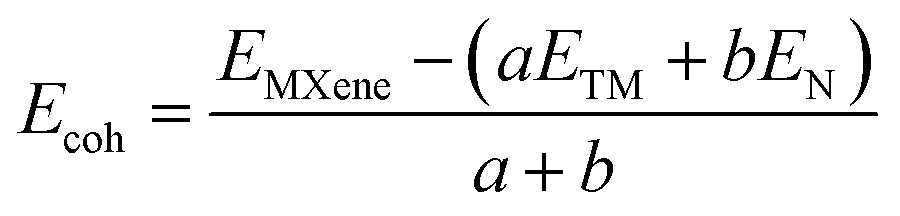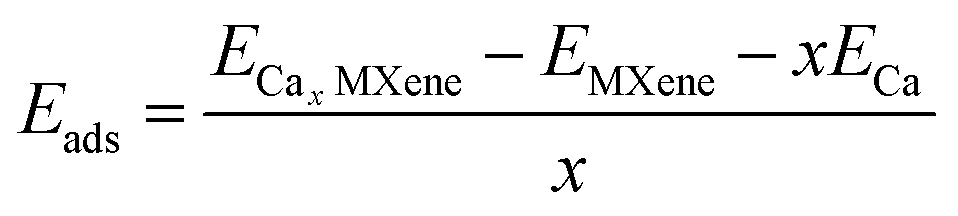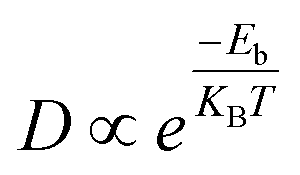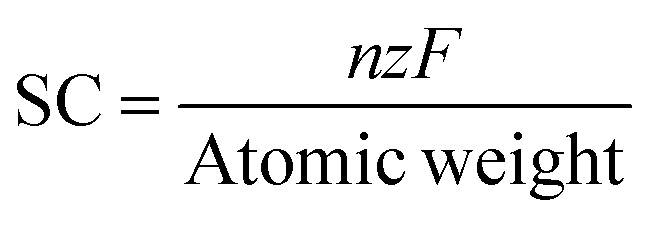DOI:
10.1039/D4NA00140K
(Paper)
Nanoscale Adv., 2024,
6, 3441-3449
Recent advances in Zr and Hf-based MXenes and their hetero-structure as novel anode materials for Ca-ion batteries: theoretical insights from DFT approach†
Received
15th February 2024
, Accepted 16th May 2024
First published on 29th May 2024
Abstract
Recently, MXenes have been widely investigated for use as electrodes in various ion storage batteries. In this study, Zr2N, Hf2N and ZrHfN were explored as potential anode materials for Ca-ion batteries. AIMD simulations predict higher structural stability for our proposed MXenes at a temperature of 300 K. The adsorption energies at the most favourable adsorption sites are 1.31, 1.33 and 1.27 eV for Zr2N, Hf2N and ZrHfN, respectively. During the adsorption process, a significant amount of charge transfer occurs from the Ca atom to the nanosheets. DOS and PDOS analyses reveal that the adsorption of Ca atoms enhances the conductivity of the nanosheets. Moreover, the low diffusion barriers are found to be 0.076, 0.073 and 0.097 eV when the Ca atom migrates from its favourable adsorption site to a nearby site on Zr2N, Hf2N and ZrHfN nanosheets, resulting in high charging rates. The theoretical capacities of Zr2N, Hf2N and ZrHfN nanosheets are 1034, 561 and 707 mA h g−1, respectively. All the results from this study suggest that our proposed nanosheets can be potential anode materials for Ca-ion batteries. Among them, the Zr2N nanosheet shows superior anodic properties for Ca-ion batteries, which is also confirmed by specific capacity, diffusion barrier and open circuit voltage calculations.
1. Introduction
Rechargeable batteries, known as ion storage batteries, play a crucial role in electronic devices by storing energy through reversible ion reduction. These batteries with different configurations are fabricated to store electric energy in various electronic devices. In order to ensure desired performances, steady operational energy sources are typically required in these devices. Long life, compact size, lightweight, high energy density, safety, environmental compatibility, low cost, and widespread consumer availability are the main characteristics of a perfect battery. Most of the ion storage batteries cannot possess the above mentioned properties owing to some limitations and challenges.1 The performance of these batteries relies on the active materials that comprise the cathode, anode, and other integrated components.2 The advancements in ion storage batteries have been made to meet the increasing demand for portable devices.3–5 Recent literature indicates that there has been a significant development in battery technology.6–10
Undoubtedly, lithium-ion batteries (LIBs) are the most popular energy storage system for various electronic devices, electric vehicles, and grid storage applications. Nevertheless, because of their toxicity, safety issues, high cost and low lithium storage capability alternatives to LIBs are being explored.11 To mitigate these kinds of issues, researchers found some earth-abundant and cheap materials such as Ca, Mg, Zn and Al, which are considered to produce highly efficient storage devices.12 Among these materials, Ca has drawn researchers' attention because of its low cost, lightweight, and chemical safety.13 The storage capability of Ca is approximately 1000 times greater than that of Li.14 Thus, the shortage of ion reservation in LIBs can be mitigated by Ca. Although Ca has rarely been employed in ion storage batteries owing to its large ionic radius, the SEP of Ca (−2.87 V) is comparable to Li's (−3.05 V), making it significant for high-voltage battery applications.13 In contrast, electrode materials and the cycling ability of calcium-ion batteries (CIBs) limit its favourable voltage.15 Therefore, finding a compatible anode material with superior energy density, high stability and a faster charging/discharging rate has been a vital step in enhancing the efficiency of CIBs.
Different types of 2D nanomaterials have been explored16–20 for battery applications. However, the performance of the storage devices is still beyond our expectations. The introduction of the transition metal carbides and nitrides termed MXenes provides new insight into various applications.21,22 As a potential storage material, MXenes have sparked significant research interest23 because of their outstanding properties including ultra large interlayer spacing, outstanding electrical conductivity, large surface area, great safety performance and high capacity.24–27 MXenes could be synthesized by selective etching of A atom from the MAX phase with hydrofluoric acid at room temperature.28 The MAX phase (Mn+1AXn) is the parent compound of MXene, where M is an early transition metal, A is an element from group A of the periodic table, X is either carbon or nitrogen and n = 1–3.29 Experimentally, Ti2C, Ti3C2, Ta4C3, Ti3CN, (Ti0.5Nb0.5)2C, (V0.5Cr0.5)3C2, Nb2C and V2C have been synthesized so far.30 Ti2C is a common MXene which is used as an anode material for ion storage devices.31,32 Additionally, Er et al. discovered Ti3C2 as a high-capacity electrode for Li-ion batteries.30 However, the capacity and stability of current MXene electrode materials have fallen short of our requirements. It has been anticipated that MXenes containing other M elements such as Sc, Zr, Hf, and Mo exist.33–35 Without functionalization, nitride MXenes are more stable than their carbide counterparts, which are typically unstable.36 Moreover, N imparts one extra electron to the crystal structure, making nitride-based MXenes more metallic than carbides.37 The preparation of nitride-based MXene has been challenging due to the high formation energy of Mn+1Nn.38 Hence, the synthesis of nitride MXenes still needs a breakthrough. Therefore, it is necessary to investigate nitride-based MXenes comprehensively to find a way out.
In this work, M2N nanosheets, comprising two M (Zr or Hf) atomic layers and one N atomic layer with the pattern of M–N–M, form Zr2N, Hf2N and ZrHfN MXenes. The interaction between Ca and our proposed MXenes was investigated using the density functional theory (DFT). The ab initio molecular dynamics (AIMD) simulation examined the thermal stability of the pristine Zr2N nanosheet. Furthermore, the band structure, density of states (DOS), charge transfer and charge density difference (CDD) maps explained the electronic properties of the nanosheets. Finally, the efficiency of these anode materials was predicted by calculating the diffusion barrier, specific capacity (SC) and open circuit voltage (OCV).
2. Computational details
Density functional theory (DFT) was employed in the Dmol3 module to investigate the anodic behaviour of the materials.16 Within the generalized gradient approximation (GGA), Perdew–Burke–Ernzerhof (PBE) was implemented to explain the exchange-correlation functional of electron–electron interactions.39,40 The long-range dispersion correction proposed by Tkatchenko and Scheffler (TS) was utilized to take into account the van der Waals interaction.41 To describe the core electrons for relativistic effects, DFT semi-core pseudopots (DSPP) were considered and a double-numeric quality basis with a polarization (DNP) function was chosen as a basis set.42 The global orbital cut-off of 5.6 Å was set for better quality. A k-points mesh of 4 × 4 × 1 was applied for geometry optimization. To avoid the interaction between surrounding layers, periodic boundary conditions were implemented with a vacuum distance of 20 Å along the z-direction. Furthermore, the AIMD simulation was performed using a Hoover thermostat and NVT ensemble at 300 k with 10 ps and 2 fs time step to explain thermal stability.43
The cohesive energies of Zr2N, Hf2N and ZrHfN MXenes were measured using the following equation,44
| | 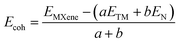 | (1) |
where, the total energy of MXene is defined as
EMXene;
ETM and
EN represent the energy of Zr or Hf and N atoms, respectively;
a and
b are the number of atoms.
To understand the adsorption strength of MXenes, adsorption energy per Ca adatom was calculated using the equation given below,16
| | 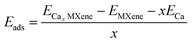 | (2) |
where,
ECax MXene is the total energy of the complex with
x number of Ca;
EMXene is the total energy of MXene;
ECa is the energy of a single Ca of a bulk Ca cluster and
x is the total number of Ca atoms adsorbed in MXene.
The average open circuit voltage (OCV) is a crucial parameter to evaluate the performance of ion batteries. It mainly depends on the anode material and the number of Ca atoms adsorb on it.45 The OCV can be obtained by,46
| | 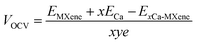 | (3) |
where,
EMXene and
ExCa-MXene represent the total energy of MXene before and after the adsorption of Ca ad atoms;
y represents the valence electron of the Ca atom (here
y = 2).
3. Results and discussion
3.1. Geometry and structural stability
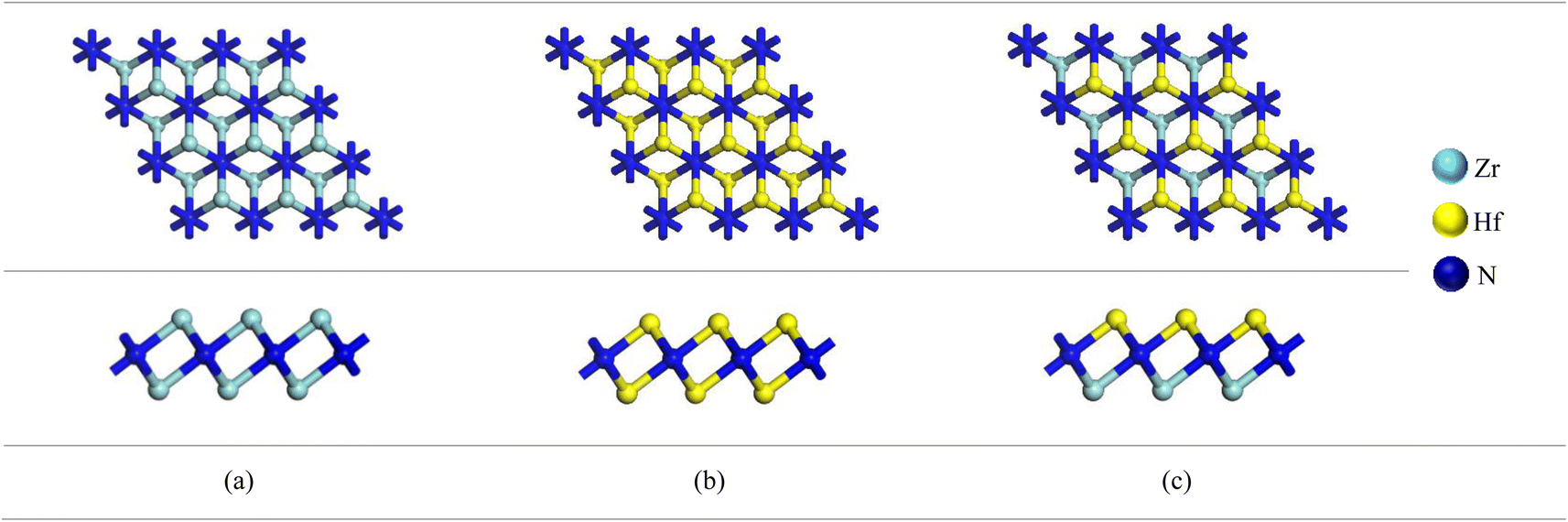
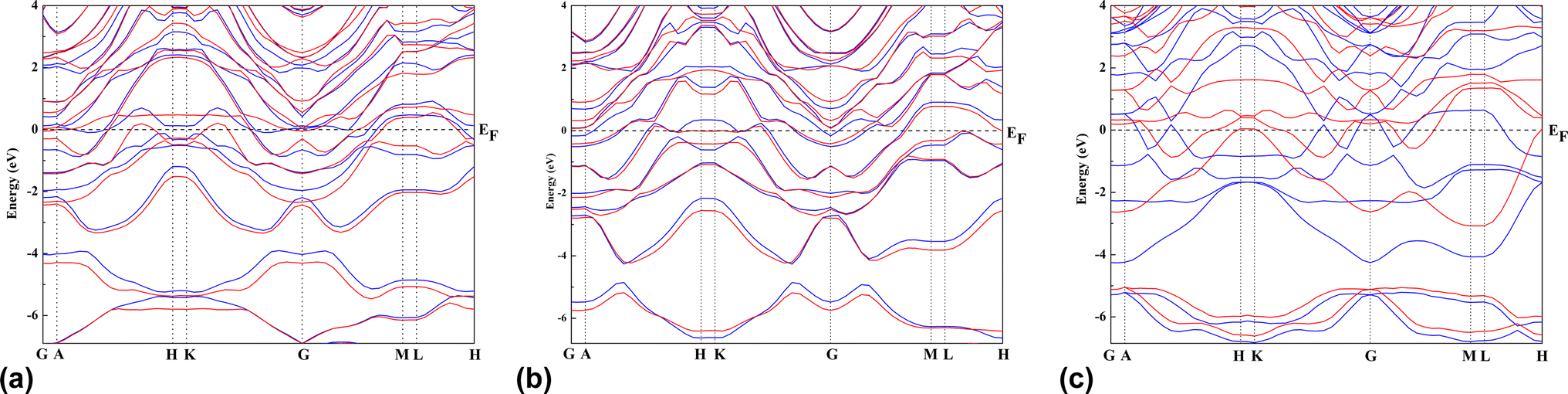
A 3 × 3 × 1 supercell of Zr2N consists of 18 Zr atoms and 16 N atoms. On the other hand, Zr atoms are replaced by Hf and Zr/Hf atoms to form Hf2N and bi-transition metal ZrHfN where the doping ratio is 52.94%. Previously, Zhang et al. investigated bi-transition metal carbide V1/2M1/2C as an anode material for Li-ion batteries.47 In this study, we have chosen Zr2N, Hf2N and ZrHfN nanosheets as anode material for Ca-ion batteries, which have been optimized and shown in Fig. 1. The lattice parameters of these optimized structures are a = b = 9.1137 Å. The bond lengths of Zr–N and Hf–N are 2.206 and 2.204 Å in Zr2N and Hf2N, respectively, while in ZrHfN, the bond lengths are 2.218 and 2.192 Å for Zr–N and Hf–N bonds. The bond lengths vary due to the chemical environment of different atoms. The band structure of a crystal structure explains the allowed energy levels of electrons. As a result, it can easily reveal whether a material is a conductor, semi-conductor, or insulator.48 Since PBE functional underestimates the band gap, the HSE06 hybrid functional was implemented to describe the band structures of Zr2N, Hf2N and ZrHfN nanosheets.49 As stated in Fig. 2, these three nanosheets show metallic behaviour which is consistent with the previous study.50,51 Hence, these nanosheets would be ideal anode materials for rechargeable batteries.
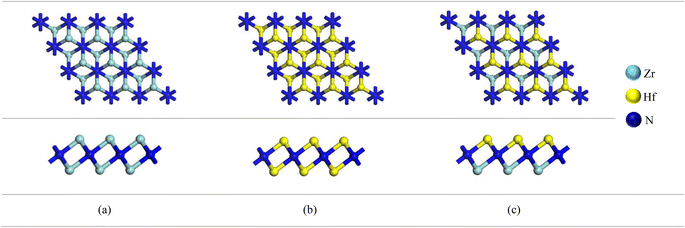 |
| | Fig. 1 The optimized structures of (a) Zr2N, (b) Hf2N and (c) ZrHfN. | |
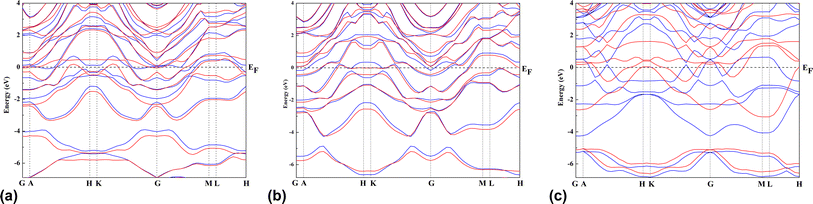 |
| | Fig. 2 The band structures of (a) Zr2N, (b) Hf2N and (c) ZrHfN where blue lines indicate up spin and red lines denote down spin. | |
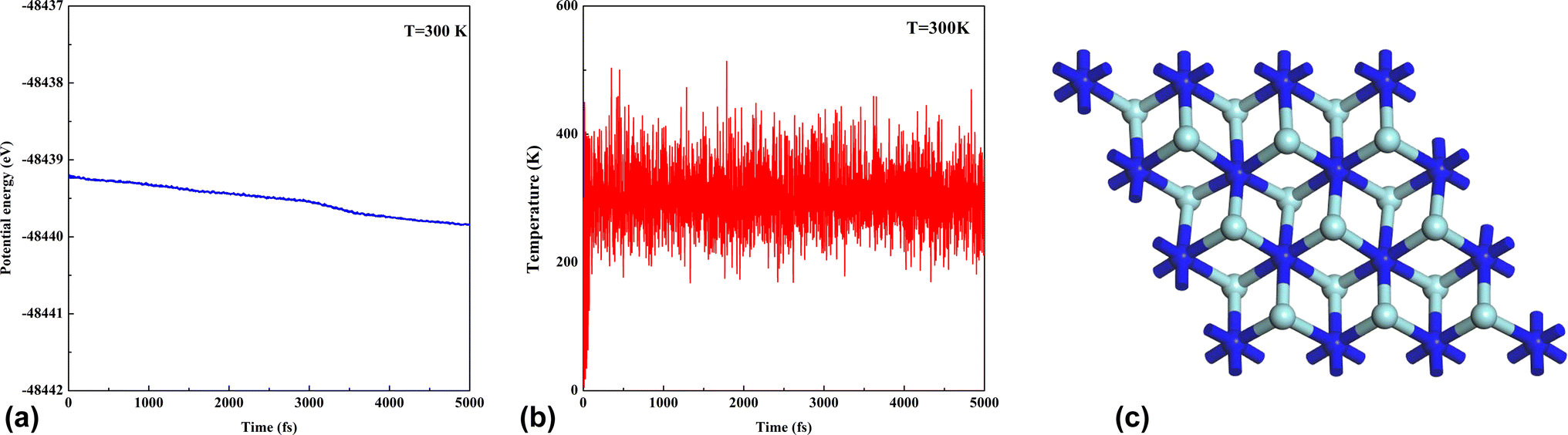
The cohesive energy is the parameter to measure the strength of the bond in the crystal structures. Shein et al.52 found that the cohesive energy of nitride MXenes was lower than that of carbide MXenes. In this study, the cohesive energy of Zr2N, Hf2N and ZrHfN are 0.13, 0.12 and 0.11 eV, respectively. These values of cohesive energy indicate that the Zr2N nanosheet has more structural stability among the three nanosheets. The thermal stability of pristine Zr2N nanosheet was investigated by ab initio molecular dynamics (AIMD) simulation at 300 K temperature. The variation of total energy and the temperature fluctuations of Zr2N are shown in Fig. 3. The AIMD simulation time was set to 2 fs. Fig. 3a depicts that the energy fluctuates with a slight decrease, which confirms the structural stability of Zr2N at 300 K. Moreover, there is no considerable deformation in the atomic structure. This calculation predicts that Zr2N has good thermal stability at the temperature of 300 K, which supports the previous study.53 Since Zr atoms are substituted by Hf and Zr/Hf to form Hf2N and ZrHfN, these two nanosheets would also be thermodynamically stable at this temperature.
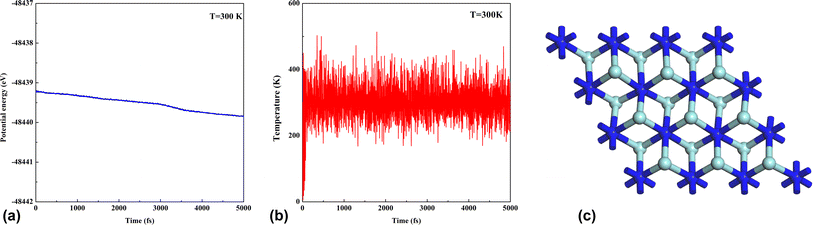 |
| | Fig. 3 The evaluation of thermal stability. The variation in (a) the potential energy, (b) temperature and (c) the structure of pristine Zr2N nanosheet at T = 300 K. | |
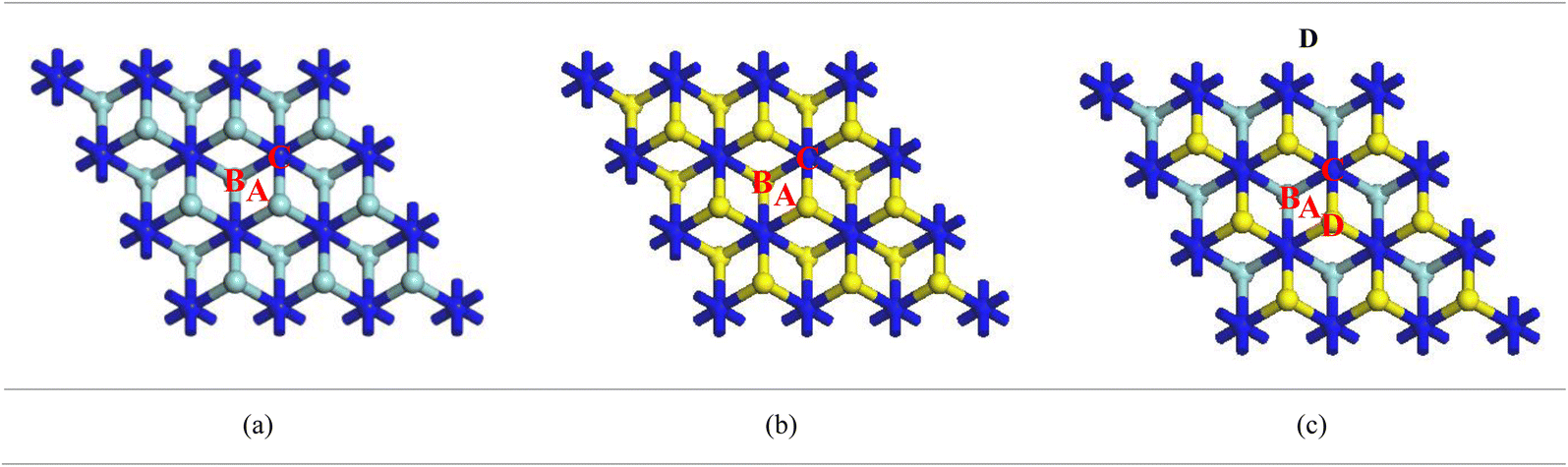
3.2. Adsorption energies at different adsorption sites
Adsorption energy is an important parameter to explain the molecular interaction of a system. Therefore, it is necessary to find the most favourable adsorption site for each nanosheet. Different adsorption sites (center, over Zr, over Hf and over N) of the Ca adatom on MXenes were investigated to explore the suitable adsorption sites and are represented in Fig. 4a–c. As per Table 1, the most favourable adsorption site for a Ca adatom is over Zr in both Zr2N and ZrHfN nanosheets. On the other hand, the Hf2N nanosheet shows better adsorptivity when the Ca atom is placed on the Hf atom. The adsorption energy basically depends on the distance between the adatom and the uppermost layer of the nanosheet.43 The top layers of Zr2N, Hf2N and ZrHfN nanosheets consist of Zr and/or Hf atoms, resulting in higher adsorption energy over Zr and Hf atoms with minimum interaction distance. The minimum distances between Ca adatom and MXenes are represented in Fig. S1.† The adsorption energies of these three nanosheets are negative and maximum energies are found to be −1.31, −1.33 and −1.27 ev for the adsorption of single Ca atom on Zr2N, Hf2N and ZrHfN, respectively. Charge transfer analysis of the system also predicts the superior adsorption behaviour of our proposed MXenes while Ca adatom is over transition metal atom (Zr or Hf). Table 1 shows that at the transition metal sites, a significant amount of charge is transferred during the adsorption of Ca and the calculated values are 1.100, 1.175 and 1.106e for Zr2N, Hf2N and ZrHfN, respectively. The positive value of charge infers that charge is transferred from the Ca atom to nanosheets. Moreover, charge density difference (CDD) maps of these three adsorption systems were investigated to obtain better insight into charge transfer as represented in Fig. S2.† The CDD map gives a visualization of charge transfer using the following equation,54| | | Δρ = ρCa−MXene − ρMXene − ρCa | (4) |
where ρMXene and ρCa represent the electronic charge distribution of MXene and Ca atoms, respectively, and ρCa-MXene denotes the charge distribution of Ca-adsorbed MXene. The red and yellow colours of the CDD map exhibit the accumulation and depletion of electrons, respectively. Looking more closely at the CDD maps, it is observed that charge is transferred from the Ca atom to nearby transition metal atoms of the nanosheet.
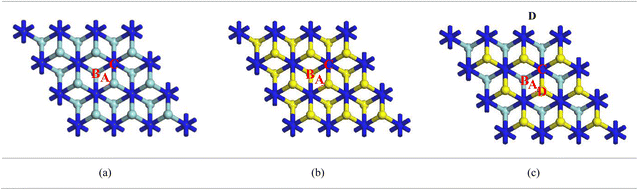 |
| | Fig. 4 Different adsorption sites of the Ca atom on (a) Zr2N, (b) Hf2N and (c) ZrHfN, where A (Center), B (over Zr/Hf), C (over N) and D (over Hf) are the adsorption sites for the Ca atom. | |
Table 1 The calculated adsorption energy (Eads), charge transfer (Q) and distance (d) in different adsorption sites
| Structures |
Adsorption site |
E
ads (eV) |
Q (e) |
d (Å) |
| Ca-Zr2N |
Center |
−1.30 |
1.010 |
3.099 |
| Over Zr |
−1.31 |
1.100 |
3.266 |
| Over N |
−1.27 |
0.996 |
3.227 |
| Ca-Hf2N |
Center |
−1.31 |
1.173 |
2.968 |
| Over Hf |
−1.33 |
1.175 |
3.172 |
| Over N |
−1.31 |
1.162 |
3.183 |
| Ca-ZrHfN |
Center |
−1.25 |
1.100 |
2.931 |
| Over Zr |
−1.27 |
1.106 |
3.211 |
| Over Hf |
−1.23 |
1.067 |
2.894 |
| Over N |
−1.25 |
1.098 |
3.205 |
3.3. Ca adsorption on Zr2N, Hf2N and ZrHfN
After finding a favourable adsorption site for Ca adatoms, the number of Ca atoms was increased on Zr2N, Hf2N and ZrHfN nanosheets. Firstly, adatoms were adsorbed over Zr, Hf and Zr atoms of Zr2N, Hf2N and ZrHfN nanosheets, respectively, as these sites showed maximum adsorption ability (as per the previous section). Secondly, Ca adatoms were distributed on both sides of the nanosheets (when the number of adatoms was more than 10) to mitigate the repulsion tendency. When more than 7 atoms are added to the upper surface and more than 14 atoms to the both sides of the nanosheets, they arrange themselves along different lines on the nanosheets. Ca adatoms distribute on the nanosheets along different lines due to their repulsion. The optimized configurations of nanosheets with Ca adatoms are represented in Fig. S3–S5.† A maximum of 36 Ca atoms were loaded on both sides of the nanosheets without observing any major structural deformation. Since adatoms accumulate on and also leave the nanosheet concerning charging and discharging, structural stability is highly recommended for ion batteries.17
The adsorption behaviour and charge transfer with respect to the number of Ca ions on these three nanosheets were explored and the data are shown in Fig. S6.† According to Fig. S6a,† the adsorption energies exhibit a decreasing trend with ion concentration because extra Ca ions on the nanosheet cause repulsion in this system. This repulsion also affects the favourable adsorption sites of atoms and forces them to rearrange randomly. The calculated adsorption energies were −1.31, −1.33 and −1.27 eV during the single atom adsorption on Zr2N, Hf2N and ZrHfN, respectively. Eventually, these adsorption energies were reduced to −0.53, −0.51 and −0.59 eV while the maximum number of Ca atoms are attached to Zr2N, Hf2N and ZrHfN, respectively. The negative values of the adsorption energy imply a favourable interaction during the adsorption process. Similar to the adsorption energy, charge transfer diminished from 1.100, 1.175 and 1.106e to 0.160, 0.208 and 0.190e for the adsorption of a maximum number of Ca atoms on Zr2N, Hf2N and ZrHfN, respectively [Fig. S6b†]. It is manifested that the maximum charge transfer occurred between Ca atoms and the Hf2N nanosheet.
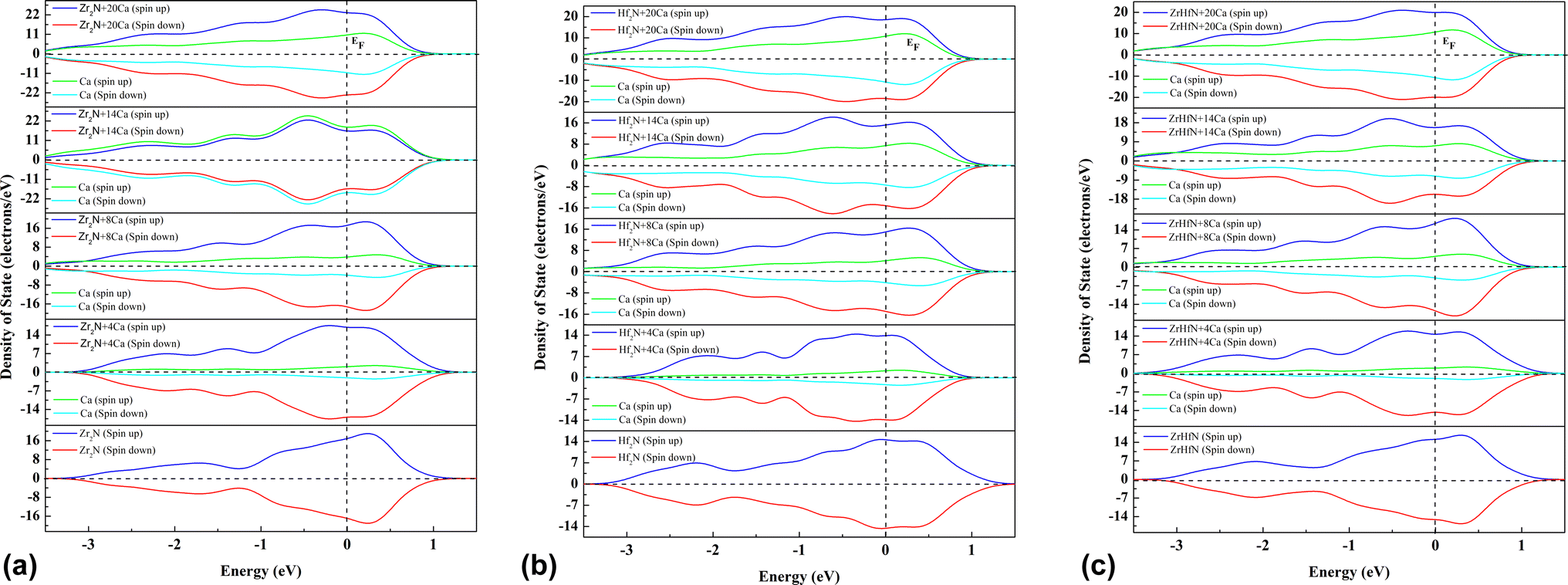
The electronic properties of Zr2N, Hf2N and ZrHfN nanosheets were examined by total density of states (TDOS) and partial density of states (PDOS) with up and down spin. The electrical conductivity of these three nanosheets is explained by looking at the DOS before and after the adsorption of ions. Fig. 5 depicts three nanosheets showing metallic behaviour due to the availability of an occupied state at the Fermi level. This metallic characteristic of nanosheets is crucial for battery applications, as an anode material should be conducted during battery operation. Looking at the DOSs, we can see that extra states are available after the Ca ion adsorption, i.e., this process makes our proposed nanosheets even more conducting, which is favourable for ion batteries. Additionally, extra states in DOS ensure the increased concentration of ca atoms.
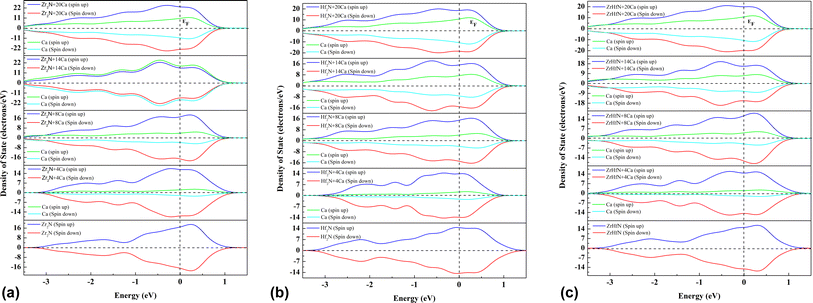 |
| | Fig. 5 Density of states of (a) Zr2N, (b) Hf2N and (c) ZrHfN. Fermi level are at zero energy which is denoted by the dotted line. | |
3.4. Ca ion diffusion on MXenes
Diffusion barrier and migration path are two crucial terms to evaluate the performance of an anode material in an ion storage battery. In this study, the barriers of Ca ions on the surface of Zr2N, Hf2N and ZrHfN were investigated by the LST/QST method. The diffusion barrier of Ca ions on electrode material affects the charging/discharging rate of ion storage batteries. Hence, the charging/discharging rate depends on how fast the Ca ion migrates from one site to another throughout the electrode material. The Arrhenius equation states that the diffusion barrier at constant temperature determines the mobility of metal atoms. The Arrhenius equation is given below,17| | 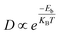 | (5) |
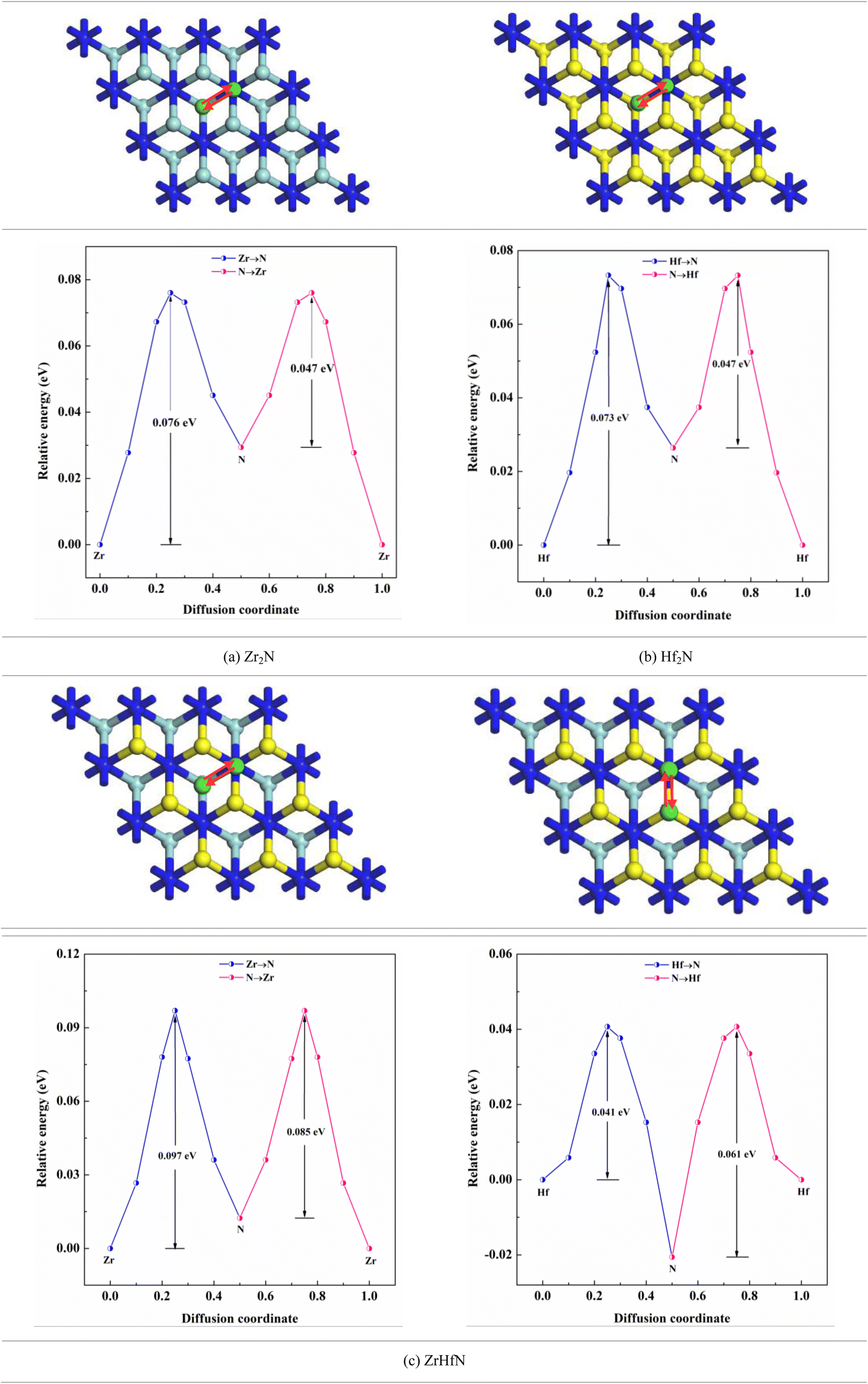
where, Eb, KB and T are the migration barrier, Boltzmann constant and ambient temperature, respectively. According to Fig. 6, two paths were chosen to calculate the energy barrier. For Zr2N and Hf2N, first path is from the transition metal site to the nearest N atom such as Zr → N and Hf → N and the second path is the reverse process; N → Zr and N → Hf. Moreover, for ZrHfN, there were four paths; Zr → N, Hf → N, N → Zr and N → Hf. The energy barriers of Ca atoms along Zr → N and N → Zr are 0.076 and 0.047 eV, respectively, for Zr2N. The migration energy of Ca atoms on Hf2N stands at 0.073 and 0.047 eV along Hf → N and N → Hf, respectively. On the other hand, the Ca atoms diffuse on ZrHfN along Zr → N, Hf → N, N → Zr and N → Hf with the energy barriers of 0.097, 0.041, 0.085 and 0.061 eV, respectively. These values show consistency with the previous theoretical value of nitride MXenes (<0.1 eV)55 and also predict that our proposed MXenes have superior diffusion properties than graphene56 and other covalent organic framework-based materials.57 The smallest diffusion barriers are found when Ca atoms move on Zr2N and Hf2N. Therefore, one of these two nanosheets as an anode material would possess fast charging/discharging properties for CIBs. However, the above data on diffusion barriers reveals that the three nanosheets have better diffusion properties for Ca atoms.
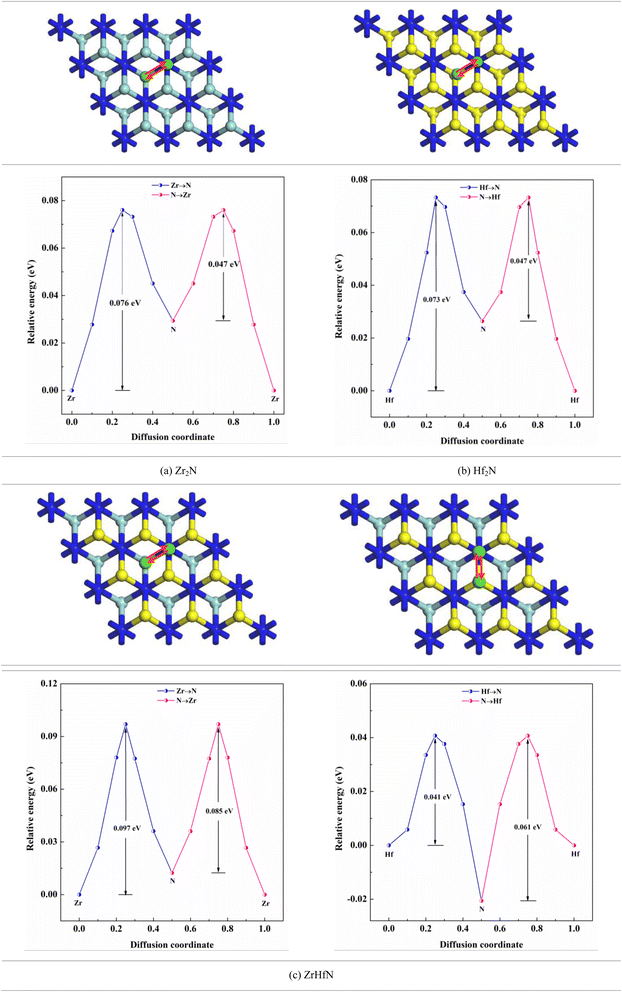 |
| | Fig. 6 Ca ion diffusion through the different paths on the nanosheets. | |
3.5. Specific capacity and OCV
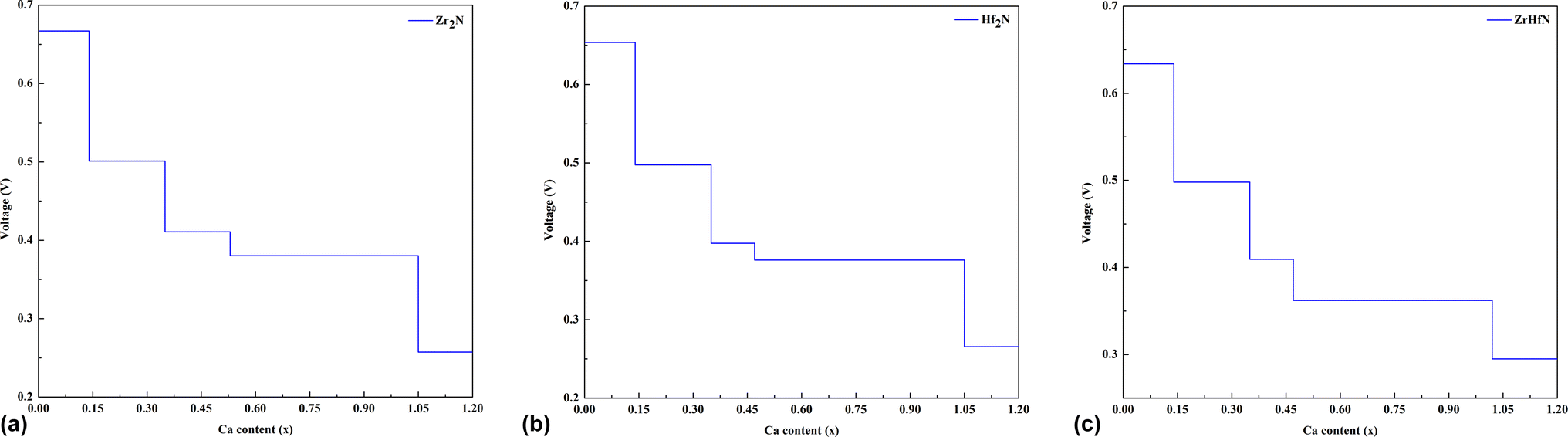
The specific capacity (SC) and the open circuit voltage (OCV) are the two important factors in examining the overall efficiency of an electrode material. The maximum capacity of an anode material has been calculated by the equation given below,58| |  | (6) |
where n and z are the valence electrons of Ca ions and the total number of ions in the electrochemical system and F represents the Faraday constant (96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 C mol−1). The maximum specific capacity depends on the highest number of alkali atoms adsorbed on the nanosheet.59 Moreover, the higher value of the specific capacity of an ion battery infers that the maximum amount of electricity can be supplied during the discharge process. In the present study, Zr2N and Hf2N adsorbed a maximum of 36 Ca atoms, while ZrHfN adsorbed 35 Ca atoms with minimum deformation. The specific capacity values of Ca ion on Zr2N, Hf2N and ZrHfN are 1034, 561 and 707 mA h g−1, respectively. Among the three nanosheets, Zr2N has the highest capacity. It is noticeable that the obtained superior capacity of Zr2N is almost 2 times greater than the previous theoretical capacity (545.9 mA h g−1).60
500 C mol−1). The maximum specific capacity depends on the highest number of alkali atoms adsorbed on the nanosheet.59 Moreover, the higher value of the specific capacity of an ion battery infers that the maximum amount of electricity can be supplied during the discharge process. In the present study, Zr2N and Hf2N adsorbed a maximum of 36 Ca atoms, while ZrHfN adsorbed 35 Ca atoms with minimum deformation. The specific capacity values of Ca ion on Zr2N, Hf2N and ZrHfN are 1034, 561 and 707 mA h g−1, respectively. Among the three nanosheets, Zr2N has the highest capacity. It is noticeable that the obtained superior capacity of Zr2N is almost 2 times greater than the previous theoretical capacity (545.9 mA h g−1).60
The OCV is the electrical potential difference between anode and cathode while no load is connected. The main objective of OCV calculation is to determine the electrical potential capability of an ion storage battery. The calculated OCVs of CIB in terms of the number of Ca ions on Zr2N, Hf2N and ZrHfN are displayed in Fig. 7. For Zr2N, Hf2N and ZrHfN nanosheets, the OCV decreases with increasing Ca concentration. When the ion concentration reaches its peak, the OCV drops to its lowest value. This reduction occurs due to the existence of a large number of Ca ions on the nanosheets, which repel each other. The OCVs for single Ca ion on Zr2N, Hf2N and ZrHfN nanosheets are 0.65, 0.68 and 0.63 eV, respectively. While the nanosheets are able to adsorb the maximum number of Ca atoms (∼36 per nanosheet), the OCVs decrease to 0.27, 0.26, and 0.29 for Zr2N, Hf2N and ZrHfN, respectively. The positive OCV infers that all the structures are favourable as anode materials for Ca-ion batteries. The previous DFT calculation predicted the possible maximum and minimum voltage of Ca-ion batteries, which were 1.20 and 0.57 V, respectively.60 These values confirm the validity of our calculated OCVs for CIB.
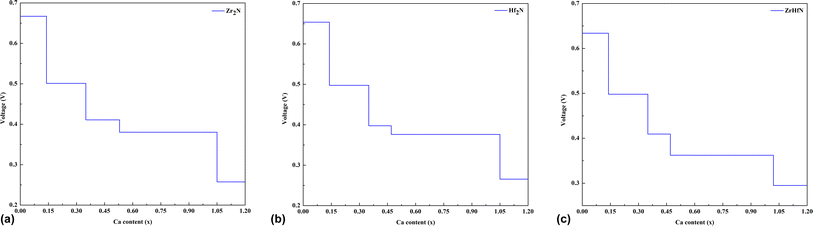 |
| | Fig. 7 The calculated OCV for different numbers of Ca on (a) Zr2N, (b) Hf2N and (c) ZrHfN. | |
4. Conclusions
First-principles calculations were implemented within DFT to investigate the performance of the transition metal nitrides (MXenes) as an anode material for CIBs. In this study, Zr and Hf replaced the transition metal atoms to form Zr2N, Hf2N and ZrHfN MXenes. All these nanosheets show metallic behaviour, which is favourable for ion storage batteries. AIMD calculations confirm that these structures have good thermal stability at 300 K temperature. To explore the adsorption behaviour of Ca atom on Zr2N, Hf2N and ZrHfN, the adsorption energies of these nanosheets were calculated. The most favourable adsorption site was found by adsorbing a single Ca atom in different adsorption sites which was over the transition metal atom (Zr or Hf) of the nanosheets. The adsorption energies at the most favourable adsorption site are −1.31, −1.33 and −1.27 eV for Zr2N, Hf2N and ZrHfN, respectively. The diffusion barrier of Zr2N, Hf2N and ZrHfN nanosheets are 0.076/0.047 along Zr → N/N → Zr, 0.073/0.047 along Hf → N/N → Hf and 0.097/0.041 and 0.085/0.061 eV along Zr → N/Hf → N and N → Zr/N → Hf, respectively. Ca atom showed ultrafast migration barrier on these nanosheets. Moreover, specific capacity was calculated as 1034, 561 and 707 mA h g−1 for Zr2N, Hf2N and ZrHfN, respectively. The results of this study infer that our proposed nanosheets would be the superior anode materials for CIBs as well as other ion storage batteries due to their promising properties. However, the Zr2N nanosheet has better characteristics as an anode material for CIBs.
Author contributions
Tanvir Ahmed: data curation; formal analysis; writing – original draft. Afiya Akter Piya: writing – review and editing (equal). Siraj Ud Daula Shamim: conceptualization (equal); investigation (equal); methodology (equal); software (equal); supervision (equal); validation (equal).
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thankfully acknowledge the Research Cell of Mawlana Bhashani Science and Technology University funded by UGC of Bangladesh (Grant No. 3631108) under the Ministry of Science and Technology (MOST) and the Bangladesh Research and Education Network (BdREN) for their computational access.
References
- Y. Liang, C. Z. Zhao, H. Yuan, Y. Chen, W. Zhang, J. Q. Huang, D. Yu, Y. Liu, M. M. Titirici, Y. L. Chueh, H. Yu and Q. Zhang, InfoMat, 2019, 6–32 CrossRef.
- F. Cheng, J. Liang, Z. Tao and J. Chen, Adv. Mater., 2011, 23(15), 1695–1715, DOI:10.1002/adma.201003587.
- Z. Liu, S. Yang, B. Sun, X. Chang, J. Zheng and X. Li, Angew. Chem., Int. Ed., 2018, 57(32), 10187–10191, DOI:10.1002/anie.201805468.
- Y. Li, H. Xu, P. H. Chien, N. Wu, S. Xin, L. Xue, K. Park, Y. Y. Hu and J. B. Goodenough, Angew. Chem., Int. Ed., 2018, 57(28), 8587–8591, DOI:10.1002/anie.201804114.
- J. Xie, H. J. Peng, J. Q. Huang, W. T. Xu, X. Chen and Q. Zhang, Angew. Chem., Int. Ed., 2017, 129, 16441–16445, DOI:10.1002/anie.201710025.
- W. Chen, Y. Jin, J. Zhao, N. Liu and Y. Cui, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 11694–11699, DOI:10.1073/pnas.1809344115.
- T. Chen, Z. Zhang, B. Cheng, R. Chen, Y. Hu, L. Ma, G. Zhu, J. Liu and Z. Jin, J. Am. Chem. Soc., 2017, 139, 12710–12715, DOI:10.1021/jacs.7b06973.
- J. Le Shi, C. Tang, J. Q. Huang, W. Zhu and Q. Zhang, J. Energy Chem., 2018, 27, 167–175, DOI:10.1016/j.jechem.2017.09.014.
- X. B. Cheng, C. Yan, X. Q. Zhang, H. Liu and Q. Zhang, ACS Energy Lett., 2018, 3, 1564–1570 CrossRef CAS.
- K. Y. Zhang, Y. Q. Fu, H. H. Liu, J. L. Yang, M. Y. Su, Y. Wang and X. L. Wu, Phys. Scr., 2023, 98, 12 Search PubMed.
- X. Deng, L. Li, G. Zhang, X. Zhao, J. Hao, C. Han and B. Li, Energy Storage Mater., 2022, 467–481 CrossRef CAS.
- Y. C. Rao, S. Yu, X. Gu and X. M. Duan, Appl. Surf. Sci., 2019, 479, 64–69 CrossRef CAS.
- M. Hayashi, H. Arai, H. Ohtsuka and Y. Sakurai, J. Power Sources, 2003, 617–620 CrossRef CAS.
- J. M. Newhouse, S. Poizeau, H. Kim, B. L. Spatocco and D. R. Sadoway, Electrochim. Acta, 2013, 91, 293–301, DOI:10.1016/j.electacta.2012.11.063.
- X. Han, C. Liu, J. Sun, A. D. Sendek and W. Yang, RSC Adv., 2018, 8, 7196–7204, 10.1039/c7ra12400g.
- S. U. Daula Shamim, M. K. Hossain, S. M. Hasan, A. Hossain and F. Ahmed, Mol. Simul., 2020, 46, 1135–1145 CrossRef CAS.
- S. U. D. Shamim, M. K. Hossain, S. M. Hasan, A. A. Piya, M. S. Rahman, M. A. Hossain and F. Ahmed, Appl. Surf. Sci., 2022, 579, 152147, DOI:10.1016/j.apsusc.2021.152147.
- Z. Xu, X. Lv, J. Chen, L. Jiang, Y. Lai and J. Li, Phys. Chem. Chem. Phys., 2017, 19, 7807–7819, 10.1039/c7cp00064b.
- S. S. Rifah, M. S. Zaman, A. A. Piya and S. U. D. Shamim, Phys. Chem. Chem. Phys., 2024, 26, 6667–6677, 10.1039/D3CP04532C.
- X.-T. Zhao, J.-Z. Guo, W.-L. Li, J.-P. Zhang and X.-L. Wu, Chinese Chem. Lett., 2024, 35, 108715, DOI:10.1016/j.cclet.2023.108715.
- M. Naguib and Y. Gogotsi, Acc. Chem. Res., 2015, 48, 128–135, DOI:10.1021/ar500346b.
- J. M. Cao, I. V. Zatovsky, Z. Y. Gu, J. L. Yang, X. X. Zhao, J. Z. Guo, H. Xu and X. L. Wu, Prog. Mater. Sci., 2023, 135, 101105 CrossRef CAS.
- W. Liu, J. Cao, F. Song, D. D. Zhang, M. Okhawilai, J. Yi, J. Q. Qin and X. Y. Zhang, Rare Met., 2023, 42, 100–110, DOI:10.1007/s12598-022-02120-z.
- M. K. Aslam and M. Xu, Nanoscale, 2020, 15993–16007 RSC.
- B. Anasori, M. R. Lukatskaya and Y. Gogotsi, Nat. Rev. Mater., 2017, 1–17 Search PubMed.
- F. Bu, M. M. Zagho, Y. Ibrahim, B. Ma, A. Elzatahry and D. Zhao, Nano Today, 2020, 30, 100803 CrossRef CAS.
- R. Fang, C. Lu, A. Chen, K. Wang, H. Huang, Y. Gan, C. Liang, J. Zhang, X. Tao, Y. Xia and W. Zhang, ChemSusChem, 2020, 1409–1419 CrossRef CAS PubMed.
- D. Er, J. Li, M. Naguib, Y. Gogotsi and V. B. Shenoy, ACS Appl. Mater. Interfaces, 2014, 6, 11173–11179, DOI:10.1021/am501144q.
- B. Anasori, Y. Xie, M. Beidaghi, J. Lu, B. C. Hosler, L. Hultman, P. R. C. Kent, Y. Gogotsi and M. W. Barsoum, ACS Nano, 2015, 9(10), 9507–9516, DOI:10.1021/acsnano.5b03591.
- D. Er, J. Li, M. Naguib, Y. Gogotsi and V. B. Shenoy, ACS Appl. Mater. Interfaces, 2014, 6, 11173–11179 CrossRef CAS PubMed.
- Q. Wan, S. Li and J. B. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 6369–6377 CrossRef CAS PubMed.
- S. Das, S. U. D. Shamim, M. K. Hossain, F. Ahmed, M. A. Hossain and M. O. Rahman, Appl. Surf. Sci., 2022, 600, 154173, DOI:10.1016/j.apsusc.2022.154173.
- M. Kurtoglu, M. Naguib, Y. Gogotsi and M. W. Barsoum, MRS Commun., 2012, 2, 133–137, DOI:10.1557/mrc.2012.25.
- Q. Hu, H. Wang, Q. Wu, X. Ye, A. Zhou, D. Sun, L. Wang, B. Liu and J. He, Int. J. Hydrogen Energy, 2014, 39(20), 10606–10612, DOI:10.1016/j.ijhydene.2014.05.037.
- M. Khazaei, M. Arai, T. Sasaki, C. Y. Chung, N. S. Venkataramanan, M. Estili, Y. Sakka and Y. Kawazoe, Adv. Funct. Mater., 2013, 23, 2185–2192 CrossRef CAS.
- Z. Xia, Q. Huang and S. Guo, FlatChem, 2019, 100129 CrossRef CAS.
- J. C. Lei, X. Zhang and Z. Zhou, Front. Phys., 2015, 276–286 CrossRef.
-
P. Urbankowski, B. Anasori, T. Makaryan, D. Er, S. Kota, P. L. Walsh, M. Zhao, V. B. Shenoy, M. W. Barsoum and Y. Gogotsi, MXenes From Discovery to Applications of Two-Dimensional Metal Carbides and Nitrides, 2023, pp. 79–94 Search PubMed.
- S. P. Huang, J. Zhang, Y. R. Ren and W. K. Chen, Appl. Surf. Sci., 2021, 569, 151002, DOI:10.1016/j.apsusc.2021.151002.
- S. Nyamdelger, T. Ochirkhuyag, D. Sangaa and D. Odkhuu, Phys. Chem. Chem. Phys., 2020, 22(10), 5807–5818, 10.1039/c9cp06472a.
- K. C. Wasalathilake, G. A. Ayoko and C. Yan, Carbon, 2018, 140, 276–285 CrossRef CAS.
- X. Wang, Y. Cai, S. Wu and B. Li, Appl. Surf. Sci., 2020, 525, 146501, DOI:10.1016/j.apsusc.2020.146501.
- K. Fan, Y. Ying, X. Li, X. Luo and H. Huang, J. Phys. Chem. C, 2019, 123(30), 18207–18214, DOI:10.1021/acs.jpcc.9b03963.
- J. Mawwa, S. U. D. Shamim, S. Khanom, M. K. Hossain and F. Ahmed, RSC Adv., 2021, 11, 32810–32823 RSC.
- L. Pei, T. Wang, R. Lu and C. Zhu, J. Power Sources, 2014, 253, 412–418, DOI:10.1016/j.jpowsour.2013.12.083.
- X. Zhang, J. Hu, Y. Cheng, H. Y. Yang, Y. Yao and S. A. Yang, Nanoscale, 2016, 8(33), 15340–15347, 10.1039/c6nr04186h.
- B. Zhang, W. Zhang, Q. Meng, L. Fan and Q. Zhang, Phys. Chem. Chem. Phys., 2019, 21(3), 1606–1613, 10.1039/c8cp06487c.
-
L. K. Lamontagne, Lect. Notes Search PubMed.
- Q. He, B. Yu, Z. Li and Y. Zhao, Energy Environ. Mater., 2019, 12979–12986 Search PubMed.
- Y. Yue, B. Wang, N. Miao, C. Jiang, H. Lu, B. Zhang, Y. Wu, J. Ren and M. Wang, Ceram. Int., 2021, 47(2), 2367–2373, DOI:10.1016/j.ceramint.2020.09.079.
- T. Jing, D. Liang, J. Hao, M. Deng and S. Cai, Phys. Chem. Chem. Phys., 2019, 21(10), 5394–5401, 10.1039/c9cp00028c.
- I. R. Shein and A. L. Ivanovskii, Comput. Mater. Sci., 2012, 65, 104–114 CrossRef CAS.
- G. Liu, S. Xu, L. Wu, J. Zhang, Q. Wang and P. Lu, Mater. Chem. Phys., 2020, 250, 123028, DOI:10.1016/j.matchemphys.2020.123028.
- S. U. D. Shamim, D. Roy, S. Alam, A. A. Piya, M. S. Rahman, M. K. Hossain and F. Ahmed, Appl. Surf. Sci., 2022, 596, 153603 CrossRef CAS.
- X. Zhao, P. Wang, E. Lv, C. Wu, K. Ma, Z. Gao, I. D. Gates and W. Yang, Appl. Surf. Sci., 2021, 569, 151050, DOI:10.1016/j.apsusc.2021.151050.
- D. Odkhuu, D. H. Jung, H. Lee, S. S. Han, S. H. Choi, R. S. Ruoff and N. Park, Carbon, 2014, 66, 39–47, DOI:10.1016/j.carbon.2013.08.033.
- B. Ball and P. Sarkar, J. Phys. Chem. C, 2020, 124(29), 15870–15878, DOI:10.1021/acs.jpcc.0c02770.
- J. Yang, J. Wang, X. Dong, L. Zhu, D. Hou, W. Zeng and J. Wang, Appl. Surf. Sci., 2021, 544, 148775, DOI:10.1016/j.apsusc.2020.148775.
- B. Tian, T. Huang, J. Guo, H. Shu, Y. Wang and J. Dai, Mater. Sci. Semicond. Process., 2020, 109, 104946, DOI:10.1016/j.mssp.2020.104946.
- A. Ponrouch, D. Tchitchekova, C. Frontera, F. Bardé, M. E. A. De Dompablo and M. R. Palacín, Electrochem. commun., 2016, 66, 75–78, DOI:10.1016/j.elecom.2016.03.004.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence and
Siraj Ud
Daula Shamim
and
Siraj Ud
Daula Shamim