
Pyrazole derivatives as selective orexin-2 receptor antagonists (2-SORA): synthesis, structure–activity–relationship, and sleep-promoting properties in rats†
Christine
Brotschi
 *,
Martin H.
Bolli
*,
John
Gatfield
,
Catherine
Roch
,
Thierry
Sifferlen
,
Alexander
Treiber
,
Jodi T.
Williams
and
Christoph
Boss
*,
Martin H.
Bolli
*,
John
Gatfield
,
Catherine
Roch
,
Thierry
Sifferlen
,
Alexander
Treiber
,
Jodi T.
Williams
and
Christoph
Boss
Idorsia Pharmaceuticals Ltd, Drug Discovery and Preclinical Development, Hegenheimermattweg 91, 4123 Allschwil, Basel-Landschaft, Switzerland
First published on 24th November 2023
Abstract
Selective orexin 2 receptor antagonists (2-SORA) such as seltorexant (15) are in clinical development for the treatment of insomnia and other conditions such as depression. Herein, we report our structure–activity–relationship (SAR) optimization efforts starting from an HTS hit (1) (N-(1-((5-acetylfuran-2-yl)methyl)-1H-pyrazol-4-yl)-5-(m-tolyl)oxazole-4-carboxamide) that was derived from an unrelated in-house GPCR-agonist program. Medicinal chemistry efforts focused on the optimization of orexin 2 receptor (OX2R) antagonistic activity, stability in liver microsomes, time dependent CYP3A4 inhibition, and aqueous solubility. Compounds were assessed for their brain-penetrating potential in in vivo experiments to select the most promising compounds for our in vivo sleep model. Our lead optimization efforts led to the discovery of the potent, brain penetrating and orally active, 2-SORA (N-(1-(2-(5-methoxy-1H-pyrrolo[3,2-b]pyridin-3-yl)ethyl)-1H-pyrazol-4-yl)-5-(m-tolyl)oxazole-4-carboxamide) 43 with efficacy in a sleep model in rats comparable to 15.
Introduction
The orexin system comprises two subtypes of G protein-coupled receptors (GPCRs), namely the orexin 1 receptor (OX1R) and the orexin 2 receptor (OX2R), which are widely distributed throughout the brain. These receptors are activated by two endogenous ligands, the neuropeptides orexin-A and -B, which are exclusively produced in the lateral hypothalamus. While orexin-A binds with similar affinity to both OX1R and OX2R, orexin-B binds preferentially to OX2R. The orexin system plays a crucial role in regulating various physiological processes, including the sleep-awake cycle,1–4 reward mechanisms,5,6 anxiety/panic responses,7 addiction,8 depression,9 and eating/feeding behavior.10Decades of dedicated research efforts focused on the orexin system11,12 and the three dual orexin receptor antagonists (DORA) suvorexant 2,13–17 lemborexant 3 (ref. 18–20) and daridorexant 4 (ref. 21 and 22) recently gained marketing approval for the treatment of insomnia (Fig. 1).
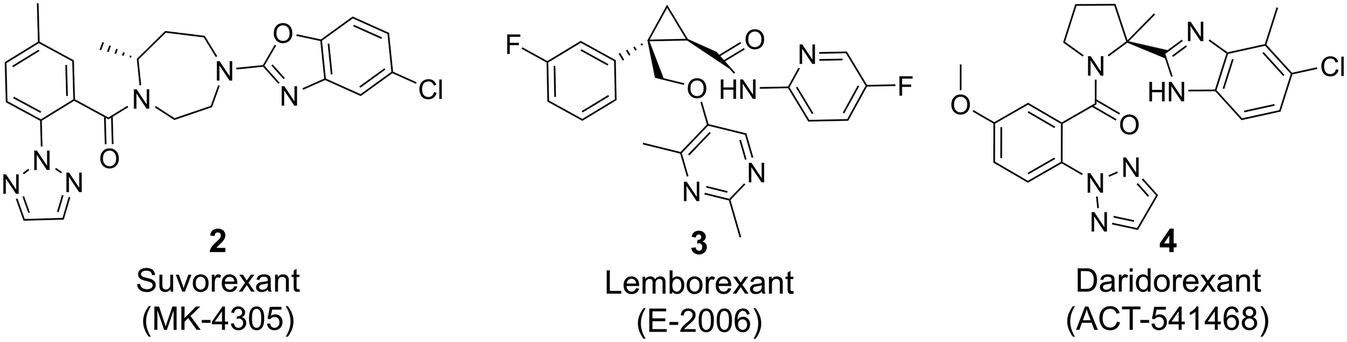 | ||
| Fig. 1 DORAs approved for insomnia. | ||
While DORAs antagonize both receptors with similar affinity, selective OXR antagonists (SORA) may either selectively antagonize OX1R (1-SORA) or OX2R (2-SORA). Several 1-SORAs have been explored for the treatment of addiction, stress, anxiety, panic, and eating disorder. One example, nivasorexant,23,24 was investigated for the treatment of binge eating but proved ineffective.25 More intense research on 2-SORAs was triggered by the disclosure that comparable to DORAs also the 2-SORA TCS-OX2-29 5 caused a reduction in latency to sleep onset and an increase in total sleep time26,27 and since then, various 2-SORAs have been evaluated in preclinical and clinical studies for the treatment of insomnia or depression.
Notable 2-SORA compounds include spiro-piperidine 6,28 LSN2424100 7,29 EMPA 8,30 MK-1064 9,31,32 MK-3697 10,33 piperidine 11,34 MK-8133 12,35 diazepam 13,36 and JNJ-10397049 14 (ref. 37 and 38) (Fig. 2). The debate whether selective antagonism of OX2R alone is sufficient to promote sleep in humans persisted until the results of 9 were published.39 This 2-SORA promoted sleep in mouse, dog, and human with efficacies similar to DORAs. The 2-SORA seltorexant (MIN-202, JNJ42847922) 15 (ref. 40 and 41) initially developed and found to be efficacious in insomnia disorders is currently investigated in major depressive disorders in phase 3 clinical studies.42 Other recently introduced 2-SORA compounds include SDM-878 16 (ref. 43) and JNJ-48816274.44 The structure of the latter compound has not been disclosed.
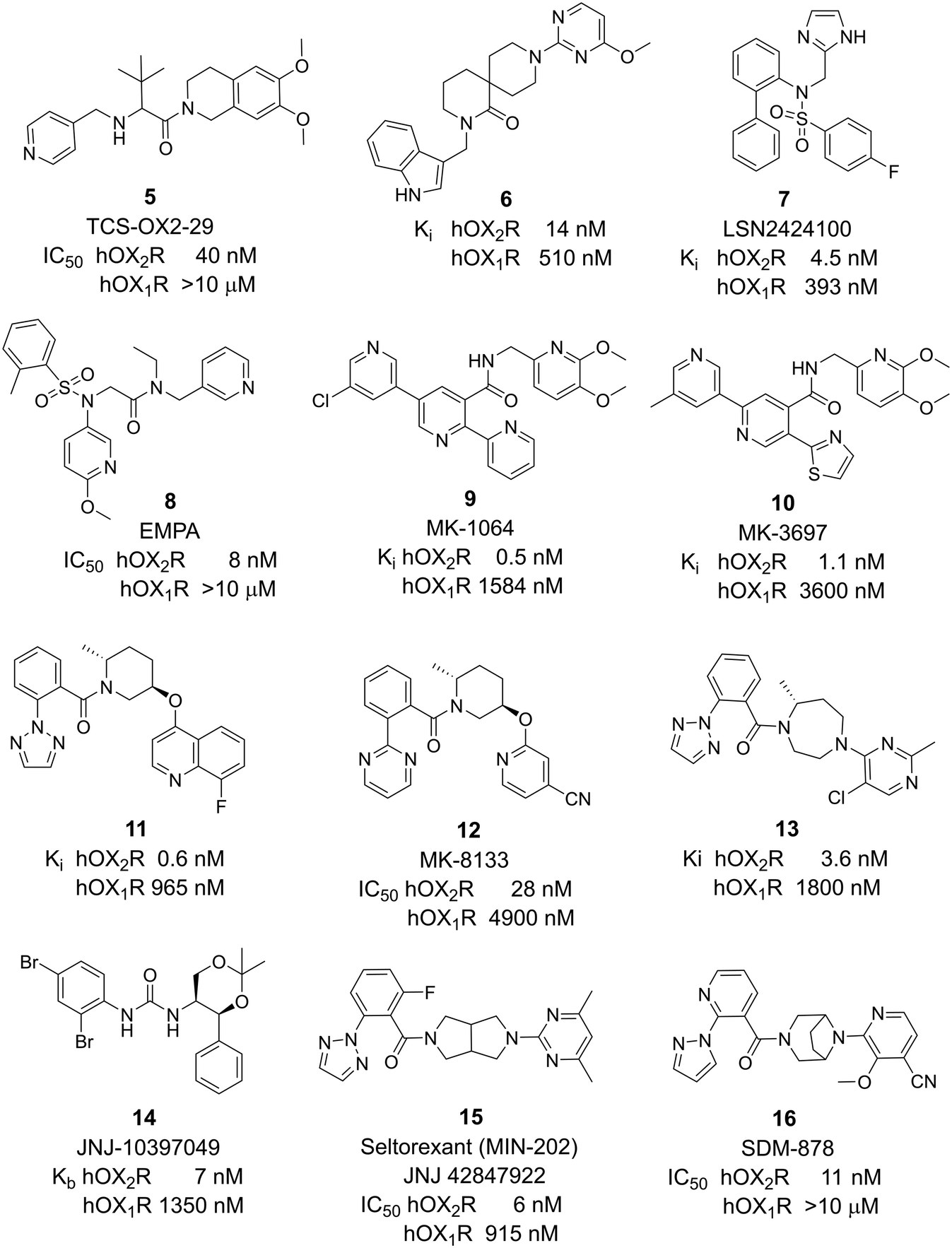 | ||
| Fig. 2 2-SORAs investigated in research, preclinical, and clinical development. Potency data as published in the literature (see references in the text). | ||
Results and discussion
An in house high-throughput screening campaign identified a cluster of compounds showing activities on OX1R and OX2R. Inhibitory activities were determined using fluorometric imaging plate reader (FLIPR) technology. Compound-mediated inhibition of the orexin-A induced Ca2+ flux was measured in CHO cells overexpressing either the human OX1 or the human OX2 receptor and the results are expressed as IC50 values. Compound 1 (ref. 45) showed a dual profile with an IC50-value of 2 μM on both OX1R and OX2R (Fig. 3). First, our efforts focused on structure–activity–relationship (SAR) studies to improve OX2R potency. In a next step, we optimized metabolic stability as measured in human liver microsomes (HLM). Bearing in mind that for the indication of a sleep drug the targeted duration of action in humans was expected to be in the range of 6 to 8 h, we aimed at shortlisting compounds with moderate in vitro stability in HLM (Clint ∼200 μL min−1 mg−1) in a first instance. We anticipated that more detailed (mechanistic) PK studies and predictions would be needed later to then identify the most suitable development candidates.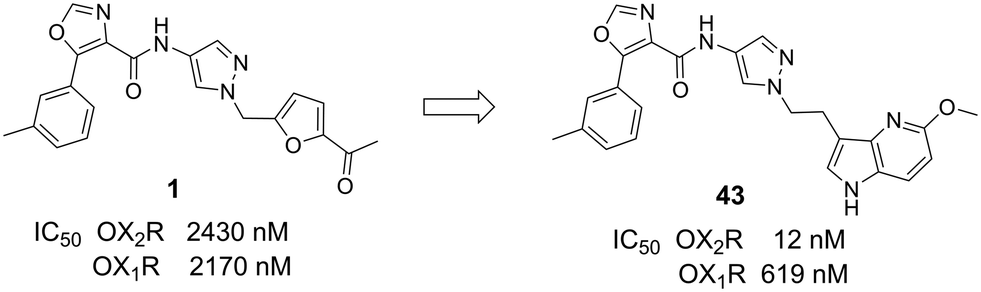 | ||
| Fig. 3 From HTS hit 1 to compound 43. | ||
Compounds were found to be substantially less stable in rat liver microsomes (RLM) when compared to HLM. Rat experiments were therefore performed at a high dose (100 mg kg−1) attempting to saturate metabolism and thus achieve exposures suitable for pharmacodynamic evaluation. Furthermore, CYP450 inhibition, including time-dependent inhibition (TDI) of CYP3A4 was monitored to reduce the risk of potential drug–drug interactions. Aqueous solubility was assessed to further guide compound prioritization. Pharmacokinetic experiments of key analogs helped us shortlist compounds with oral bioavailability. Brain penetration properties were assessed in in vivo experiments to then select the most promising compounds for our in vivo sleep model. These efforts eventually led to the identification of pyrazole 43, which is structurally distinct from known 2-SORAs. Pyrazole 43 was found to be a potent, orally bioavailable, brain penetrating 2-SORA with efficacy in a sleep model in rats comparable to 15.
For SAR optimization purposes, the hit structure was dissected into three parts, namely part A, B, and C (Fig. 4).
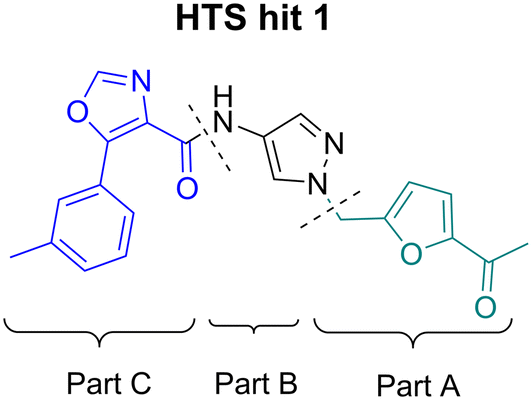 | ||
| Fig. 4 SAR investigation on part A, B, and C of HTS hit 1. | ||
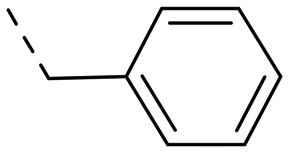
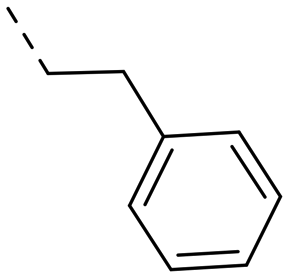
We started our SAR investigation with part A by keeping part B (4-aminopyrazole) and part C (biaryl-carboxamide) of the HTS hit 1 constant. The unsubstituted benzyl (17), the unsubstituted phenethyl (18), or the phenoxy ether analog 19 were equally potent on both orexin receptors (OXRs) and showed affinities comparable to compound 1 (Table 1).
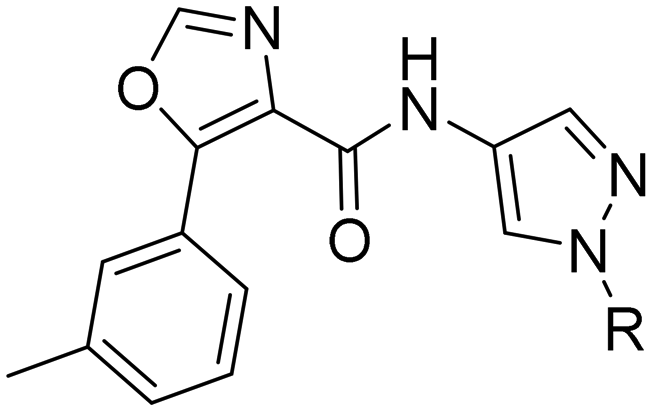
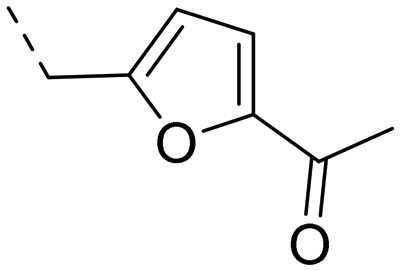
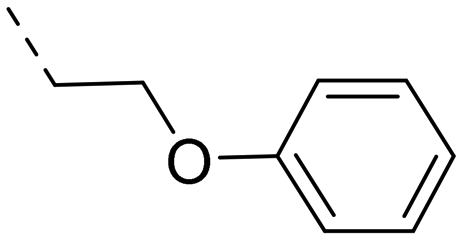
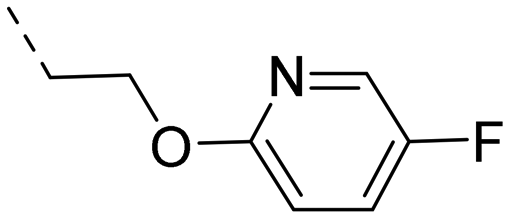
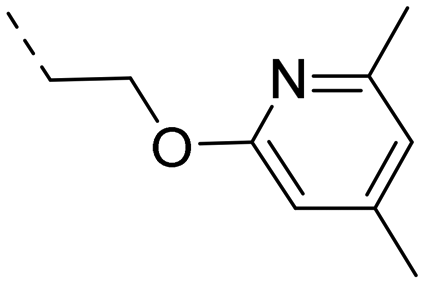
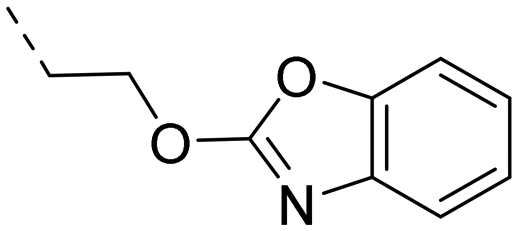
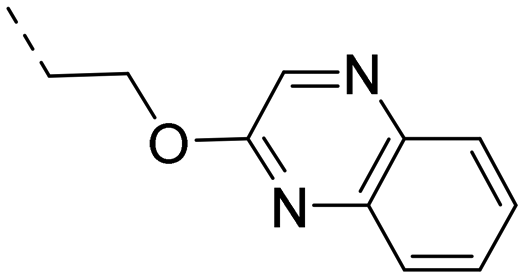
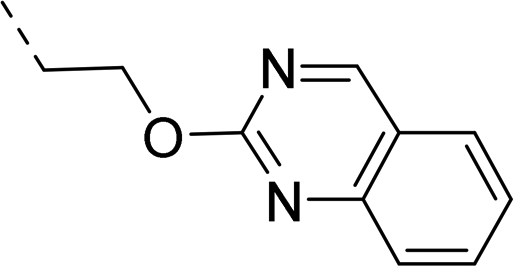
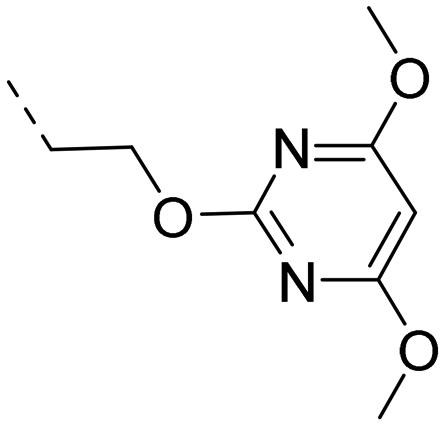
2-Pyridyls 20 and 21 were also tolerated by both receptors. Compound 21 showed a slight increase on OX2R potency. Heteroaryls, such as benzoxazole (22), quinoxaline (23) and quinazoline (24) were tolerated by both receptors but did not show improved potency. Interestingly, pyrimidine analog 25 was found to be OX2R selective with a potency of 34 nM and 1567 nM on OX2R and OX1R, respectively.
The promising potency profile of compound 25 prompted us to investigate the influence of the linking atom X (Table 2). Potency was not altered when the oxygen in 25 was replaced by nitrogen (26). The C-analog 27 was slightly less potent on OX2R and only about 10-fold selective over OX1R. By comparing the intrinsic clearance in liver microsomes, we found the N- and C-analogs 26 and 27, to be slightly more stable in HLM and RLM than the O-analog 25. Inhibitory activity towards CYP3A4 were 48 μM, 17 μM, and >50 μM for 25, 26 and 27, respectively. However, a potential for drug–drug interaction was detected by measuring time-dependent inhibition (TDI) of CYP3A4. After prolonged incubation (30 min), IC50-values decreased by >10-, 6.4- and 6.6-fold (“shift”) for compounds 25, 26 and 27, respectively.
| X | O | N | C | |
|---|---|---|---|---|
| a IC50 values in nM; data are the geometric mean of at least three independent measurements determined by FLIPR assay (see ESI† for details). b Intrinsic clearance [μL min−1 mg−1] with human liver microsomes (HLM) and rat liver microsomes (RLM), at 1 μM. c IC50-value of cytochrome 3A4 inhibition in μM. d Time-dependent inhibition on CYP3A4, values are the observed shift of inhibition before and after pre-incubation (30 min). e Solubility [μg mL−1] as measured in, Fasted State Simulated Intestinal Fluid (FaSSIF) at pH 6.8, and Fed State Simulated Intestinal Fluid (FaSSIF) at pH 5. | ||||
|
Compd | 25 | 26 | 27 |
| OX2Ra | 34 | 29 | 61 | |
| OX1Ra | 1567 | 1477 | 729 | |
| HLM/RLMb | 294/>1250 | 141/860 | 155/882 | |
| CYP3A4c | 48 | 17 | >50 | |
| CYP3A4 shiftd | >10 | 6.4 | 6.6 | |
| Solubilitye | 12/59 | 11/103 | 62/266 | |
| FaSSIF/FeSSIF | ||||
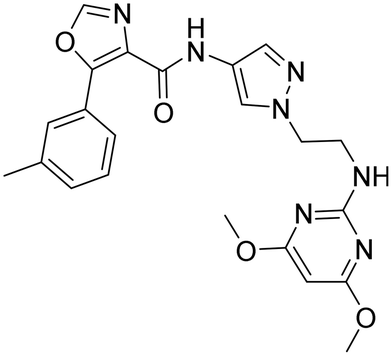
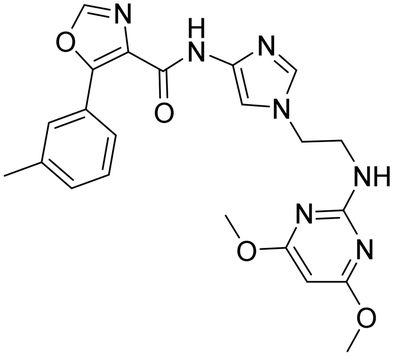
Aqueous solubility was low for 25 and 26, and slightly improved for 27. We found that the N-analog 26 had the best overall profile and therefore we kept the N-pyrimidine for the next round of optimization constant and focused our SAR investigation on part B by replacing the 4-aminopyrazole by a 4-amino-imidazole moiety (28). Unfortunately, potency of 28 was reduced when compared to 26 (Table 3) and the CYP3A4 shift was increased.
|
|
|
|
|
|---|---|---|---|
| a IC50 values in nM; data are the geometric mean of at least three independent experiments and were determined by FLIPR assay (see the ESI† for details). b Intrinsic clearance [μL min−1 mg−1] with human and rat liver microsomes, at 1 μM. c Time-dependent inhibition on CYP3A4, values are the observed shift of inhibition before and after pre-incubation. | |||
| Compd | 26 | 28 | 29 |
| OX2Ra | 29 | 106 | 4 |
| OX1Ra | 1477 | >3883 | 22 |
| HLM/RLMb | 141/860 | 191/>1250 | 1161/>1250 |
| CYP3A4 shiftc | 6.4 | >9.1 | 2.0 |
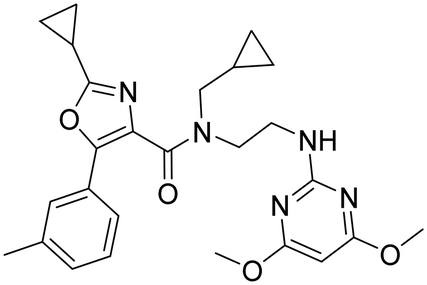
Omitting the pyrazole moiety (part B) and modifying the biaryl carboxamide (part A) led to compound 29 which had a markedly improved potency, but a clearly reduced OX2R selectivity when compared to 26. The reduced shift in the TDI of CYP3A4 (shift of 2) was encouraging. However, as one could expect from the higher intrinsic clearance in RLM (Clint > 1250 vs. 860 μL min−1 mg−1) also the in vivo clearance in rats was higher for 29 than for 26 (Table 4). In the corresponding oral rat PK experiment, plasma exposure of 29 (cmax and AUC) was much lower than for pyrazole 26 and compound 29 showed a clearly lower bioavailability (1%) than pyrazole 26 (14%) indicating that omitting the pyrazole had a negative impact on pharmacokinetic properties. Along these lines, a poor pharmacokinetic profile of a series of DORAs containing a diamino-ethyl linker due to extensive oxidative metabolism was reported by Merck.46
| iva | pob | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compd | Dose [mg kg−1] | CL [mL min−1 kg−1] | Vss [L kg−1] | T 1/2 [h] | Dose [mg kg−1] | AUC0–∞ [ng h mL−1] | C max [ng mL−1] | T max [h] | F [%] |
| a iv = intravenous, formulation: HPbCD 30%/H2O/5% DMSO. b Oral, as a suspension in 5% DMSO, 95% aq. gelatine (7.5%). | |||||||||
| 26 | 1.0 | 40 | 1.4 | 0.6 | 10 | 588 | 242 | 0.5 | 14 |
| 29 | 1.0 | 53 | 6 | 4.3 | 10 | 25 | 8.14 | 0.5 | 1 |
In a permeability assay using human MDR1-overexpressing MDCK1 cells, both compounds showed high permeability in vitro in both directions (26: Papp AB = 34.3, Papp BA = 43.9; 29: Papp AB = 35.3, Papp BA = 47.6 [10−6 cm s−1]) and hence an efflux ratio of close to 1 indicating that none of compounds is a substrate of MDR1. However, low plasma exposure of 29 in the rat lead to low brain levels. In the corresponding PK experiment performed at 100 mg kg−1, the brain concentration 3 h after compound administration reached only 21 nmol g−1 for 29 while it was at 750 nmol g−1 for 26 (Table 8). These findings combined with the fact that compound 29 showed a nearly 10-fold higher intrinsic clearance in HLM made this sub-series clearly less attractive for further SAR studies.
Next, we revisited part A and replaced the amino-pyrimidine unit by a N-linked indole. Various substitution patterns were analyzed for their impact on potency, metabolic stability, and TDI of CYP3A4 (Table 5).
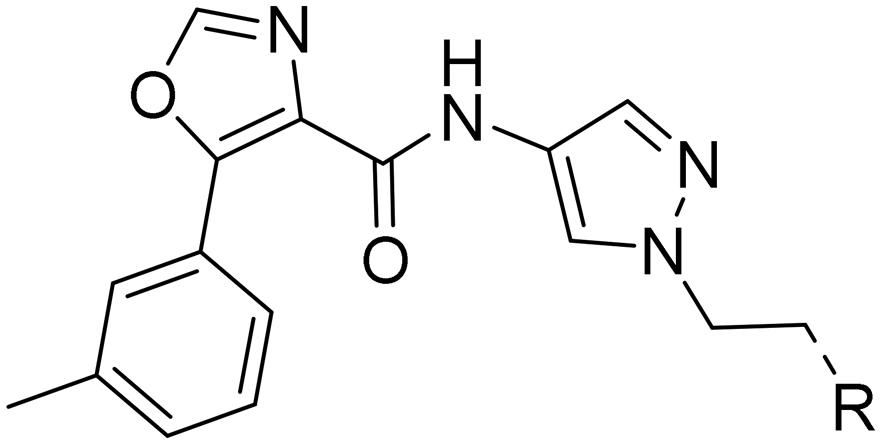
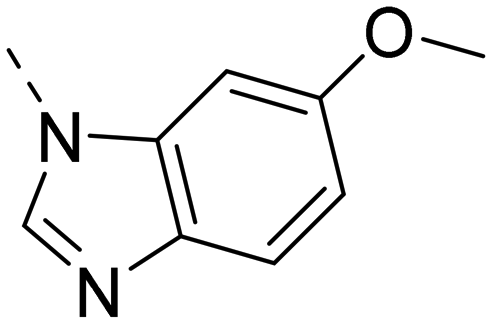
| R |
|
|
|
|
|
|---|---|---|---|---|---|
| a IC50 values in nM; data are the geometric mean of at least three independent experiments and were determined by FLIPR assay (see the ESI† for details). b Intrinsic metabolic clearance [μL min−1 mg−1] in human liver microsomes, at 1 μM. c Time-dependent inhibition of CYP3A4, values are the observed shift of the IC50-value before and after pre-incubation (30 min). nd = not determined. | |||||
| Compd | 35 | 36 | 37 | 38 | 39 |
| OX2Ra | 42 | 16 | 390 | 6 | 50 |
| OX1Ra | 179 | 675 | 1355 | 879 | 1253 |
| HLMb | 350 | 266 | nd | 284 | 404 |
| CYP3A4 shiftc | 25 | 9.6 | nd | 9.2 | 3.3 |
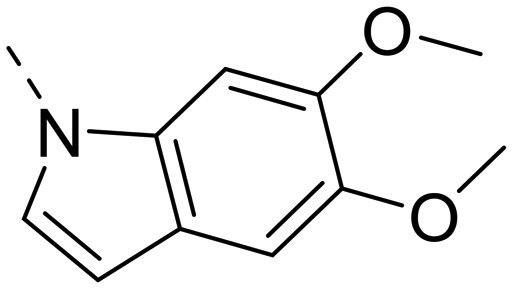
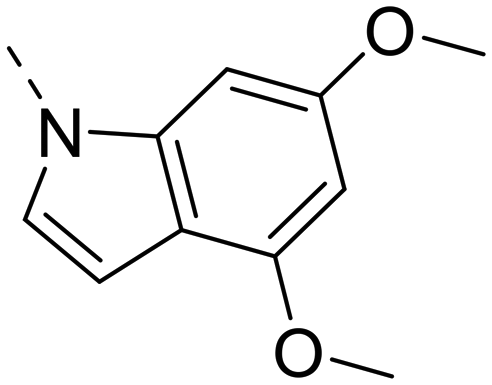
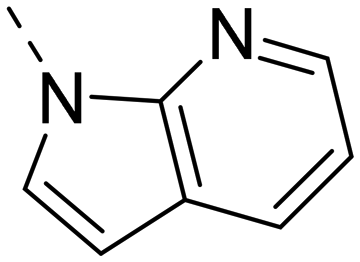
Unsubstituted indole 30 showed an OX2R potency of 103 nM and a weaker OX1R affinity of 497 nM. A 5-methoxy substituent (32) was not well tolerated by both OXR, however a 6-methoxy substituent (33) increased potency solely on OX2R to 7 nM, resulting in a OX2R selective compound. Adding a second methoxy substituent at the 5-position (35) led to a slight decrease in OX2R potency and an increase in OX1R potency, resulting in a rather dual compound when compared to 33. In contrast, when a second methoxy group was added at the 4-position (36), no major differences in OXR affinities were observed when compared to 33. The 7-azaindole (37) was slightly less potent when compared to indole 30. However, the addition of a 6-methoxy group (38) to the 7-azaindole resulted in a single digit nanomolar compound on OX2R, with similar potencies as observed for indole 33. Moving the nitrogen to position 5 (39) resulted in a decrease in OX2R affinity. Benzimidazole 31 was as potent on OX2R, but ∼2-fold less potent on OX1R when compared to its indole analog 30. On the other hand, the affinity profiles of indole 33 and benzimidazole 34 were rather similar. The intrinsic clearance in HLM of indoles and benzimidazoles were in the range of 200–400 μL min−1 mg−1, with little impact of the substitution pattern on in vitro stability (30vs.32 or 33vs.34). Interestingly, the two benzimidazoles 31 and 34 showed a clearly reduced TDI on CYP3A4 when compared to their indole analogs 30 and 33, respectively. While compound 31 was not potent enough, the IC50-value of 12 nM on OX2R for compound 34 made this an interesting compound for further investigation (see below).
Next, we turned our attention to C-3-linked indoles and 4-aza-indoles (Table 6). The C-linked indole 40 was clearly less potent on OX2R when compared to its N-linked indole analog 32. In line with observations made with compounds 32 and 33 (Table 5), moving the methoxy group to the 5-position (41) resulted in a highly potent (2 nM) and selective 2-SORA.
|
|
||||
|---|---|---|---|---|
| R |
|
|
|
|
| a IC50 values in nM; data are the geometric mean of at least three independent experiments and were determined by FLIPR assay (see the ESI† for details). b Intrinsic metabolic clearance [μL min−1 mg−1] with human microsomes (HLM) and rat liver microsomes (RLM), at 1 μM. c Time-dependent inhibition on CYP3A4, values are the observed shift of inhibition before and after pre-incubation. d Solubility [μg mL−1] as measured in Fasted State Simulated Intestinal Fluid (FaSSIF) at pH 6.8, and Fed State Simulated Intestinal Fluid (FaSSIF) at pH 5. nd = not determined. | ||||
| Compd | 40 | 41 | 42 | 43 |
| OX2Ra | 1487 | 2 | 32 | 12 |
| OX1Ra | 1880 | 393 | 2051 | 619 |
| HLM/RLMb | nd | 46/298 | 153/566 | 92/366 |
| CYP3A4 shiftc | 8.8 | 6.9 | 12 | 6.7 |
| Solubilityd | nd | 33/167 | nd | 34/393 |
| FaSSIF/FeSSIF | ||||
Methylation of the N-indole (42) decreased the compound's potency on both OXRs. Compared to indole 41, 4-azaindole (43) was less potent on OX2R, resulting in a less selective compound towards OX2R. The intrinsic clearance for 41 and 43, in HLM was 46 and 92 μL min−1 mg−1, respectively. While the 3-linked indoles 41 and 43 showed lower shifts in the TDI of CYP3A4 than their N-linked analogs 33 and 38, respectively, IC50-values for CYP3A4 inhibition still markedly decreased with prolonged incubation time.
Compounds 41 and 43 have a similar aqueous solubility profile, with reasonable solubility in FaSSIF and FeSSIF.
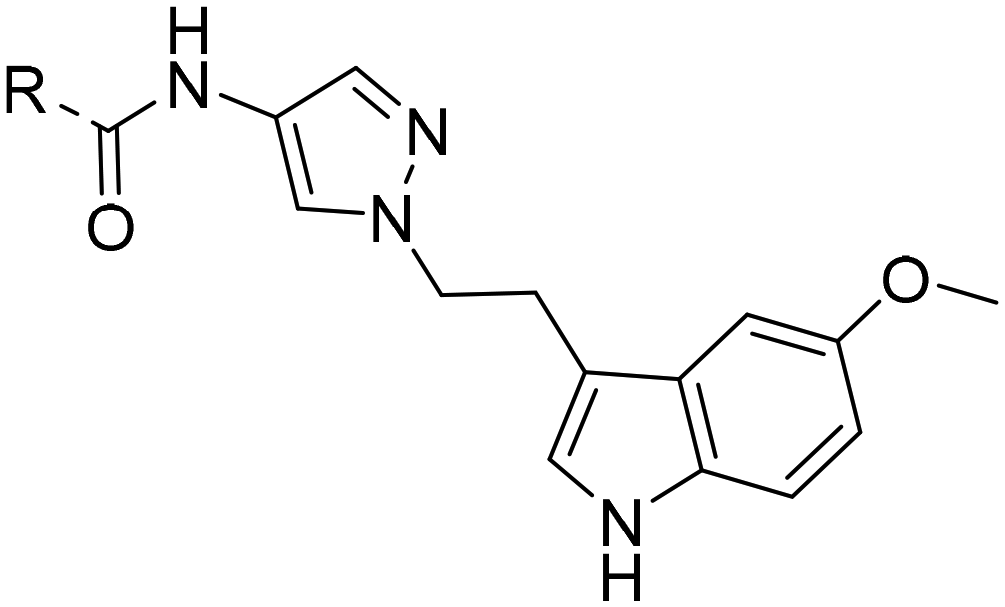
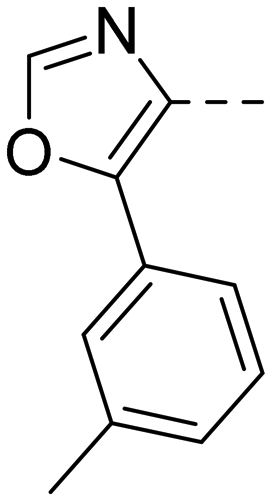
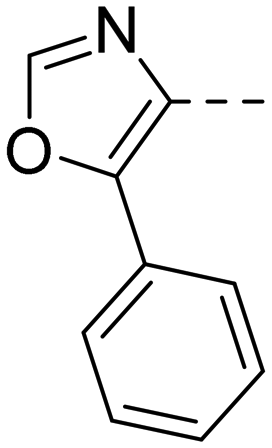
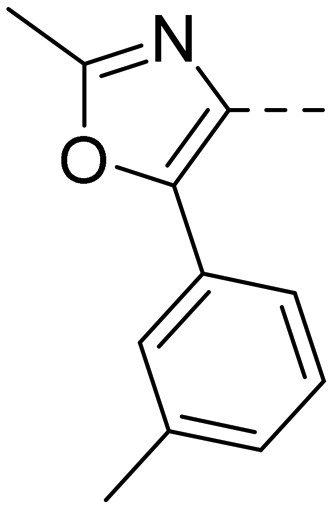
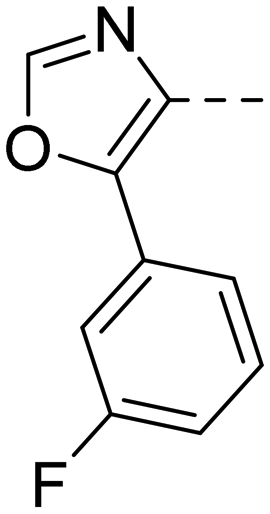
After optimizing part A and part B, we explored the SAR of part C (Table 7). Small variations of the 5-phenyl-oxazole-4-carboxamide such as in compounds 44, 46, or 47 had little impact on potency and microsomal stability. However, a 2-methyl-substituent on the oxazole ring (45) was not well tolerated by the OX2R. 2-Triazolyl-phenyl-carboxamides as in 48 or 49, which are often found in DORA's (e.g.2, 4) and in 2-SORA compounds (11, 13, and 15) were not tolerated by either of the two OXRs. Interestingly, such carboxamides in combination with an “inverted” pyrazole-linker in the part B, resulted in potent DORAs.47 Unfortunately, none of the modifications in part C led to improved CYP3A4 shift values.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| R |
|
|
|
|
|
|
|
| a IC50 values in nM; data are the geometric mean of at least three independent experiments and were determined by FLIPR assay (see the ESI† for details). b Intrinsic metabolic clearance [μL min−1 mg−1] with human microsomes (HLM) and rat liver microsomes (RLM), at 1 μM. c Time-dependent inhibition on CYP3A4, values are the observed shift of inhibition before and after pre-incubation. nd = not determined. | |||||||
| Compd | 41 | 44 | 45 | 46 | 47 | 48 | 49 |
| OX2Ra | 2.1 | 2.4 | 1772 | 4.3 | 4.9 | 4631 | >6820 |
| OX1Ra | 393 | 835 | 1033 | 719 | 1027 | >7289 | >4458 |
| HLMb | 46 | 55 | nd | 59 | 97 | nd | nd |
| CYP3A4 shiftc | 4.2 | 5.1 | nd | 7.9 | 8.7 | nd | nd |
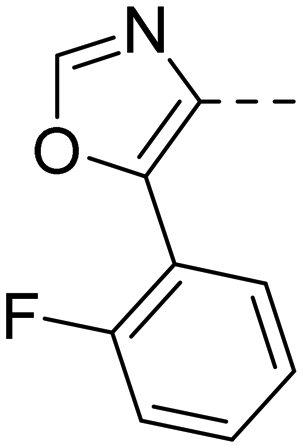
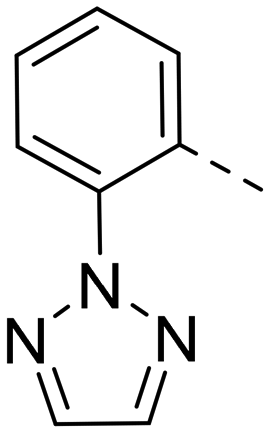
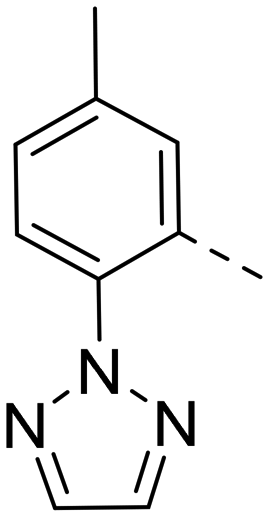
Based on the above compound characterization and SAR studies, pyrimidine 26, benzimidazole 34, and the two indoles 41 and 43 appeared interesting candidates for further profiling. Permeability data measured in the MDR1-MDCK1 assay indicated that none of the compounds 26, 41 and 43 is a substrate of the P-gp efflux pump (Table 8). The brain penetration potential of 26, 34, 41, 43 was then assessed in the Wistar rat. Plasma and brain concentrations were measured 3 h after oral administration of 100 mg kg−1 of the compound and results are reported in Table 8.
| Compd | OX2Ra | OX1Ra | HLMb | [P]c | [B]c | [B/P]d | MDR1 effluxe |
|---|---|---|---|---|---|---|---|
| a IC50 values in nM; data are the geometric mean of at least three independent experiments and were determined by FLIPR assay (see the ESI† for details). b Intrinsic metabolic clearance [μL min−1 mg−1] with human microsomes (HLM) at 1 μM. c Brain penetration experiment was performed in rats (n = 3) by sampling plasma (P) and brain (B), 3 h following administration of 100 mg kg−1 p.o.; [P] in ng mL−1, [B] in ng g−1. d Brain/plasma ratios were calculated assuming a brain density around 1 g mL−1. e Multidrug resistant protein 1 (MDR1) efflux ratio (Papp (B-A)/Papp (A-B)). nd = not determined. | |||||||
| 26 | 29 | 1477 | 141 | 156 | 335 | 2.2 | 1.3 |
| 29 | 4 | 22 | 1161 | 46 | 10 | 0.2 | 1.3 |
| 34 | 12 | 733 | 419 | 368 | 21 | 0.06 | nd |
| 41 | 2.0 | 449 | 46 | 1859 | 3372 | 1.8 | 1.2 |
| 43 | 9.5 | 713 | 92 | 5053 | 5213 | 1.0 | 2.7 |
| 15 | 2.5 | 530 | 95 | 3465 | 1286 | 0.37 | 10.4 |
Of the four compounds tested, compound 26 showed the lowest plasma concentrations and, despite the good brain penetration indicated by a [B/P] ratio of 2.2, was not further characterized in vivo. Also compound 34 reached low plasma concentrations and even lower brain concentrations. The low brain to plasma ratio of 0.06 hinted that 34 is a likely substrate of the rat P-gp. Whether this is the case for the human transporter was not evaluated. Compared to 26 and 34, indole analog 41 and aza-indole 43 showed clearly higher plasma and brain concentrations, with total brain concentrations corresponding to 7.6 and 11.8 μM, respectively. The brain to plasma ratio of 1.8 and 1.0 indicated that compounds 41 and 43 are not rat P-gp substrates, correlating with the efflux ratios of 1.2 and 2.7 measured in the in vitro assay containing the human MDR1 transporter protein. The efflux ratio of 10 indicated that compound 15 is a substrate of the human MDR1 transporter. In the rat, the total brain concentration of compound 15 was equivalent to 3.2 μM and was 2.4- and 3.7-fold lower than that of compound 41 and 43, respectively.
For compound 41 and 43 the selectivity profile was assessed in a curve-shift FLIPR experiment. Compound 41 showed an OX2 selectivity profile of 388-fold over OX1R (Kb hOX1/hOX2) with Kb (hOX1) of 504 nM and Kb (hOX2) of 1.3 nM. Similarly, compound 43 was shown to be an OX2 selective compound with a selectivity ratio of 311-fold with Kb (hOX1) of 1744 nM and Kb (hOX2) of 5.6 nM.
Next, 41, and 43 were tested in vivo for their effect on the sleep/wake stages in freely moving male Wistar rats implanted with radiotelemetry transmitters for continuous recording of electroencephalographic/electromyographic (EEG/EMG) signals. In addition, 15 (ref. 40 and 41) was tested under the same experimental conditions as a reference compound. All three compounds were tested at 100 mg kg−1 and were administered orally at the beginning of the night active phase. Results are shown in Fig. 5.
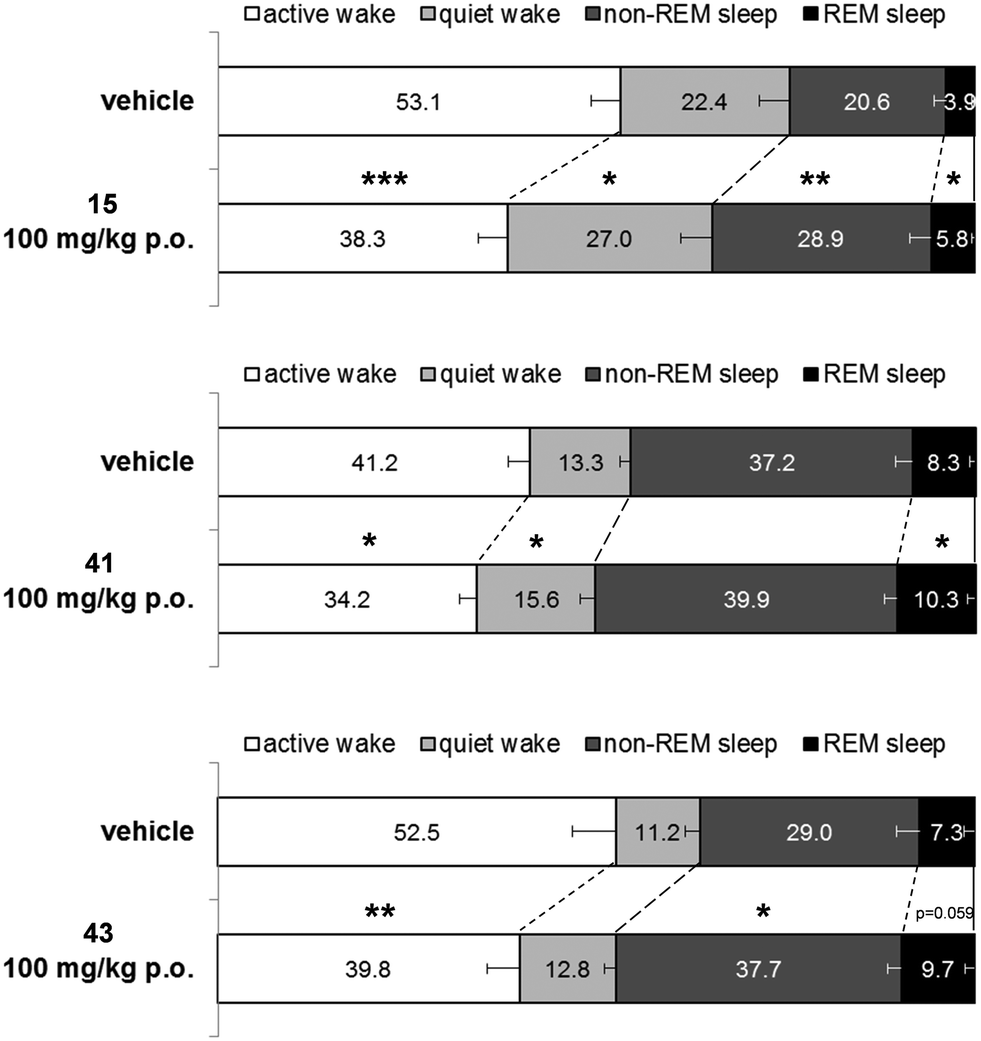 | ||
| Fig. 5 Effect of 15, 41, and 43 on the time spent in sleep and wake stages (percentage of total time) during the 6 h night active period post administration in male Wistar rats. Rats were administered a single oral dose of vehicle (PEG 400) or compound. Data are expressed as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 (n = 7/8 per group). | ||
Over the 6 h night (active) period following administration, 15 decreased significantly the time spent in active wake by 54 min compared to vehicle treated rats (p = 0.0002, paired t-test). However, time spent in quiet wake was increased by 17 min (p = 0.0454, paired t-test). In parallel to the overall decrease in wakefulness, 15 increased significantly both the time spent in rapid eye movement (REM) sleep and non-REM sleep by 7 min and 30 min, respectively, compared to matched-vehicle treated rats (p = 0.0118 and p = 0.0011, respectively, paired t-test). As observed previously with dual orexin receptor antagonists, the relative proportion of time spent in non-REM and REM sleep over the total sleep time stayed the same. Animals treated with 15 spent 82% of their sleeping time in non-REM sleep which is very similar to the time spent in non-REM sleep for the vehicle treated rats (84%, p = 0.2672; paired t-test).
Under the same experimental conditions, compound 41 decreased the time spent awake over the 6 h period following oral administration to a lesser extent than 15. The decrease in active wake time was of 25 min and the increase in quiet wake time of 9 min (p = 0.0218 and p = 0.0327 respectively compared to matched vehicle treated rats, paired t-test). Due to an overall limited decrease of wakefulness, the increase of time spent in non-REM sleep and REM sleep was small (9 and 7 min respectively) and only reached statistical significance for REM sleep (p = 0.0422 compared to matched vehicle treated rats, paired t-test).
Compound 43 decreased significantly the time spent in active wake compared to vehicle treated rats (−46 min; p = 0.0083, paired t-test) but did not significantly impact the time spent in quiet wake (+6 min; p = 0.1982, paired t-test). Consequently, both the time spent in non-REM and REM sleep increased compared to vehicle treated rats (+31 and +9 min respectively). The increase reached statistical significance only for the non-REM sleep time (p = 0.0122 and p = 0.0590 for non-REM and REM sleep, respectively, paired t-test). Similar to what was observed with 15, the relative proportion of time spent in non-REM and REM sleep over the total sleep time stayed stable after 100 mg kg−1 of compound 43 (p = 0.6599, paired t-test) with 80 to 81% of the sleeping time in non-REM sleep.
Since the herein described optimized orexin receptor antagonists originated from an in house HTS campaign, it was important to rule out that the optimized compounds have agonistic activities on the initial target. Compounds 41 and 43 were measured and no agonistic activities could be detected (EC50 > 25 μM).
The synthesis of the compounds 1, and 17 to 49 are described in the ESI† or in patent WO2012110986A1.48 As a prototypical example, the synthesis of 43 is depicted in Scheme 1. Hydrazine 51 was synthesized from commercially available 6-methoxypyridin-3-amine 50, in acidic conditions and the presence of NaNO2 and SnCl4. Aza-indole 52 was formed in presence of sulfuric acid at elevated temperature from hydrazine 51 and commercially available 2-(3-chloropropyl)-1,3-dioxolane. Bromination under Appel conditions49 with CBr4 and PPh3 afforded intermediate 53, which reacted with 4-nitro-pyrazole in the presence of Cs2CO3 to nitro-pyrazole analog 54. Reduction of the nitro group to give amine 55 was achieved by hydrogenation (Pd/C). Compound 43 was obtained by amide coupling of amine 55 with carboxylic acid 59 using TBTU and N,N-diisopropylethylamine (DIPEA). Carboxylic acid 59 was synthesized in a two-step procedure from commercially available 3-methylbenzoyl chloride 56 and ethyl 2-isocyanoacetate 57 in presence of Et3N and DMAP to form oxazole-carboxylic ester 58, which was hydrolyzed under basic conditions to carboxylic acid 59.
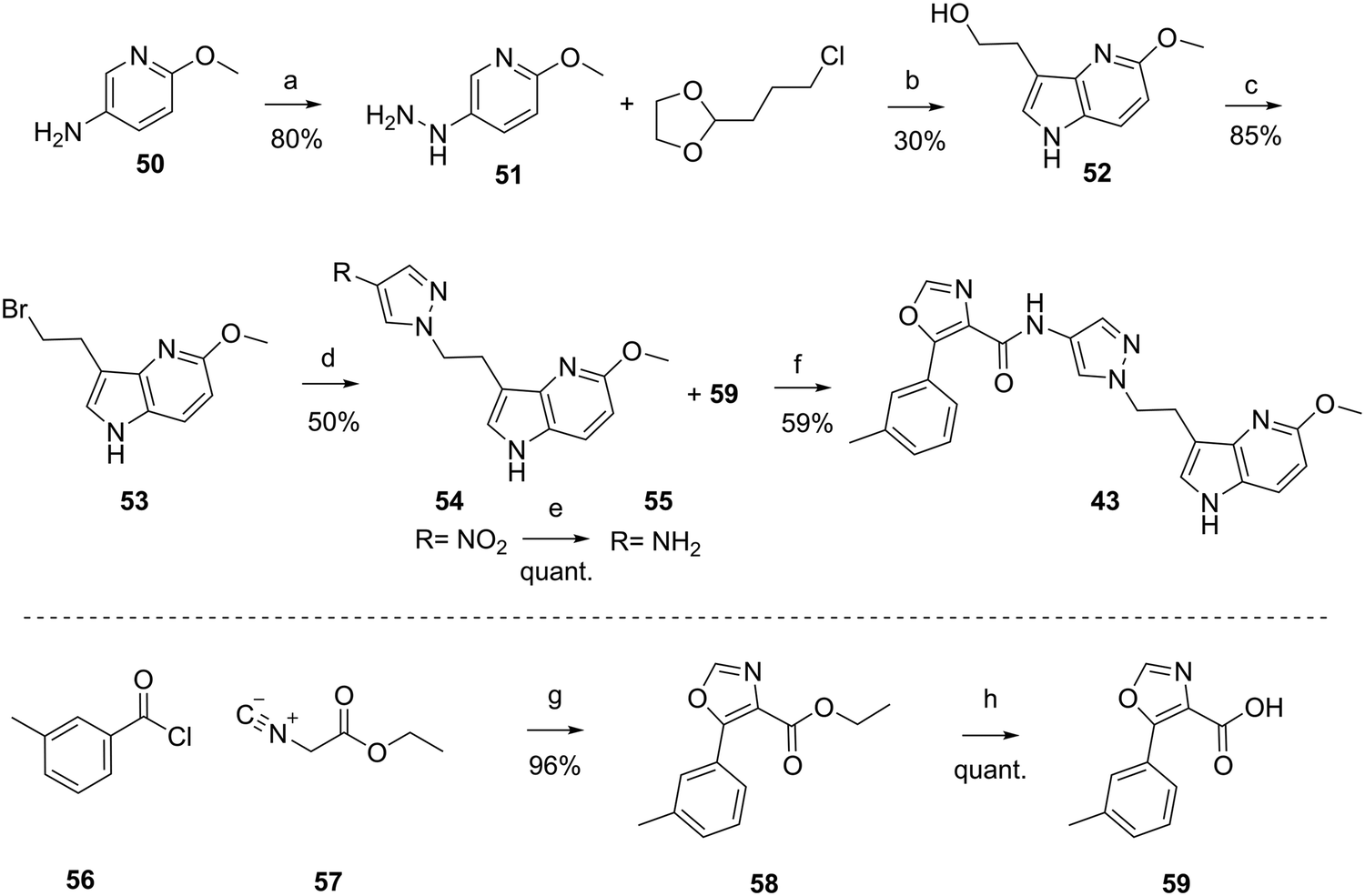 | ||
| Scheme 1 Synthesis of C-3-linked aza-indole 43. Reagents and conditions: (a) 1) NaNO2 in 6 N aq. HCl, −5 °C to 0 °C in 40 min; 2) SnCl2·2H2O in 6 N aq. HCl, 0 °C, 3 h, 80%; (b) 4% aq. H2SO4, 100 °C, 16 h, 30%; (c) CBr4, PPh3, DCM, rt, 2 h, 85%; (d) 4-nitro-1H-pyrazole, Cs2CO3 in MeCN/DMF, 110 °C, 0.5 h, 50%; (e) Pd/C, H2, EtOH, rt, 16 h, 100%; (f) 59, TBTU, DIPEA, DCM, rt, 1 h, 59%; (g) DMAP, Et3N, THF, reflux, 1.5 h, 96%; (h) 4 N aq. NaOH in THF, rt 16 h, 100%. Quant. = quantitative, PPh3 = triphenylphosphine, DMAP = 4-dimethylaminopyridine. | ||
In summary, our approach to identify novel orally active 2-SORAs started from an in-house HTS hit 1, originally characterized as a potent agonist in an unrelated GPCR agonist program.45 Our optimization efforts led to the discovery of pyrazole-derivatives as potent 2-SORAs that are devoid of this agonistic activity.
Conclusion
Compound 43 was found to be a potent, orally active, and brain penetrating 2-SORA, which showed sleep-promoting effects comparable to the 2-SORA clinical compound 15. Compound 43 serves as a viable pharmacological tool compound to further investigate the role of OX2R in the regulation of sleep architecture as well as its contribution to other disorders. Additional studies to assess the compound's potential for development are warranted.Animal welfare and ethical conduct
Animal studies were carried out in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the National Institutes of Health (Bethesda, MD) and were approved by the local Basel-Landschaft cantonal veterinary office (Switzerland).Author contributions
Supervision: CBo, MB; conceptualisation: all authors; investigation: CBr, AT, JG, CR; formal analysis: CBr, JG, AT, CR, MB; methodology; JG, AT, CR; writing: CBr, CR, MB.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
For expert technical support, the authors gratefully thank Jeanine Thanner, Stéphanie Bazire, Katalin Menyhart, Celia Müller-Grandjean, Thomas Sasse, Manon Kiry, Gunther Schmidt, Marco Tschanz, Stephane Delahaye, Isabelle Weber, Fabienne Drouet, Eric Soubieux, Rolf Wuest, Julia Friedrich, Stephanie Hertzog, Päivi Aänismaa, Cédric Fischer, Hélène Massinet, Hélène Roellinger, Markus Gude, and Bibia Heidmann.References
- C. Brisbare-Roch, J. Dingemanse, R. Koberstein, P. Hoever, H. Aissaoui, S. Flores, C. Mueller, O. Nayler, J. van Gerven, S. L. de Haas, P. Hess, C. Qiu, S. Buchmann, M. Scherz, T. Weller, W. Fischli, M. Clozel and F. Jenck, Nat. Med., 2007, 13, 150–155 CrossRef CAS PubMed.
- A. L. Gotter, C. J. Winrow, J. Brunner, S. L. Garson, S. V. Fox, J. Binns, C. M. Harrell, D. Cui, K. L. Yee, M. Stiteler, J. Stevens, A. Savitz, P. L. Tannenbaum, S. J. Tye, T. McDonald, L. Yao, S. D. Kuduk, J. Uslaner, P. J. Coleman and J. J. Renger, BMC Neurosci., 2013, 14, 90 CrossRef PubMed.
- A. J. Roecker, C. D. Cox and P. J. Coleman, J. Med. Chem., 2016, 59, 504–530 CrossRef CAS PubMed.
- C. Muehlan, C. Roch, C. Vaillant and J. Dingemanse, J. Sleep Res., 2023, e13902 CrossRef PubMed.
- S. Vaseghi, S. Zarrabian and A. Haghparast, Neurosci. Biobehav. Rev., 2022, 133, 104516 CrossRef PubMed.
- C. Baimel, S. E. Bartlett, L. C. Chiou, A. J. Lawrence, J. W. Muschamp, O. Patkar, L. W. Tung and S. L. Borgland, Br. J. Pharmacol., 2015, 172, 334–348 CrossRef CAS PubMed.
- P. L. Johnson, W. Truitt, S. D. Fitz, P. E. Minick, A. Dietrich, S. Sanghani, L. Traskman-Bendz, A. W. Goddard, L. Brundin and A. Shekhar, Nat. Med., 2010, 16, 111–115 CrossRef CAS PubMed.
- M. H. James, S. V. Mahler, D. E. Moorman and G. Aston-Jones, Curr. Top. Behav. Neurosci., 2017, 33, 247–281 Search PubMed.
- H. Fagan, E. Jones and D. S. Baldwin, CNS Drugs, 2023, 37, 1–12 CrossRef CAS PubMed.
- T. Sakurai, A. Amemiya, M. Ishii, I. Matsuzaki, R. M. Chemelli, H. Tanaka, S. C. Williams, J. A. Richarson, G. P. Kozlowski, S. Wilson, J. R. Arch, R. E. Buckingham, A. C. Haynes, S. A. Carr, R. S. Annan, D. E. McNulty, W. S. Liu, J. A. Terrett, N. A. Elshourbagy, D. J. Bergsma and M. Yanagisawa, Cell, 1998, 92, 1 CrossRef PubMed page following 696.
- C. Wang, Q. Wang, B. Ji, Y. Pan, C. Xu, B. Cheng, B. Bai and J. Chen, Front. Mol. Neurosci., 2018, 11, 220 CrossRef PubMed.
- A. Bonifazi, F. Del Bello, G. Giorgioni, A. Piergentili, E. Saab, L. Botticelli, C. Cifani, E. Micioni Di Bonaventura, M. V. Micioni Di Bonaventura and W. Quaglia, Med. Res. Rev., 2023, 43, 1607–1667 CrossRef CAS PubMed.
- C. D. Cox, M. J. Breslin, D. B. Whitman, J. D. Schreier, G. B. McGaughey, M. J. Bogusky, A. J. Roecker, S. P. Mercer, R. A. Bednar, W. Lemaire, J. G. Bruno, D. R. Reiss, C. M. Harrell, K. L. Murphy, S. L. Garson, S. M. Doran, T. Prueksaritanont, W. B. Anderson, C. Tang, S. Roller, T. D. Cabalu, D. Cui, G. D. Hartman, S. D. Young, K. S. Koblan, C. J. Winrow, J. J. Renger and P. J. Coleman, J. Med. Chem., 2010, 53, 5320–5332 CrossRef CAS PubMed.
- J. M. Uslaner, W. J. Herring and P. J. Coleman, ACS Pharmacol. Transl. Sci., 2020, 3, 161–168 CrossRef CAS PubMed.
- W. J. Herring, E. Snyder, K. Budd, J. Hutzelmann, D. Snavely, K. Liu, C. Lines, T. Roth and D. Michelson, Neurology, 2012, 79, 2265–2274 CrossRef CAS PubMed.
- P. J. Coleman, A. L. Gotter, W. J. Herring, C. J. Winrow and J. J. Renger, Annu. Rev. Pharmacol. Toxicol., 2017, 57, 509–533 CrossRef CAS PubMed.
- E. Snyder, J. Ma, V. Svetnik, K. M. Connor, C. Lines, D. Michelson and W. J. Herring, Sleep Med., 2016, 19, 93–100 CrossRef PubMed.
- J. Yardley, M. Karppa, Y. Inoue, K. Pinner, C. Perdomo, K. Ishikawa, G. Filippov, N. Kubota and M. Moline, Sleep Med., 2021, 80, 333–342 CrossRef PubMed.
- Y. Yoshida, Y. Naoe, T. Terauchi, F. Ozaki, T. Doko, A. Takemura, T. Tanaka, K. Sorimachi, C. T. Beuckmann, M. Suzuki, T. Ueno, S. Ozaki and M. Yonaga, J. Med. Chem., 2015, 58, 4648–4664 CrossRef CAS PubMed.
- P. Murphy, M. Moline, D. Mayleben, R. Rosenberg, G. Zammit, K. Pinner, S. Dhadda, Q. Hong, L. Giorgi and A. Satlin, J. Clin. Sleep Med., 2017, 13, 1289–1299 CrossRef PubMed.
- C. Boss, J. Gatfield, C. Brotschi, B. Heidmann, T. Sifferlen, M. von Raumer, G. Schmidt, J. T. Williams, A. Treiber and C. Roch, ChemMedChem, 2020, 15, 2286–2305 CrossRef CAS PubMed.
- E. Mignot, D. Mayleben, I. Fietze, D. Leger, G. Zammit, C. L. A. Bassetti, S. Pain, D. S. Kinter, T. Roth and Investigators, Lancet Neurol., 2022, 21, 125–139 CrossRef CAS PubMed.
- J. T. Williams, C. Brotschi, M. Bolli, C. Roch, T. Sifferlen, M. Steiner, A. Treiber, J. Gatfield and C. Boss, J. Med. Chem., 2023 Search PubMed , submitted.
- P. Kaufmann, M. Ort, G. Golor, R. Kornberger and J. Dingemanse, Prog. Neuropsychopharmacol. Biol. Psychiatry, 2021, 108, 110166 CrossRef CAS PubMed.
- Idorsia Media Release May 10, 2022. Phase 2a study results in binge eating disorder. https://ml-eu.globenewswire.com/Resource/Download/f62bfcbf-18c1-408c-9bd1-d9417476b684.
- M. Hirose, S. Egashira, Y. Goto, T. Hashihayata, N. Ohtake, H. Iwaasa, M. Hata, T. Fukami, A. Kanatani and K. Yamada, Bioorg. Med. Chem. Lett., 2003, 13, 4497–4499 CrossRef CAS PubMed.
- I. Fraser, L. Aluisio, S. Yun, J. Shelton, B. Lord, S. Sutton, X. Jiang, C. A. Dvorak, C. Dugovic, P. Bonaventure, N. I. Carruthers and T. Lovenberg, In vivo neurochemical characterization of a selective orexin-2 receptor antagonist, Poster 167.17, in Society for Neuroscience Meeting, San Diego, CA, 2010 Search PubMed.
- C. Betschart, S. Hintermann, D. Behnke, S. Cotesta, M. Fendt, C. E. Gee, L. H. Jacobson, G. Laue, S. Ofner, V. Chaudhari, S. Badiger, C. Pandit, J. Wagner and D. Hoyer, J. Med. Chem., 2013, 56, 7590–7607 CrossRef CAS PubMed.
- T. E. Fitch, M. J. Benvenga, C. D. Jesudason, C. Zink, A. B. Vandergriff, M. M. Menezes, D. A. Schober and L. M. Rorick-Kehn, Front. Neurosci., 2014, 8, 5 Search PubMed.
- P. Malherbe, E. Borroni, L. Gobbi, H. Knust, M. Nettekoven, E. Pinard, O. Roche, M. Rogers-Evans, J. G. Wettstein and J. L. Moreau, Br. J. Pharmacol., 2009, 156, 1326–1341 CrossRef CAS PubMed.
- S. P. Mercer, A. J. Roecker, S. Garson, D. R. Reiss, C. Meacham Harrell, K. L. Murphy, J. G. Bruno, R. A. Bednar, W. Lemaire, D. Cui, T. D. Cabalu, C. Tang, T. Prueksaritanont, G. D. Hartman, S. D. Young, C. J. Winrow, J. J. Renger and P. J. Coleman, Bioorg. Med. Chem. Lett., 2013, 23, 6620–6624 CrossRef CAS PubMed.
- A. J. Roecker, S. P. Mercer, J. D. Schreier, C. D. Cox, M. E. Fraley, J. T. Steen, W. Lemaire, J. G. Bruno, C. M. Harrell, S. L. Garson, A. L. Gotter, S. V. Fox, J. Stevens, P. L. Tannenbaum, T. Prueksaritanont, T. D. Cabalu, D. Cui, J. Stellabott, G. D. Hartman, S. D. Young, C. J. Winrow, J. J. Renger and P. J. Coleman, ChemMedChem, 2014, 9, 311–322 CrossRef CAS PubMed.
- A. J. Roecker, T. S. Reger, M. C. Mattern, S. P. Mercer, J. M. Bergman, J. D. Schreier, R. V. Cube, C. D. Cox, D. Li, W. Lemaire, J. G. Bruno, C. M. Harrell, S. L. Garson, A. L. Gotter, S. V. Fox, J. Stevens, P. L. Tannenbaum, T. Prueksaritanont, T. D. Cabalu, D. Cui, J. Stellabott, G. D. Hartman, S. D. Young, C. J. Winrow, J. J. Renger and P. J. Coleman, Bioorg. Med. Chem. Lett., 2014, 24, 4884–4890 CrossRef CAS PubMed.
- I. T. Raheem, M. J. Breslin, J. Bruno, T. D. Cabalu, A. Cooke, C. D. Cox, D. Cui, S. Garson, A. L. Gotter, S. V. Fox, C. M. Harrell, S. D. Kuduk, W. Lemaire, T. Prueksaritanont, J. J. Renger, C. Stump, P. L. Tannenbaum, P. D. Williams, C. J. Winrow and P. J. Coleman, Bioorg. Med. Chem. Lett., 2015, 25, 444–450 CrossRef CAS PubMed.
- S. D. Kuduk, J. W. Skudlarek, C. N. DiMarco, J. G. Bruno, M. H. Pausch, J. A. O'Brien, T. D. Cabalu, J. Stevens, J. Brunner, P. L. Tannenbaum, S. L. Garson, A. T. Savitz, C. M. Harrell, A. L. Gotter, C. J. Winrow, J. J. Renger and P. J. Coleman, Bioorg. Med. Chem. Lett., 2015, 25, 2488–2492 CrossRef CAS PubMed.
- A. J. Roecker, S. P. Mercer, J. M. Bergman, K. F. Gilbert, S. D. Kuduk, C. M. Harrell, S. L. Garson, S. V. Fox, A. L. Gotter, P. L. Tannenbaum, T. Prueksaritanont, T. D. Cabalu, D. Cui, W. Lemaire, C. J. Winrow, J. J. Renger and P. J. Coleman, Bioorg. Med. Chem. Lett., 2015, 25, 4992–4999 CrossRef CAS PubMed.
- C. Dugovic, J. E. Shelton, L. E. Aluisio, I. C. Fraser, X. Jiang, S. W. Sutton, P. Bonaventure, S. Yun, X. Li, B. Lord, C. A. Dvorak, N. I. Carruthers and T. W. Lovenberg, J. Pharmacol. Exp. Ther., 2009, 330, 142–151 CrossRef CAS PubMed.
- F. Micheli, M. Antolini, R. Di Fabio, A. Pellacani and A. Pozzan, Bioorg. Med. Chem. Lett., 2010, 20, 6405–6407 CrossRef CAS PubMed.
- A. L. Gotter, M. S. Forman, C. M. Harrell, J. Stevens, V. Svetnik, K. L. Yee, X. Li, A. J. Roecker, S. V. Fox, P. L. Tannenbaum, S. L. Garson, I. D. Lepeleire, N. Calder, L. Rosen, A. Struyk, P. J. Coleman, W. J. Herring, J. J. Renger and C. J. Winrow, Sci. Rep., 2016, 6, 27147 CrossRef CAS PubMed.
- M. A. Letavic, P. Bonaventure, N. I. Carruthers, C. Dugovic, T. Koudriakova, B. Lord, T. W. Lovenberg, K. S. Ly, N. S. Mani, D. Nepomuceno, D. J. Pippel, M. Rizzolio, J. E. Shelton, C. R. Shah, B. T. Shireman, L. K. Young and S. Yun, J. Med. Chem., 2015, 58, 5620–5636 CrossRef CAS PubMed.
- P. Bonaventure, J. Shelton, S. Yun, D. Nepomuceno, S. Sutton, L. Aluisio, I. Fraser, B. Lord, J. Shoblock, N. Welty, S. R. Chaplan, Z. Aguilar, R. Halter, A. Ndifor, T. Koudriakova, M. Rizzolio, M. Letavic, N. I. Carruthers, T. Lovenberg and C. Dugovic, J. Pharmacol. Exp. Ther., 2015, 354, 471–482 CrossRef CAS PubMed.
- K. Recourt, P. de Boer, R. Zuiker, R. Luthringer, J. Kent, P. van der Ark, I. Van Hove, J. van Gerven, G. Jacobs, L. van Nueten and W. Drevets, Transl. Psychiatry, 2019, 9, 240 CrossRef PubMed.
- S. Maehara, N. Yuge, C. Higashi, T. Ota, J. Furukawa and T. Takeuchi, Neuropsychopharmacol. Rep., 2020, 40, 182–189 CrossRef CAS PubMed.
- V. L. Revell, C. Della Monica, J. Mendis, H. Hassanin, R. J. Halter, S. R. Chaplan and D. J. Dijk, Neuropsychopharmacology, 2022, 47, 719–727 CrossRef CAS PubMed.
- D. Bur, O. Corminboeuf, S. Cren, H. Fretz, C. Grisostomi, X. Leroy, J. Pothier and S. Richard-Bildstein, WO2009077954, 2009.
- P. J. Coleman, C. D. Cox and A. J. Roecker, Curr. Top. Med. Chem., 2011, 11, 696–725 CrossRef CAS PubMed.
- A. Futamura, D. Nozawa, Y. Araki, Y. Tamura, S. Tokura, H. Kawamoto, Y. Tokumaru, S. Kakihara, T. Aoki and N. Ohtake, Bioorg. Med. Chem., 2017, 25, 5203–5215 CrossRef CAS PubMed.
- M. Bolli, C. Boss, C. Brotschi, M. Gude, B. Heidmann, T. Sifferlen and J. T. Williams, WO2012110986, 2012.
- V. S. C. de Andrade and M. C. S. de Mattos, Curr. Org. Synth., 2015, 12, 309–327 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3md00573a |
| This journal is © The Royal Society of Chemistry 2024 |