
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBall-milling for efficient synthesis of pyridine-containing iron(II) photosensitizers†
Enita
Rastoder
a,
Thierry
Michel
b,
Frédéric
Lamaty
 *a and
Xavier
Bantreil
*ac
*a and
Xavier
Bantreil
*ac
aIBMM, Univ Montpellier, CNRS, ENSCM, Montpellier, France. E-mail: frederic.lamaty@umontpellier.fr; xavier.bantreil@umontpellier.fr
bLaboratoire Charles Coulomb, UMR 5221 CNRS, Université de Montpellier, Montpellier 34095, France
cInstitut Universitaire de France (IUF), France
First published on 20th February 2024
Abstract
Photoredox catalysis is becoming more and more prevalent in the 21st century as a new tool for organic and polymer synthesis. In addition, this domain clearly fits the expectations of the twelve principles of green chemistry. However, access to metal containing photosensitizers is not always straightforward and can require long reaction times, the use of toxic solvents and multi-step synthesis. These are definitely drawbacks that could be overcome with the use of novel technologies. In this report, we develop a one-pot two-step synthesis of iron(II) photosensitizers using ball-milling. Overall reaction times were drastically reduced, no solvent was needed during the reaction, and ten complexes could be isolated in high yields (73–99%). Using a transparent milling jar, the formation of the complexes could be followed using in situ Raman spectroscopy.
Introduction
The use of coordination compounds as catalysts is nowadays extremely important in the academic and industrial world to perform rapid and efficient organic transformations. The tremendous improvements inherent to catalysis are evident by the many Nobel prizes in this field.1–4 Among the different applications of catalysis, photoredox catalysis has emerged, providing new opportunities to diversify the organic chemist's toolbox.5 More recently, combining photoredox catalysts with more classical metal-based catalysis has allowed scientists to go one step further in the complexity of the transformations that can be promoted.6 Photoredox catalysis is not limited to the synthesis of small molecules of interest but has also been applied in polymerization reactions.7–9 Among the different catalysts that present photoredox properties upon light irradiation, iron complexes have attracted strong interest given the price, the natural availability of the metal, and the high efficiency of the complexes.10Mechanochemistry, and more specifically ball-milling, has emerged in the past few years as an alternative activation method that could allow reactions to be facilitated while reducing the amount of solvent used during the reaction.11–17 In some cases, the use of solvent-free/solventless milling resulted in a change of selectivity,18,19 or in the isolation of compounds that could not be obtained otherwise.20 Mechanochemistry has also been applied successfully to the synthesis of coordination complexes and corresponding ligands, but examples remain limited.21 In this area, we reported the efficient mechanosynthesis of metallic complexes featuring NHC (N-Heterocyclic Carbene) ligands,22–27 involved in catalysis28,29 or as anticancer agents,23 salen-like ligands,30 and sydnone-based bidentate ligands.31 In this work, we pursued our interest in facilitating the synthesis of coordination complexes, focusing on the synthesis of photoredox catalysts. In particular, pyridine-containing iron(II) catalysts have been shown to efficiently initiate radical and cationic polymerization.32–34 However, their classical synthesis required long reaction times, step-wise synthesis and the use of refluxing toluene. Based on previous reports involving a grinding approach for the controlled access to polymorphs of Schiff bases,35–38 the auto-assembly of supramolecular iron complexes39 and the synthesis of first-row transition-metal bis(imino)pyridine complexes,40 we thought that a mechanochemical approach could allow access to such valuable photoredox complexes more efficiently (Fig. 1).
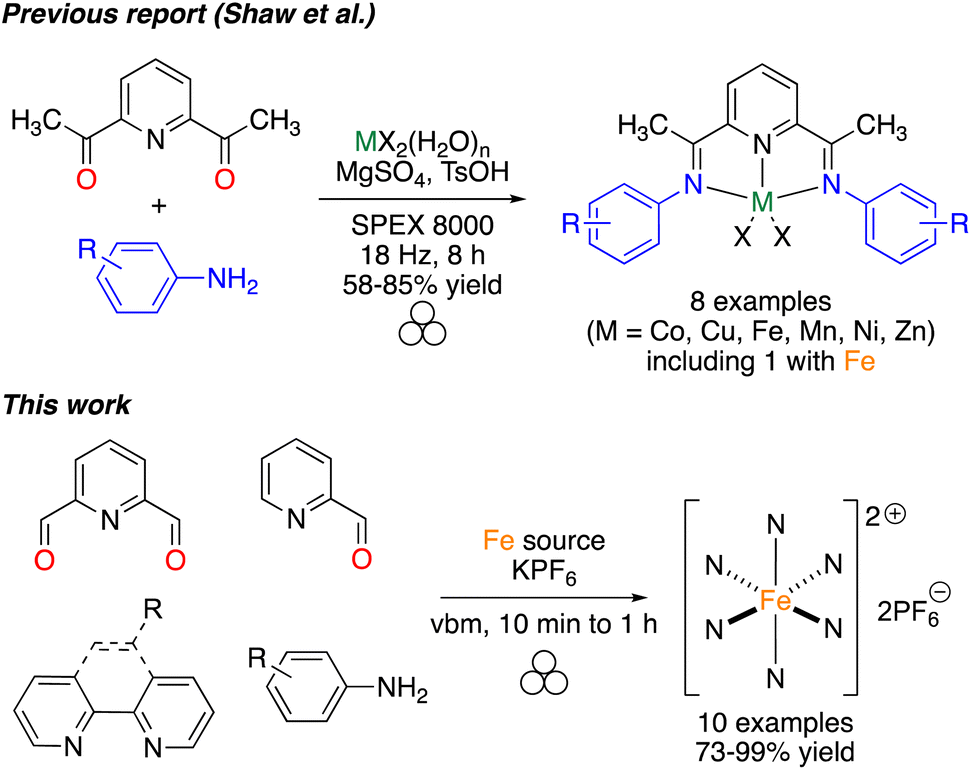 | ||
| Fig. 1 Synthetic strategy for the synthesis of iron complexes. | ||
Results and discussion
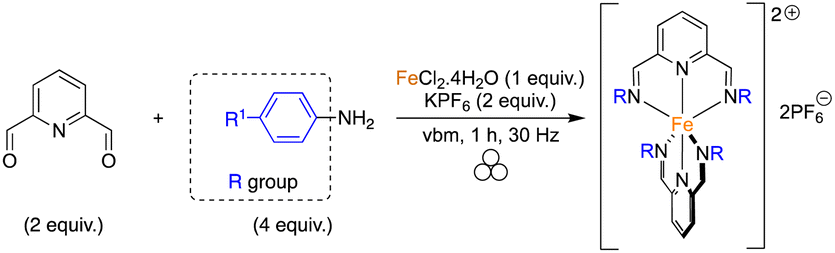
As shown in Fig. 1, different pyridine-containing molecules were chosen because they are good σ-donor as well as π-acceptor ligands and thus form strong M–L bonds.41 In addition, with these types of highly π-conjugated ligands, the corresponding complexes should absorb light in the visible region of the electromagnetic spectrum. We first focused on ligands involving diimines formed from 2,6-pyridinedicarboxaldehyde (2,6-PDCA) and aromatic amines. The corresponding complexes, in which two tridentate ligands bind to the metal center to form the octahedral 18-electron metallic complex, are usually prepared in a stepwise manner, i.e. preparation of the ligand (using refluxing toluene in a Dean–Stark apparatus) and then coordination to the metal center. In our mechanochemical approach, a Teflon reactor (15 mL) and one stainless steel ball (10 mm diameter) were utilized in a vibratory ball-mill (vbm).In the first experiments, we attempted the formation of the ligand through imine formation. Several reports already described the easy mechanochemical formation of imine bonds.30,42,43 However, in our case, and probably due to partial hydrolysis of the imines, full conversion was never obtained. We thus decided to directly add an iron(II) source that could, due to the coordination reaction which is favored entropically (chelate effect) and kinetically, enhance the formation of the tridentate ligand. Indeed, the templating effect of a metal-ion was already demonstrated for the preparation of macrocyclic complexes in solution.44,45 Reaction at 25 Hz using FeSO4·7H2O as the iron(II) source did not give satisfactory results as the products obtained from the milling jar were insoluble in common solvents (H2O, acetone, DMSO, chloroform), thus preventing 1H NMR analysis (Table 1, entry 1). Switching to FeCl2·4H2O furnished a compound that was soluble in H2O (Table 1, entry 2). However, hydrolysis of the diimine occurred in D2O and inaccurate conversions were measured by 1H NMR. An anion metathesis with KPF6, which was shown previously to be highly efficient in the solid state,23,27,46 was thus performed to change the solubility of the final complex. Gratifyingly, the addition of potassium hexafluorophosphate after 1 h of milling, and a further 30 min of milling, allowed a final compound soluble in organic solvents to be obtained and an 85% conversion was measured (Table 1, entry 3). Besides, increasing the vibrational frequency from 25 to 30 Hz furnished a conversion of 93% (Table 1, entry 4). Finally, a one-pot mechanochemical method, where all the reagents were added together at the same time and ground for 1 h at 30 Hz, was realized and furnished a complete conversion into desired complex 1a (Table 1, entry 5). The pure complex 1a was isolated straightforwardly through a dissolution in acetone of the mixture, precipitation in diethyl ether and washing with water and diethyl ether. Contrary to what was reported with 2,6-diacetylpyridine, the use of p-toluenesulfonamide, as a catalyst, and MgSO4, as an additive to trap the water generated during imine formation, was not compulsory.40 Moreover, our protocol allowed us to work under stoichiometric conditions, thus facilitating the isolation of the final complex.
|
|
||
|---|---|---|
| Entry | Conditions | Conv.b (%) |
| a Standard reaction conditions: the iron(II) source (1 equiv.), 2,6-pyridinedicarboxaldehyde (2 equiv.), para-toluidine (4 equiv.), and KPF6 (2 equiv.) were milled at 25–30 Hz for 1 h to 1.5 h. Milling load = 20 mg mL−1. Total mass of reagents: 290 mg. b Determined by 1H NMR. c n.d. = not determined, insoluble mixture. d Hydrolysis of the ligand was observed. e All the reagents added at the start of the reaction. | ||
| 1 | FeSO4·7H2O, 25 Hz, 1 h | n.d.c |
| 2 | FeCl2·4H2O, 25 Hz, 1 h | 44d |
| 3 | FeCl2·4H2O, KPF6 (added after 1 h), 25 Hz, 1.5 h | 85 |
| 4 | FeCl2·4H2O, KPF6 (added after 1 h), 30 Hz, 1.5 h | 93 |
| 5 | FeCl2·4H2O, KPF6, 30 Hz, 1 he | 99 |
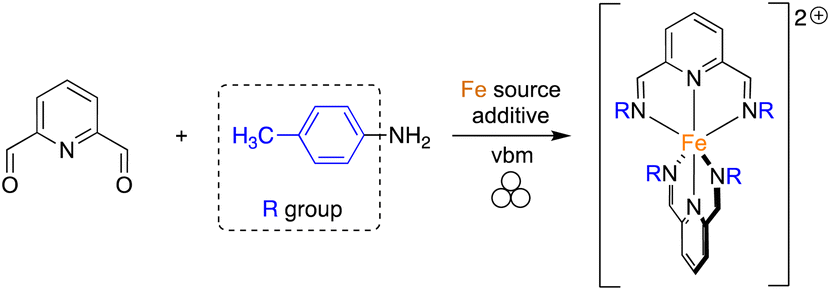
With this result in hand, the one-pot method was applied to the synthesis of complexes 1a–d using various amines (Table 2). The first successful compound (1a) was formed from p-toluidine with a yield of 86% (Table 2, entry 1). The one-pot synthesis was carried out with no addition of solvents in only an hour using vbm at 30 Hz. In contrast, the solution-based synthesis33 followed a 2-step route where the diimine was formed first, via a reflux in toluene for 4 h. Only after recrystallization of the diimine was the complexation carried out in ethanol for 2 h giving an overall yield of 65%. Maintaining the same mechanochemical procedure, iron(II) complexes 1b and 1c, synthesized from p-anisidine and 4-(dimethylamino)aniline, were obtained with full conversion and respective isolated yields of 82% and 88% (Table 2, entries 2 & 3). Once again, via the solution-based method, the imines were formed first in refluxing toluene, then complexation was performed in ethanol with overall yields of 71% and 86%, respectively. Finally, complex 1d was obtained from 4-bromoaniline in 73% yield (Table 2, entry 4). Bulky anilines such as 2,4,6-trimethylaniline and 2,6-diisopropylaniline were also tested. However, although the formation of the imines was effective, the coordination to the metal center remained unsuccessful, probably because of the steric hindrance of the diamine ligands.
|
|
|||
|---|---|---|---|
| Entry | R1 | Compound (yield %) | In lit. (yield %) |
| a Reaction conditions: 2,6-pyridinedicarboxaldehyde (2 equiv.), amine (4 equiv.), FeCl2·4H2O (1 equiv.), KPF6 (2 equiv.), milling load = 20 mg mL−1, vbm, 30 Hz, 1 h. Total mass of reagents: 290 mg. b Not reported. | |||
| 1 | CH3 | 1a (86) | (1) 4 h reflux in toluene (91) |
| (2) 2 h in ethanol at r.t. (71) | |||
| 2 | OCH3 | 1b (82) | (1) 2 h reflux in toluene (76) |
| (2) 2 h in ethanol at r.t. (93) | |||
| 3 | N(CH3)2 | 1c (88) | (1) 3 h reflux in toluene (96) |
| (2) 2 h in ethanol at r.t. (90) | |||
| 4 | Br | 1d (73) | n.r.b |
As already reported, Raman spectroscopy is to date one of the most efficient techniques for the in situ monitoring of mechanochemical reactions.47–49 We thus explored if this technique could be useful to monitor the formation of complex 1a. First, Raman spectra of pure substrates (i.e. aniline, 2,6-pyridinedicarboxaldehyde), intermediate ligand L1 and final complex 1a were acquired using a portable Raman spectrometer with a laser excitation wavelength at 785 nm (Fig. 2a). The formation of L1 could be unambiguously observed with the disappearance of the stretching vibration band of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond around 1690 cm−1 and the appearance of the stretching vibration band of the CN bond around 1600 cm−1.50 In addition, the band around 1000 cm−1 corresponds to the pyridine core breathing mode.51,52 Upon complexation with iron, the signals are reduced and difficult to assign (Fig. 2a, green curve). However, we attempted the in situ monitoring for the formation of 1a from 2,6-pyridinedicarboxaldehyde, using a home-made set-up and a transparent PMMA milling jar.53 Interestingly, the formation of the ligand L1 was found to be almost instantaneous, as after 2 min of milling, only traces of the remaining 2,6-pyridinedicarboxaldehyde appear as shown by the tiny band around 1700 cm−1 (Fig. 2b). Formation of complex 1a also seems to start fairly rapidly with the appearance of a broad band around 1450 cm−1. Upon prolonged milling (up to 60 min), the complete disappearance of the bands belonging to the intermediate ligand and the appearance of broad bands clearly showed the formation of the final complex.
O bond around 1690 cm−1 and the appearance of the stretching vibration band of the CN bond around 1600 cm−1.50 In addition, the band around 1000 cm−1 corresponds to the pyridine core breathing mode.51,52 Upon complexation with iron, the signals are reduced and difficult to assign (Fig. 2a, green curve). However, we attempted the in situ monitoring for the formation of 1a from 2,6-pyridinedicarboxaldehyde, using a home-made set-up and a transparent PMMA milling jar.53 Interestingly, the formation of the ligand L1 was found to be almost instantaneous, as after 2 min of milling, only traces of the remaining 2,6-pyridinedicarboxaldehyde appear as shown by the tiny band around 1700 cm−1 (Fig. 2b). Formation of complex 1a also seems to start fairly rapidly with the appearance of a broad band around 1450 cm−1. Upon prolonged milling (up to 60 min), the complete disappearance of the bands belonging to the intermediate ligand and the appearance of broad bands clearly showed the formation of the final complex.
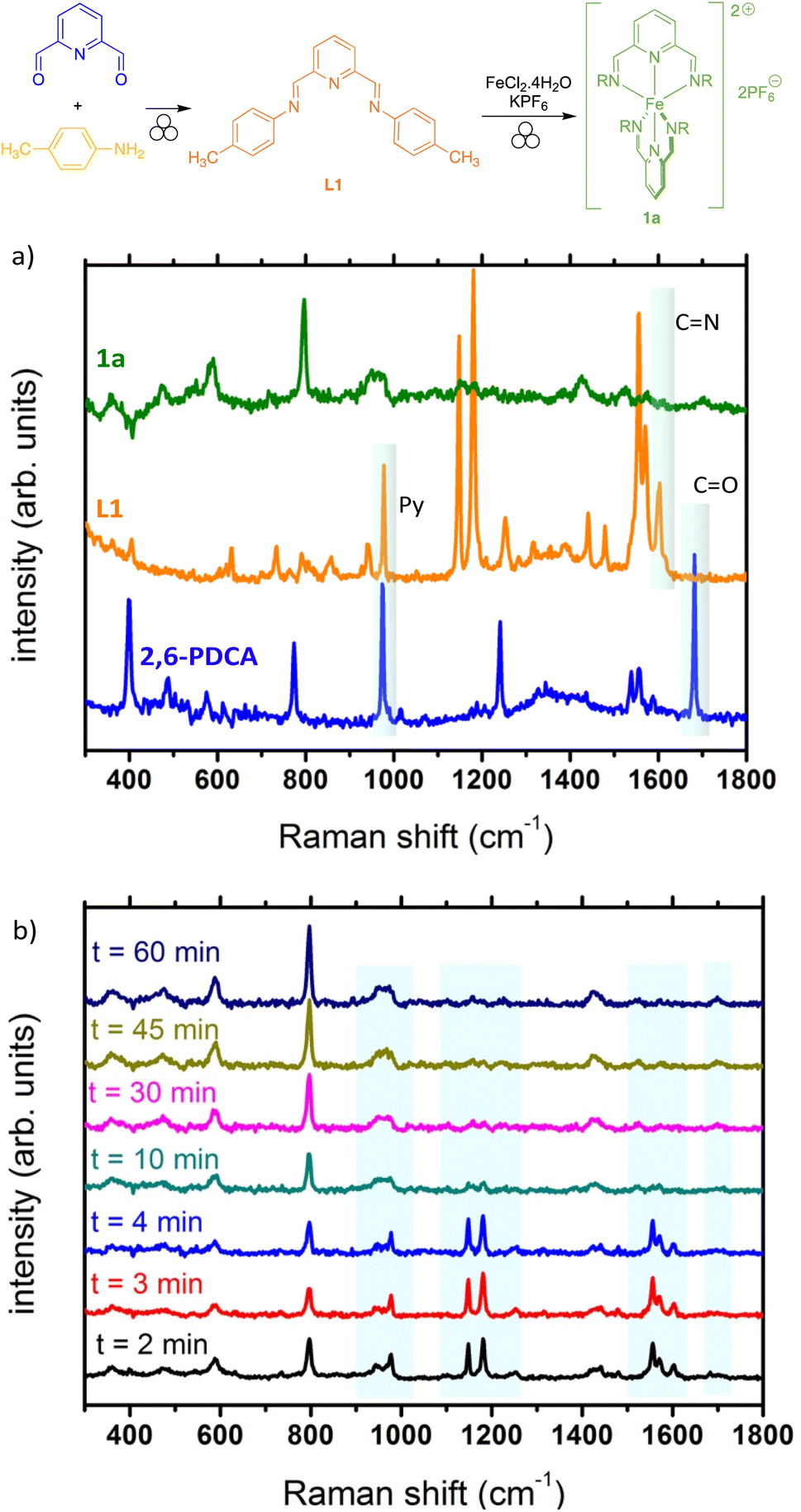 | ||
| Fig. 2 Raman spectroscopy for reaction monitoring. (a) Spectra of pure compounds (vertically shifted for clarity): 2,6-pyridinedicarboxaldehyde (blue), ligand (orange), and complex 1a (green). (b) In situ monitoring of the mechanosynthesis of complex 1a. | ||
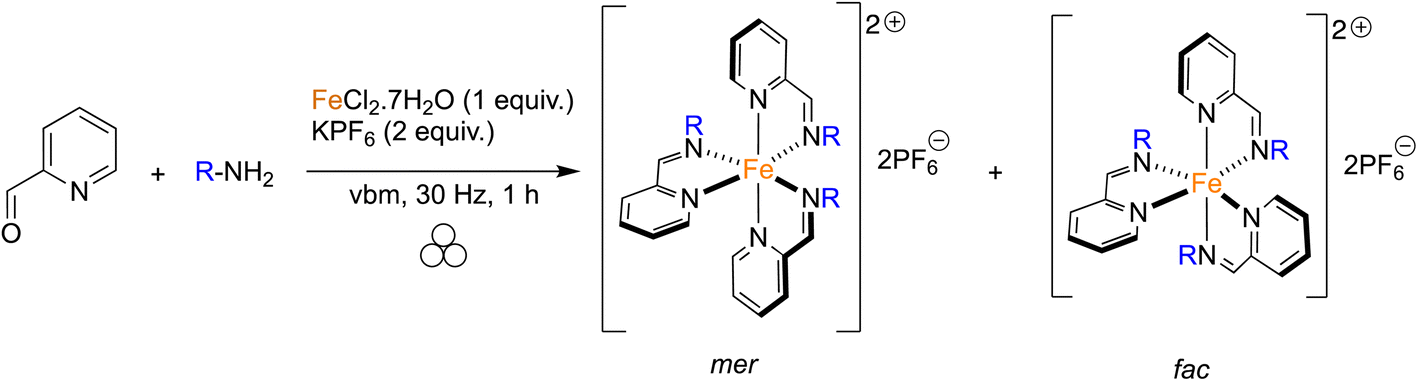
Given the good results obtained for the one-pot mechanosynthesis of 1a–d, a second family of iron complexes was targeted starting from 2-pyridinecarboxaldehyde, various amines, iron(II) chloride and potassium hexafluorophosphate (Table 3). In this case, three bidentate ligands bind to the metal center in order to give an octahedral complex. The assembly of the ligands around the metal can take two different forms which gives rise to the facial (fac) and meridional (mer) geometric stereoisomers.54 The ligands of the fac isomer are equivalent because of a C3 axis of symmetry. In contrast, the mer isomer does not have any planes or axes of symmetry. With this in mind, the ratio between the two isomers could be determined by 1H NMR. When p-toluidine was used as an amine, using a vbm agitated at 30 Hz for 1 h, product 2a was formed with a mer/fac ratio of 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and a yield of 73% (Table 3, entry 1). Even when the reaction time was increased (up to 3 h), the mer/fac ratio remained the same. It was suggested that π-stacking, trans-influence and steric hindrance between the ligands play an important role in the formation of the mer compound as a major isomer.54 In the literature, 1H NMR reports reveal the presence of both isomers in a slightly different mer/fac ratio of 5:1.32,34 Complex 2a was formed in solution following a 2-step synthesis. The imine was obtained via a seven-hour-long reflux in toluene and later coordinated to iron(II) in ethanol for one hour allowing an overall yield of 44%. Using the same one-pot protocol, compound 2b was obtained from p-anisidine with a mer/fac ratio of 12:1 in an 88% yield (Table 3, entry 2). Interestingly, the solution-based method, which followed room temperature stirring for 18 h, gave the same yield but with an 8:1 mer/fac ratio. Finally, complex 2c was synthesized from 4-(dimethylamino)aniline with a 96% yield (Table 3, entry 3), whereas in solution an overall yield of only 43% was reported. Whether mechanochemical or classic solution conditions were applied, only the mer isomer was formed in this case. From 2a to 2c the mer/fac ratio increased in favor of the mer configuration, which seems related to the steric hindrance and electron-donating properties of the aniline para substituent. Indeed, the –N(CH3)2 is a bulkier and more electron-donating group than the –OCH3 and –CH3. These properties also seem to be related to the catalytic activity of the complexes in the photopolymerization of acrylates, since among complexes 2a–c, 2c exhibited the best catalytic activity.33 Once again, the use of bulky anilines (2,6-diisopropylaniline, 2,4,6-trimethylaniline and 1-naphthylamine) was revealed to be unfruitful as no complex was observed.
:1 and a yield of 73% (Table 3, entry 1). Even when the reaction time was increased (up to 3 h), the mer/fac ratio remained the same. It was suggested that π-stacking, trans-influence and steric hindrance between the ligands play an important role in the formation of the mer compound as a major isomer.54 In the literature, 1H NMR reports reveal the presence of both isomers in a slightly different mer/fac ratio of 5:1.32,34 Complex 2a was formed in solution following a 2-step synthesis. The imine was obtained via a seven-hour-long reflux in toluene and later coordinated to iron(II) in ethanol for one hour allowing an overall yield of 44%. Using the same one-pot protocol, compound 2b was obtained from p-anisidine with a mer/fac ratio of 12:1 in an 88% yield (Table 3, entry 2). Interestingly, the solution-based method, which followed room temperature stirring for 18 h, gave the same yield but with an 8:1 mer/fac ratio. Finally, complex 2c was synthesized from 4-(dimethylamino)aniline with a 96% yield (Table 3, entry 3), whereas in solution an overall yield of only 43% was reported. Whether mechanochemical or classic solution conditions were applied, only the mer isomer was formed in this case. From 2a to 2c the mer/fac ratio increased in favor of the mer configuration, which seems related to the steric hindrance and electron-donating properties of the aniline para substituent. Indeed, the –N(CH3)2 is a bulkier and more electron-donating group than the –OCH3 and –CH3. These properties also seem to be related to the catalytic activity of the complexes in the photopolymerization of acrylates, since among complexes 2a–c, 2c exhibited the best catalytic activity.33 Once again, the use of bulky anilines (2,6-diisopropylaniline, 2,4,6-trimethylaniline and 1-naphthylamine) was revealed to be unfruitful as no complex was observed.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Compound | R | mer/fac ratio | Yield (%) | Lit. (yield %) |
| a Reaction conditions: 2-pyridinecarboxaldehyde (3 equiv.), amine (3 equiv.), FeCl2·4H2O (1 equiv.), KPF6 (2 equiv.), milling load = 20 mg mL−1, vbm, 30 Hz. Total mass of reagents: 290 mg. | |||||
| 1 | 2a |
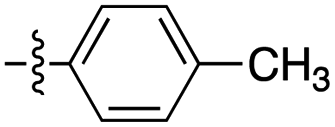
|
6:1 |
73 | (1) 7 h reflux in toluene (63) |
| (2) 1 h in ethanol at r.t. (70) | |||||
| 2 | 2b |
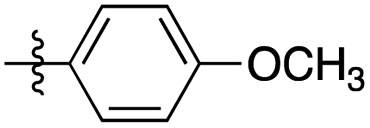
|
12:1 |
88 | 18 h stirring in ethanol at r.t. (88) |
| 3 | 2c |
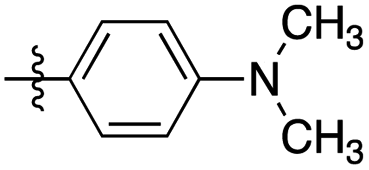
|
>20:1 |
96 | (1) 5 h reflux in toluene (71) |
| (2) 2 h in ethanol at r.t. (60) | |||||
Seeing that polyamines could coordinate efficiently to iron, we realized the mechanosynthesis of complexes featuring 2,2′-bipyridine-like ligands, which were also found to have interesting photoredox properties (Scheme 1).33 In this case, FeSO4·7H2O was used as the metal source because it allowed higher conversions than FeCl2·4H2O. Since the ligands were employed as such without further transformations, the reaction times could be lowered to obtain complete conversion. When 2,2′-bipyridine was taken as the ligand, only 10 minutes of agitation at 30 Hz was needed, and compound 3a was yielded in 83%. In solution, a one-hour reflux in water was required to give a 93% yield.33 Interestingly, powder X-ray diffraction of 3a, directly taken from the milling jar without further treatment, showed a high degree of crystallinity (Fig. 3). Likewise, 3b, featuring 1,10-phenanthroline, was readily synthesized in 15 minutes of ball milling (90% yield), while synthesis in refluxing water for 1 h furnished an 82% yield.33 Of note, the synthesis of similar complexes was previously reported using an agate mortar and pestle for application as spin crossover materials.55 Finally, when 1,10-phenanthrolin-5-amine was employed as a ligand, 3c was formed in 30 minutes with quantitative yield. The solution synthesis of 3c required the use of a boiling mixture of chloroform/water for 1 h, and furnished a poor yield of 19%.58 Compared to 2,2′-bipyridine and 1,10-phenanthroline, 1,10-phenanthrolin-5-amine does not possess a C2 symmetry and thus, a mer/fac isomerism could occur. However, 1H NMR revealed the equivalency of all the ligands, meaning that only the fac isomer was formed. As shown below, it is noteworthy that the conversion of the slightly colored reagents (Scheme 1a) could be witnessed easily as complexes 3a–c are red solids (Scheme 1b).
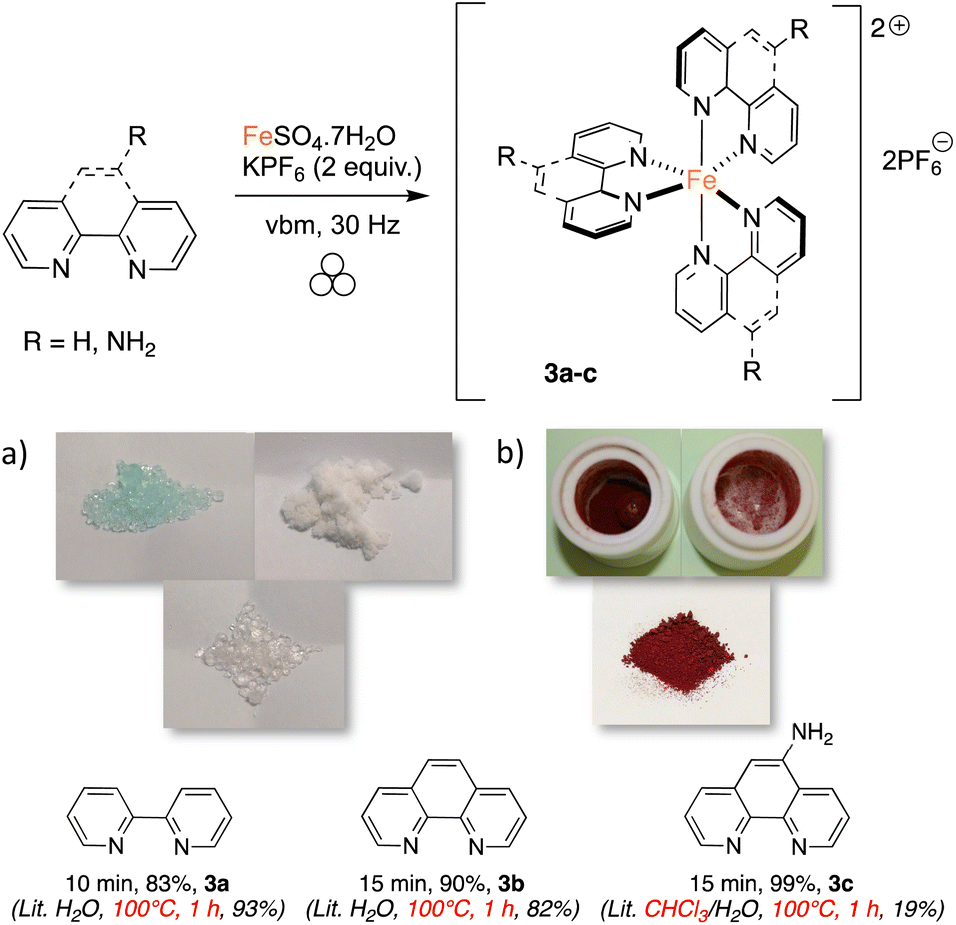 | ||
| Scheme 1 Mechanosynthesis of iron complexes featuring bipyridine-like ligands. (a) White and slightly colored reagents (FeSO4·7H2O, KPF6 and 2,2′-bipyridine); (b) reaction mixture (red complex 3a). | ||
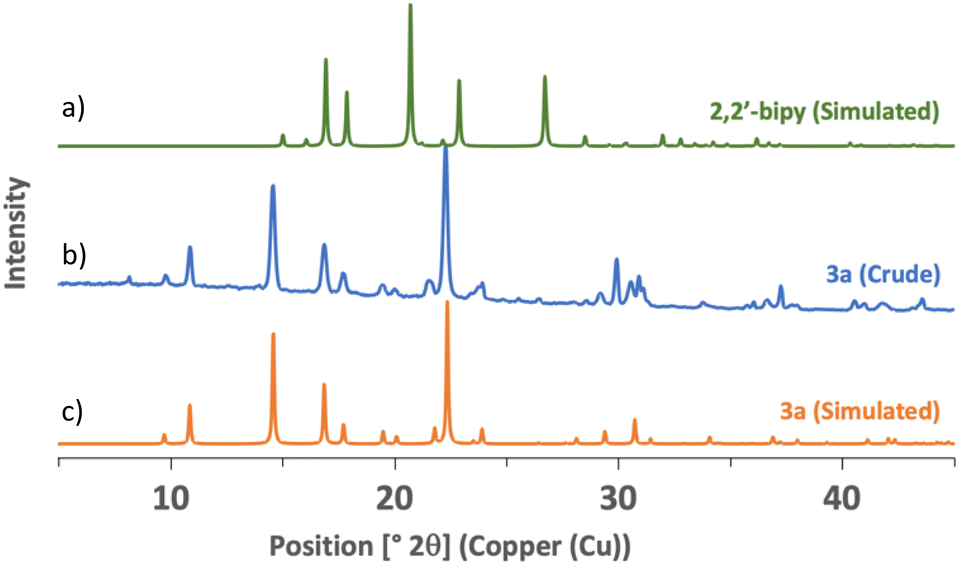 | ||
| Fig. 3 PXRD patterns of (a) 2,2′-bipyridine – simulated from ref. 56; (b) complex 3a – sample directly taken from the milling jar; and (c) complex 3a – simulated from ref. 57. | ||
Experimental
Commercial reagents were used as received. Analyses were performed at the “Plateforme Technologique Laboratoire de Mesures Physiques” (IBMM, Université de Montpellier). 1H NMR spectra were recorded on a Bruker Avance III HD 400 MHz spectrometer and are reported in ppm using deuterated solvent for calibration (acetone-d6 at 2.05 ppm). Data are reported as s = singlet, d = doublet, t = triplet, q = quadruplet, qt = quintuplet, sept = septuplet, m = multiplet; coupling constant in Hz; integration. 13C NMR spectra were recorded on a Bruker Avance III HD 101 MHz spectrometer and are reported in ppm using a deuterated solvent for calibration (acetone-d6 at 29.84 and 206.26 ppm). 19F NMR spectra were recorded on a Bruker Avance III HD 376 MHz spectrometer and are reported in ppm. 31P NMR spectra were recorded on a Bruker Avance III HD 162 MHz spectrometer and are reported in ppm. X-ray powder patterns were recorded in the Bragg–Brentano q–q reflection geometry, on a Bruker D8 Discover diffractometer equipped with a primary monochromator (CuKα1, λ = 1.54056 Å) and LYNXEYE_XE_T detector. Measurements were performed at room temperature in the 5–45° 2θ range, using a step size of 0.033° and a counting time per step of 2.5 s. The milling treatments were carried out in a vibrating Retsch Mixer Mill 200 or 400 (vbm) operated at 25 Hz or 30 Hz. Milling load (ML) is defined as the ratio between the mass of the reactant over the free volume of the jar and was fixed at 20 mg mL−1. Raman measurements were conducted on an iRaman® analyser (Opton Laser International), equipped with a TE cooled linear array detector (2048 pixels) and a non-contact fiber-optic Raman probe (working distance: 5.4 mm). The excitation wavelength was 785 nm. Ex situ measurements (pure samples) consisted of one spectrum with a radiation time of 500 ms. In situ measurements (using a PMMA milling jar) were performed every 30 s, consisting of one spectrum with a radiation time of 500 ms.General procedure A: synthesis of complexes 1a–d
FeCl2·4H2O (1 equiv.), the corresponding amine (4 equiv.), KPF6 (2 equiv.) and 2,6-pyridinedicarboxaldehyde (2 equiv.) were introduced in a 15 mL Teflon grinding jar with one stainless steel ball (10 mm diameter). The jar was closed, sealed with Parafilm, placed in the vibratory ball-mill and subjected to grinding for 1 h at 30 Hz. The solid was dissolved in a minimum amount of acetone and then precipitated in ether. After washing with water and ether, the product was dried in vacuo.General procedure B: synthesis of complexes 2a–c
FeCl2·4H2O (1 equiv.), the corresponding amine (3 equiv.), KPF6 (2 equiv.) and 2-pyridinecarboxaldehyde (3 equiv.) were introduced in a 15 mL Teflon grinding jar with one stainless steel ball (10 mm diameter). The jar was closed, sealed with Parafilm, placed in the vibratory ball-mill and subjected to grinding for 1 h at 30 Hz. The solid was dissolved in a minimum amount of acetone and then precipitated in ether. After washing with water and ether, the product was dried in vacuo.General procedure C: synthesis of complexes 3a–c
FeSO4·7H2O (1 equiv.), the corresponding bipyridine (3 equiv.) and KPF6 (2 equiv.) were introduced in a 15 mL Teflon grinding jar with one stainless steel ball (10 mm diameter). The jar was closed, sealed with Parafilm, placed in the vibratory ball-mill and subjected to grinding for a specific time at 30 Hz. The solid was dissolved in a minimum amount of acetone and then precipitated in ether. After washing with water and ether, the product was dried in vacuo.Conclusions
In summary, a mechanochemical route was developed as an efficient alternative for the formation of three families of iron complexes. By solventless milling, the complexes could be synthesized rapidly (less than 1 h of milling), efficiently, and with improved total conversions and yields compared to solution chemistry. In addition, easy treatment involving crystallization and filtration allowed the isolation of the compounds. Although the first step of imine formation remained incomplete in the sequential route, the one-pot procedure readily allowed full conversion to be reached. Finally, Raman spectroscopy revealed a useful technique for the in situ monitoring of the reactions. Further studies should be performed to study the influence of the synthetic method on the photophysical properties of the complexes.Author contributions
Xavier Bantreil: conceptualization (equal); writing – original draft (lead); formal analysis (lead); writing – review and editing (equal). Frédéric Lamaty: conceptualization (equal); writing – original draft (supporting); writing – review and editing (equal). Thierry Michel: resources (equal); writing – review and editing (equal). Enita Rastoder: resources (equal).Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Université de Montpellier, Centre National de la Recherche Scientifique (CNRS), Ecole Nationale Supérieure de Chimie de Montpellier (ENSCM) and Agence Nationale de la Recherche (grant no. ANR-16-CE07-0009-01) are acknowledged for funding. We thank D. Granier for fruitful discussions on PXRD.Notes and references
- C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef CAS PubMed.
- R. R. Schrock, Adv. Synth. Catal., 2007, 349, 41–53 CrossRef CAS.
- R. H. Grubbs, Adv. Synth. Catal., 2007, 349, 34–40 CrossRef CAS.
- Y. Chauvin, Adv. Synth. Catal., 2007, 349, 27–33 CrossRef CAS.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Chem. Rev., 2022, 122, 1485–1542 CrossRef CAS PubMed.
- A.-H. Bonardi, F. Dumur, G. Noirbent, J. Lalevee and D. Gigmes, Beilstein J. Org. Chem., 2018, 14, 3025–3046 CrossRef CAS PubMed.
- N. Corrigan, S. Shanmugam, J. Xu and C. Boyer, Chem. Soc. Rev., 2016, 45, 6165–6212 RSC.
- J. Lalevee, M.-A. Tehfe, F. Morlet-Savary, B. Graff, F. Dumur, D. Gigmes, N. Blanchard and J.-P. Fouassier, Chimia, 2012, 66, 439–441 CrossRef CAS PubMed.
- P. Xiao, J. Zhang, D. Campolo, F. Dumur, D. Gigmes, J. P. Fouassier and J. Lalevee, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 2673–2684 CrossRef CAS.
- T. Friščić, C. Mottillo and H. M. Titi, Angew. Chem., Int. Ed., 2020, 59, 1018–1029 CrossRef PubMed.
- D. Tan and F. García, Chem. Soc. Rev., 2019, 48, 2274–2292 RSC.
- J. Howard, Q. Cao and D. L. Browne, Chem. Sci., 2018, 9, 3080–3094 RSC.
- N. R. Rightmire and T. P. Hanusa, Dalton Trans., 2016, 45, 2352–2362 RSC.
- G.-W. Wang, Chem. Soc. Rev., 2013, 42, 7668–7700 RSC.
- L. Takacs, Chem. Soc. Rev., 2013, 42, 7649–7659 RSC.
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413–447 RSC.
- J. L. Howard, M. C. Brand and D. L. Browne, Angew. Chem., Int. Ed., 2018, 57, 16104–16108 CrossRef CAS PubMed.
- J. G. Hernández and C. Bolm, J. Org. Chem., 2017, 82, 4007–4019 CrossRef PubMed.
- F. Cuccu, L. De Luca, F. Delogu, E. Colacino, N. Solin, R. Mocci and A. Porcheddu, ChemSusChem, 2022, 15, e202200362 CrossRef CAS PubMed.
- A. Beillard, X. Bantreil, T.-X. Métro, J. Martinez and F. Lamaty, Chem. Rev., 2019, 119, 7529–7609 CrossRef CAS PubMed.
- A. Beillard, E. Golliard, V. Gillet, X. Bantreil, T.-X. Métro, J. Martinez and F. Lamaty, Chem.–Eur. J., 2015, 21, 17614–17617 CrossRef CAS PubMed.
- A. Beillard, F. Quintin, J. Gatignol, P. Retailleau, J.-L. Renaud, S. Gaillard, T.-X. Métro, F. Lamaty and X. Bantreil, Dalton Trans., 2020, 49, 12592–12598 RSC.
- A. Beillard, X. Bantreil, T.-X. Métro, J. Martinez and F. Lamaty, Green Chem., 2018, 20, 964–968 RSC.
- A. Beillard, T.-X. Métro, X. Bantreil, J. Martinez and F. Lamaty, Chem. Sci., 2017, 8, 1086–1089 RSC.
- A. Beillard, X. Bantreil, T.-X. Métro, J. Martinez and F. Lamaty, New J. Chem., 2017, 41, 1057–1063 RSC.
- A. Beillard, X. Bantreil, T.-X. Métro, J. Martinez and F. Lamaty, Dalton Trans., 2016, 45, 17859–17866 RSC.
- F. Quintin, J. Pinaud, F. Lamaty and X. Bantreil, Organometallics, 2020, 39, 636–639 CrossRef CAS.
- A. Beillard, T.-X. Métro, X. Bantreil, J. Martinez and F. Lamaty, Eur. J. Org Chem., 2017, 4642–4647 CrossRef CAS.
- P. Milbeo, F. Quintin, L. Moulat, C. Didierjean, J. Martinez, X. Bantreil, M. Calmès and F. Lamaty, Tetrahedron Lett., 2021, 63, 152706 CrossRef CAS.
- N. Pétry, T. Vanderbeeken, A. Malher, Y. Bringer, P. Retailleau, X. Bantreil and F. Lamaty, Chem. Commun., 2019, 55, 9495–9498 RSC.
- J. Zhang, D. Campolo, F. Dumur, P. Xiao, J. P. Fouassier, D. Gigmes and J. Lalevée, ChemCatChem, 2016, 8, 2227–2233 CrossRef CAS.
- J. Zhang, D. Campolo, F. Dumur, P. Xiao, J. P. Fouassier, D. Gigmes and J. Lalevée, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 2247–2253 CrossRef CAS.
- J. Zhang, D. Campolo, F. Dumur, P. Xiao, J. P. Fouassier, D. Gigmes and J. Lalevée, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 42–49 CrossRef CAS.
- D. Cinčić, I. Brekalo and B. Kaitner, Cryst. Growth Des., 2012, 12, 44–48 CrossRef.
- V. Stilinović, D. Cinčić, M. Zbačnik and B. Kaitner, Croat. Chem. Acta, 2012, 85, 485–493 CrossRef.
- M. Zbačnik, I. Nogalo, D. Cinčić and B. Kaitner, CrystEngComm, 2015, 17, 7870–7877 RSC.
- J. Schmeyers, F. Toda, J. Boy and G. Kaupp, J. Chem. Soc., Perkin Trans. 2, 1998, 989–994 RSC.
- C. Giri, P. K. Sahoo, R. Puttreddy, K. Rissanen and P. Mal, Chem.–Eur. J., 2015, 21, 6390–6393 CrossRef CAS PubMed.
- T. E. Shaw, L. Mathivathanan and T. Jurca, Organometallics, 2019, 38, 4066–4070 CrossRef CAS.
- S. Xu, J. E. T. Smith and J. M. Weber, Inorg. Chem., 2016, 55, 11937–11943 CrossRef CAS.
- M. Ferguson, N. Giri, X. Huang, D. Apperley and S. L. James, Green Chem., 2014, 16, 1374–1382 RSC.
- G. Kaupp, J. Schmeyers and J. Boy, Tetrahedron, 2000, 56, 6899–6911 CrossRef CAS.
- M. G. B. Drew, C. V. Knox and S. M. Nelson, J. Chem. Soc., Dalton Trans., 1980, 942–948 RSC.
- M. G. B. Drew, A. H. bin Othman, S. G. McFall, P. D. A. McLlroy and S. M. Nelson, J. Chem. Soc., Dalton Trans., 1977, 1173–1180 RSC.
- N. Pétry, F. Luttringer, X. Bantreil and F. Lamaty, Faraday Discuss., 2023, 241, 114–127 RSC.
- S. Lukin, K. Užarević and I. Halasz, Nat. Protoc., 2021, 16, 3492–3521 CrossRef CAS PubMed.
- D. Gracin, V. Štrukil, T. Friščić, I. Halasz and K. Užarević, Angew. Chem., Int. Ed., 2014, 53, 6193–6197 CrossRef CAS PubMed.
- H. Kulla, M. Wilke, F. Fischer, M. Rollig, C. Maierhofer and F. Emmerling, Chem. Commun., 2017, 53, 1664–1667 RSC.
- W. Dai, F. Shao, J. Szczerbiński, R. McCaffrey, R. Zenobi, Y. Jin, A. D. Schlüter and W. Zhang, Angew. Chem., Int. Ed., 2016, 55, 213–217 CrossRef CAS PubMed.
- N. Valley, L. Jensen, J. Autschbach and G. C. Schatz, J. Chem. Phys., 2010, 133, 054103 CrossRef PubMed.
- M. Pagliai, L. Bellucci, M. Muniz-Miranda, G. Cardini and V. Schettino, Phys. Chem. Chem. Phys., 2006, 8, 171–178 RSC.
- See the ESI† for details.
- S. L. Dabb and N. C. Fletcher, Dalton Trans., 2015, 44, 4406–4422 RSC.
- J. H. Askew and H. J. Shepherd, Chem. Commun., 2018, 54, 180–183 RSC.
- L. L. Merritt and E. Schroeder, Acta Crystallogr., 1956, 9, 801–804 CrossRef CAS.
- S. Dick, Z. Kristallogr. - New Cryst. Struct., 1998, 213, 370 Search PubMed.
- L. G. Bachas, L. Cullen, R. S. Hutchins and D. L. Scott, J. Chem. Soc., Dalton Trans., 1997, 1571–1578 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, description of the compounds, and copies of the 1H and 13C NMR spectra of the compounds. See DOI: https://doi.org/10.1039/d3mr00033h |
| This journal is © The Royal Society of Chemistry 2024 |