
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of short DNA and RNA fragments by resonant acoustic mixing (RAM)†
James D.
Thorpe
a,
Julian
Marlyn
a,
Stefan G.
Koenig
 b and
Masad J.
Damha
*a
b and
Masad J.
Damha
*a
aDepartment of Chemistry, McGill University, Montreal, QC H3A0B8, Canada. E-mail: masad.damha@mcgill.ca
bSynthetic Molecule Process Chemistry, Genentech Inc., South San Francisco, CA, USA
First published on 28th March 2024
Abstract
We demonstrate the first use of Resonant Acoustic Mixing (RAM) without bulk solvent for the synthesis of short oligonucleotide fragments. Using the modified H-phosphonate approach, DNA, RNA, and 2′-modified nucleotides were successfully coupled to 3′-protected nucleosides in high yields (63–92%) while reducing solvent volume by 90%. In addition to synthesizing protected phosphodiester (PO) dimers and trimers, we also synthesized protected phosphorothioate (PS) dimers in good yields (63–65%). Using phosphoramidite chemistry, we were similarly able to reduce the solvent volume by 90% while coupling DNA phosphoramidites (58–92%) and RNA phosphoramidites (55–95%) with 3′-protected nucleosides in high yields followed by traditional oxidation with iodine in solution. Both strategies were successfully scaled up to multi-gram quantities which was facilitated by the use of RAM, offering the potential for larger scale-up, up to hundreds of kilograms continuously.
Introduction
Oligonucleotide (ON) therapeutics are being approved at an unprecedented rate, with more than a dozen reaching the market in the last seven years alone.1 Given the high degree of chemical modifications2,3 in approved ON therapeutics, the current gold-standard for RNA and DNA synthesis remains solid-phase oligonucleotide synthesis (SPOS) using well-established phosphoramidite chemistry.4 However, the environmental impact of SPOS is well documented, with applications of excess amounts of reagents and solvents and inefficient use of starting materials.5,6 While some enzymatic methods for ON synthesis have recently emerged,5,6 they generally suffer from poor recognition of chemically modified nucleoside triphosphates (NTPs) by polymerases and difficulty in synthesizing the required NTPs. Furthermore, while they offer the ability to carry out the reactions in aqueous media and can often forego the more common protecting group strategies of SPOS, these methods have not yet reached the same scale as chemical methods for oligonucleotide synthesis. In 2016, the American Chemical Society (ACS) Green Chemistry Institute Pharmaceutical Roundtable (GCIPR) identified the development of greener processes for promising active pharmaceutical ingredients (APIs, e.g., oligonucleotides) as a critical unmet need, and as a result, a GCIPR focus team was created to address this important area.5,6A recently introduced and scalable mechanochemical methodology that uniquely eliminates the need for bulk solvent, as well as milling media, is resonant acoustic mixing (RAM) (Table 1).7–11 RAM shakes materials at a low acoustic frequency (58–62 Hz), with energy input modulated through changes in the vertical acceleration of the reaction vessel usually measured in units of g (acceleration on Earth due to gravity), up to 100 g. This allows the system to maximize the amplitude while simultaneously minimizing the power consumption. Compared to other mixing technologies such as ultrasonic, paint shaker, or vibration ball mills (VBM) (Table 1), RAM operates at an intermediate frequency, but with relatively large displacements and low power usage.8 The unique conditions occurring at the resonant frequency of the system allow for highly efficient mixing by generating intense mixing zones with diameters of approximately 50 μm.12 In principle, this allows for simpler scale up of RAM processes and lower maintenance costs due to less wear-and-tear on the instruments.
| Instrument | RAM | Ultrasonic | Paint mixer | VBM |
|---|---|---|---|---|
| Displacement (cm) | 1–4 | ≪1 | 5–10 or more | 1–2 |
| Frequency (Hz) | 58–62 | 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
10–15 | 15–30 |
Our group previously demonstrated the use of VBM for the synthesis of short DNA fragments while significantly reducing solvent consumption during reactions.13 Inspired by recent publications using RAM for carrying out a range of chemical reactions,7–11 we describe herein the first use of RAM for the synthesis of di- and trinucleotides with a range of different chemistries (i.e., DNA, RNA and 2′-modified RNA). This method reduces the solvent consumption by up to 90% during monomer coupling and oxidation/sulfurization of internucleotide linkages while being readily scaled up to multi-gram quantities.
Results and discussion
We carried out the reactions in a commercially available LabRAM I instrument from Resodyn corporation (Fig. 1). This instrument is typically used with a mixing vessel as shown with a capacity of up to 500 grams. For our purposes this was much too large of a scale initially, and we had a custom-designed sample holder built that could hold up to 33 plastic or glass vials (Fig. 1). This allowed us to conduct smaller scale reactions and screen a range of conditions simultaneously. | ||
| Fig. 1 RAM instrumentation. Left: Resodyn's LabRAM I instrument. Right: Our custom-built sample holder compatible with the LabRAM I. | ||
First, we set out to synthesize short DNA fragments via the modified H-phosphonate approach.13 The initial reaction involved coupling of H-phosphonate 1a with 2a in the presence of an acid chloride activator, followed by reaction with N-phenylthiophthalimide (PTP, a sulfur-transfer reagent) to yield the desired fully-protected phosphorothioate triester 3a (Scheme 1) which could later be deprotected to yield the desired phosphodiester (PO) internucleotide linkage.14 The sulfur transfer step was necessary to protect the internucleotide linkage as previous studies have shown the instability of H-phosphonate diesters;15 we also encountered difficulties in isolating H-phosphonate diesters.
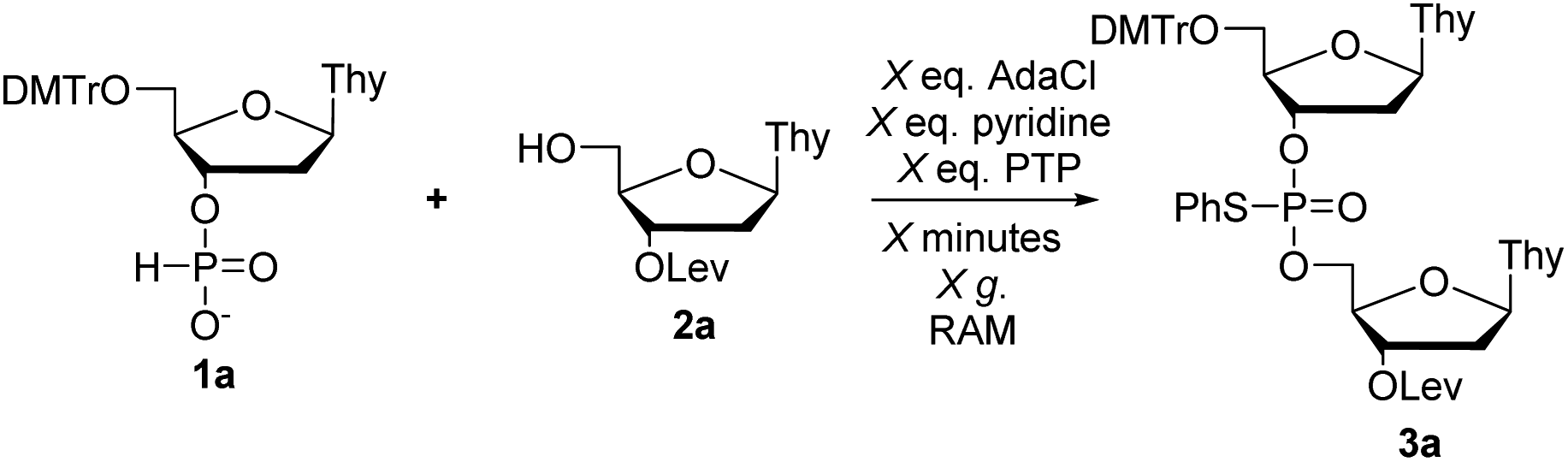 | ||
| Scheme 1 Optimization of H-phosphonate coupling and sulfur-transfer by RAM. | ||
Variables to optimize included the nature of the activator, amounts of reagents (activator, pyridine, PTP), times, and acceleration (Table 2). We were able to conveniently monitor the reactions by analyzing the 31P NMR spectra of crude products before any further isolation steps (Fig. 2). A selection of the optimization reactions is discussed below.
| Entry | Eq. AdaCl | Eq. PTP | Eq. pyr | Time (min) | η (μL mg−1) | Conversion (%) |
|---|---|---|---|---|---|---|
| a All reactions carried out with 0.2 mmol of 2a at 60 g and the stoichiometry of all reagents are in relation to this. Conversion determined by integration of peaks in 31P NMR spectra. b Reaction carried out with PvCl as the activator. | ||||||
| 1 | 5 | 1.1 | 10 | 60 | 0.33 | 29 |
| 2 | 3 | 1.1 | 10 | 60 | 0.40 | 44 |
| 3 | 3 | 1.1 | 15 | 60 | 0.60 | 47 |
| 4 | 3 | 1.1 | 15 | 15/15 | 0.60 | 90 |
| 5 | 3 | 1.1 | 15 | 15/5 | 1.12 | 88 |
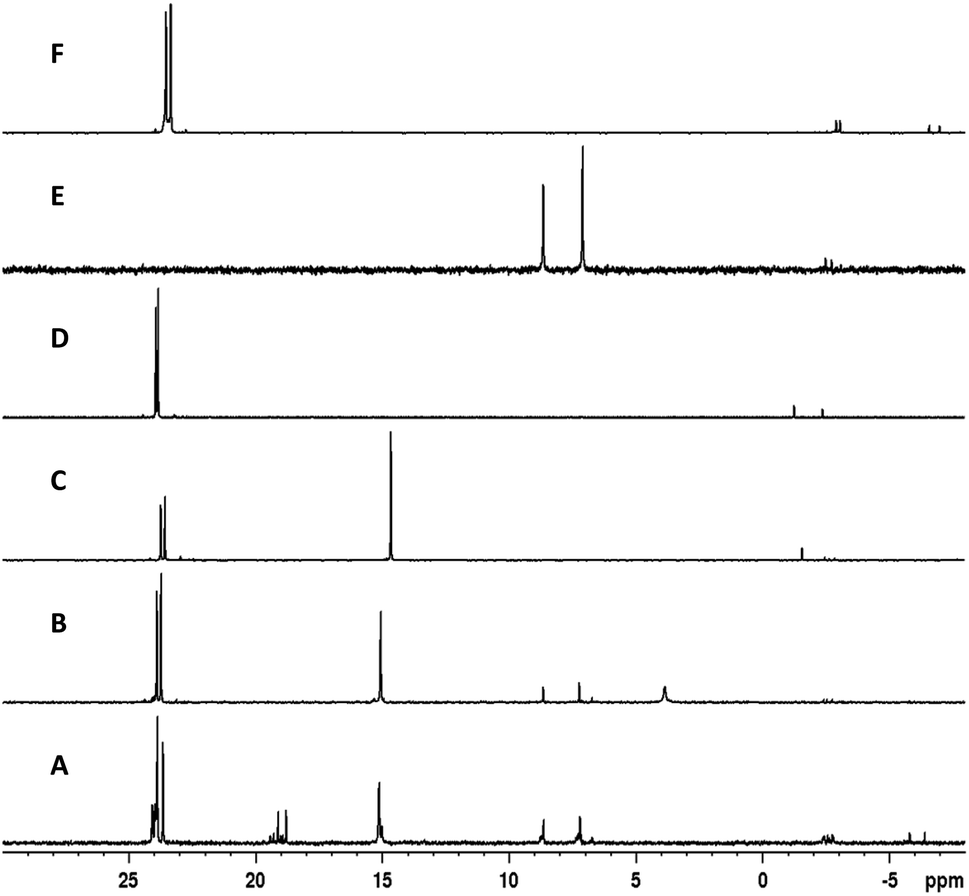 | ||
| Fig. 2 Crude 31P NMR spectra of experiments from Table 2. (A) Entry 1. (B) Entry 2. (C) Entry 3. (D) Entry 4. (E) Entry 5, before sulfur transfer. (F) Entry 5, after sulfur transfer. | ||
Beginning with similar conditions optimized for VBM but attempting the sulfur-transfer step simultaneously to the coupling, we started with 5 eq. of adamantyl chloride (AdaCl), 1.1 eq. of PTP, and 10 eq. of pyridine at 60 g for 60 minutes (entry 1). The crude 31P NMR displayed formation of the product (Fig. 2A) around 24 ppm, but also formation of numerous side products. Surprisingly, reducing the equivalents of AdaCl from five to three, reduced the formation of side products and produced a cleaner spectrum (entry 2, Fig. 2B), however, this resulted in incomplete conversion of the H-phosphonate diester (minor peaks at 7–9 ppm) to the phosphorothioate triester. Increasing the equivalents of pyridine from 10 to 15 (entry 3, Fig. 2C) also resulted in a cleaner spectrum, albeit with one major unidentified product around 15 ppm. Satisfied with the equivalents of all reagents, we set out to determine if the formation of side-products could be controlled by reaction time.
Reducing the reaction times to 15 minutes for coupling followed by 15 minutes for sulfur transfer completely suppressed the formation of the side-product at 15 ppm, with only minor unidentified impurities around −2 ppm (entry 4, Fig. 2D). Finally, we also attempted the reaction with pivaloyl chloride (PvCl) as the coupling reagent (15 min, entry 5) and analyzed the reaction mixture by 31P NMR (Fig. 2E) which showed two minor peaks around −2.5 ppm and H-phosphonate diester at 7–9 ppm. We then performed the sulfur-transfer step with PTP for 5 minutes and analyzed the 31P NMR again (Fig. 2F). The H-phosphonate diester peaks disappeared completely and the peaks for phosphorothioate triester 3a were the major product at 24 ppm. However, the peaks at −2 ppm remained and some new minor peaks emerged at −7 ppm. Nonetheless, the crude reaction product was relatively clean, and we were satisfied with the optimized conditions. The parameter η now reported in several mechanochemistry publications illustrates the liquid/weight (solvent to reagent) ratio of a chemical reaction. The optimized conditions were achieved with η = 1.02–1.12 μL mg−1 (depending on the MW of 1) falling within the range of liquid-assisted RAM (LA-RAM) reactions.10,11 This value is significantly lower than typical solution-phase reactions which are characterized by η > 10 μL mg−1.16 Our optimized conditions were conducted with ∼90% less solvent than a comparable solution-phase reaction. It should be noted that the use of the liquid PvCl for optimized reactions increased the value of η as compared to reactions using AdaCl which achieved η = 0.56–0.6 μL mg−1 (entry 4, Table 2). Although not shown here, increasing the acceleration from 60 g to 90 g had no effect on the reaction.
Having established optimal conditions for coupling and sulfur-transfer of H-phosphonates by RAM, we set out to expand the strategy to other nucleobases and purify the products to assess the yield. Thus, we repeated the reaction with H-phosphonates 1a–d and 0.2 mmol of 2a to yield dimers 3a–d in excellent yield (81–90%, Scheme 2). These yields corresponded to an average yield of >90% per step which represents an improvement over VBM17 while reducing the equivalents of activator. Although AdaCl was effective as an activator as well, PvCl seemed to be more efficient, likely due to improved mixing when using a liquid under these conditions. The products were analyzed by 31P NMR (Fig. 3) and HRMS and confirmed the identity of the products.
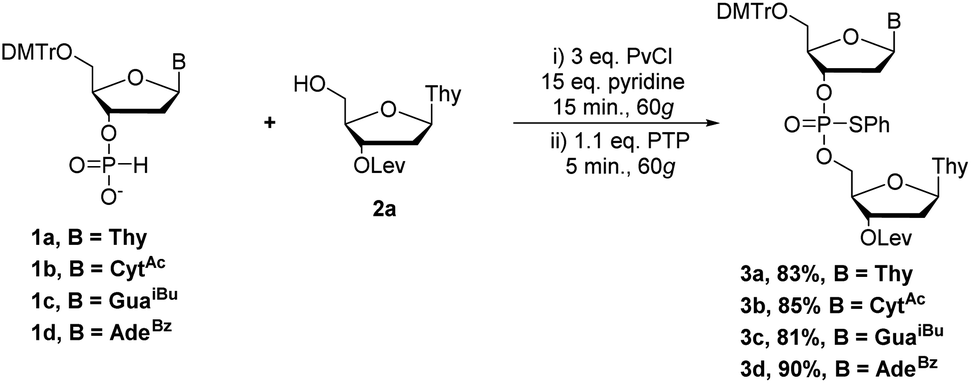 | ||
| Scheme 2 Optimized RAM conditions for H-phosphonate coupling and sulfur-transfer. | ||
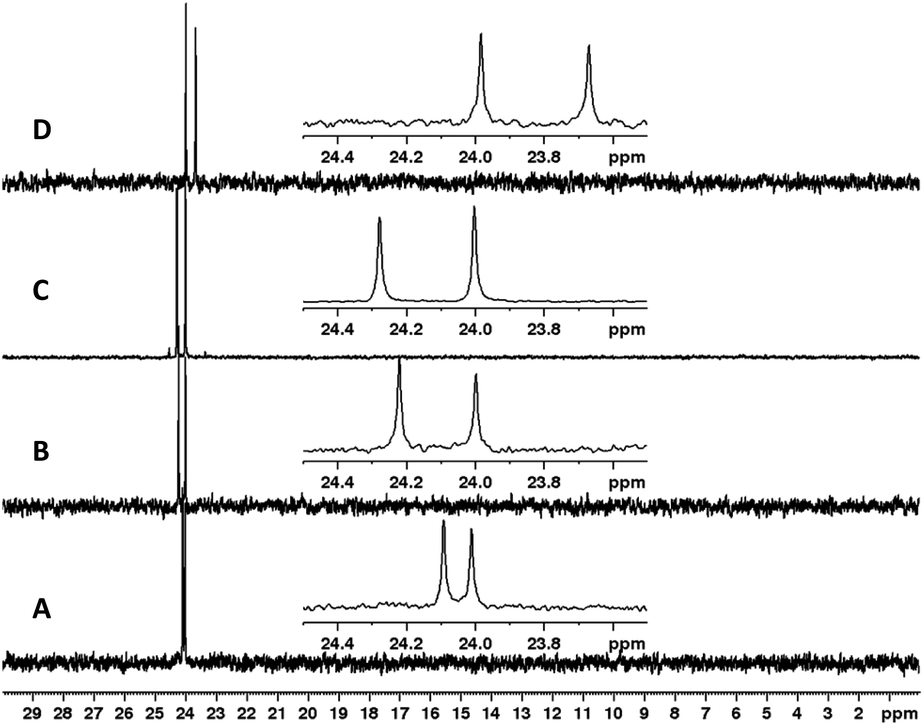 | ||
| Fig. 3 31P NMR (CDCl3) spectra of dimers 3a–d synthesized by RAM. (A) 3a. (B) 3b. (C) 3c. (D) 3d. | ||
Although the reactions carried out above were limited by the size of the vials used in the same holder, we wanted to see if scaling up the reaction from 0.2 mmol to 1 mmol was possible. The mixing vessel provided with the LabRAM I was still too large for our purposes and its composition also incompatible with some of the reagents used in our reactions (pyridine). We settled on using polytetrafluoroethylene (PTFE) jars that were chemically compatible with all our reagents and could be used for multigram reactions. The larger scale reactions were found to be similarly efficient under the exact same conditions and dimer 3a was isolated in 74% yield. We were able to further scale up the reaction and doubled the scale from 1 mmol to 2 mmol to synthesize dimer 3d in 77% unoptimized yield from 1d and 2a. While the isolated yield was still slightly lower than the 0.2 mmol scale reactions which may be due to a lower filling degree, the first-pass crude 31P NMR spectra of the scaled-up reactions of 3a and 3d were quite clean (Fig. 4).
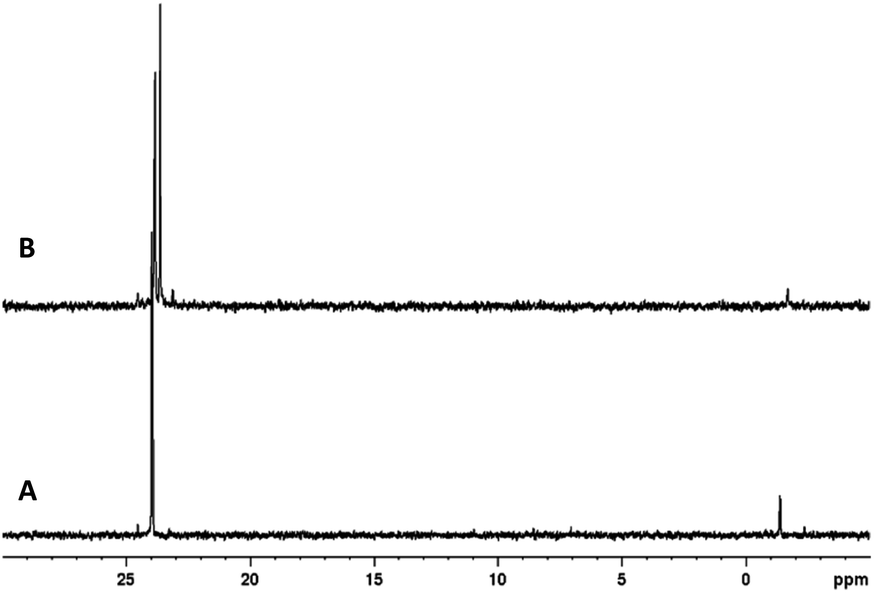 | ||
| Fig. 4 Crude 31P NMR spectra of scaled up RAM reactions. (A) 3a (1 mmol). (B) 3d (2 mmol). | ||
Having scaled up the reaction successfully, we also set out to synthesize a longer sequence. Rather than using RAM, dimer 3d was detritylated under traditional conditions in a solution of trifluoroacetic acid (TFA) in dichloromethane to yield 4 in 87% yield (Scheme 3). Preliminary results have indicated that detritylation is also possible by RAM within 45 minutes and is just as efficient as the same solution-phase reaction. Subsequent coupling of 4 with 1b by RAM under the same conditions previously optimized yielded mixed-base trimer 5 in good yield (79%). Analysis of the 31P NMR spectrum of 5 is consistent with the diastereomeric mixture expected for this compound (ESI†). HRMS analysis confirmed the identity of 5. Having a satisfactory strategy for the synthesis of DNA dimers and trimers, we turned our attention to expanding the substrate scope to include other nucleosides commonly used in ON therapeutics: 2′OH nucleosides (RNA), 2′F nucleosides (2′F-RNA), 2′OMe nucleosides (2′OMe-RNA), and phosphorothioate (PS) internucleotide linkages.
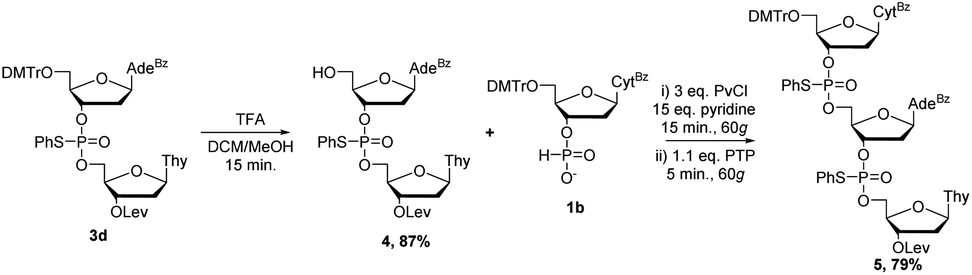 | ||
| Scheme 3 Synthesis of a nucleotide trimer by RAM. | ||
Beginning with 2′-tert-butyl(dimethyl)silyl (TBDMS) protected nucleoside, we attempted to couple 1e with 2a under the same conditions previously optimized for DNA couplings (Scheme 4). Analysis of the crude 31P NMR of the reaction mixture showed a similar spectrum as observed previously with almost exclusive formation of the desired dimer 6a. Purification of the reaction mixture yielded 6a in a 70% yield (Scheme 4). We then set out to couple 1e with 2b, which was protected at the 2′-position with a triisopropylsilyl (TIPS)18 group, to yield fully protected RNA dimer 6b in a 64% yield after purification. Although the yields were slightly lower than the DNA couplings, this was expected with nucleosides bearing bulky protecting groups at the C2′ position.
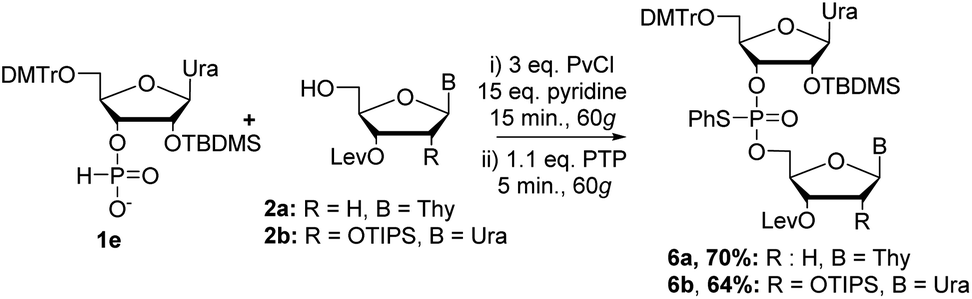 | ||
| Scheme 4 RNA coupling by RAM. | ||
We next moved on to nucleosides modified at the C2′-position (Schemes 5 and 6). Coupling of 2′-F 1f and 2′-OMe 1g to 2a required further optimization from the conditions used for the DNA and RNA couplings as evidenced by the crude 31P NMR spectra (ESI†). Increasing the equivalents of the activator from three to five equivalents, as well as increasing the reaction time during coupling to 30 minutes, effectively formed the H-phosphonate diester. Prolonged reaction time with PTP during the sulfur transfer step led to a complicated 31P NMR spectrum of the crude mixture but reducing the sulfurization time from 5 minutes to 2.5 minutes (ESI†) improved the formation of the desired product 6c (84% yield). Using these conditions, 1f was also effectively coupled with 2′-OMe nucleoside 2c to yield 6d in 73% yield (Scheme 5).
 | ||
| Scheme 5 2′F nucleoside coupling by RAM. | ||
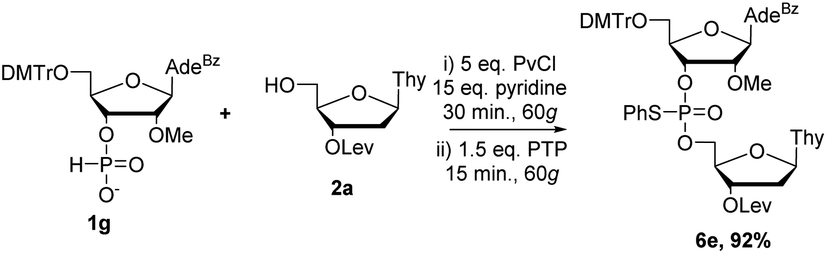 | ||
| Scheme 6 2′OMe nucleoside coupling by RAM. | ||
Separate optimization of coupling of 2′OMe nucleoside 1g was also required. As with 1f, increasing the equivalents of the activator from three to five and coupling time from 15 to 30 minutes was needed to ensure efficient formation of the H-phosphonate diester. In contrast to the sulfur transfer step of 1f, 1g required increasing equivalents of PTP to 1.5 as well as increasing reaction time to 15 minutes. Encouragingly, under these conditions we were able to isolate 6e in 92% yield (Scheme 6).
Having established conditions for coupling the four most common nucleosides employed in ON therapeutics (DNA, RNA, 2′F, and 2′OMe) we set about to develop conditions for the synthesis of compounds containing PS linkages. Reese had previously used a reagent similar to PTP,19 but bearing a cyanoethyl group, rather than phenyl, appended to the sulfur atom which could then be deprotected under basic conditions to yield the desired PS linkage. We had difficulty in synthesizing this reagent and turned our attention to synthesizing an alternative sulfur transfer reagent that could provide transfer of the desired S-cyanoethyl moiety to the internucleotide linkage. Instead, we chose an electrophilic sulfurization reagent (7),20–22 synthesized from p-toluene thiosulfonate and 3-bromopropionitrile in 81% yield (Scheme 7).
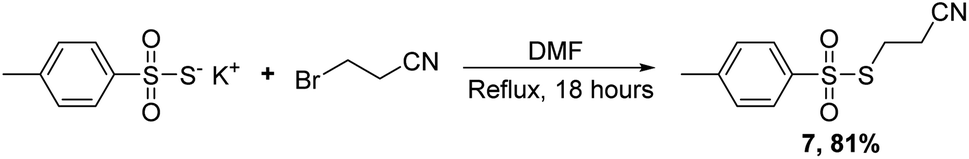 | ||
| Scheme 7 Synthesis of alternative sulfur transfer reagent 7. | ||
Coupling of 1a with 2a followed by sulfur transfer with a mixture of 7 and pyridine (1:5 ratio) provided 8a in 65% yield after purification (Scheme 8). To demonstrate the feasibility of the synthesis of PS backbones in conjunction with modified nucleosides, we coupled 1f with 2a followed by sulfur transfer with 7 and obtained 8b in 63% yield after purification. However, under these conditions, we still required the increased coupling time and equivalents of activator for the coupling step, although performing the sulfur transfer reaction for five minutes did not lead to any noticeable degradation. Analysis of the 31P NMR spectra of these compounds bearing S-cyanoethyl linkages showed a 2–3 ppm downfield shift as compared to the dimers with SPh backbones (ESI†).
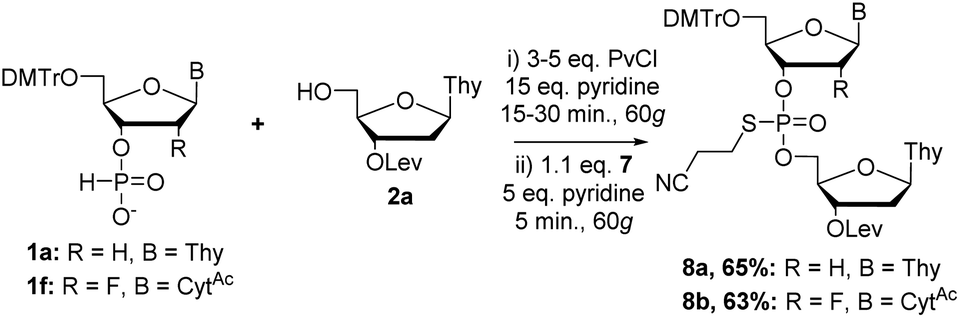 | ||
| Scheme 8 Synthesis of PS backbone protected dimers by RAM. | ||
In addition to H-phosphonate couplings, we also investigated the use of RAM for a phosphoramidite coupling strategy. Initially, couplings were attempted neat as a mixture of 2a, 2 eq. of 3′-phosphoramidite 9a–d, and 3.0 eq. of 5-(ethylthio)-1H-tetrazole (ETT), shaken at 60 g for 45 minutes in a flame dried glass vial (Scheme 9). Analysis of the resulting mixtures by 31P NMR showed that without solvent, reactions did not proceed cleanly and resulted in an indistinguishable mixture of products after oxidation with 1.0 M iodine in 9:1 2,6-lutidine/H2O.
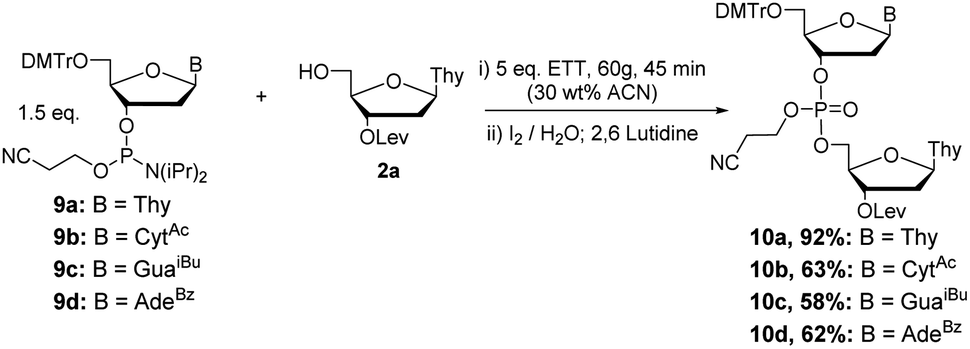 | ||
| Scheme 9 Optimized conditions for phosphoramidite coupling and oxidation by RAM. | ||
To remedy this, we sought to determine the minimum solvent requirement for successful couplings via RAM. Starting with a standard solution phase concentration of 0.22 M in dry acetonitrile (ACN), the quantity of solvent was reduced by 50% until TLC analysis of the crude showed incomplete couplings (presence of limiting 2a). Coupling reactions were able to tolerate concentrations up to 1.32 M, representing a sixfold decrease in solvent. The reported reactions were achieved with η = ∼0.3 μL mg−1, falling between a neat (η = 0 μL mg−1) and slurry (η = 2–12 μL mg−1) type regime. The quantity of ETT was further optimized, with 31P NMR showing 5.0 eq. of activator giving the best results. 4,5-Dicyanoimidazole (DCI) was also tested, giving similar results (data not shown). The subsequent oxidation was accomplished via RAM or manual mixing with a slight excess of 1.0 M I2 in 2,6-lutidine/water (9:1 ratio). With adequate conditions determined for pursuing phosphoramidite couplings, the dTpT dimer 10a was synthesized in scales ranging from 0.2 to 3 mmol in 60–92% yield (Scheme 9).
Couplings with the other three bases produced dimers 10b–d in 58–63% yields (Scheme 9). Additionally, couplings with 2′TBDMS protected ribonucleotides 11a–d (Scheme 10) under the same conditions previously optimized yielded the desired products 12a–d in good yield (55–95%).
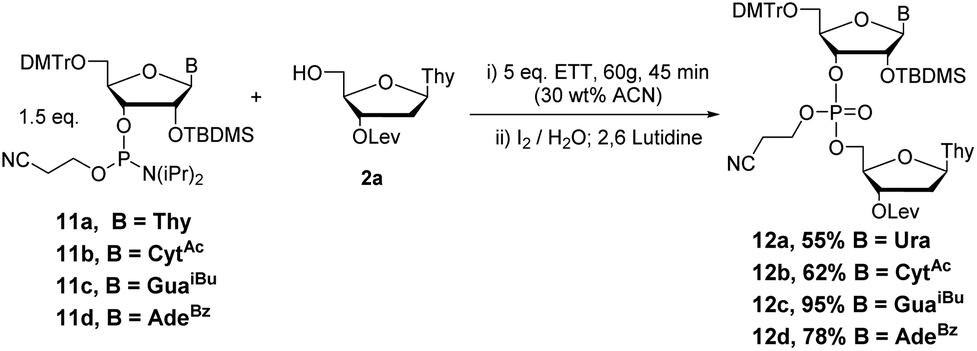 | ||
| Scheme 10 Optimized conditions for RNA phosphoramidite coupling and oxidation by RAM. | ||
Conclusions
In summary, we have demonstrated the use of resonant acoustic mixing (RAM) for the synthesis of short oligonucleotide fragments with a range of different ON chemistries. In comparison with traditional SPOS or in liquid-phase oligonucleotide synthesis,23 our approach reduces the solvent consumption by up to 90% during coupling and oxidation/sulfurization reactions while being readily scaled up to multi-gram quantities. Although current purification by column chromatography is solvent-intensive, the combination of RAM with ionic tags24 or other soluble supports25 that allow precipitation of products might eliminate chromatography completely and further improve the process mass intensity (PMI) of oligonucleotide synthesis at general scales. The instrument used in this work operates at a scale of up to 500 grams, but commercially available RAM instruments can operate up to hundreds of kilograms continuously. This work represents the first application of RAM for nucleic acid chemistry and broadens the applicability of RAM for organic transformations.7–11Author contributions
James D. Thorpe: synthesis, analysis, investigation, writing of original draft, editing; Julian Marlyn: synthesis, analysis, investigation, writing, editing; Stefan G. Koenig: conceptualization, guidance, administration, review, editing; Masad J. Damha: conceptualization and chemistry, resources, supervision, writing, review, editing.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This investigation was supported by research grants to M. J. D. from the American Chemical Society (ACS) Green Chemistry Institute Pharmaceutical Roundtable (GCIPR); (https://www.acsgcipr.org/) and a Discovery Grant from the National Science and Engineering Council of Canada. The activities of the GCIPR reflect its members' shared belief that the pursuit of green chemistry and engineering is imperative for business and environmental sustainability. We thank Tomislav Friščić and Cameron Lennox for assistance and providing their RAM facilities at the early stage of this project. We also wish to thank Jale Muslehiddinoglu for the careful and insightful review of our manuscript and Sam Minter for design and construction of the customized sample holder.Notes and references
- M. Egli and M. Manoharan, Nucleic Acids Res., 2023, gkad067 Search PubMed.
- A. Khvorova and J. K. Watts, Nat. Biotechnol., 2017, 35, 238–248 CrossRef CAS PubMed.
- L. K. McKenzie, R. El-Khoury, J. D. Thorpe, M. J. Damha and M. Hollenstein, Chem. Soc. Rev., 2021, 50, 5126–5164 RSC.
- S. Beaucage and M. Caruthers, Tetrahedron Lett., 1981, 22, 1859–1862 CrossRef CAS.
- E. Moody, R. Obexer, F. Nickl, R. Spiess and S. Lovelock, Science, 2023, 380, 1150–1154 CrossRef CAS PubMed.
- D. J. Wiegand, J. Rittichier, E. Meyer, H. Lee, N. J. Conway, D. Ahlstedt, Z. Yurtsever, D. Rainone, E. Kuru and G. Church, bioRxiv, 2023, preprint, 2023.2006.2029.547106, DOI:10.1101/2023.06.29.547106.
- C. Lennox, T. Borchers, L. Gonnet, C. Barrett, S. Koenig, K. Nagapudi and T. Friscic, Chem. Sci., 2023, 14, 7475–7481 RSC.
- L. Gonnet, C. B. Lennox, J. L. Do, I. Malvestiti, S. G. Koenig, K. Nagapudi and T. Friščić, Angew. Chem., 2022, 134, e202115030 CrossRef.
- F. Effaty, L. Gonnet, S. G. Koenig, K. Nagapudi, X. Ottenwaelder and T. Friščić, Chem. Commun., 2023, 59, 1010–1013 RSC.
- H. M. Titi, J.-L. Do, A. J. Howarth, K. Nagapudi and T. Friščić, Chem. Sci., 2020, 11, 7578–7584 RSC.
- L. Gonnet, T. H. Borchers, C. B. Lennox, J. Vainauskas, Y. Teoh, H. M. Titi, C. J. Barrett, S. G. Koenig, K. Nagapudi and T. Friščić, Faraday Discuss., 2023, 241, 128–149 RSC.
- K. S. Hope, H. J. Lloyd, D. Ward, A. A. Michalchuk and C. R. Pulham, New Trends in Research of Energetic Materials, 2015, pp. 134–143 Search PubMed.
- C. B. Reese and H. Yan, J. Chem. Soc., Perkin Trans. 1, 2002, 2619–2633 RSC.
- C. B. Reese and L. Zard, Nucleic Acids Res., 1981, 9, 4611–4626 CrossRef CAS PubMed.
- F. Westheimer, S. Huang and F. Covitz, J. Am. Chem. Soc., 1988, 110, 181–185 CrossRef CAS.
- J.-L. Do and T. Friščić, ACS Cent. Sci., 2017, 3, 13–19 CrossRef CAS PubMed.
- J. D. Thorpe, D. O'Reilly, T. Friščić and M. J. Damha, Chem.–Eur. J., 2020, 26, 8857–8861 CrossRef CAS PubMed.
- M. Hassler, Y. Q. Wu, N. M. Reddy, T. H. Chan and M. J. Damha, Tetrahedron Lett., 2011, 52, 2575–2578 CrossRef CAS.
- C. Reese, J. Chem. Soc., Perkin Trans. 1, 1999, 1477–1486 RSC.
- N. Iwamoto, D. C. Butler, N. Svrzikapa, S. Mohapatra, I. Zlatev, D. W. Sah, S. M. Standley, G. Lu, L. H. Apponi and M. Frank-Kamenetsky, Nat. Biotechnol., 2017, 35, 845–851 CrossRef CAS PubMed.
- J. Michalski, T. Modro and J. Wieczorkowski, J. Chem. Soc., 1960, 1665–1670 RSC.
- W. K.-D. Brill, Tetrahedron Lett., 1995, 36, 703–706 CrossRef CAS.
- S. Katayama and K. Hirai, Synthesis of Therapeutic Oligonucleotides, 2018, pp. 83–95 Search PubMed.
- R. A. Donga, S. M. Khaliq-Uz-Zaman, T.-H. Chan and M. J. Damha, J. Org. Chem., 2006, 71, 7907–7910 CrossRef CAS PubMed.
- H. Lönnberg, Beilstein J. Org. Chem., 2017, 13, 1368–1387 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4mr00009a |
| This journal is © The Royal Society of Chemistry 2024 |