H. D. Neff-LaFord, M. Jonas, M. E. Anderson, J. R. Setter and P. D. Senter, Reducing hydrophobicity of homogeneous antibody-drug conjugates improves pharmacokinetics and therapeutic index, Nat. Biotechnol., 2015, 33, 733–735 CrossRef CAS PubMed.
- A. D. Hobson, M. J. McPherson, W. Waegell, C. A. Goess, R. H. Stoffel, X. Li, J. Zhou, Z. Wang, Y. Yu, A. Hernandez Jr, S. H. Bryant, S. L. Mathieu, A. K. Bischoff, J. Fitzgibbons, M. Pawlikowska, S. Puthenveetil, L. C. Santora, L. Wang, L. Wang, C. C. Marvin, M. E. Hayes, A. Shrestha, K. A. Sarris and B. Li, Design and Development of Glucocorticoid Receptor Modulator Agonists as Immunology Antibody-Drug Conjugate Payloads, J. Med. Chem., 2022, 65, 4500–4533 CrossRef CAS PubMed.
- A. D. Hobson, M. J. McPherson, M. E. Hayes, C. Goess, X. Li, J. Zhou, Z. Wang, Y. Yu, J. Yang, L. Sun, Q. Zhang, P. Qu, S. Yang, A. Hernandez Jr, S. H. Bryant, S. L. Mathieu, A. K. Bischoff, J. Fitzgibbons, L. C. Santora, L. Wang, L. Wang, M. M. Fettis, X. Li, C. C. Marvin, Z. Wang, M. V. Patel, D. L. Schmidt, T. Li, J. T. Randolph, R. F. Henry, C. Graff, Y. Tian, A. L. Aguirre and A. Shrestha, Discovery of ABBV-3373, an Anti-TNF Glucocorticoid Receptor Modulator Immunology Antibody Drug Conjugate, J. Med. Chem., 2022, 65, 15893–15934 CrossRef CAS PubMed.
- B. L. McRae, A. D. Levin, M. E. Wildenberg, P. J. Koelink, P. Bousquet, I. Mikaelian, A. S. Sterman, S. Bryant, G. D'Haens, R. Kamath, J. Salfeld and G. R. van den Brink, Fc Receptor-mediated Effector Function Contributes to the Therapeutic Response of AntiTNF Monoclonal Antibodies in a Mouse Model of Inflammatory Bowel Disease, J. Crohns Colitis, 2016, 10, 69–76 CrossRef PubMed.
- S. R. Trevino, J. M. Scholtz and C. N. Pace, Amino acid contribution to protein solubility: Asp, Glu, and Ser contribute more favourably than the other hydrophilic amino acids in RNase Sa, J. Mol. Biol., 2007, 366, 449–460 CrossRef CAS PubMed.
- S. Fekete, J. L. Veuthey, A. Beck and D. Guillarme, Hydrophobic interaction chromatography for the characterization of monoclonal antibodies and related products, J. Pharm. Biomed. Anal., 2016, 130, 3–18 CrossRef CAS PubMed.
- Y. Wang, S. Fan, W. Zhong, X. Zhou and S. Li, Development and Properties of Valine-Alanine based Antibody-Drug Conjugates with Monomethyl Auristatin E as the Potent Payload, Int. J. Mol. Sci., 2017, 25, 1860 CrossRef PubMed (19 pages).
- X. Lu and R. M. Murphy, Asparagine Repeat Peptides: Aggregation Kinetics and Comparison with Glutamine Repeats, Biochemistry, 2015, 54, 4784–4794 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3md00473b |
| This journal is © The Royal Society of Chemistry 2024 |