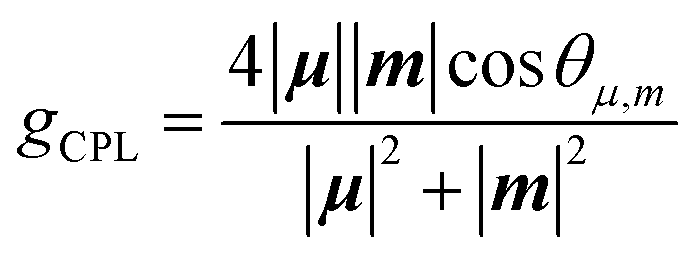Completely solvent-free synthesis of double heterohelicenes and their further ring fusion using mechanochemical reaction†
Received
3rd April 2024
, Accepted 4th May 2024
First published on 7th May 2024
Abstract
In this study, we developed a simple and efficient method for synthesizing double heterohelicenes (DHHs) composed of two heteroacenes bearing an NH group, such as benzo[b]phenoxazine (BPO) and dibenzo[b,i]phenoxazines (DBPO), using mechanochemical oxidative C–N coupling reactions, allowing complete solvent-free synthesis from commercially available compounds. Our new synthetic method afforded more than 1 g of DHH, which has a high dissymmetry factor for circularly polarized luminescence (gCPL) of >1 × 10−2, in a one-pot mechanochemical reaction using BPO as a reactant. In addition, mechanochemical oxidative coupling also allows for further fusion reactions of DHHs, leading to semi- or fully planarized molecules, which have not been previously achieved through solution-phase reactions. We isolated semi-planarized heterohelicenes 5 and 6 and determined their structures using single-crystal X-ray analysis. Compounds 5 and 6 exhibited enhanced electron donor properties compared to DHHs 3 and 4. The enantiomers of 6 exhibited clear CPL emissions with a |gCPL| value of 2 × 10−3. The magnitudes of the transition magnetic dipole moment (TMDM) of 5 and 6 increased compared to those of 3 and 4. Transition moment density analysis revealed that large TMDM densities appeared on the newly formed C–C bonds, providing a unique molecular design guideline for enhancing the magnitude of the TMDM without expanding the molecular structure.
Introduction
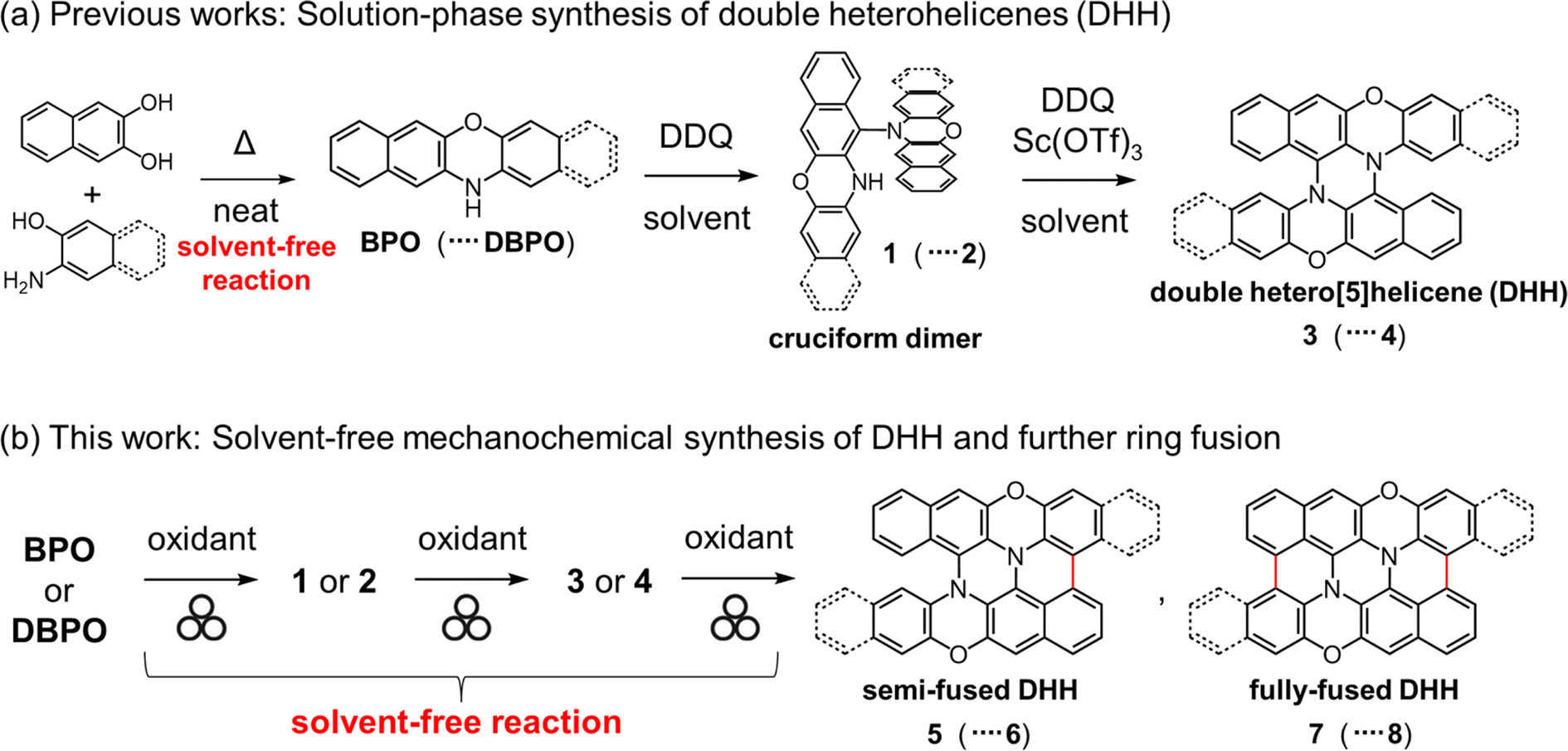
The incorporation of heteroatoms into the structures of carbon-based π-conjugated molecules is a valuable approach for controlling their physical properties. In particular, heteroatom-embedded polyaromatic hydrocarbons (PAHs) are gaining increasing interest as luminescent materials for organic light-emitting diodes (OLEDs) and photoredox catalysts.1–4 Heterohelicenes are promising compounds for circularly polarized luminescence (CPL) materials5,6 and chiral electron donors or acceptors.7 In 2015, we reported a facile method for the synthesis of double heterohelicenes composed of two heteroacenes bearing an NH group.8 This method affords double heterohelicenes in a one-pot manner from readily available NH-containing heteroacenes, such as N,N′-dihydrodiazapentacene (DHDAP),8 benzo[b]phenoxazine (BPO),9 dibenzo[b,i]phenoxazine (DBPO),8,9 phenothiazine,10 benzo[b]phenothiazine (BPT),11 and phenoselenazine,12via their cruciform dimers by successive oxidative C–N bond formation (Scheme 1a). However, one drawback of this method is that it requires large amounts of reaction solvents, owing to the low solubility of the starting materials and intermediate cruciform dimers. For example, the solution synthesis of 3 from 0.815 g of BPO requires 600 mL of CH2Cl2 as the reaction solvent.9 Therefore, it is desirable to make this method more environmentally compatible and to facilitate large-scale synthesis.
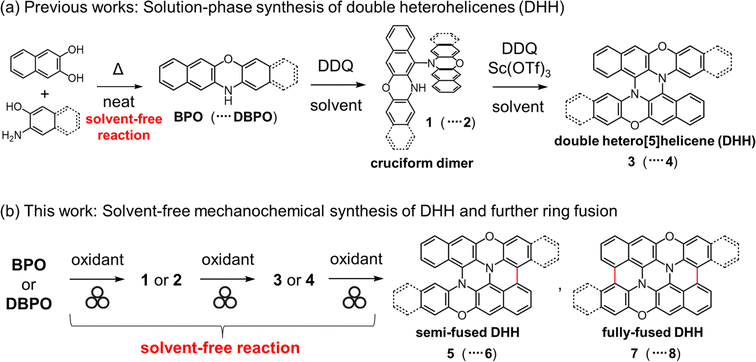 |
| | Scheme 1 (a) Previous synthetic method of double heterohelicenes in solution and (b) solvent-free mechanochemical synthesis of double heterohelicenes and further fused compounds. | |
Recently, mechanochemical synthesis has gained increasing attention in the field of organic chemistry.13–16 The biggest advantage of mechanochemical synthesis over conventional solution-phase synthesis is that it requires minimal or no reaction solvents. Therefore, mechanochemical reactions can make the reaction protocol simpler and safer, and can be applied to compounds with very low solubility.17 There have been several recent reports on the application of mechanochemical reactions to oxidative coupling reactions of aromatic compounds.18–27 In 2018, Borchardt et al. reported the mechanochemical Scholl reaction, an oxidative cyclodehydrogenation forming aryl–aryl C–C bonds.18 They established the mechanochemical synthesis of fully planarized PAHs, such as hexa-peri-hexabenzocoronene (HBC) and its larger analogs, from non-planar dendritic oligophenylene precursors using ball mills as reactors. The study by Borchardt et al. demonstrated the usefulness of mechanochemistry for planarization of non-planar molecules by intramolecular oxidative fusion reactions. Our double heterohelicene synthesis method involves the folding of cruciform dimers by intramolecular C–N bond formation. Therefore, mechanochemistry using a ball mill, which can apply local pressure to the reactants, may facilitate the conversion of cruciform dimers to double heterohelicenes. Importantly, NH-containing heteroacenes, such as BPO, DBPO, and BPT, are obtained by solid-state condensation of commercially available compounds on multigram scales. Hence, if the solvent-free conversion of NH-containing heteroacenes to double heterohelicenes is realized, all the reaction steps can be carried out using solvent-free reactions, leading to large-scale synthesis of double heterohelicenes in an economic and ecological manner. Furthermore, mechanochemical reactions may initiate intramolecular oxidative C–C bond formation in double heterohelicenes, affording semi-planarized or fully planarized compounds that cannot be obtained by oxidation in solution (Scheme 1b). In this study, we successfully accomplished the mechanochemical one-pot conversion of BPO and DBPO to double heterohelicenes (3 and 4) via cruciform dimers (1 and 2), in much higher yields compared to conventional solution-phase synthesis. Furthermore, we synthesized novel semi-planarized heterohelicenes 5 and 6 and characterized their structures and electronic properties.
Results and discussion
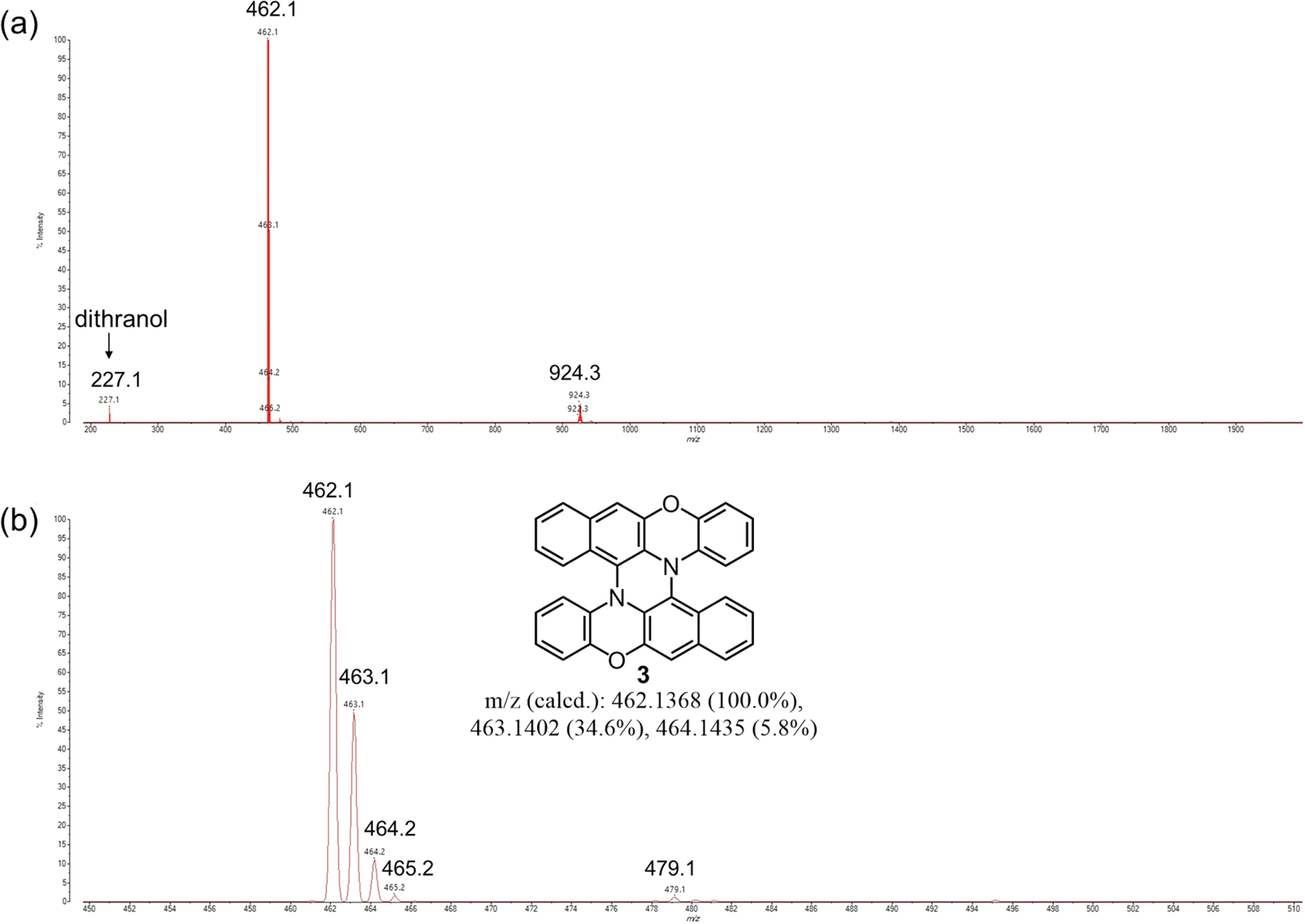
First, we studied the mechanochemical conversion of the cruciform dimer of BPO (1) to double hetero[5]helicene 3 by intramolecular oxidative C–N coupling. As an initial trial, we applied conditions similar to the mechanochemical Scholl reaction reported by Borchardt et al. to 1 (Table 1, entry 1).18 In a 45 mL zirconium dioxide (ZrO2) jar of a planetary mill, twenty-two zirconium dioxide (ZrO2) balls (10 mm ϕ), 1 (200 mg), FeCl3 (840 mg, 12 eq.), and NaCl (8.96 g) as a bulking material were placed under an ambient condition, and the mixture was milled at 600 rpm for 30 min. After milling, the reaction mixture was extracted with CH2Cl2. MALDI-TOF MS measurements of the reaction crude confirmed the formation of double helicene 3, but peaks corresponding to the monochlorinated derivative of 3 and dimer of 1 were also observed (Fig. S1†). The Rf values in TLC analysis of 3 and the chlorinated derivative were very similar, and complete separation of these products by silica gel chromatography was difficult. The crude yield of 3 was about 6% in this condition. After testing various conditions (Table S1†), we found that the combination of DDQ and Sc(OTf)3, the same oxidants used in the solution-phase synthesis of double heterohelicenes, afforded double heterohelicenes in good yields, suppressing chlorination under mechanochemical conditions as follows. In the 50 mL stainless-steel jar of a mixer mill, 200 mg of 1, DDQ (1.1 eq., 108 mg), and Sc(OTf)3 (233 mg, 1.1 eq.) were placed with twelve stainless-steel balls (10 mm ϕ) and milled at a vibration frequency of 30 Hz for 1 h (Table 1, entry 2). The MALDI-TOF MS of the crude product before column chromatography showed that this reaction condition gave almost no chlorinated products, and gave the target double helicene 3 very cleanly (Fig. 1). The isolation yield of 3 was 70% after silica gel column chromatography. The reaction using only DDQ as an oxidant without Sc(OTf)3 also afforded 3, but the yield decreased to 23%, and a significant amount of 1 remained unreacted, confirming the importance of Sc(OTf)3 in the intramolecular C–N coupling of the cruciform dimer (Table 1, entry 3). Sc(OTf)3 is likely to enhance the oxidation power of DDQ by coordinating with DDQ as a Lewis acid.28 By using a planetary mill, the reaction of 1 (200 mg) using DDQ/Sc(OTf)3 as the oxidants gave 3 in 72% yield without chlorination reactions (Table 1, entry 4). The use of 500 mg of 1 as a reactant slightly increased the yield (83%) using a planetary mill (Table 1, entry 5, Fig. S2†). Considering these results, we concluded that the combination of DDQ/Sc(OTf)3 is the best reagents for the mechanochemical conversion of the cruciform dimers to double heterohelicenes.
Table 1 Reaction conditions and yields of synthesis of 3 from 1. (Mill type: p = planetary, m = mixer)
| Entry |
Reactant |
Weight of reactant |
Mill type |
Milling material |
Oxidant |
Milling speed |
Total reaction time |
Yield of 3 |
|
Contains small amount of chlorinated 3.
|
| 1 |
1
|
0.20 g |
p |
ZrO2 |
FeCl3 + NaCl |
600 rpm |
30 min |
ca. 6%a |
| 2 |
1
|
0.20 g |
m |
Steel |
DDQ/Sc(OTf)3 |
30 Hz |
60 min |
70% |
| 3 |
1
|
0.20 g |
m |
Steel |
DDQ |
30 Hz |
60 min |
23% |
| 4 |
1
|
0.20 g |
p |
ZrO2 |
DDQ/Sc(OTf)3 |
600 rpm |
30 min |
72% |
| 5 |
1
|
0.50 g |
p |
ZrO2 |
DDQ/Sc(OTf)3 |
600 rpm |
60 min |
83% |
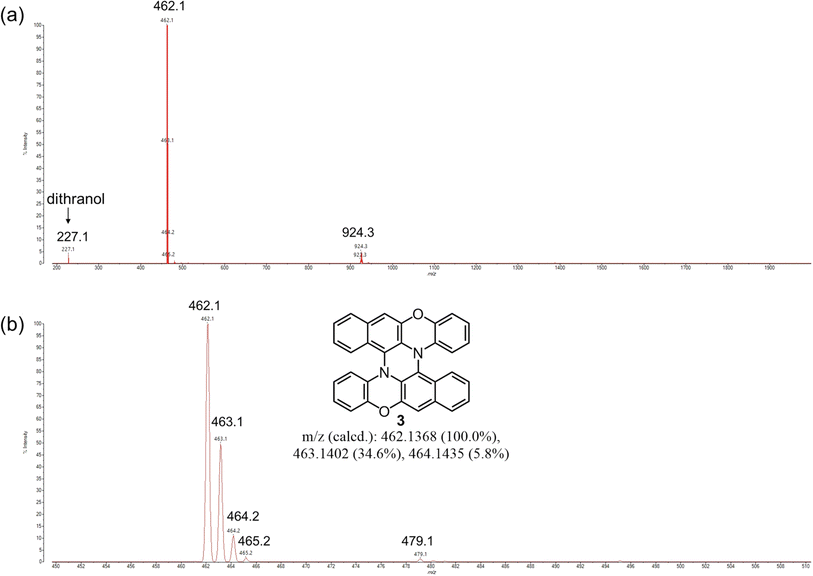 |
| | Fig. 1 MALDI-TOF MS of the crude product (before chromatography) on the reaction of 1 using DDQ/Sc(OTf)3 as oxidants and a mixer mill (Table 1, entry 2). Mass range: (a) 200–2000, (b) 450–510. | |
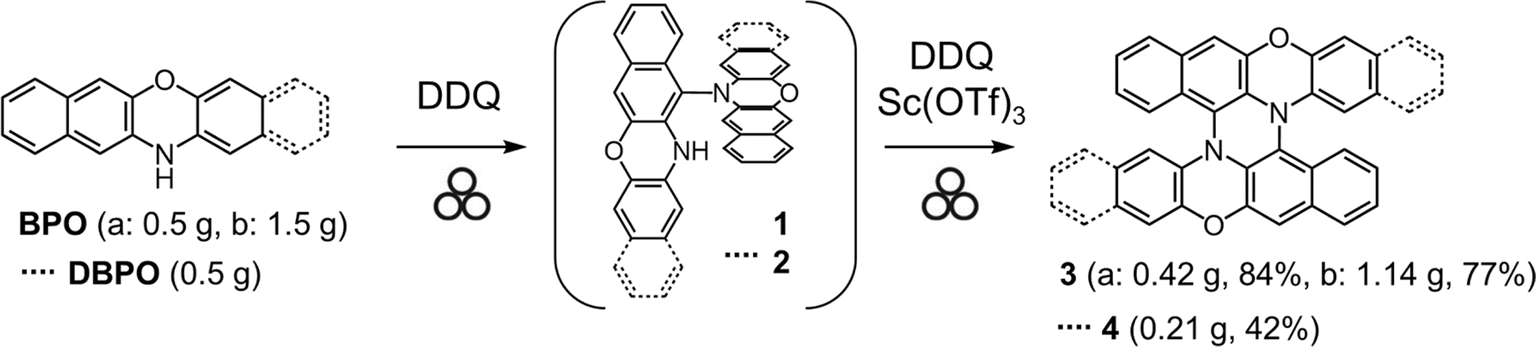
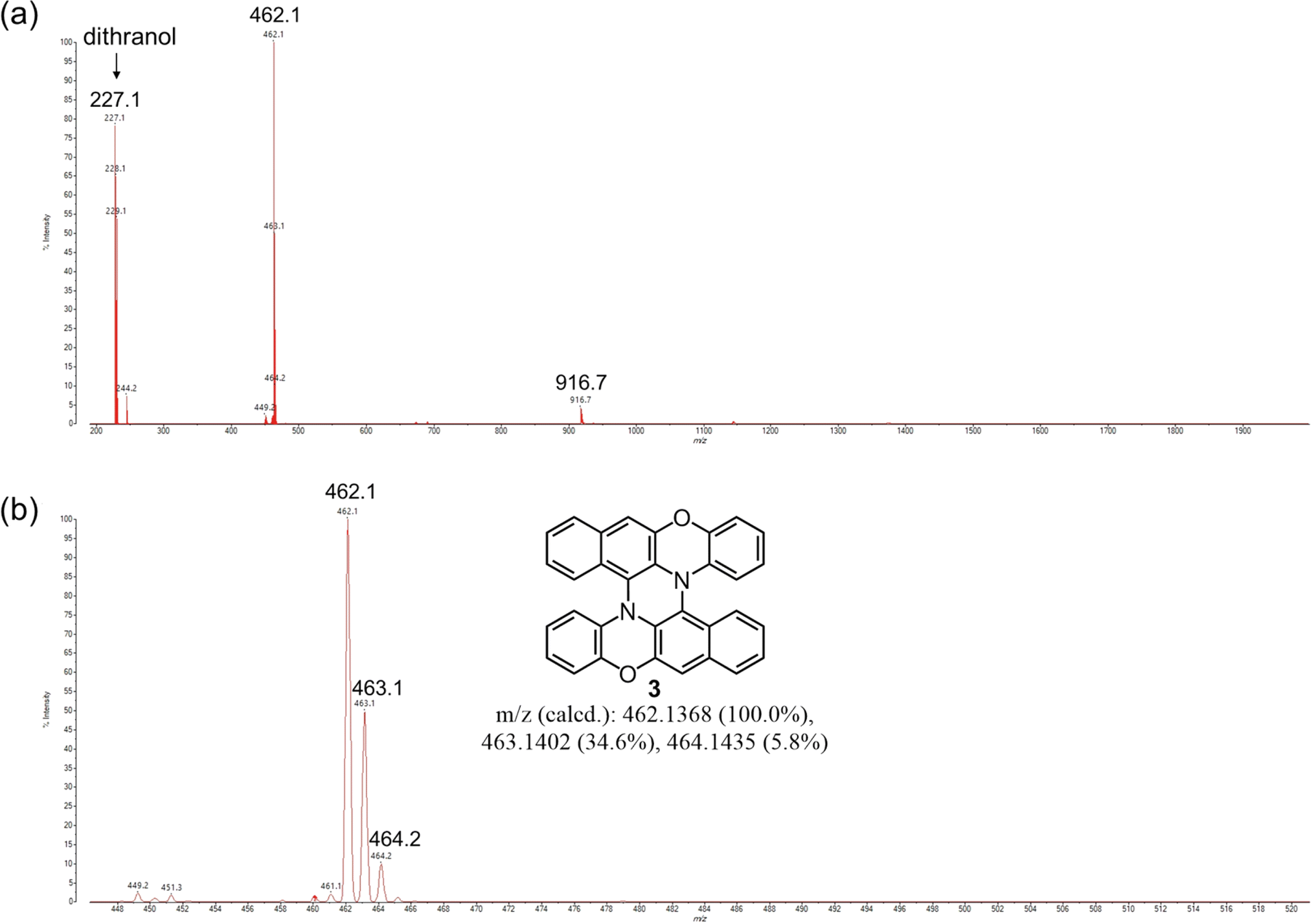
Next, we attempted to synthesize 3 directly from BPO by a mechanochemical reaction. In solution-phase reactions, BPO and DBPO selectively dimerize to form 1 and 2, respectively, by oxidation with DDQ. Based on the results of the solution-phase reactions, we attempted mechanochemical oxidation of BPO with DDQ. Surprisingly, the reactions afforded dimer 1 in very high yields by using either a mixer mill or planetary mill. The reaction using a planetary mill gave 1 in 79% yield, and double heterohelicene 3 was also obtained in 12% yield, while the reaction using a mixer mill gave 1 in 86% yield and 3 in 1% yield (See ESI† page 5 and 6). These results indicate that a planetary mill can easily afford intramolecular oxidative coupling of cruciform dimers compared to a mixer mill. When BPO was reacted with DDQ/Sc(OTf)3, severe oligomerization of BPO occurred (Fig. S3†). Therefore, we attempted the one-pot mechanochemical synthesis of 3 from BPO by tandem oxidation reactions using DDQ in the first step and DDQ/Sc(OTf)3 in the second step (Scheme 2). In a 45 mL ZrO2 jar of a planetary mill, BPO (500 mg) and DDQ (292 mg, 0.6 eq.) were placed with twenty-two ZrO2 balls (10 mm ϕ) and milled at 600 rpm for 30 min (Table 2, entry 1). After milling, the jar was opened, and the formation of 1 and the consumption of BPO were confirmed by MALDI-TOF MS and TLC. Next, DDQ and Sc(OTf)3 (0.6 eq.) were added to the reaction jar and the jar was set on the ball mill again. The mixture was milled at 600 rpm for another 60 min. The MALDI-TOF MS of the crude product indicated that double heterohelicene 3 was formed very cleanly (Fig. 2). After purification by silica gel chromatography, 3 was obtained in the two steps in 84% yield. This yield was twice higher than that of the one-pot reaction in solution (42%).9 The reaction at a rotational speed of 800 rpm resulted in the formation of small amounts of overreacted products, in addition to the formation of 3 in 61% yield (Table 2, entry 2, Fig. S4†). When 1.5 g of BPO was reacted at 600 rpm, 1.14 g of 3 (77% yield) was obtained in a one-pot manner (Table 2, entry 3). The one-pot mechanochemical reaction of BPO using a mixer mill also afforded 3 (Table 2, entry 4), but the yield decreased to 55% from that obtained using a planetary mill on the same scale (84%). MALDI-TOF MS of the crude product of the reaction (Table 2, entry 4) revealed the formation of certain amounts of oligomers formed by the successive addition of BPO to 1 or 3 (Fig. S5†), confirming the superiority of a planetary mill over a mixer mill in the intramolecular oxidative C–N coupling. When DBPO was reacted using a planetary mill, double heterohelicene 4 was obtained in 42% yield (Table 2, entry 5). This yield was much higher than that of the one-pot reaction of DBPO in solution (10%).8
 |
| | Scheme 2 One-pot mechanochemical synthesis of double hetero[5]helicenes 3 and 4 from heteroacenes. | |
Table 2 Reaction conditions and yields of synthesis of double hetero[5]helicenes 3 and 4 from heteroacenes. (Mill type: p = planetary, m = mixer)
| Entry |
Reactant |
Weight of reactant |
Mill type |
Milling material |
Oxidant |
Milling speed |
Total reaction time |
Yield of 3 or 4 |
| 1 |
BPO
|
0.50 g |
p |
ZrO2 |
DDQ → DDQ/Sc(OTf)3 |
600 rpm |
30 + 60 min |
84% |
| 2 |
BPO
|
0.50 g |
p |
ZrO2 |
DDQ → DDQ/Sc(OTf)3 |
800 rpm |
30 + 30 min |
61% |
| 3 |
BPO
|
1.50 g |
p |
ZrO2 |
DDQ → DDQ/Sc(OTf)3 |
600 rpm |
30 + 60 min |
77% |
| 4 |
BPO
|
0.50 g |
m |
Steel |
DDQ → DDQ/Sc(OTf)3 |
30 Hz |
30 + 120 min |
55% |
| 5 |
DBPO
|
0.50 g |
p |
ZrO2 |
DDQ → DDQ/Sc(OTf)3 |
600 rpm |
30 + 30 min |
42% |
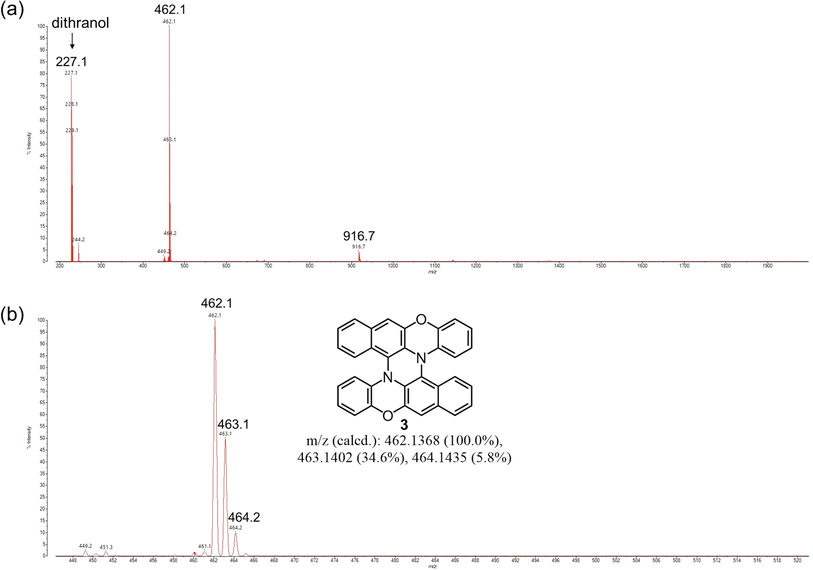 |
| | Fig. 2 MALDI-TOF MS of the crude product (before chromatography) on the reaction of BPO using DDQ → DDQ/Sc(OTf)3 as oxidants and a planetary mill (Table 2, entry 1). Mass range: (a) 200–2000, (b) 448–520. | |
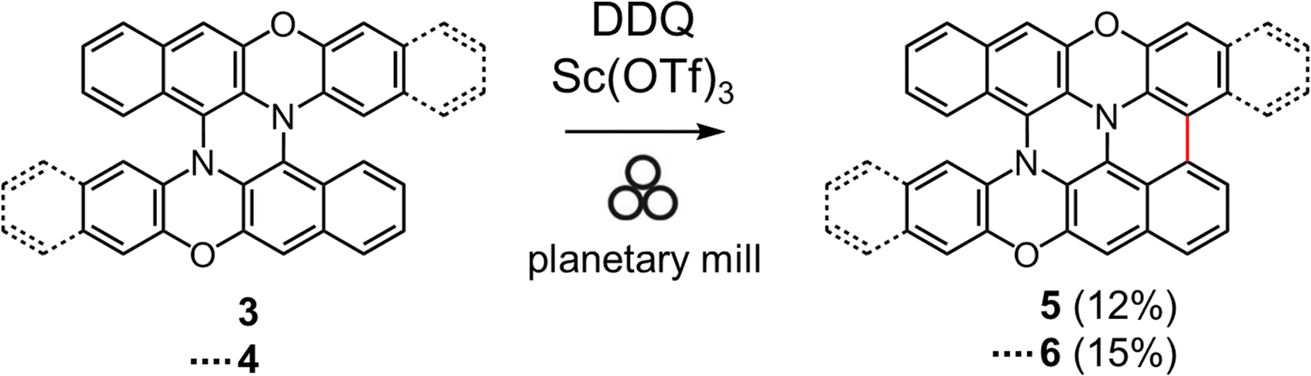
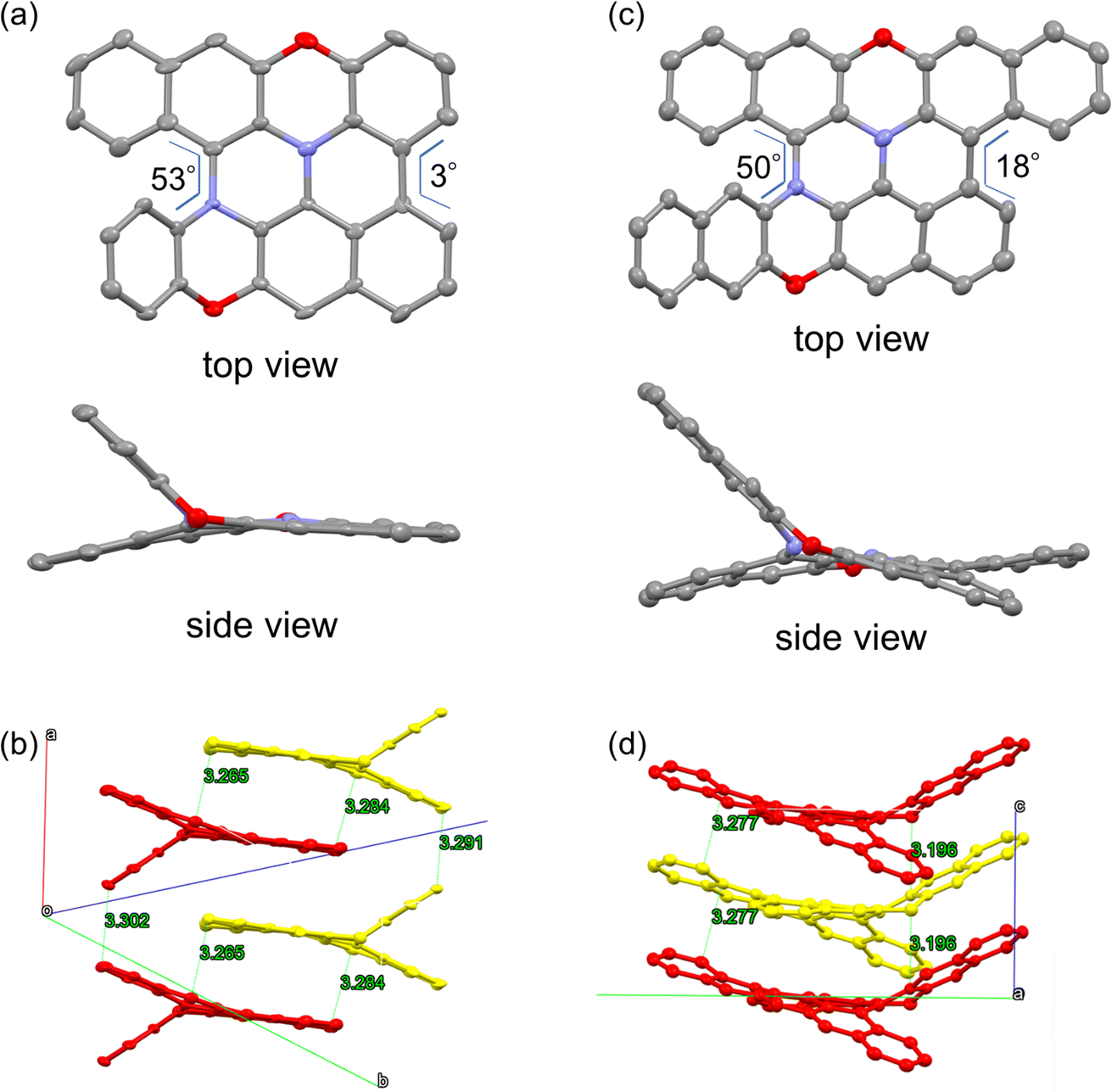
Encouraged by the success of the mechanochemical synthesis of double heterohelicenes 3 and 4, we aimed at further ring-fusion of double heterohelicenes via oxidative intramolecular C–C bond formation (Scheme 3). Double heterohelicene 3 (300 mg) was milled with DDQ, Sc(OTf)3 (1.1 eq.), and twenty-two ZrO2 balls (10 mm ϕ) in a 45 mL ZrO2 jar of a planetary mill at 850 rpm for 30 min. After the reaction, the reaction solid was extracted with CH2Cl2 and monitored by MALDI-TOF MS and TLC. MALDI-TOF MS exhibited the most intense peak at m/z = 460, corresponding to 5, indicating the formation of an intramolecular C–C bond (Fig. S6†). MALDI-TOF MS also showed a small peak corresponding to doubly fused product 7 (m/z = 458). TLC analysis showed a new red spot below an orange spot of 3. The red spot on the TLC plate was isolated as a red-brown solid by silica gel chromatography. X-ray single crystal analysis of a red-brown solid recrystallized from CH2Cl2/hexane solution revealed that the crystal contained a racemate of semi-planarized hetero[5]helicene 5 (Fig. 3a). Half of the molecule was almost planarized by ring fusion and the torsion angle of the newly formed bay region was 3°. The torsion angle of the intact hetero[5]helicene moiety was 53°, which was larger than that of 3 (48°). The (P)- and (M)-isomers of 5 were stacked alternately by facing each other's planarized parts to form a one-dimensional columnar array (Fig. 3b). The isolation yield of 5 was 12%. Doubly fused product 7 could not be isolated by silica gel chromatography, probably because of the very low solubility of 7. When DBPO was reacted under the same conditions, the semi-planarized product 6 was obtained in 15% yield as a purple-black solid. Recrystallization of 6 from CH2Cl2/ethanol solution yielded single crystals, and the structure was clarified by X-ray crystal analysis (Fig. 3c). In 6, a carbo[4]helicene moiety was formed by ring fusion. The torsion angle at the carbo[4]helicene moiety was 18°. The torsion angle of the intact hetero[5]helicene of 6 was 50°, which was larger than that of 4 (46°). The (P)- and (M)-isomers of 6 were alternately aligned to form a one-dimensional columnar array similar to that of 5 (Fig. 3d). Fully planarized product 8 was not obtained, similar to 7, probably because of its very low solubility. An increase in the amount of oxidant or the reaction time for the oxidative ring fusion reaction of 3 and 4 resulted in the formation of insoluble byproducts, probably due to oligomerization or polymerization, and the yields of 5 and 6 did not improve.
 |
| | Scheme 3 Mechanochemical synthesis of 5 and 6. | |
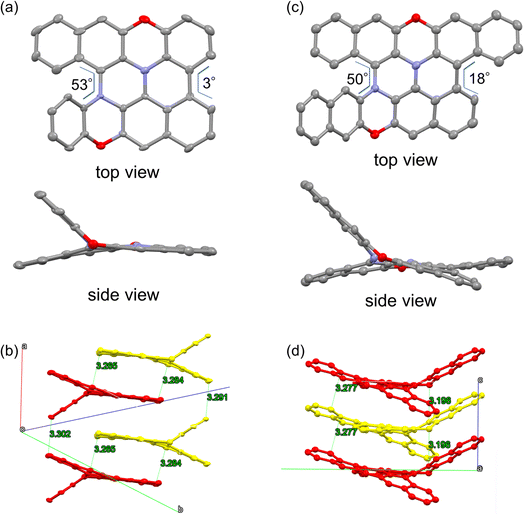 |
| | Fig. 3 ORTEP drawings of (a) 5 and (c) 6, and packing structures of (b) 5 and (d) 6. Hydrogen atoms are omitted for clarity. Thermal ellipsoids are set at 50% probability. Yellow and red in (b) and (d) represent the (P)- and (M)-isomers, respectively. | |
DFT calculations were performed for semi- and fully planarized compounds to investigate the effects of ring fusion on their electronic structures. Fig. 4a shows the frontier Kohn–Sham MOs of 3, 5, and 7. In the HOMO of 3, the two carbon atoms that react upon the formation of 5 have opposite MO signs, and the ring fusion of 3 is thought to result in elevation of the HOMO level. Actually, the calculated HOMO levels of 5 was 0.22 eV higher than that of 3, and the HOMO of 7 was 0.32 eV higher than that of 5. Therefore, the ring fusion of the helicene moieties in 3 increased its electron-donating properties. The degree of change in the LUMO level was relatively small compared with the change in the HOMO level upon ring fusion. The LUMO level of 5 was decreased by 0.13 eV from that of 3, and the LUMO of 7 was decreased by 0.02 eV from that of 5. The modest alteration in the LUMO energy level can be explained by the absence of the MO coefficient at one of the carbon atoms of the reaction sites in the LUMO of 3. The TD-DFT-calculated oscillator strength (f) for the HOMO–LUMO transition of 3 was only 0.0002. This is because the HOMO and LUMO of 3 belong to the same irreducible representation (a) and the components of the electronic transition dipole moment perpendicular to the C2 axis become zero.9 On the other hand, the oscillator strength for the HOMO–LUMO transition of 5 increased to 0.0288, owing to the symmetry lowering from the C2 to C1 symmetric structure. Further ring fusion of 5 to 7 resulted in a completely planar C2h symmetric structure, and the HOMO–LUMO transition became completely symmetry-forbidden (f = 0). DFT calculations for the DBPO series (4, 6, and 8) showed similar trends to those of the BPO series (3, 5, and 7), as shown in Fig. 4b.
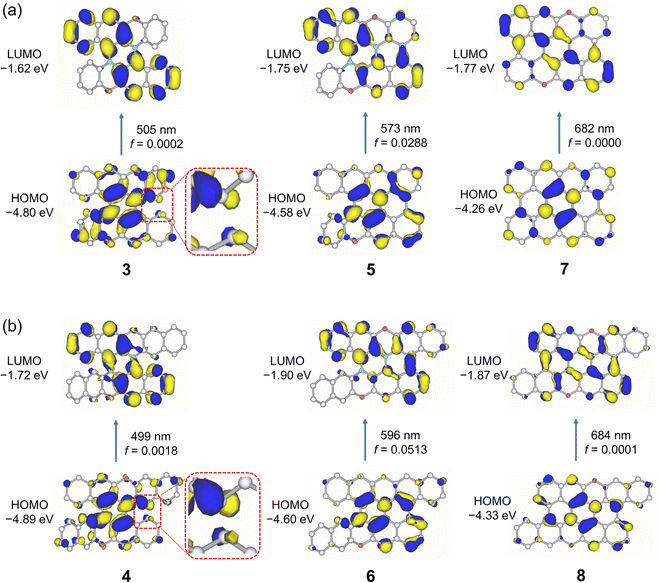 |
| | Fig. 4 Frontier Kohn–Sham molecular orbitals and DFT calculated oscillator strengths of (a) 3, 5, and 7 and (b) 4, 6, and 8 calculated at the B3LYP-GD3BJ/6-311G(2d,p) level of theory. | |
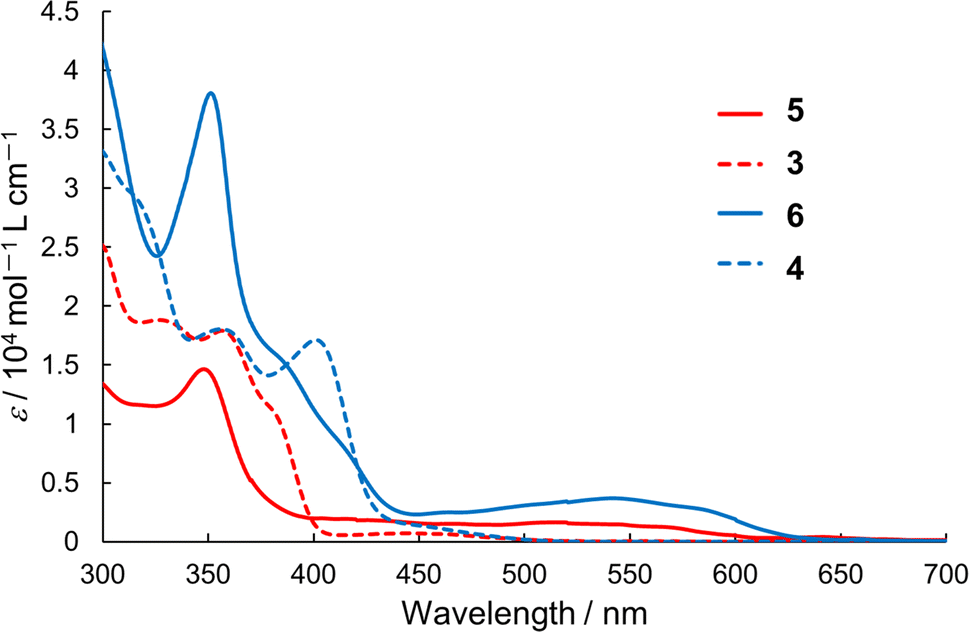
We measured the UV-vis absorption spectra of semi-planarized heterohelicenes 5 and 6 in 1.0 × 10−5 M CH2Cl2 solutions and compared them with those of 3 and 4 (Fig. 5). The solution of 5 exhibited the lowest energy transition band around 520 nm, which was attributed to the HOMO–LUMO transition. This lowest energy band was red-shifted by 2990 cm−1 from that of 3 (450 nm), and its molar extinction coefficient was more than twice that of 3. The observed red shift and increase in the molar extinction coefficient were in accordance with the results of the TD-DFT calculations. Similarly, the lowest energy absorption band of 6 was more intense and red-shifted than that of 4.
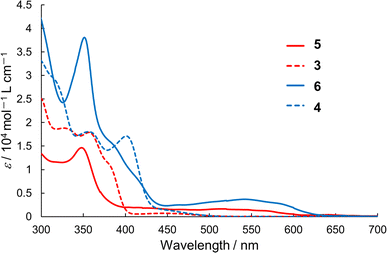 |
| | Fig. 5 UV-vis absorption spectra of 3–6 in dichloromethane. | |
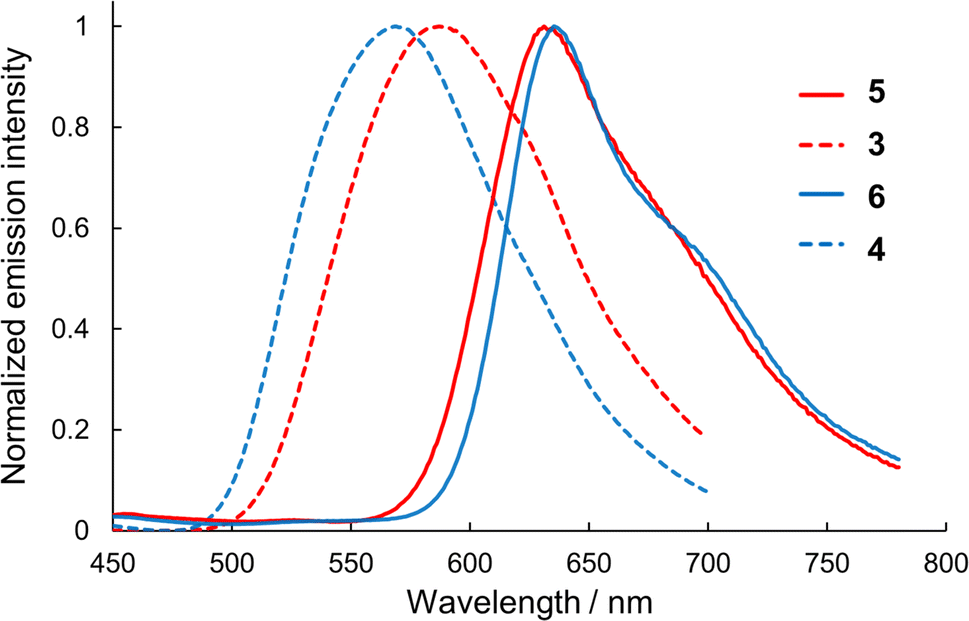
Semi-planarized helicenes 5 and 6 in CH2Cl2 exhibited red emission upon UV irradiation. Fig. 6 shows the emission spectra of 5 and 6 in CH2Cl2 at 298 K. The emission peaks of 5 and 6 (631 nm and 638 nm, respectively) were red-shifted by 1190 cm−1 and 1900 cm−1, respectively, compared to those of 3 and 4 (587 nm and 569 nm), owing to the decrease in the HOMO–LUMO gap in the semi-planarized helicenes. The emission quantum yields of 5 and 6 in CH2Cl2 at 298 K were 1.1 and 0.7%, which decreased from the precursor double helicenes (3.5 and 3.8% for 3 and 4, respectively). Emission lifetimes of 5 and 6 in CH2Cl2 were 5.17 ns and 1.75 ns (Fig. S10†), respectively, and the radiative and nonradiative rate constants (kr and knr) were determined from the emission lifetimes and emission quantum yields as summarized in Table 3. For both 5 and 6, the values of kr slightly increased from those of 3 and 4, respectively, probably owing to symmetry lowering. However, the significant increase in knr exceeded the increase in kr, resulting in the decreased emission quantum yields of 5 and 6 compared to those of 3 and 4.
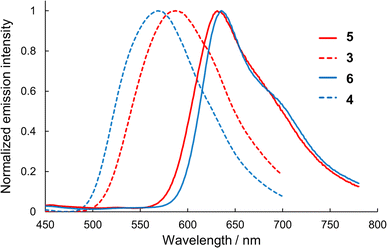 |
| | Fig. 6 Emission spectra of 3–6 in dichloromethane. | |
Table 3 Photophysical properties of 3–6 in dichloromethane
|
|
λ
em/nm |
Φ/% |
τ/ns |
k
r/s−1 |
k
nr/s−1 |
|
3
|
587 |
3.5 |
21.1 |
1.7 × 106 |
4.6 × 107 |
|
4
|
569 |
3.8 |
13.4 |
2.8 × 106 |
7.2 × 107 |
|
5
|
631 |
1.1 |
5.17 |
2.1 × 106 |
1.9 × 108 |
|
6
|
638 |
0.7 |
1.75 |
4.0 × 106 |
5.7 × 108 |
We measured the cyclic voltammograms (CV) and differential pulse voltammograms (DPV) of 5 and 6 to investigate the effect of ring fusion on their electrochemical properties (Fig. S11 and S12†). Both 5 and 6 exhibited two sets of reversible one-electron oxidation processes in the CVs. The first oxidation potentials of −0.13 V (for 5) and −0.07 V (for 6) were negatively shifted compared to those of 3 and 4 by 0.18 V and 0.22 V, respectively, indicating the increased electron donating properties of 5 and 6 (Table S5†). The second oxidation potentials were 0.44 V for both 5 and 6, which were negatively shifted compared to those of 3 and 4 by 0.24 V and 0.28 V, respectively. According to the oxidation potentials, semi-planarized helicene 5 is thought to be a stronger electron donor than the double heterohelicenes composed of two DHDAP,8 which have four amino nitrogen atoms and show the lowest first oxidation potential (E1 = −0.10 V) among the double heterohelicenes we have reported.
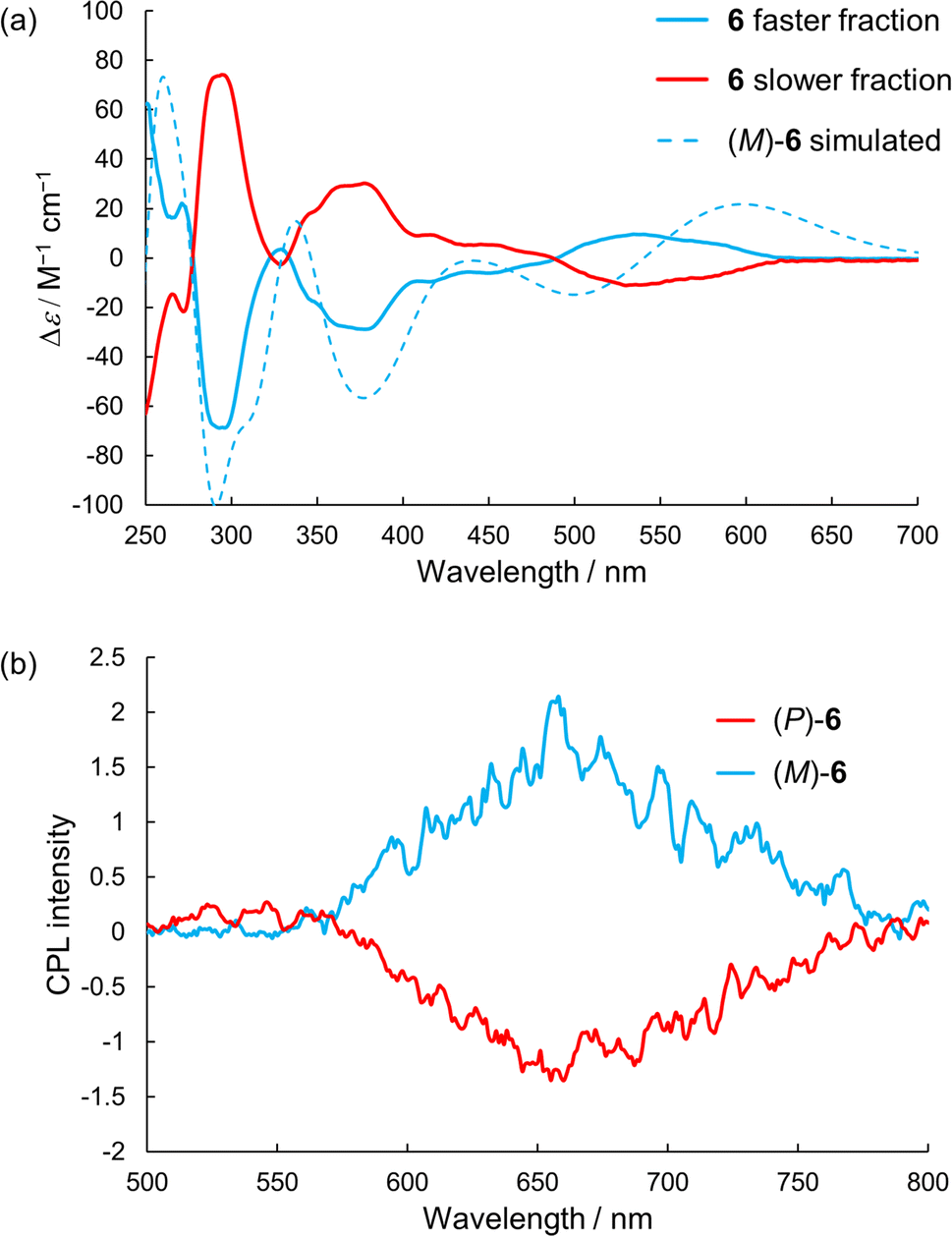
We attempted optical resolution of semi-planarized helicenes 5 and 6 using chiral column chromatography. Unfortunately, the optical resolution of 5 was unsuccessful because of its low solubility in the mobile phase. In contrast, we successfully isolated the enantiomers of 6 using a chiral column for a medium-pressure liquid chromatography system (See ESI†). As shown in Fig. 7a, the faster and slower elution fractions of 6 exhibited mirror-image CD spectra. The faster elution fraction exhibited the first positive Cotton effect. The TD-DFT-simulated spectrum for the (M)-enantiomer exhibited the first positive Cotton effect and matched well with the spectrum of the faster eluting enantiomer. The sign of CD signal of 6 was consistent to that of 4; (M)-isomer shows the first positive Cotton effect. The enantiomers of 6 exhibited clear CPL emissions with a peak at approximately 650 nm, as shown in Fig. 7b. The signs of CPL matched those of the lowest energy bands in the CD spectra for each enantiomer, similar to the case of 4. The |gCPL| value of 6 was 2 × 10−3 (at 650 nm). The |gCPL| value of 6 was smaller than that of 4 (2.3 × 10−2), but 6 is a rare example of a CPL emitter in the longer wavelength region with a moderately high |gCPL| value, despite its smaller molecular skeleton compared with those of the previous examples.29,30 The decrease in the |gCPL| value of 6 was explained as follows. The dissymmetry factor for the CPL (gCPL) is theoretically described as eqn (1):
| | 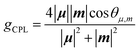 | (1) |
where
μ and
m are the transition electronic dipole moment (TEDM) and transition magnetic dipole moment (TMDM) for the de-excitation transition, respectively, and
θμ,m is the angle between the
μ and
m vectors. In
4, the magnitude of
μ for the HOMO–LUMO transition becomes relatively small (|
μ| = 17.9 × 10
−20 esu cm) because the transition is nearly symmetry-forbidden, and the angle between
μ and
m becomes parallel (
θμ,m is 0°). Therefore, the calculated |
gCPL| value of
4 is very large (|
gCPL| = 0.31). On the other hand, in
6, |
μ| for the HOMO–LUMO transition increased to 214 × 10
−20 esu cm because of symmetry lowering, and
θμ,m became far from parallel (
θμ,m = 83.7°), resulting in a decreased |
gCPL| value of 4.6 × 10
−3 in
6 (Table S6 and Fig. S14
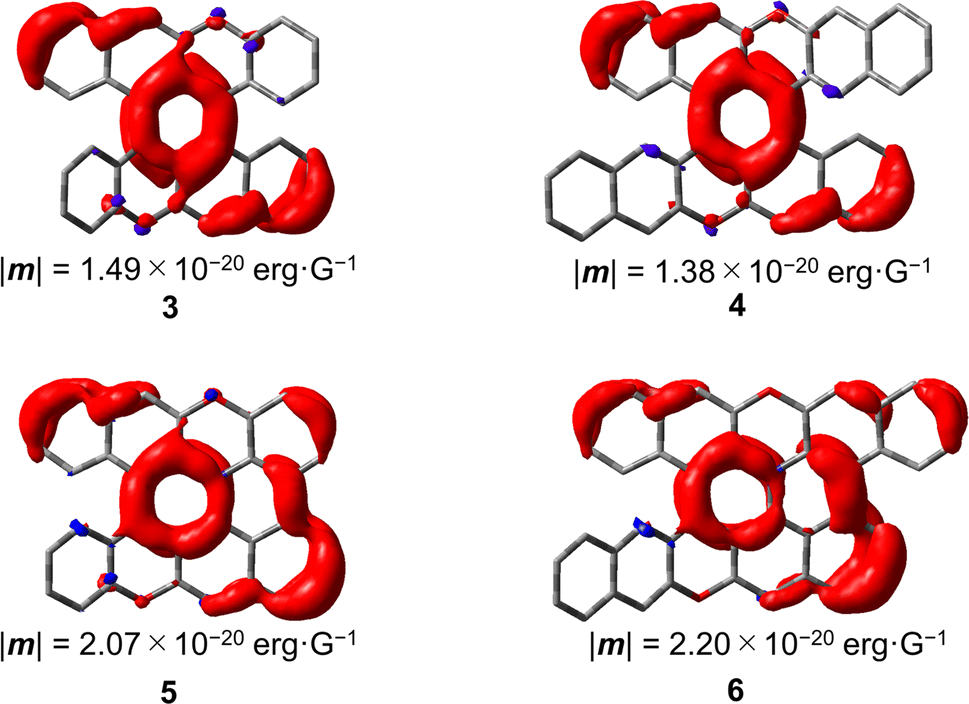
†). Interestingly, the magnitude of |
m| increased by 1.6 times from
4 (1.38 × 10
−20 erg G
−1) to
6 (2.20 × 10
−20 erg G
−1) upon ring fusion of one helicene moiety. Similarly, the magnitude of |
m| of
5 (2.07 × 10
−20 erg G
−1) also increased by 1.4 times to that of
3 (1.49 × 10
−20 erg G
−1). In general, enhancing the magnitude of |
m| leads to an increase in the |
gCPL| values of the organic CPL emitters. However, the relationship between the molecular structure and the magnitude of |
m| is not fully understood. To obtain insight into the enhanced TMDM in
5 and
6, we performed density mapping of TMDM that visualizes the origin of the magnitudes of TMDM.
31,32 The total TMDM density (ρ
totalm) is expressed as the density form of TMDM, whose space integrals give |
m|.
32Fig. 8 shows the distribution of the TMDM density in
3–6. In
3 and
4, the TMDM densities were distributed on the central six-membered rings and the outer edges of the naphthalene moieties fused to the central six-membered rings. In
5 and
6, the TMDM densities were further extended through the newly formed C–C bonds compared with those in
3 and
4. Thus, the extension of the TMDM densities
via new C–C bonds is attributed to the increase in the magnitude of |
m| in
5 and
6. Very recently, Novoa, Mota, Cuerva, and Miguel
et al. proposed a general rationalization for improving the magnitude of the TMDM of helical-conjugated systems and found a linear relationship between |
m| and the inner area of the helix, analogous to classical solenoids, that is, |
m| can be increased by increasing the number of helical turns or extending the helix diameter.
33 On the other hand, the results of this study indicated that modification of the outer regions of a helicene moiety can also increase the magnitude of the TMDM without increasing the length of the helix, and this study will provide another molecular design to enhance |
m|.
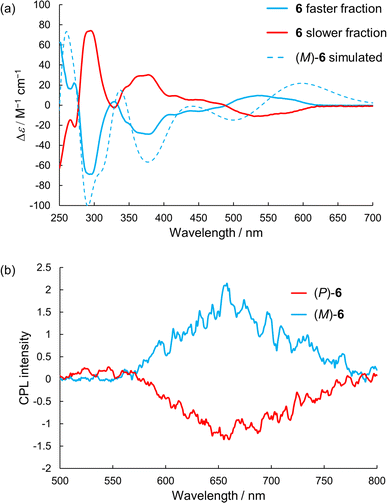 |
| | Fig. 7 (a) CD and (b) CPL spectra of 6 in dichloromethane. Simulated CD spectrum was calculated at the B3LYP-GD3BJ/6-311G(2d,p) level of theory. | |
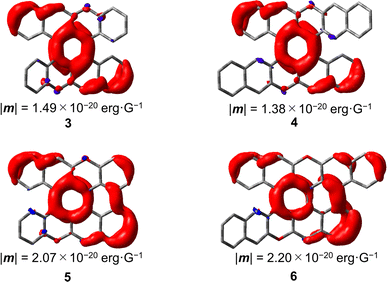 |
| | Fig. 8 TMDM densities for the S1 → S0 transitions of 3–6 calculated at the B3LYP-GD3BJ/6-311G(2d,p) level of theory. | |
Conclusion
In summary, we developed a facile and efficient synthetic method for double heterohelicenes using mechanochemical oxidative C–N coupling reactions. Owing to the development of this method, double heterohelicenes can now be synthesized using only solvent-free reactions from commercially available compounds. Using this mechanochemical reaction, more than 1 g of the double hetero[5]helicene 3, which exhibits a high gCPL value, can be synthesized from BPO in a one-pot manner. In addition, mechanochemical oxidative coupling enabled further ring fusion of double hetero[5]helicenes to afford semi-planarized heterohelicenes and fully planarized hetero-nanographenes that have not been obtained using conventional solution-phase synthesis. We successfully isolated semi-planarized heterohelicenes 5 and 6, and clarified their structures using X-ray single-crystal analysis. Compounds 5 and 6 showed red-shifted absorption and emission spectra and increased electron-donating properties compared to the double hetero[5]helicenes 3 and 4. We succeeded in achieving optical resolution of 6, and the enantiomers of 6 exhibited clear CPL emission at 650 nm with a |gCPL| value of 2 × 10−3. The magnitudes of the transition magnetic moment (m) of 5 and 6 increased compared with those of 3 and 4 upon C–C bond formation. We believe that this study will lead to the application of heterohelicenes as organic functional materials through the development of efficient and economical synthesis methods for this class of compounds.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research (17H04874, 18K05264, 20H02726) from the Japan Society for the Promotion of Science (JSPS), a Grant-in-Aid for Transformative Research Areas (A) “Condensed Conjugation” (JSPS KAKENHI Grant Number JP20H05866) from MEXT, Ogasawara Toshiaki Memorial Foundation, Shorai Foundation for Science and Technology, The Foundation for the Promotion of Ion Engineering, Izumi Science and Technology Foundation, ENEOS Tonen General Research Grants, and Iwatani Naoji Foundation. Computation time for the theoretical calculations was provided by Research Center for Computational Science, Okazaki, Japan.
Notes and references
- T. Hatakeyama, K. Shiren, K. Nakajima, S. Nomura, S. Nakatsuka, K. Kinoshita, J. Ni, Y. Ono and T. Ikuta, Adv. Mater., 2016, 28, 2777–2781 CrossRef CAS PubMed.
- M. Mamada, M. Hayakawa, J. Ochi and T. Hatakeyama, Chem. Soc. Rev., 2024, 53, 1624–1692 RSC.
- S. Dadashi-Silab, X. Pan and K. Matyjaszewski, Chem.–Eur. J., 2017, 23, 5972–5977 CrossRef CAS PubMed.
- U. H. F. Bunz and J. Freudenberg, Acc. Chem. Res., 2019, 52, 1575–1587 CrossRef CAS PubMed.
- W.-W. Yang and J.-J. Shen, Chem.–Eur. J., 2022, 28, e202202069 CrossRef CAS PubMed.
- Y. Xu, Q. Wang, X. Song, Y. Wang and C. Li, Chem.–Eur. J., 2023, 29, e202203414 CrossRef CAS PubMed.
- F. Tani, M. Narita and T. Murafuji, Chempluschem, 2020, 85, 2093–2104 CrossRef CAS PubMed.
- D. Sakamaki, D. Kumano, E. Yashima and S. Seki, Angew Chem. Int. Ed. Engl., 2015, 54, 5404–5407 CrossRef CAS PubMed.
- D. Sakamaki, S. Tanaka, K. Tanaka, M. Takino, M. Gon, K. Tanaka, T. Hirose, D. Hirobe, H. Yamamoto and H. Fujiwara, J. Phys. Chem. Lett., 2021, 9283–9292 CrossRef CAS PubMed.
- D. Sakamaki, D. Kumano, E. Yashima and S. Seki, Chem. Commun., 2015, 51, 17237–17240 RSC.
- S. Tanaka, D. Sakamaki, N. Haruta, T. Sato, M. Gon, K. Tanaka and H. Fujiwara, J. Mater. Chem., 2023, 11, 4846–4854 CAS.
- D. Sakamaki, Y. Inoue, K. Shimomura, D. Taura, E. Yashima and S. Seki, Tetrahedron Lett., 2023, 114, 154294 CrossRef CAS.
- A. Stolle, T. Szuppa, S. E. S. Leonhardt and B. Ondruschka, Chem. Soc. Rev., 2011, 40, 2317–2329 RSC.
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413–447 RSC.
- J. L. Howard, Q. Cao and D. L. Browne, Chem. Sci., 2018, 9, 3080–3094 RSC.
- K. Kubota, T. Seo, K. Koide, Y. Hasegawa and H. Ito, Nat. Commun., 2019, 10, 111 CrossRef PubMed.
- T. Seo, N. Toyoshima, K. Kubota and H. Ito, J. Am. Chem. Soc., 2021, 143, 6165–6175 CrossRef CAS PubMed.
- S. Grätz, D. Beyer, V. Tkachova, S. Hellmann, R. Berger, X. Feng and L. Borchardt, Chem. Commun., 2018, 54, 5307–5310 RSC.
- S. Grätz, M. Oltermann, E. Troschke, S. Paasch, S. Krause, E. Brunner and L. Borchardt, J. Mater. Chem. A, 2018, 6, 21901–21905 RSC.
- S. Grätz, M. Oltermann, C. G. Vogt and L. Borchardt, ACS Sustainable Chem. Eng., 2020, 8, 7569–7573 CrossRef.
- A. Krusenbaum, S. Grätz, S. Bimmermann, S. Hutsch and L. Borchardt, RSC Adv., 2020, 10, 25509–25516 RSC.
- G. Báti, D. Csókás, T. Yong, S. M. Tam, R. R. S. Shi, R. D. Webster, I. Pápai, F. García and M. C. Stuparu, Angew Chem. Int. Ed. Engl., 2020, 59, 21620–21626 CrossRef PubMed.
- T. Yong, G. Báti, F. García and M. C. Stuparu, Nat. Commun., 2021, 12, 1–8 CrossRef PubMed.
- J. Stanojkovic, R. William, Z. Zhang, I. Fernández, J. Zhou, R. D. Webster and M. C. Stuparu, Nat. Commun., 2023, 14, 803 CrossRef CAS PubMed.
- G. Báti, S. Laxmi and M. C. Stuparu, ChemSusChem, 2023, 16, e202301087 CrossRef PubMed.
- G. Báti, D. Csókás and M. C. Stuparu, Chem.–Eur. J., 2024, 30, e202302971 CrossRef PubMed.
- M. Xuan, C. Schumacher, C. Bolm, R. Göstl and A. Herrmann, Adv. Sci., 2022, 9, e2105497 CrossRef PubMed.
- S. Fukuzumi and K. Ohkubo, Chem.–Eur. J., 2000, 6, 4532–4535 CrossRef CAS PubMed.
- C. M. Cruz, S. Castro-Fernández, E. Maçôas, J. M. Cuerva and A. G. Campaña, Angew Chem. Int. Ed. Engl., 2018, 57, 14782–14786 CrossRef CAS PubMed.
- S. Míguez-Lago, I. F. A. Mariz, M. A. Medel, J. M. Cuerva, E. Maçôas, C. M. Cruz and A. G. Campaña, Chem. Sci., 2022, 13, 10267–10272 RSC.
- H. Kubo, D. Shimizu, T. Hirose and K. Matsuda, Org. Lett., 2020, 22, 9276–9281 CrossRef CAS PubMed.
- H. Kubo, T. Hirose, T. Nakashima, T. Kawai, J.-Y. Hasegawa and K. Matsuda, J. Phys. Chem. Lett., 2021, 12, 686–695 CrossRef CAS PubMed.
- R. G. Uceda, C. M. Cruz, S. Míguez-Lago, L. Á. de Cienfuegos, G. Longhi, D. A. Pelta, P. Novoa, A. J. Mota, J. M. Cuerva and D. Miguel, Angew Chem. Int. Ed. Engl., 2024, 63, e202316696 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Masayuki
Gon
*a,
Masayuki
Gon