
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-tube B7-H3 detection based on isothermal exponential amplification and dendritic hybridization chain reaction†
Xiangyun
Chen
a,
Chun
Xuan
a,
Jingtao
Lin
a,
Zhongquan
Pan
a,
Xiaoliang
Wu
a,
Pin
Wu
a,
Zhenchang
Liang
*c,
Luxin
Yu
 *b and
Cailing
Qiu
*a
*b and
Cailing
Qiu
*a
aDalang Hospital of Dongguan, Dongguan 523770, China. E-mail: youling85@126.com
bGuangdong Provincial Key Laboratory of Medical Molecular Diagnostics, The First Dongguan Affiliated Hospital, Guangdong Medical University, Dongguan 523808, China. E-mail: yuluxin2006@163.com
cZhongshan City Shiqisuhuazan Hospital, Zhongshan, 528400, China. E-mail: lzhchang206@sina.com
First published on 15th March 2024
Abstract
We have developed a one-tube fluorescence strategy for the detection of B7-H3 based on a proximity hybridization-mediated protein-to-DNA signal transducer, isothermal exponential amplification (EXPAR), and dendritic hybridization chain reaction (D-HCR). In this assay, a protein signal transducer was employed to convert the input protein to output single-stranded DNA with a nicking site. Antibody-conjugated DNA1 was first hybridized with the output DNA (DNA3). The binding of antibodies conjugated DNA1 and DNA2 to the same protein was able to increase the local concentrations, resulting in strand displacement between DNA3 and DNA2. DNA3 with a nicking endonuclease recognition sequence at the 5′ end then hybridized with hairpin probe 1 to mediate EXPAR in the presence of nicking endonuclease and DNA polymerase. A large number of single-strand DNA were produced in the circle of nicking, polymerization, and strand displacement. The resulting ssDNA products were further amplified by D-HCR to produce many large-molecular concatemers. The resulting DNA products can be monitored in real-time fluorescence signaling. Our proposed assay can realize one-tube detection due to the same reaction temperature of the protein-to-DNA signal transducer, EXPAR, and DHCR. This assay has a linear range from 100 fg mL−1 to 1 μg mL−1 with a detection limit down to 100 fg mL−1. This work shows a good performance in clinical specimen detection.
Introduction
B7-H3 (CD276) is one of the B7 family expressed in many tissues and on antigen-presenting cells. B7-H3 has been reported to be an enhancer of the T-cell response and a checkpoint molecule, as a target for cancer immunotherapy.1,2 The expression of B7-H3 is reported to be related to several diseases such as breast cancer, leukaemia, and systemic lupus erythematosus.3,4 Sensitive and rapid detection of B7-H3 has a major impact in diverse areas, such as clinical diagnosis, therapy progress monitoring, prognosis estimation, and disease prevention.Recently, many detection methods have been developed for B7-H3 assay. Enzyme-linked immunosorbent assay (ELISA) is one of the most commonly used methods for soluble B7-H3 detection.5,6 ELISA needs to establish a standard cure for every test. This approach is time-consuming, laborious, and high-cost. Flow cytometry (FCM) was applied to detect the expression of B7-H3 on the cell surface.7,8 However, FCM is hard to use for B7-H3 quantitative analysis. Several novel protein detection techniques based on advanced materials have been developed recently.9–12 However, these methods are still a long way from clinical application due to their limitations such as stability and inconvenience. Therefore, continuing efforts have been made to seek ideal tools for fast, sensitive, cost-effective, and easy-to-use B7-H3 detection.
Chris Le and his coworkers developed a signal translator for non-nucleic acid target detection by converting the input target molecule into a sequence-specific output DNA signal.13–16 The output DNA signal can be further amplified by DNA signal amplification methods. The signal translator was recently applied in proteins and small molecular detection with high specificity.17,18
Isothermal amplification has attracted a great deal of research interest because of its simplicity, portability, short assay time, and cost-effectiveness.19,20 Isothermal exponential amplification (EXPAR) is an isothermal nucleic acid amplification technique with excellent amplification efficiency.21,22 However, the single-stranded DNA produced by EXPAR is not only difficult to operate, but also easily leads to a high background signal. Therefore, dual amplification approaches coupled with EXPAR emerged recently for nucleic acid, protein, and small molecular detections. Detection strategies based on EXPAR and the hybridization chain reaction (HCR) were developed for the detection of 17 β-estradiol, BRCA1 gene, influenza A (H7N9) virus DNA and thrombin.23–27 Detection approaches based on EXPAR and catalytic hairpin assembly (CHA) have widely been applied in microRNA and protein assays.17,28
In this work, we developed a one-tube fluorescence strategy based on a protein-to-DNA signal transducer and a dual isothermal nucleic acid amplification for the detection of B7-H3. We employed a protein-to-DNA signal transducer to convert the B7-H3 protein signal into DNA signal. The output DNA was first amplified by EXPAR to generate a great number of ssDNA. The resulting ssDNA was further amplified by D-HCR. All of the reaction components can be completed in one tube. The resulting DNA products can be monitored in real-time fluorescence signaling. This detection method might shorten analysis time, not produce aerosol contamination, and reduce detection costs.
Experimental
Chemicals and materials
Recombinant Human B7 Homolog 3 (B7-H3) protein (CD276), and anti-B7-H3 monoclonal antibody were purchased from Sigma-Aldrich (Steinheim, Germany). Recombinant Human B7-H2, recombinant human B7-H4, recombinant human B7-H5, and recombinant human B7-H6 were obtained from SinoBiological, (Beijing, China). The Bst DNA polymerase, SYBR Green I, and Nb.BbvCI nicking endonuclease were purchased from New England Biolabs (New England, USA). SYBR Green I was purchased from Solarbio (Guangzhou, China), NuSeive GTG agarose was obtained from Lonza (Basel, Switzerland). Enzyme-linked immunosorbent assay (ELISA) kit for soluble B7-H3 detection was purchased from Ruixin Biotechnology, (Quanzhou, China). Other chemicals were purchased from standard commercial sources and were of analytical grade. The 0.22 μm MILLEX®GP filter unit was purchased from Merck Millipore (Merck Millipore, USA). Ultrapure water (18.2 MΩ cm−1) was used to prepare the buffer solution. The oligonucleotide sequences are listed in Table 1.| Oligonucleotide | Sequences |
|---|---|
| a The loop portion of each hairpin is underlined. ▲ Represents the nicking endonuclease site of Nb.BbvCI. | |
| DNA1 (12-nt) | 5′-NH2-C6-TTTTTTTTTTTTTTTTTCGTACGTAGG-3′ |
| DAN2 (12-nt) | 5′-CCTACGTACGAATTTTTTTTTTTTTTT-C6-NH2-3′ |
| DNA3 | 5′-GATACGGC▲TGAGG CCTACGTACGAA-3 |
| Hairpin 1 (H1) |
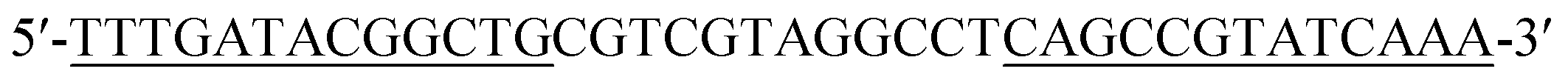
|
| Hairpin probe 2 (DH2) |
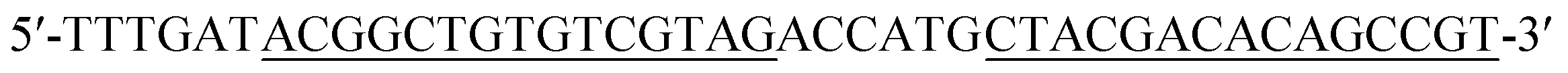
|
| Hairpin probe 3 (DH3) |
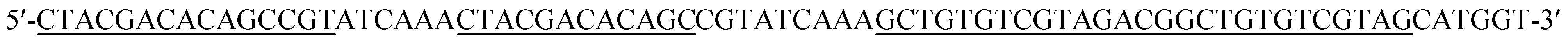
|
Preparation of DNA1/DNA2-B7-H3 antibody conjugates
DNA1/DNA2-B7-H3 antibody conjugates were constructed according to our previous work with minor modifications.17 The amine-modified DNA1 and DNA2 probes were conjugated with anti-B7-H3 monoclonal antibody through the 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide/N-hydroxysuccinimide (EDC/NHS) self-assembled activity, respectively. Firstly, 20.6 mg EDC and 11.5 mg NHS were mixed in 1 mL of water for 10 min. 10 μL of this mixed solution was added to 200 μL solution containing 13.5 mg mL−1 anti-B7-H3 antibody, 10 μM DNA1 or 10 μM DNA2 in carbonate buffer (pH 11.0), and reacted for 2 hours. The solution was then centrifuged for 5 min at 4000 rpm, followed by rinsing for two times with 200 μL of water for 5 min at 4000 rpm. Finally, the supernatant was abandoned and the precipitate was resuspended to a final volume of 200 μL with rCutSmar buffer. Final protein concentrations were determined with a Pierce BCA protein assay kit.Construction of protein-to-DNA signal transducer
The protein-to-DNA signal transducer consisted by anti-B7-H3 antibody-conjugated DNA1, anti-B7-H3 antibody-conjugated DNA2, and signal-output element (DNA3). Briefly, 100 μL rCutSmar buffer containing 2.5 μM anti-B7-H3 antibody-DNA1 and 2 μM DNA3 was heated to 55 °C for 5 min, and allowing the solution to cool down to 25 °C slowly for 2 hours to form anti-B7-H3 antibody conjugated DNA1/DNA3 duplex.Fluorescence detection of B7-H3
Proximity hybridization-mediated isothermal exponential amplification and D-HCR was performed in 100 μL rCutSmar buffer containing 300 nM anti-B7-H3 antibody conjugated DNA1/DNA3 complex, 300 nM anti-B7-H3 antibody conjugated DNA2, 200 nM H1, 30 U Bst polymerase large fragment, 20 U Nb.BbvCI nicking endonuclease, 100 μM dNTPs, 300 nM DH2, 300 nM DH3, 2 μL of 10 × SYBR Green I, and different concentrations of B7-H3 at 37 °C for 120 min. Meanwhile, the fluorescence signal from SYBR Green I staining was monitored at an interval of 2 min by a fluorospectrophotometer FL6500 (PerkinElmer, USA).Gel electrophoresis analysis
Two kinds of agarose gel electrophoresis were used to verify small or large amplification fragments, respectively. For small fragment amplification products detection, 4% GTG agarose gel (3% NuSeive GTG agarose +1% agarose) prepared in 1 × TBE buffer (89 mmol L−1 Tris; 89 mmol L−1 boric acid; 2 mmol L−1 EDTA pH 8.3) at 65 V constant voltage for about 75 min. After the gel was stained for 30 min in a 4S GelRed solution, it was photographed by FluorChem FC3 Chemiluminescent imaging system (ProteinSimple, USA). To analyze large fragments, 2% agarose gel was prepared in 1 × TAE buffer (40 mM Tris-acetate, 1 mM EDTA, pH8.0) at 90 V constant voltage for about 30 min. After the gel was stained for 30 min in a 4 S GelRed solution, it was photographed by FluorChem FC3 Chemiluminescent imaging system (ProteinSimple, USA).Results
One-tube B7-H3 detection
The protein-to-DNA signal transducer (Scheme 1) consists of target-recognition elements (antibody-conjugated DNA1 and antibody-conjugated DNA2) and an output-signal element (DNA3). B7-H3 antibody conjugated DNA1 is firstly hybridized to DNA3 to form a stable DNA1/DNA3 duplex. DNA3 is designed to have a nicking endonuclease recognition site near its 5′ end (green region). DNA2 sequence is designed to be complementary with DNA1 at the 5′ terminus. In the presence of B7-H3, the binding of the same B7-H3 to the two antibodies coupled with DNA1 and DNA2 assembles DNA1 and DNA2 together, resulting in increasing their local effective concentrations. This process raises the melting temperature for intramolecular hybridization (DNA1 and DNA2), favouring the strand displacement reaction between DNA2 and DNA3. As a result, the output DNA3 is released for further isothermal signal amplification.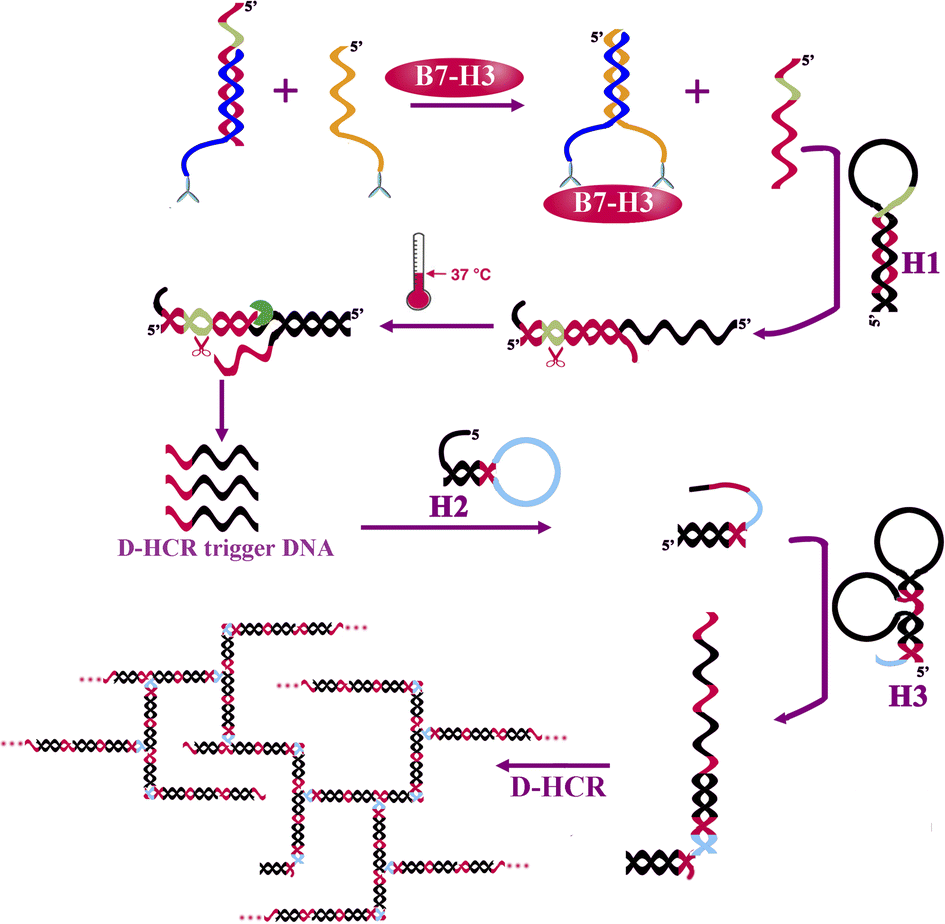 | ||
| Scheme 1 Schematic illustration of the one-tube B7-H3 detection based on protein-to-DNA signal transducer, isothermal exponential amplification, and dendritic hybridization chain reaction. | ||
The releasing output DNA3 was further amplified by EXPAR and DHCR with three kinds of hairpin probes. DNA3 recognizes and partially hybridizes with H1 to form a partial complementary double-stranded DNA containing a complete nicking endonuclease recognition site near its 5′ end. EXPAR is initiated in the presence of nicking endonuclease, polymerase, and dNTPs to produce a large number of single-strand DNA (DHCR trigger DNA) in the circle of nicking, polymerization, and strand displacement. The resulting ssDNA recognizes and partially hybridizes with DH2 causing the hairpin probe to undergo a conformational change and leading to stem separation and forming an DH2-DNA3 intermediate. Then the DH2-DNA3 intermediate catalyzes the dynamic assembly of DH3 to produce H2-H3 duplex accompanying the release of ssDNA. DH3 containing two domains that are complimentary with DH2 catalyzes the dynamic assembly of DH2, forming a dendritic structure for the next cycle for the DH2–DH3 self-assemble reaction. Meanwhile, the releasing ssDAN can further initiate another DHCR reaction for signal amplification. Finally, a great number of dendritic duplex DNA concatemers are synthesized. The amplification products can be detected by a fluorospectrophotometer.
The hairpin probe retains its original stem-loop structure in the absence of B7-H3 protein. Because the hairpin probes were designed to be more stable in hairpin structure than in heterodimer structure. The release of output DNA3 by competing DNA2 and dsDNA products is extremely limited.
The reaction of protein-to-DNA signal transducer, EXPAR, and DHCR can react in one tube.
Feasibility study
In this work, EXPAR and D-HCR were used to amplify the output DNA and produce a large number of dendritic duplex DNA concatemers for B7-H3 detection. We first analyzed the real-time fluorescence response of the 100 ng mL−1 B7-H3, sample without nicking endonuclease, sample without polymerase, and negative control (NC) for the products obtained from the dual isothermal amplification. As presented in Fig. 1(A), the fluorescence signal of B7-H3 was about 10 times higher than NC at 120 min, which indicated dramatic isothermal amplification occurred as expected. The limited fluorescence signal was observed in the sample without nicking endonuclease and the sample without polymerase due to the partially complementary sequences structure of hairpin probes (stem structure of DH2 and DH3).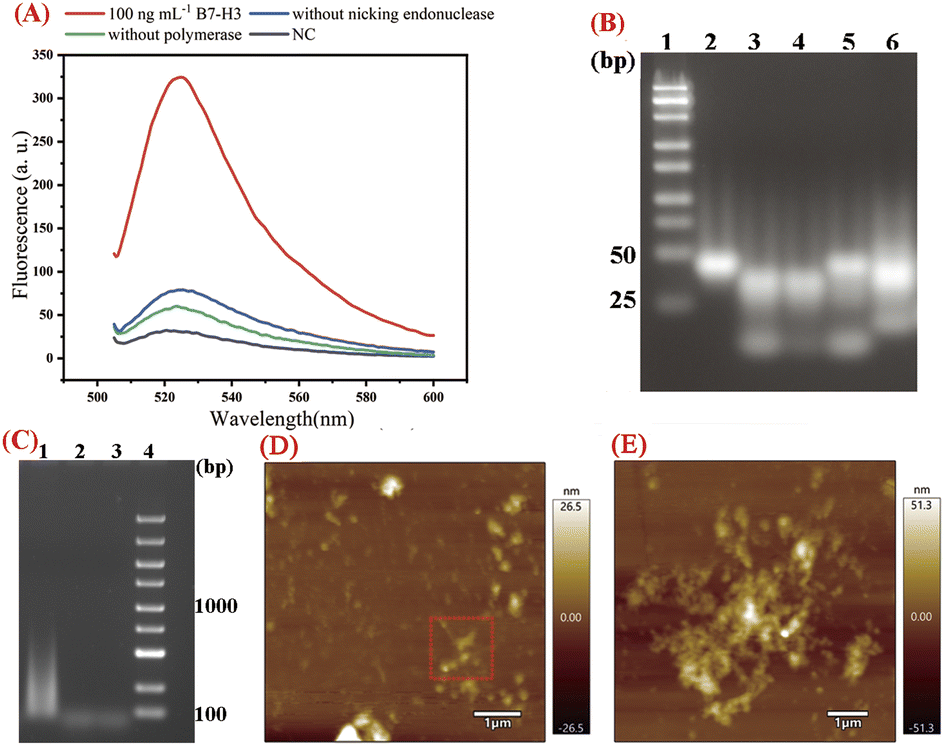 | ||
| Fig. 1 Feasibility evaluation of the one-tube B7-H3 detection based on isothermal exponential amplification and D-HCR. (A) Real-time fluorescence curves during isothermal amplification with 100 ng mL−1 B7-H3, without nicking endonuclease, without polymerase and NC. (B) Agarose gel electrophoresis analysis of isothermal reaction products. 1: DNA marker (25–500 bp). 2: Ab-DNA1 + DNA3, 3: Ab-DNA1 + DNA3 + Ab-DNA2 + target for 120 min, 4: Ab-DNA1 + DNA3 + Ab-DNA2 + H1 + target for EXPAR for 0 min, 5: Ab-DNA1 + DNA3 + Ab-DNA2 + H1 + target for EXPAR for 120 min, 6: all reaction components for 120 min. (C) Agarose gel electrophoresis analysis of DHCR products. 1: all reaction components with 100 ng mL−1 B7-H3, 2. Without DH2, 3. NC. (D) AFM characterization of D-HCR. (E) Enlarged AFM characterization of D-HCR. | ||
We then conducted agarose gel electrophoresis for the reaction products obtained from different stages of isothermal reaction. 4% GTG agarose gel (3% NuSeive GTG agarose +1% agarose) was applied for the analysis of EXPAR reaction products. As shown in Fig. 1(B), the last band at the bottom of lane 3 indicated that DNA3 was released by the hybridization of DNA1 and DNA2. The last band at the bottom of lane 5 displayed the products of EXPAR. The smear-like bands in land 6 indicated the big molecular DNA products of DHCR. 2% agarose gel electrophoresis was used to further verify the reaction products of DHCR. As displayed in Fig. 1(C), a lot of smear bands were observed ranging from 100 bp to 1000 bp in Lane 1, indicating numerous DHCR concatemers were produced during the process of DHCR. No such smear-like bands were observed in Lane 2 and Lane 3.
To verify the D-HCR products more intuitively, AFM characterization was applied to confirm the nanostructure of D-HCR products. As displayed in Fig. 1(D and E), numerous dendritic structures can be seen in the products of D-HCR. These results indicated protein-to-DNA signal transducer, EXPAR, and DHCR work properly as expected for B7-H3 detection.
Optimization of experiment conditions
The performance of our proposed assay is closely related to the concentrations of DH2 and DH3, reaction time, and reaction temperature. The main experiment conditions of this assay were optimized systematically to detect B7-H3 with high sensitivity and selectivity.In this work, the concentrations of DH2 and DH3 were the key factors that affected the analytical performance of the assay because partially complemented stem structures in DH2 and DH3 could produce background fluorescence signals. The concentrations of DH2 and DH3 were studied based on the fluorescence response and F/F0 response toward the detection of 100 ng mL−1 B7-H3 and corresponding negative control (NC). As displayed in Fig. 2(A), the fluorescence intensity at 524 nm increases as the rising of DH2 and DH3 concentrations in the range from 10 nM to 500 nM. The fluorescence response of 500 nM DH2 and DH3 was almost the same as 300 nM DH2 and DH3. However, the fluorescence response of NC with 500 nM DH2 and DH3 is higher than the NC with 300 nM DH2 and DH3 due to increasing the background signal. The F/F0 response of different concentrations of DH2 and DH3 also verified this view (Fig. 2(B)). F/F0 response elevated gradually with the increasing of DH2 and DH3 concentrations ranging from 10 nM to 300 nM. Over 300 nM, the fluorescence response increases very slowly but the background signal increases greatly, leading to a decrease of the F/F0 ratios. Therefore, 300 nM DH2 and DH3 were selected as the optimal concentrations in this assay.
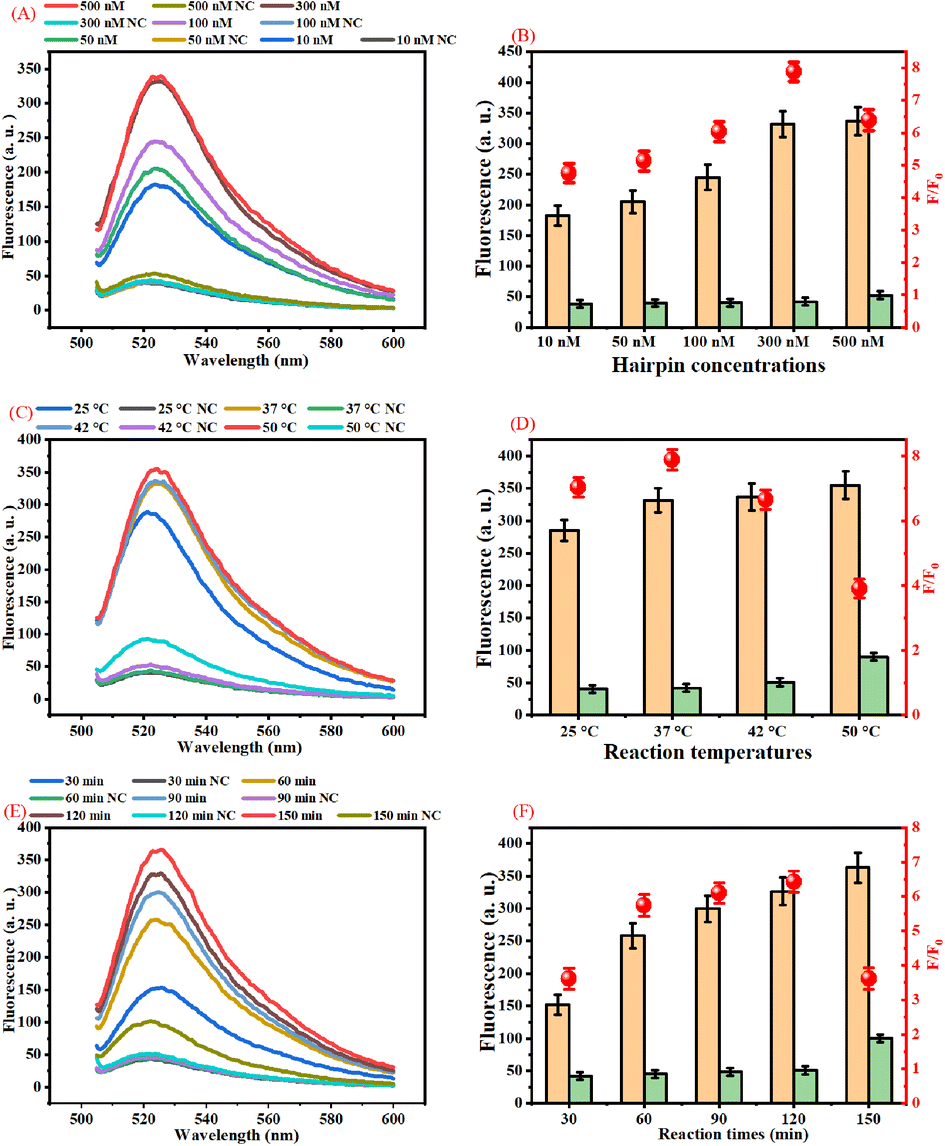 | ||
| Fig. 2 Optimization of the experiment conditions. (A) Real-time fluorescence curves during isothermal amplification with different concentrations of DH2 and DH3. (B) Effect of DH2 and DH3 concentrations on the F/F0 ratio with 100 ng mL−1 B7-H3. (C) Real-time fluorescence curves during isothermal amplification with different reaction temperatures. (D) Effect of reaction temperature on the F/F0 ratio with 100 ng mL−1 B7-H3 of the proposed assay. (E) Real-time fluorescence curves during isothermal amplification with different reaction times. (F) Effect of reaction times on the F/F0 ratio with 100 ng mL−1 B7-H3 of the proposed assay. | ||
The reaction temperature affects the amount of duplex DNA concatemers generated in the presence of B7-H3. As presented in Fig. 2(C), the fluorescence intensity increases as the reaction temperature increases from 25 °C to 37 °C. Over 37 °C the fluorescence intensity of 100 ng mL−1 B7-H3 increases slowly, but the fluorescence intensity of the negative control increased significantly. As shown in Fig. 2(D), the best F/F0 ratio was acquired at 37 °C. Therefore, 37 °C was chosen as the best reaction temperature in this assay.
In this work, the reaction time is one of the most important factors that affect the analytical performance of the assay. Usually, the products of dendritic duplex DNA concatemers increased with the increase of the reaction time due to the increasing the products of DNA3 and DHCR products. As displayed in Fig. 2(E), the fluorescence intensity increases as the reaction time increases from 30 min to 150 min. However, the background of 150 min is higher than 120 min. The F/F0 response of different reaction times also manifests this point. Fig. 2(F) showed that the highest F/F0 ratio was acquired at the reaction time of 120 min. Based on these results, 120 min was employed as the optimal reaction time for this assay.
The lengths of the stem in DH2 and DH3 (Table S1 (ESI)†) were optimized to obtain the best performance. As displayed in Fig. S2 (ESI†), 15 nt of the stem in DH2 and DH3 acquired the highest F/F0 ratio.
Analytical performances
We have examined the sensitivity and dynamic range of the one-tube B7-H3 assay using different concentrations of B7-H3 protein under optimal conditions. As shown in Fig. 3(A), the fluorescence intensity increased when the B7-H3 protein was increased from 10 fg mL−1 to 10 μg mL−1, indicating a great number of dendritic duplex DNA concatemers were produced. According to the fit linear model and equation y = 20.45 + 42.81x, the limit of detection (LOD) is calculated to be 100 fg mL−1 based on three times the standard deviation corresponding to three measurements of negative control samples (NC).29,30 Based on the molecular weight of B7-H3 protein (49.83 kDa), the LOD is calculated to be 0.2 fM. Generally, human basal serum B7-H3 concentrations is about (25.17 ± 8.58) ng mL−1. This one-tube assay holds great promise in monitoring of B7-H3 with its superior detection sensitivity. As illustrated by Fig. 3(B), a good linear relationship between the fluorescence and the logarithm of B7-H3 concentrations was from 100 fg mL−1 to 1 μg mL−1. The sensitivity of our proposed assay is superior to traditional protein methods or protein-to-DNA transducer-based approaches (Table S2 (ESI)†). The significant improvement of the sensitivity can be attributed to (1) exponential isothermal amplification can convert trace target DNA into numerous trigger DNA. (2) trigger DNA can be further amplified by the DHCR.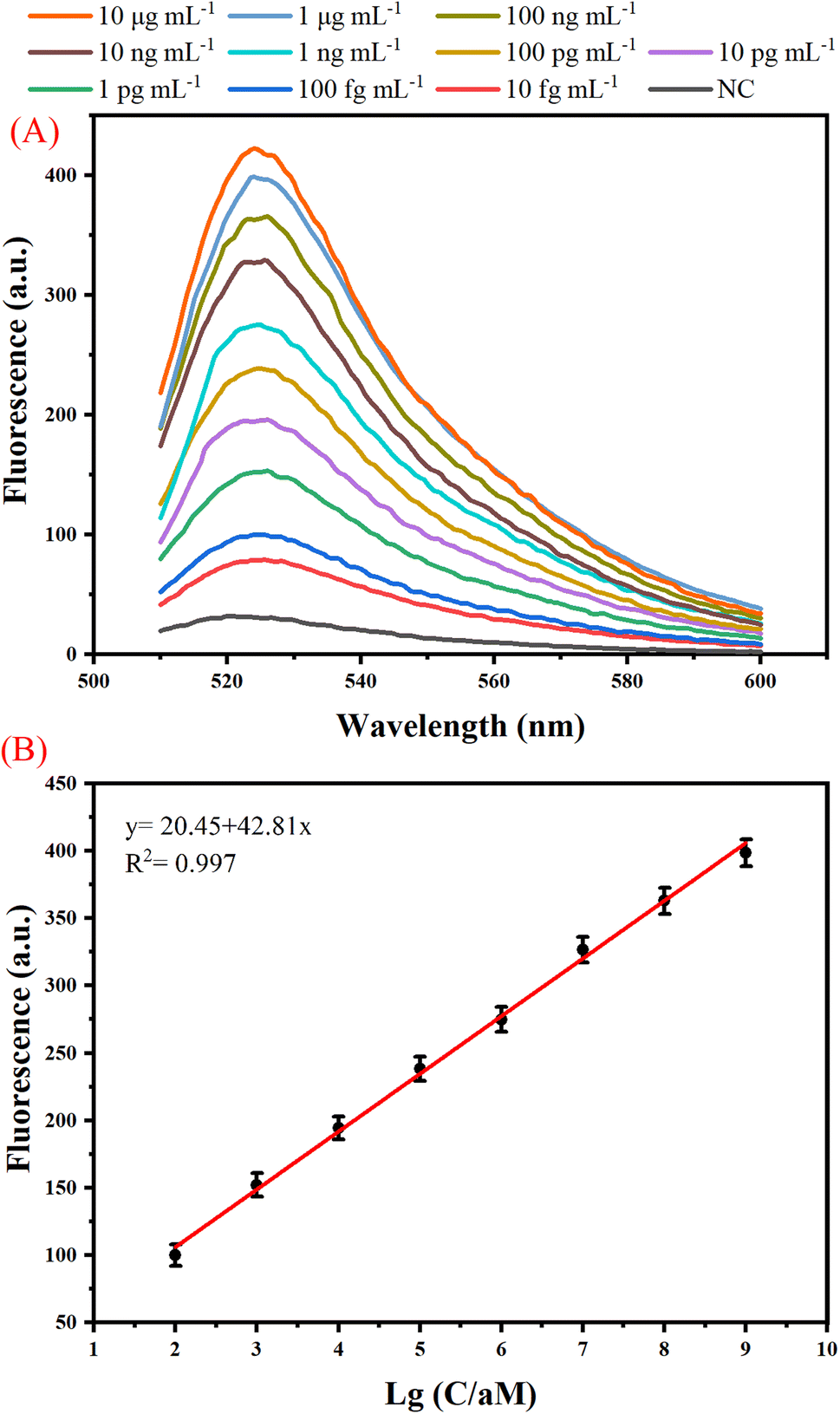 | ||
| Fig. 3 Analytical performances of the proposed one-tube B7-H3 assay. (A) Real-time fluorescence curve detected from the isothermal reaction for target at different concentrations in the range from 10 fg mL−1 to 10 μg mL−1. (B) Relationship between fluorescence and the log of B7-H3 concentrations. Error bars represent standard deviation, n = 3. | ||
The specificity of the one-tube B7-H3 assay was studied by testing the responses of the assay to other kinds of B7 family proteins including B7-H1, B7-H2, B7-H4, B7-H5, and B7-H6 which are similar to B7-H3. As shown in Fig. 4 (A), a distinct enhancement of fluorescence signal was detected only in 100 ng mL−1 B7-H3, whereas all other B7 family proteins at a concentration of 10 μg mL−1 did not yield an obvious fluorescence signal. The photo images and bar chart results also manifested that our proposed B7-H3 detection has good specificity to distinguish targets from similar proteins (Fig. 4(B)). These results indicated that our constructed one-tube B7-H3 approach exhibited excellent selectively responsive to B7-H3.
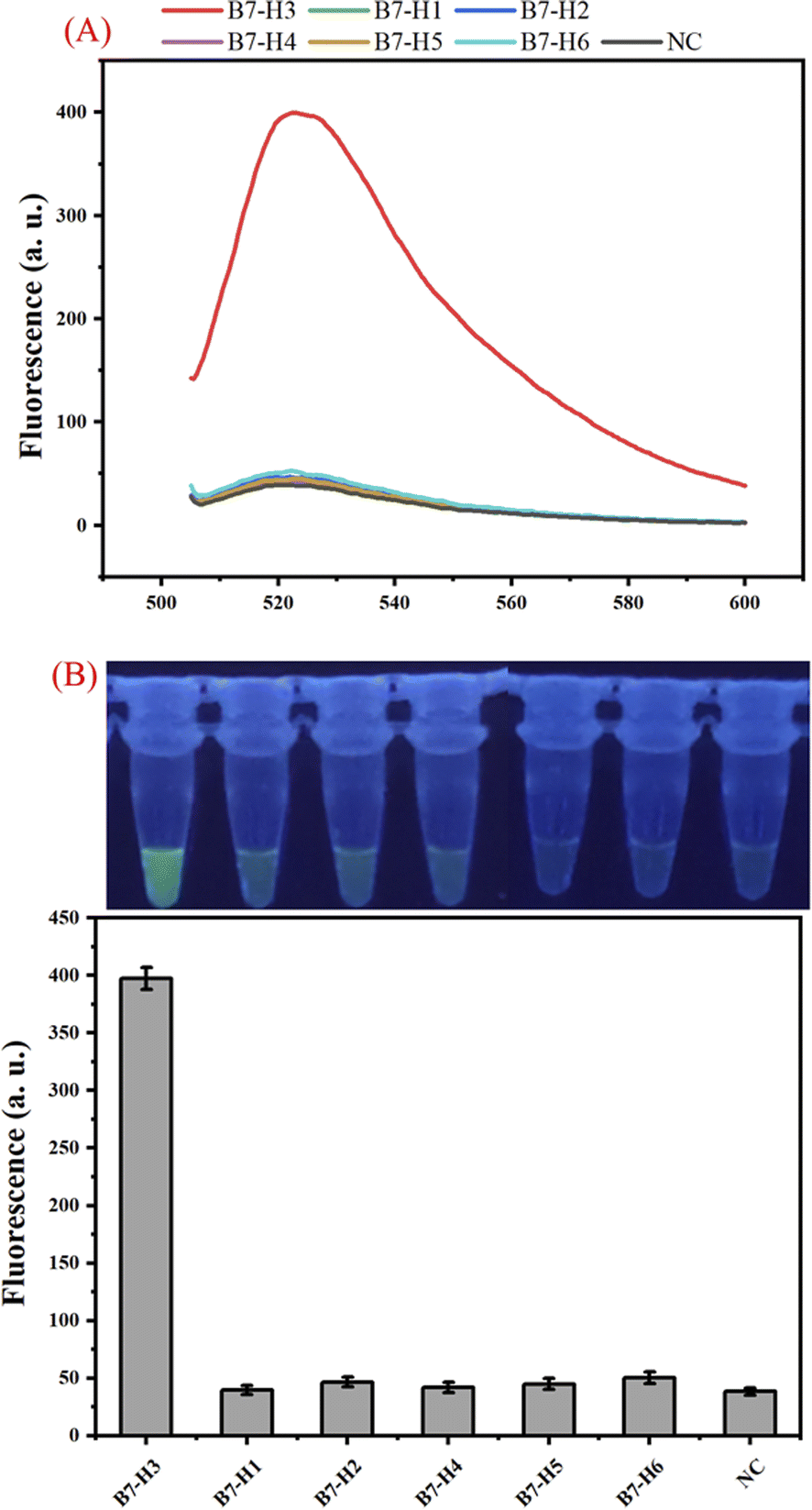 | ||
| Fig. 4 Specificity of the proposed one-tube B7-H3 assay. (A) Real-time fluorescence curve detected from B7-H3, B7-H1, B7-H2, B7-H4, B7-H5, B7-H6, and NC. (B) Photo images and bar chart of the assay for specificity analysis with B7-H3 and other kinds of B7 family proteins which are similar to B7-H3. | ||
To demonstrate whether our developed B7-H3 assay could be applied to real sample analysis, a spiking test was carried out in 20% human serum and saliva samples. Aliquotes of the serum and saliva samples were spiked with different concentrations of B7-H3 protein. As displayed in Table S3 (ESI),† the recovery values and the relative standard deviations were in the range of 90.57–112.2% and 2.62–8.28%, respectively. As displayed in Fig. 5(A), the fluorescence of B7-H3 in serum and saliva samples is the same as the samples in the reaction buffer. These results indicated that this assay has the potential to be applied to detect real samples.
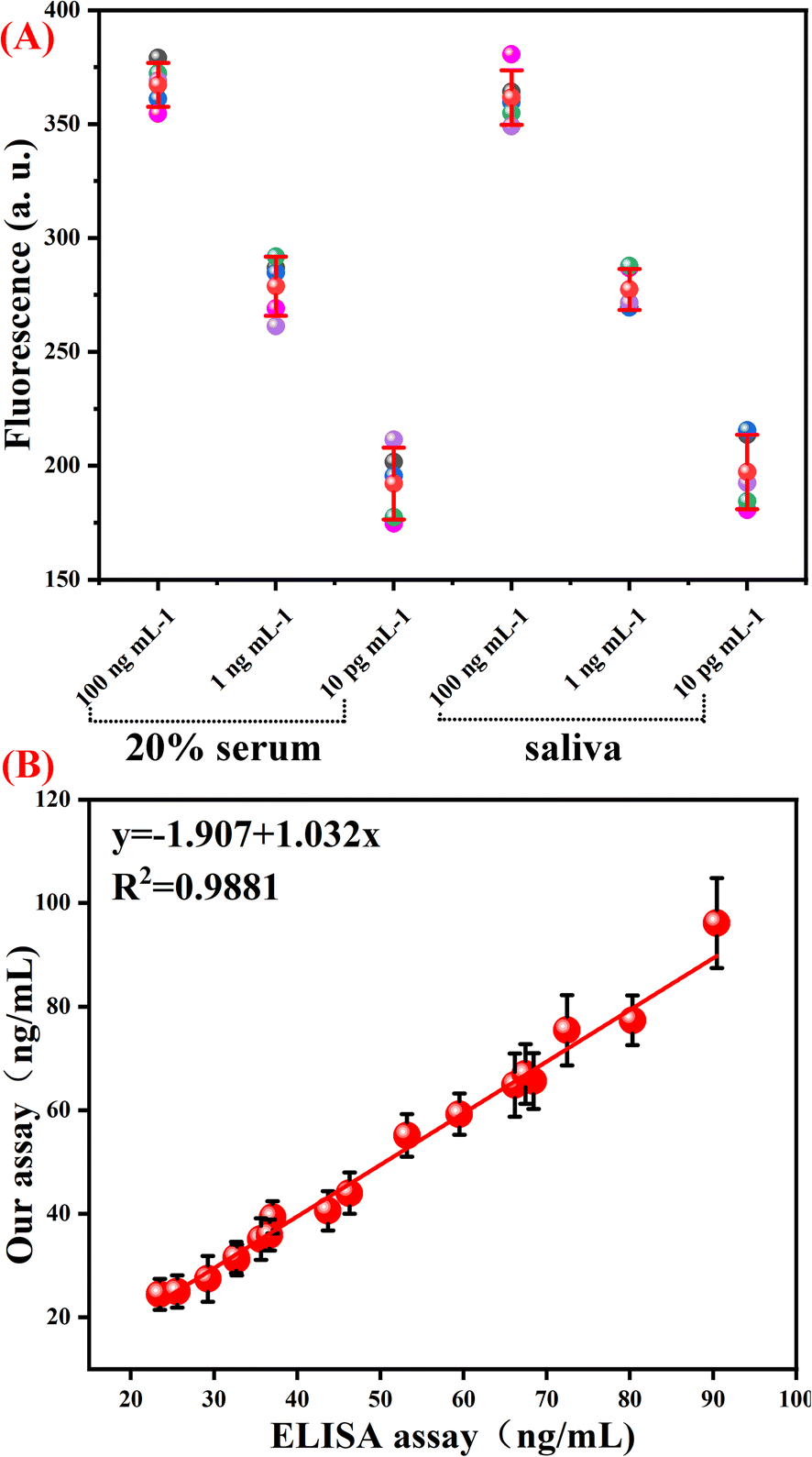 | ||
| Fig. 5 Spiking tests and real sample performance of the proposed B7-H3 assay. (A) The fluorescence response of our B7-H3 assay performed in serum and saliva samples. (B) The scatter plot and linear regression of our proposed one-tube B7-H3 assay and the referencing ELISA method. | ||
Finally, this assay was applied to detect soluble B7-H3 in 18 clinical samples (6 samples of Systemic Lupus Erythematosus (SLE) and 12 samples of non-SLE control). Enzyme-linked immunosorbent assay (ELISA) was employed as a referencing method for soluble B7-H3 detection in clinical serum. As shown in Fig. 5(B), the detection results of our proposed one-tube B7-H3 assay are similar to the referencing method at the same concentration. The correlation coefficient is 0.9981. These results demonstrated that our constructed one-tube B7-H3 assay can be used for clinical sample detection.
The protein-to-DNA signal transducer (anti-B7-H3 antibody conjugated DNA1/DNA3 duplex) and other reaction components were stable within 15 days (Fig. S1, ESI†).
Conclusions
In this word, we have constructed a one-tube B7-H3 assay based on a dual isothermal amplification. A protein-to-DNA signal transducer was employed to convert the B7-H3 signal into output ssDNA. The releasing ssDNA was further amplified by a dual isothermal amplification strategy and detected by a fluorospectrophotometer. Compared to traditional protein detection methods, our proposed B7-H3 approach is more sensitive and convenient. The assay can be conducted in one step within 2 hours. Because this method is a one-tube test, there is no risk of laboratory aerosol contamination. The unit test costs of this assay are comparable to ELISA. By combining these advantages, this proposed sensing method can detect B7-H3 as low as 100 fg mL−1. In addition, this strategy can discriminate B7-H3 from other B7 family proteins. Due to its high sensitivity and specificity, this method performs well in clinical specimen detection. We expect this method can be expanded as a universal sensor platform for various biomolecules by changing the protein-to-DNA transducer.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The work is supported by the Dongguan Science and Technology of Social Development Program (20211800905322).Notes and references
- A. I. Chapoval, J. Ni, J. S. Lau, R. A. Wilcox, D. B. Flies, D. Liu, H. Dong, G. L. Sica, G. Zhu, K. Tamada and L. Chen, Nat. Immunol., 2001, 2, 269–274 CrossRef CAS PubMed.
- S. Yang, W. Wei and Q. Zhao, Int. J. Biol. Sci., 2020, 16, 1767–1773 CrossRef CAS PubMed.
- L. Shao, Q. Yu, R. Xia, J. Zhang, S. Gu, D. Yu and Z. Zhuang, Pathol., Res. Pract., 2021, 224, 153461 CrossRef CAS PubMed.
- J. Sun, H. Lai, D. Shen, P. Wu, J. Yang, Z. Sun and Y. Guo, J. Immunol. Res., 2017, 2017, 5728512 Search PubMed.
- X. Wei, G. Zhang, H. Yuan, X. Ding, S. Li, X. Zhang and J. Hou, J. Urol., 2011, 185, 532–537 CrossRef CAS PubMed.
- G. Zhang, J. Hou, J. Shi, G. Yu, B. Lu and X. Zhang, Immunology, 2008, 123, 538–546 CrossRef CAS PubMed.
- J. Yim, J. Koh, S. Kim, S. G. Song, H. K. Ahn, Y. A. Kim, Y. K. Jeon and D. H. Chung, Eur. J. Cancer, 2020, 133, 74–85 CrossRef CAS PubMed.
- W. Zhang, L. Zhang, J. Qian, J. Lin, Q. Chen, Q. Yuan, J. Zhou, T. Zhang, J. Shi and H. Zhou, Bioengineered, 2021, 12, 11987–12002 CrossRef CAS PubMed.
- Y. Gao, Y. Zeng, X. Liu and D. Tang, Anal. Chem., 2022, 94, 4859–4865 CrossRef CAS PubMed.
- X. Huang, L. Lu, Q. Lin, Q. Wei and D. Tang, Biosens. Bioelectron., 2023, 239, 115608 CrossRef CAS PubMed.
- X. Pei, B. Zhang, J. Tang, B. Liu, W. Lai and D. Tang, Anal. Chim. Acta, 2013, 758, 1–18 CrossRef CAS PubMed.
- R. Zeng, M. Qiu, Q. Wan, Z. Huang, X. Liu, D. Tang and D. Knopp, Anal. Chem., 2022, 94, 15155–15161 CrossRef CAS PubMed.
- F. Li, Y. Lin and X. C. Le, Anal. Chem., 2013, 85, 10835–10841 CrossRef CAS PubMed.
- F. Li, H. Zhang, C. Lai, X. F. Li and X. C. Le, Angew. Chem., Int. Ed., 2012, 51, 9317–9320 CrossRef CAS PubMed.
- F. Li, H. Zhang, Z. Wang, X. Li, X. F. Li and X. C. Le, J. Am. Chem. Soc., 2013, 135, 2443–2446 CrossRef CAS PubMed.
- H. Zhang, F. Li, X. F. Li and X. C. Le, Methods, 2013, 64, 322–330 CrossRef CAS PubMed.
- Z. Tang, W. Zhao, Y. Deng, Y. Sun, C. Qiu, B. Wu, J. Bao, Z. Chen and L. Yu, Analyst, 2022, 147, 1709–1715 RSC.
- Y. Yu, G. Su, H. Zhu, Q. Zhu, Y. Chen, B. Xu, Y. Li and W. Zhang, Int. J. Nanomed., 2017, 12, 5903–5914 CrossRef CAS PubMed.
- R. Zeng, J. Xu, L. Lu, Q. Lin, X. Huang, L. Huang, M. Li and D. Tang, Chem. Commun., 2022, 58, 7562–7565 RSC.
- K. Zhang, S. Lv, Q. Zhou and D. Tang, Sens. Actuators, B, 2020, 307, 127631 CrossRef CAS.
- R. Zeng, L. Zhang, L. Su, Z. Luo, Q. Zhou and D. Tang, Biosens. Bioelectron., 2019, 133, 100–106 CrossRef CAS PubMed.
- H. Gong, X. Hu, R. Zeng, Y. Li, J. Xu, M. Li and D. Tang, Sens. Actuators, B, 2022, 369, 132307 CrossRef CAS.
- Y. Yu, Z. Chen, W. Jian, D. Sun, B. Zhang, X. Li and M. Yao, Biosens. Bioelectron., 2015, 64, 566–571 CrossRef CAS PubMed.
- Z. Tang, W. Zhao, Y. Sun, Y. Deng, J. Bao, C. Qiu, X. Xiao, Y. Xu, Z. Xie, J. Cai, X. Chen, M. Lin, G. Xu, Z. Chen and L. Yu, Langmuir, 2022, 38, 12050–12057 CrossRef CAS PubMed.
- H. Chai, J. Zhu, Z. Guo, Y. Tang and P. Miao, Biosens. Bioelectron., 2023, 220, 114900 CrossRef CAS PubMed.
- Y. Jiang, X. Chen, N. Feng and P. Miao, Anal. Chem., 2022, 94, 14755–14760 CrossRef CAS PubMed.
- P. Miao and Y. Tang, Anal. Chem., 2020, 92, 12700–12709 CrossRef CAS PubMed.
- H. Liu, T. Tian, Y. Zhang, L. Ding, J. Yu and M. Yan, Biosens. Bioelectron., 2017, 89, 710–714 CrossRef PubMed.
- S. Lv, Y. Tang, K. Zhang and D. Tang, Anal. Chem., 2018, 90, 14121–14125 CrossRef CAS PubMed.
- Z. Qiu, J. Shu and D. Tang, Anal. Chem., 2017, 89, 5152–5160 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3na01025b |
| This journal is © The Royal Society of Chemistry 2024 |