DOI:
10.1039/D4NA00023D
(Paper)
Nanoscale Adv., 2024,
6, 3426-3440
Hierarchical hollow porous structures of nickel-doped λ-MnO2 anodes for Li-ion energy storage systems†
Received
8th January 2024
, Accepted 13th May 2024
First published on 14th May 2024
Abstract
Metal oxides with hollow porous structures are attractive and promising anode candidates for Li-ion batteries due to their high surface area, high loading capacity, and low density. In this work, hierarchical hollow porous structures of nickel (Ni)-doped λ-MnO2 were prepared via a facile, and cost-effective approach, where different amounts of Ni were introduced into MnO2 structures to tailor their physical and chemical properties. When the prepared Ni-doped MnO2 hollow structures were studied as anode materials for Li-ion batteries, the electrode showed excellent electrochemical properties, such as stable cyclability and admirable rate capability. Moreover, Ni doping significantly enhances the diffusion properties of the active materials. The material was also investigated as an anode in another high power and energy Li-ion storage device, namely, a Li-ion hybrid capacitor, which exhibited excellent comprehensive electrochemical performance in terms of good specific cell capacity of 25 mA h g−1 at a high current density of 5 A g−1 and achieved a maximum power density of 29 W kg−1 (with energy density of 30 W h kg−1) with a long cycle life. These results indicate that the Ni-doped MnO2 is suitable for application as an anode material and give considerable insight into future Li-energy storage applications.
1. Introduction
Li-ion rechargeable batteries (LIBs) are one of the common sources in the quest to meet the ever-growing demand for high energy and power density applications owing to their long stability, light weight, and safety characteristics, which are favorable for portable electronic devices such as laptops, smartphones, and tablets.1–3 The overall performance of the battery is influenced by the electrode materials, hence the physical and chemical properties of the anode material play pivotal roles in delivering high specific capacities. In general, commercial graphite is used as an anode for LIBs owing to its low cost, structural stability, and long-term stable cycle performance. However, the low theoretical capacity of graphite (372 mA h g−1) cannot meet the requirement of high energy and high power densities. Consequently, alternative anode materials with high theoretical capacity have been explored, such as Si, Sn, Al, Ge, metal oxides (MnO2, Co3O4, Fe2O3, NiO, etc.), metal nitrides, and metal sulfides, and are employed as successful anode materials for LIBs.4–7 Currently, transition metal oxide-based materials are widely used in different potential applications in several fields, including nanoscale electronics, energy storage, catalysis, sensors, and water treatment.8–10 Among various metal oxides, MnO2 has attracted many researchers owing to its low cost, abundance on the Earth's crust, and non-toxic nature.11 Moreover, MnO2 exhibits various crystal morphologies as α, β, γ, and δ-type, with unique combinations of physical and chemical properties with high theoretical capacities, low redox potentials, and low polarization properties, making it a typical anode material for the advancement of LIBs and supercapacitors.12–15 However, the practical applicability of MnO2 anodes is hampered by their low electronic conductivity, poor cyclability, and large volume changes during Li-ion electrochemical conversion reactions. Thus, hollow and porous nanostructures are considered to enhance the electrochemical properties of MnO2 by affording a large surface area, low density, and an essential buffer volume during redox reactions. Several strategies have been proposed to synthesize hollow porous MnO2, such as hard template, soft templates, carbonization of Mn-based meta–organic frameworks (MOF), and hydrothermal approaches.16–18 However, the removal of the templates by acid/base etching, the carbonization temperatures, and conditions may change the structures of the materials and significantly increase the preparation costs. Therefore, the synthesis of MnO2 hollow porous structures is still challenging due to the lack of easy template removal process. Although the hollow porous nanostructures of MnO2 significantly enhance Li-ion electrode kinetics, it is not yet possible to fulfill high power demands without compromising energy density owing to insufficient electrochemically active sites and intrinsically limited electronic conductivity, hindering its practical application. Researchers have developed several methods to overcome the low conductivity of MnO2 by employing carbon frameworks, nitrogen doping, mixed metal oxides, transition metal doping (Co, Ni, V, Ti), etc.17,19–23 to greatly enhance the electrical conductivity of the host material by improving its electrochemical performance. Recent reports from the literature also confirm that using transition metal doping in MnO2 is an effective way to stabilize its cycles against higher applied current densities. Yao et al. reported Ni-doped MnO2 distorted structures to improve Na+ storage capacity. The prepared Ni–MnO2 delivered a specific capacity of 379 F g−1 at 1 A g−1 and exceptional rate performance of 281 F g−1 at 20 A g−1, and enhanced the kinetics of Na+ diffusion.22 Dong et al. synthesized Ni-doped MnO2 by using a facile sol–gel method and studied its electrochemical properties for supercapacitors. The prepared Ni-doped MnO2 delivered excellent specific capacitance of 74.36% after 2000 charge–discharge cycles.24 Asif et al. developed a Ni-doped MnO2/CNT nanoflake architecture cathode for magnesium/lithium hybrid ion batteries, which delivered capacity retention of 87% at a current density of 1 A g−1 after 3000 cycles.25 All these reports suggested that Ni doping into MnO2 greatly enhances the intrinsic electronic conductivity along with improving the kinetics and diffusion properties of the host materials. Hence, better electrochemical properties and enhanced storage capacities could be possible by using hollow porous architectures with transition metal doping. Given these benefits, Ni-doped MnO2 particles with extra added hollow structural features, such as hollow particle interiors, high surface area, and structural robustness, allow its use as a promising electrode material for the evolving Li-ion energy storage technology, such as the Li-ion hybrid capacitor (LIHC), which can deliver higher energy and power densities than typical LIBs,17 and could progress the new hybrid technology to the advanced level.
In this regard, Ni–MnO2 hollow porous structures are synthesized via a facile modified chemical approach followed by heat treatment. Operating at room temperature, an easy water-washable template, and low-temperature annealing are the main advantages of this process. By using this approach, we successfully achieved the desired hollow porous nanostructures, which are highly favorable for Li+ energy storage applications.
2. Results and discussion
Fig. 1 shows the schematic presentation of the step-by-step formation process for the hollow particles. In the first stage, Li2O2 particles are neutralized with the nitrate ions of Mn(NO3)2·6H2O, resulting in the formation of LiNO3. The oxygen evolved during the reaction was consumed to oxidize the Mn(II) cations to MnOx. In stage II, the partially consumed Li2O2 particles are covered with the in situ formed primary MnO2 layers precipitated through heterogeneous nucleation on the Li2O2 solid particles. During the reaction, the thickness of the MnO2 primary layer on the surface of the consumed Li2O2 particles increases rapidly to roughly 50 to 100 nm in size. Stage III shows the complete MnO2 encapsulated consumed Li2O2 hybrid particle. The desired amount of Ni(NO3)2·6H2O precursor is added to the reaction mixture in stage IV, here also expecting the same neutralization reaction between Li2O2 with the acidic nitrates from the nickel precursor while the Ni(II) ions are doped into the MnO2 matrix or NiOOH particles are deposited on the outer surface. The remaining deposits of Li2O2 are completely dissolved with water and alcohol washing, resulting in hollow particles of the δ-MnO2 phase,17 along with minor NiOOH (obtained from the oxidation of Ni(II) ions) and Ni phases. In the final stage, heating the as-obtained powder at 500 °C leads to the formation of the λ phase of MnO2. Moreover, the annealing at high temperatures supports Ni doping and helps to attain crystallization of the hollow particles, leading to the production of nanoparticles with meso-/nano-porous walls (see stages V and VI in Fig. 1).
 |
| | Fig. 1 Step-by-step schematic representation of the synthesis of hollow porous Ni-doped λ-MnO2 particles. | |
The XRD patterns of the synthesized materials, hMO, 10Ni-hMO, 20Ni-hMO, and 30Ni-hMO, are shown in Fig. 2a and are indexed to the 3D-spinel structures of λ-MnO2 (JCPDS card No. 440992) respective to the (111), (311), (400), (511) and (440) planes. No other marginal impurity peaks were observed, indicating no other possible side reactions. A zoomed view of the (400) peak of all of the prepared materials is shown in Fig. 2b, where the peaks are slightly shifted towards the left, indicating the successful doping of Ni into the spinel crystalline structure of MnO2. The inset of Fig. 2b shows the zoomed view of the (311) plane of 30Ni-hMO, which shows a small bump at 2θ = 37.8°, which could be related to the (111) plane of NiO, attributed to the formation of NiO, due to the higher amounts of Ni (30 mol%) precursor during the synthesis process. The surface morphology of 20Ni-hMO was analyzed by FE-SEM and the images are depicted in Fig. 3a and b, showing randomly arranged hollow particles with an average diameter ranging from ∼100 to 300 nm. All these hollow particles have uniform wall thicknesses of ∼80 nm; each particle consists of meso/nanosized voids. The surface of the hollow particles is abundantly covered with a combination of thin nanoparticles/flakes with an average thickness of ∼10–20 nm. Fig. 3c and d show the hollow particles of the 10Ni-hMO and 30Ni-hMO samples; it is evident that the 10% Ni-doped sample has a similar morphology to that of the 20Ni-hMO sample. However, as seen in Fig. 3d, an increase in the dopant Ni content to 30% results in the formation of some irregular particles (marked with colored arrows), possibly due to the presence of a minor amount of NiO (confirmed by the XRD results) along with the Ni-doped MnO2 hollow structures. All these features of hollow particles are favorable for the faster Li+ diffusions/kinetics of the active materials. The SEM-EDX spectra of the 20, 10, and 30% Ni-doped MnO2 samples are shown in Fig. 3e–g, respectively. The spectra contain Mn, Ni, and O peaks, and the elemental compositions from the spectra are depicted in the respective inset tables, revealing that the Ni contents in the 20, 10 and 30% doped samples are approximately 16.86, 7.91 and 27.61%, respectively, close to the amounts of Ni added in the respective precursors, confirming the presence of the expected amounts of Ni along with the MnO2 particles in all of the doped samples.
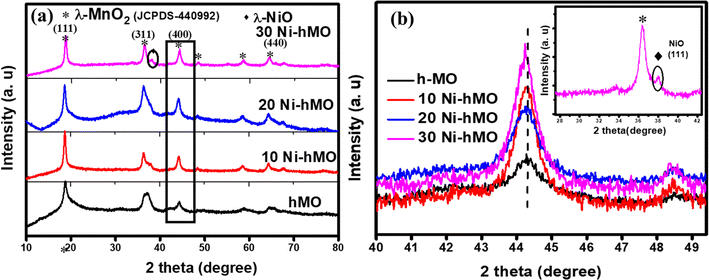 |
| | Fig. 2 XRD patterns for (a) h-MO,10Ni-hMO, 20Ni-hMO, and 30Ni-hMO. (b) Closer view of (440) peak in (a). | |
 |
| | Fig. 3 (a) SEM image of an aggregated group of 20Ni-hMO hollow particles arranged in three-dimensional micro-bundles. (b) SEM image of a close view of the marked region in (a) in which the hollow interior of the semi-formed particles can be seen and the colored arrow marks show the thickness of the wall of the specific particles. (c) SEM image of 10Ni-hMO hollow particles; colored arrows show the selected open-type hollow particles. (d) SEM image of 30Ni-hMO; the colored arrows mark the distorted shape of the hollow particles. (e–g) SEM-EDX spectra of the 20Ni-hMO, 10Ni-hMO, and 30Ni-hMO samples, respectively. | |
The SEM-EDX elemental mapping of 20Ni-hMO is shown in Fig. S1,† where Fig. S1a and b† show the SEM image and the respective all-layered element mappings containing Mn, Ni, and O. Fig. S1c–e† show the elemental mapping images of Mn, O, and Ni, respectively. This result explains the uniform distribution of Ni throughout the MnO2, which indicates the successful doping of Ni into the MnO2 matrix. The FE-SEM images of the bare hollow MnO2 structures (hMO) are shown in Fig. S2a and b;† from the images, it is evident that the hollow particles have a similar morphology and structural features to those of the Ni-doped samples shown in Fig. 3, indicating that doping did not have much effect on the hollow morphology of the samples. TEM, selected area electron diffraction (SAED), and energy dispersive X-ray scanning transmission electron microscopy (STEM-EDX) were used for the in-depth study of the morphology and structure of the samples. Fig. 4a shows the TEM images of the 20Ni-hMO sample; the micron-sized superstructure comprises an overlaid group of hollow nanoparticles (particle size ranging from 300 to 500 nm), and each particle's hollow interior is marked with colored arrows; for clarity, one specific hollow region is marked with colored cone-shaped dash lines. The wall thickness of the specific hollow particle (50–80 nm) is uniform with nanoparticles/flakes and permeable meso-/nano-pores on the outer shell, as shown in Fig. 4b. To obtain detailed structural information about the 20Ni-hMO sample, high-resolution transmission electron microscopy (HR-TEM) characterization was performed on the marked region in Fig. 4b (shown in Fig. 4c). As displayed in Fig. 4c, the major portion of the outer surface was composed of lattice fringes with an interplanar distance of about 0.46 nm, which corresponds to the (111) plane of λ-MnO2 (PDF no. 42–1169). Interestingly, lattice fringe spacing of 0.209 nm was also observed, matching well with the cubic phase (111) plane of the Ni.
 |
| | Fig. 4 TEM images of Ni-doped manganese oxide: (a) micron-sized superstructures comprised of 20Ni-hMO hollow particles (represented by colored arrows). (b) High-magnification TEM view of the marked portion of the selected particle in image (a). (c) HR-TEM image of the specific portion selected from the marked rectangle area in image (b); the red color circle shows the mesoporous void and the permeable nature of the hollow particle shell. (d) SAED pattern of a combined group of hollow particles selected in image (a). | |
The highlighted circles discern the surface defects that exist on the surface of the sample, which may contribute to the highly active surface, as they can alter the electronic structure of the Ni-doped MnO2 to afford suitable binding strengths for the electrolyte ions dissociated during electrochemical reactions. The selected area electron diffraction (SAED) pattern (see Fig. 4d) shows that the diffraction rings can be well indexed to the main planes of λ-MnO2, as was noticed in the previous XRD results. Further, the distinct dots with clear rings indicate the good crystallinity of the material, which could be attributed to the appearance of well-formed nanoparticles on the surface of the specific hollow particles. A magnified STEM view of the randomly arranged hollow particles of 20Ni-hMO (shown in Fig. S3†) and their corresponding EDX results show the uniform distribution and the existence of Ni in the 20Ni-hMO sample, indicating that effective doping of Ni into the MnO2 hollow structures occurred. The mass% of Ni content shown in the EDX spectra and in the table (see Fig. S3e†) is near to the amount of Ni added to the 20Ni-hMO sample.
Fig. 5 depicts the N2 adsorption–desorption isotherms for all of the prepared samples. As per IUPAC, isotherms are categorized into four types, I, II, III, and IV. The isotherms in Fig. 5a show the initial N2 uptake at low relative pressure (p/po) 0.2, which is accredited to the small number of micro-pores, while the type IV hysteresis loop, which was observed from 0.2 to 0.9, is related to the meso-pores in the prepared samples. The loop above 0.9 indicates that the mesopores are inter-crystalline pores owing to the aggregation of nanocrystals.17,26 The BJH pore size distribution curves are shown in Fig. 5b, where hMO shows wide mesopores between 3 and 6 nm, whereas the Ni-doped hMO materials show meso-/nano-pores of 5–20 nm. The determined surface area, pore volume, and pore diameters for all materials are summarized in Fig. 5c. From the results it was observed that surface area and pore volume increase with doping concentration owing to the presence of extra pores formed by Ni doping. Similar results have been reported in the literature.27,28
 |
| | Fig. 5 (a) N2 adsorption–desorption isotherms of the h-MO and Ni-doped h-MO samples. (b) Pore size distribution of the samples. (c) The surface area, pore volume, and diameter are summarized in the form of a table. | |
XPS was employed to investigate the surface chemical composition of the 20Ni-hMO, and the resultant plots are displayed in Fig. 6. The survey scan spectrum of 20Ni-hMO is shown in Fig. 6a, indicating that the material has a combination of Mn, Ni, and O elements and no other impurity peaks are observed, confirming the presence of Ni and MnO2 in the sample. The high-resolution Mn 2p spectrum is depicted in Fig. 6b, where an 11.7 eV spin-separation is observed from the main peaks at 640.2 and 651.9 eV, attributed to Mn 2p2/3 and Mn 2p1/3, respectively. The peak fitting method was used to split the peaks to check their chemical states. The peaks at binding energies of 640.17 and 651.89 eV are related to the Mn3+ and Mn4+, whereas the other two peaks at binding energies of 642.28 and 652.15 eV represent the Mn3+.29 The higher intensity of Mn3+ in MnO2 can initiate a large active surface that leads to better electrochemical performance.28,29Fig. 6c shows the high-resolution spectra of O1s, where the peak is split into two peaks at binding energies of 527.62 and 529.18 eV, which are related to the Mn–O and Mn–O–H bonds in the metal oxide.30 The high-resolution Ni 2p spectra are depicted in Fig. 6d, where the peaks at binding energies of 852.77 and 870.38 eV are responsible for Ni 2p3/2 and Ni 2p1/3 and the other two satellite peaks observed at binding energies of 878.09 and 859.12 eV indicate the successful doping of Ni into MnO2.31 The XPS peak values for Mn and Ni are shown in Table S1 of the ESI.† The results indicate a Ni to Mn ratio of ca. 0.23, with a Ni mass percentage of 15.9, which is close to the nominal value. Generally, XPS is a surface analysis technique and is restricted to a few nanometers (∼5 nm) depth from the sample's outer surface. Alternatively, ICP-OES is a more suitable technique for measuring the contents of certain elements in a sample. Thus, comparing the molar Ni/Mn ratio of 0.23 (see Table S2† for the ICP-OES results) with the XPS molar ratio, it is evident that the Ni ions mostly reside up to a certain nanometer length from the outer surface of the sample. The majority of the Ni content being located on the outer surface of the particles was also confirmed by the SEM-EDAX spectra and mapping results shown in Fig. 3e and S1.† The smaller ionic radii of the Ni2+ ions (low spin, 0.56 Å, and high spin, 0.60 Å) compared to the Mn3+ and Mn4+ oxidation states with large ionic radii [Mn3+ (low spin, 0.72 Å; high spin, 0.785 Å, and Mn4+ (0.67 Å)] was helpful for the successful incorporation of nickel ions into the MnO2 framework. To analyze the bonding states of Ni in the samples, XPS measurements were conducted for fresh Ni-doped electrode materials. As shown in Fig. S4,† the obtained Ni 2p spectra for all three samples showed prominent peaks located at 854.6 eV and 872.6 eV with two satellite peaks at about 861.135 and 880.05 eV, belonging to Ni2p3/2 and Ni2p1/2, respectively. These values are consistent with NiO. Additional peaks appeared at 851.01 and 852.1 eV, and small intensity peaks located at 856 and 858.79 eV, explaining the presence of Ni0 and Ni2O3, respectively.31,32 These results indicate that all three Ni-doped samples have a certain amount of NiO on the surface of the particles, and the area percentage of NiO is larger value for the 30% Ni-doped MnO2 sample (see Table S3†), which was also confirmed by the XRD results (inset of Fig. 2b). Considering the higher Ni0 content in the 20% Ni-doped sample (see Table S3†), it can be inferred that Ni was successfully doped into the MnO2 framework rather than forming other oxides on the particle's surface. The sample doped with 30% Ni has a higher Ni0 content compared to the 10% Ni-doped sample, and the presence of more NiO on the particle surface may decrease its electrochemical activity.
 |
| | Fig. 6 XPS spectrograms for 20Ni-hMO: (a) XPS survey scan, (b) Mn 2p, (d) O 1s, and (d) Ni 2p. | |
2032R-button-type coin cells were used to evaluate the electrochemical performance of all the prepared materials. To evaluate the performance of the prepared materials, different electrochemical tests were employed. To compare the electrochemical performances of all materials, all cells were operated under the same conditions. The redox behavior of the electrodes was analyzed by employing CV and the results for the first three cycles for hMO and 20Ni-hMO are shown in Fig. 7a and b. The CV curves for the other electrodes made using 10Ni-hMO and 30Ni-hMO are depicted in Fig. S5.† The first cathodic scans in Fig. 7a and b comprised two small peaks at 1.0 V and 2.68 V in hMO and 1.3 V and 2.68 V in 20Ni-hMO, representing the underlying reduction peaks of MnO2 to Mn2+ and the simultaneous formation of an SEI layer on the surface of the active material. These peaks were not observed in the following cycles, indicating the thick and stable SEI layer formation. The other peak at 0.2 V in both materials indicates the reduction of Mn and in later cycles the peak is shifted to higher voltage of 0.27 V owing to the structural reformation resulting from the Li2O formation, during conversion reactions.33 In the anodic scan, two predominant oxidation peaks are observed at 1.3 V and 2.4 V for hMO and 1.27 and 2.0 V for 20Ni-hMO, signifying the two-step conversion reaction of Mn into MnO2. From Fig. S5a,† the 10Ni-hMO and 30Ni-hMO electrodes also showed similar redox behavior with no other marginal impurity peaks. Further, for the 30Ni-hMnO (Fig. S5b†), the first cathodic peak was observed at 0.41 V owing to the reduction of the small amount of NiO present in the sample (see the XRD in Fig. 2b),34 or the larger BET surface area noticed for this sample in this work resulting in the simultaneous formation of a larger portion of Li2O and an SEI layer that could result in more positive side shifting of the reduction peak. From Fig. 7 and S5,† the shifting of the oxidation peaks towards lower potentials indicates the improved wettability and enhanced Li-ion insertion reaction kinetics. This CV peak shifting increases with increasing Ni-doping concentration reveals the improved electronic conductivity in the respective materials.35,36 The Nyquist plot analysis of cells made with fresh electrodes provides valuable insights into the charge transfer resistance and Li-ion diffusion of Ni-doped hollow MnO2 samples. The Nyquist plot of the fresh electrode cells shows semicircles for the electrodes in the high-to medium-frequency region, followed by an inclined line in the low-frequency region (Fig. S6a†). Notably, the semicircle diameter corresponds to the charge transfer resistance (Rct), and the slope of the inclined line represents Li-ion diffusion in the solid electrode material.37,38 Remarkably, 20Ni-hMO boasts a lower Rct value and steeper inclined line than all the other electrodes. This compellingly indicates that Ni doping triggers faster Li-ion diffusion in doped hollow MnO2 than bare MnO2 hollow particles. The table in Fig. S6b† distinctly shows that 30Ni-hMO has higher Rct values than the other doped electrodes. This increase in Ni dopant content to 30 wt% in hollow MnO2 leads to high resistance to charge transfer across the electrode/electrolyte interface, which may be the cause of the significant decline in its electrochemical behavior compared to its counterparts. The results suggest that Ni doping significantly improves the electrochemical performance of hollow MnO2 by increasing Li-ion diffusion while reducing charge transfer resistance.
 |
| | Fig. 7 Cyclic voltammograms for the first three cycles for (a) h-MO and (b) 20Ni-hMO at a scan rate of 0.1 mV s−1. | |
Electrochemical cyclability tests were carried out for all of the prepared materials, hMO, 10Ni-hMO, 20Ni-hMO, and 30Ni-hMO, at the applied current of 200 mA g−1 in the voltage range of 0.01–3.0 V, and the results are depicted in Fig. 8a. Among all the materials, 20Ni-hMO delivered the highest initial specific capacities of 2000/1454 mA h g−1, with an initial coulombic efficiency (ICE) of 72.7%. In the second cycle, the anode delivered capacities pf 1429/1366 mA h g−1 with coulombic efficiency of 95.6%. The irreversible capacity loss of 571 mA h g−1 is related to the formation of solid electrolyte interphase (SEI) layer on the surface of active material. After 50 cycles the specific capacities are steadily increased and reached 1636/1610 mA h g−1 with 98.4 CE%. The other materials hMO, 10Ni-MO, and 30Ni-hMO showed initial discharge/charge capacities of 1742/1201, 1979/1255(1370), and 1718/1135 mA h g−1 with 68.9, 63.4, and 66.0% of ICE. The initial CE of 20Ni-hMO is much better than the other three materials, which could be due to the improvement in the electrical conductivity from Ni doping. Despite the high Ni-doing concentration in 30Ni-hMO, it showed a lower CE, which could be due to the formation of NiO in the MnO2 synthesis process resulting from the high Ni concentration in the precursor (see inset of Fig. 2b). In the second cycle, hMO, 10Ni-hMO, and 30Ni-hMO showed specific capacities with CEs of 1278/1220 (95.4%), 1323/1260 (95.2), 1193/1141 (95.6) mA h g−1, respectively. After 50 cycles, the delivered specific capacities for these three electrodes were 1489/1456, 1530/1504, and 1288/1253 mA h g−1, respectively, with CE of more than 98%. The specific capacities for all the prepared electrodes are significantly higher than the theoretical capacity of MnO2 (1230 mA h g−1), which could be accredited to the presence of organic and inorganic compounds, such as LiF, Li2CO3, and ROCO2Li, produced during the formation of the SEI layer on the active material's surface. Moreover, the hollow nanoporous structure with three-dimensional arrangement of fine nanoparticles could generate a stable Li2O matrix and SEI layer. We assumed that this overall inorganic–organic matrix contributes to increasing extra specific capacity. Additionally, the porous morphology with the large active surface area and addition of conductive Ni into the MnO2 framework could provide more Li-ion active sites as the cycling goes on, which is also favorable for attaining higher specific capacities than the theoretical capacities. Similar trends have been reported in previous literature.36–40 The first three discharge/charge profiles of 20Ni-hMO are displayed in Fig. 8b, where the broad discharge plateau at 0.6 V followed by a long plateau at 0.2 V in the first discharge is related to the reduction of MnO2 whereas the charging process showed two plateaus at 1.3 V and 2.2 V, indicating the oxidation of Mn into MnO2, which is consistent with the CV results. The discharge/charge profiles for hMO, 10Ni-hMO, and 30Ni-hMO are displayed in Fig. S7a–c,† respectively, and are well matched with the literature reports, suggesting that similar lithium intercalation behavior is taking place in the λ-MnO2 structures. The rate capability results of the prepared materials hMO, 10Ni-hMO, 20Ni-hMO, and 30Ni-hMO were studied at applied currents ranging from 100 to 5000 mA g−1 in the voltage window of 0.01–3.0 V and the resultant specific capacity vs. cycle number plots results are depicted in Fig. 8c. For all the electrodes, the specific capacities decreased with increasing applied currents. Among them, 20Ni-hMO showed better rate results compared to the other three. 20Ni-hMO delivered discharge capacities of 2001, 1490, 1377, 1327, 1250, 1171, 1087, 1003 mA h g−1 (mentioned for every first cycle) at applied currents of 200, 300, 500, 1000, 2000, 3000, 4000 and 5000 mA g−1, respectively, and regained its full capacity of 1473 mA h g−1 at 200 mA g−1, even after severe rate testing, indicating the stable structural firmness against slow and fast Li-ion reactions. On the other hand, the hMO, 10Ni-hMO, and 30Ni-hMO materials delivered discharge capacities of 1934/1816/1791, 1293/1434/1212, 1240/1317/1105,1159/1240/1030, 1015/1141/894, 692/1045/813, 608/954/742, and 522/874/656 mA h g−1 at applied currents of 200, 300, 500, 1000, 2000, 3000, 4000 and 5000 mA g−1, respectively, and regained discharge capacities of 1292/1415/1175 mA h g−1 after applying 200 mA g−1. The rate capability results indicate that the Ni dopant plays a crucial role in significantly increasing specific capacities by enduring at high current densities, possibly by providing essential electrical conductivity to hMO particles. The Ni doping together with the unique hollow structural features are responsible for the boosted electrode performance. The long cyclability results at higher currents of 500 mA g−1 (for five cycles), 2000 mA g−1 (for 50 cycles), and 5000 mA g−1 (for 145 cycles) are evaluated and specific capacity vs. cycle number plots are depicted in Fig. 8d. The four prepared electrodes showed declined capacities in the initial cycles at 500 mA g−1, and later stabilized at 2000 mA g−1 and 5000 mA g−1. The hMO, 10Ni-hMO, 20Ni-hMO and 30Ni-hMO materials delivered capacities of 1550/1778/1788/1333 mA h g−1 at 500 mA g−1, 844/1069/1114/811 mA h g−1 at 2000 mA h g−1 and 527/918/1036/670 mA h g−1 at 5000 mA h g−1. 20Ni-hMO showed twice the discharge capacity compared to hMO, indicating the critical role of Ni doping. Increasing the Ni-doping concentration to 30% in MnO2 gave a lower capacity than the 10% and 20% Ni-doped samples, implying that excessive doping induces the formation of insulating NiO (see XRD plot in the inset of Fig. 2b) on the outer surface, which in turn reduces the electrochemical activity and may also cause the collapse of the hollow structures in some portions (see the SEM image in Fig. 3d). After cycled at 5000 mA g−1 (145 cycles, Fig. 8d), the materials delivered 453/447/889/439 mA h g−1, respectively, indicating the higher stability of 20Ni-hMO than the other materials and suggesting the better structural stability and electrochemical activity of the 20Ni-hMO electrode.
 |
| | Fig. 8 Electrochemical characterization of h-MO and Ni-doped hollow MnO2 electrodes. (a) Cycle performance at 200 mA h g−1 and (b) charge–discharge profiles for the 1st, 2nd, and 3rd cycles for the 20Ni-hMO. (c) Rate performances of the electrodes at different current densities and (d) cycle performance at high currents of 500, 2000, and 5000 mA h g−1. | |
To further investigate the kinetics of Li+ ions across the electrode/electrolyte interface, EIS analysis was performed for the hMO, 10Ni-hMO, 20Ni-hMO, and 30Ni-hMO electrodes after 50 galvanostatic charge–discharge cycles at 200 mA g−1. The resultant Nyquist plots and the respective equivalent circuit models are shown in Fig. 9a. All electrodes displayed a depressed semicircle in the middle- and high-frequency ranges, which was attributed to the charge transfer resistance at the electrode/electrolyte interface. The inclined line in the low-frequency region is related to the kinetics of lithium diffusion into the active material.41,42 The solution resistance (Rs), SEI layer resistance (RSEI), and charge transfer resistance (Rct) for all the electrodes are summarized in Table S4.† The 20Ni-hMO electrode showed a lower Rct of 20.8 Ω, compared to the other three electrodes, which could be due to the higher conductivity provided by Ni doping and the structural stability provided by the hollow microstructures leading to faster Li+ diffusion into the active material. The following equations were used to calculate the diffusion coefficient of Li+ in the prepared electrodes:
| | | Zre = Rs + RSEI + Rct + σwω−1/2 | (1) |
| | | D = R2 × T2/2A2 × F4 × C2 × σw2 | (2) |
where
D is the diffusion coefficient,
R is the universal gas constant,
T is the absolute temperature,
A is the electrode area,
C is the molar concentration of Li
+, and
F is the Faraday constant. From
Fig. 9b, it is clear that
Zre is linearly correlated with
ω−1/2 in the low-frequency region. The Warburg factor (
σw) was determined from the slope of the
Zrevs. ω−1/2 plots. The lower slope values yield high diffusion coefficients. The calculated Warburg coefficient values for hMO, 10Ni-hMO, 20Ni-hMO, and 30Ni-hMO are 45, 44, 25, and 36 Ω s
−1/2, respectively. The diffusion coefficient (
D) values calculated from
eqn (3) are summarized in Table S3.
† The
D values of the 20Ni-hMO electrode were higher than those for the other electrodes, signifying faster Li
+ kinetics in the electrode material due to shorter Li
+ pathways and more ionic channels and electrons due to the Ni dopants in hMO. According to the XRD, SEM, XPS, CV, and ICP-OES results, the most likely pathway for the solid-state conversion reaction of the Ni–MnO
2 is presented in
Fig. 10. After a few subsequent cycles, the outer surface layers of the hollow particles appeared to show the presence of secondary (Mn
3O
4) and primary phase (MnO
2) particles along with minor NiO, Ni, and Li
2O phases. This is the first attempt in the literature to provide insight into the possible formation of Mn
3O
4 and NiO phases during the Ni–MnO
2 lithiation/delithiation reaction, contributing to the understanding of metal oxide anode electrode chemistry. Additionally, the hollow particles exhibit superior electrochemical performance owing to the robust hollow region, electrolyte-viable porous walls, and other electrochemically active surface elemental compounds.
 |
| | Fig. 9 EIS results: (a) Nyquist plots for the h-MO and Ni-doped MnO2 electrodes measured after 50 charge–discharge cycles; the inset shows the model circuit. (b) Fitting results for the ZI (ohm) vs. w−1/2 plots. | |
 |
| | Fig. 10 Proposed mechanism for the solid-state conversion reaction of nickel-doped MnO2. | |
To better understand the morphological changes of the active material, SEM was carried out for the 20Ni-hMO sample after 50 cycles at 200 mA g−1. For the sample preparation, the cycled 20Ni-hMO cell was carefully disassembled in an Ar-filled glove box, and the anode electrode was collected. Then the electrode was washed with a mixture of ethylene carbonate and diethyl carbonate (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) to remove the surface SEI layer on the active material. Then the electrode was air-dried and the sample was carefully collected for SEM analysis. The resultant SEM images, corresponding elemental mapping, and SEM-EDX spectra are depicted in Fig. S8.† In the SEM image from Fig. S8a,† the 20Ni-hMO particles are aggregated to some extent due to large volume changes during the conversion reactions. Still, some of the particles retained their hollow features after 50 cycles (see the colored circles and marked with arrows in SEM images), indicating the good structural stability of the 20Ni-hMO material. The SEM-EDX elemental mapping is shown in Fig. S8b,† with Fig. S8c–f† showing the all-elements layered mapping, O, Mn, and Ni respectively. The SEM-EDX spectra of Fig. S8b,† as shown in Fig. S8g,† also confirm the peaks of Mn, O, and Ni. Furthermore, we compared our results with the results for different structures of MnO2 reported in the literature, and summarized it in the ESI (Table S5).†
:1, v/v) to remove the surface SEI layer on the active material. Then the electrode was air-dried and the sample was carefully collected for SEM analysis. The resultant SEM images, corresponding elemental mapping, and SEM-EDX spectra are depicted in Fig. S8.† In the SEM image from Fig. S8a,† the 20Ni-hMO particles are aggregated to some extent due to large volume changes during the conversion reactions. Still, some of the particles retained their hollow features after 50 cycles (see the colored circles and marked with arrows in SEM images), indicating the good structural stability of the 20Ni-hMO material. The SEM-EDX elemental mapping is shown in Fig. S8b,† with Fig. S8c–f† showing the all-elements layered mapping, O, Mn, and Ni respectively. The SEM-EDX spectra of Fig. S8b,† as shown in Fig. S8g,† also confirm the peaks of Mn, O, and Ni. Furthermore, we compared our results with the results for different structures of MnO2 reported in the literature, and summarized it in the ESI (Table S5).†
Lithium-ion hybrid capacitors (LIHSs) integrate Li-ion occur on the anode side, similar to LIB. Simultaneously, the adsorption/desorption of the cation (PF6−) occurs at the activated carbon (AC) cathode surface, identical to carbon-based supercapacitors. This method efficiently enhances energy density while maintaining high power density.43 The electrochemical cycling performance of the AC half-cells vs. Li+/Li using LiPF6 in 1:1 (EC: DEC) as the electrolyte is shown in Fig. S9† at 100 mA g−1 for different voltage ranges. All cells exhibited steady cycle life with specific capacities of 20–80 mA h g−1 (see Fig. S9a†). The AC cells cycled within the voltage ranges of 1–4 V and 2–4.5 V and delivered better specific capacities of about 60 mA h g−1. The first three galvanostatic charge–discharge curves are shown in Fig. S9b and c.† The supercapacitive characteristic was evaluated by the typical linear correlation of the discharged curves of the AC cells with a pretty stable cycle life, as shown in Figs. S9a–c,† owing to the inherent non-faradaic capacitive properties of the AC cathode. For LIHS cells, we optimized the n/p ratio to 1.2 and selected the potential range 1–4.2 V. The half-cells made with 20Ni-hMO and AC individually displayed stable discharge profiles within the potential ranges of 0.1–1 V and 2–4 V (vs. Li/Li+), respectively. Thus, the potential range of 1–4.2 V for the LIHS cells could cover the working voltage range for both anode and cathodes. The cycle performance of the 20Ni-hMO//AC at 300 mA g−1 was is in Fig. 11a. The cell delivered capacities of about 60 mA h g−1 for initial cycles and later stabilized at 45 mA h g−1 after 200 cycles with notable coulombic efficiency (CE) values (82% for the first cycle and 99.5% after 200 cycles). Fig. 11b displays the rate performance of the 20Ni-hMO//AC, LIHS cell; the cell exhibited high capacities of 70 mA h g−1 at 150 mA g−1, close to 30, 25, and 20 mA h g−1 even at high current densities of 3000, 5000 and 10000 mA g−1, respectively, and a capacity of 10 mA h g−1 even at a high current of 15000 mA g−1. These are excellent rate capability performance results and after cycling at high current densities, the cell regained its capacity of 60 mA h g−1 at 300 mA g−1, demonstrating a subsequent stable 50 cycles with minor capacity loss. In the charge–discharge curves shown in Fig. 11c, redox behavior was seen at low current densities of 2–3.5 V, owing to the kinetics of the faradaic lithiation in the 20Ni-hMO anode. At high current densities, a progressive decrease in discharge time observed with good liner relation at high currents means excellent power performance. This indicates that at high current rates the cell capacitive behavior relies on non-faradaic kinetics, possibly due to the adsorption of PF6− on the AC cathode. To evaluate the durability of our electrode materials for high-rate applications, such as electric vehicles, military, and space applications, and concerning safety, it is essential to study the specific capacity and cycling stability at a high current rate. To this end, we tested the cyclic performance of the 20Ni-hMO//AC cell at 10 A g−1 (see Fig. 11d). The 20Ni-hMO//AC hybrid cell tested at 10 A g−1 demonstrated excellent cyclability for over 5000 charge–discharge cycles with stable specific capacities of about 20 mA h g−1. The capacity retention of 20Ni-hMO//AC is as high as 83.5% after 1000 cycles and 65.2% after 5000 cycles, with CE close to unity, and we noted that our LIHS could complete fast charge–discharge within 25 s at 10000 mA g−1 (Fig. 11c), which is comparable with that of SCs. Here, we believe the initial capacity loss at high current density (10 A g−1) is attributed to the delay in electrode wetting for our hollow nanostructured 20Ni-hMO anode material. This leads to a non-homogeneous distribution of current density and the unstable formation of a SEI film. After a few cycles, the SEI film stabilizes, which helps stabilize the capacities for further cycles.
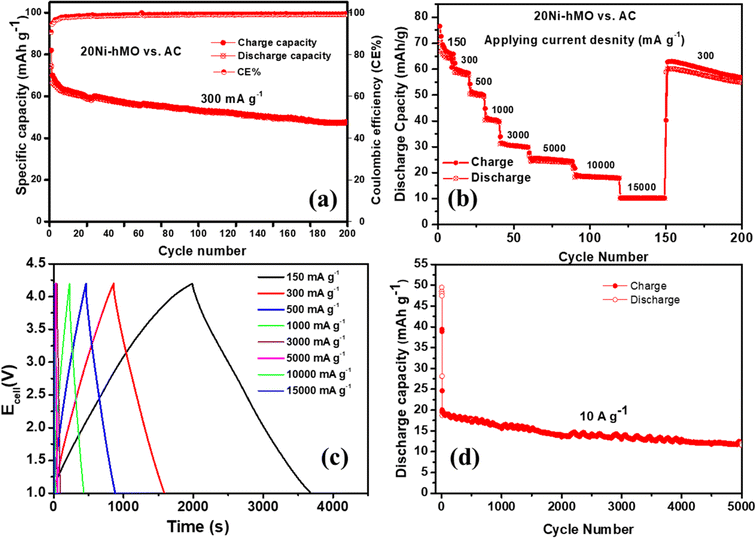 |
| | Fig. 11 Electrochemical performance of 20Ni-hMO//AC LiHC cell: (a) cycling performance at 300 mA g−1, (b) rate capability tested at various current densities, and (c) the galvanostatic charge–discharge curves in the voltage range of 1–4.2 V corresponding to (b). (d) Cycling performance at 10000 mA g−1. | |
The decline in capacity with prolonged cycling is likely due to structural and volume changes caused by the anode electrode during cycling. However, it is highlighted that the noticed cycling performance shown by our LIHS cell is better than those of previously reported similar LIC devices.44–48
The Ragone plot of the 20Ni-hMO//AC cell is illustrated in Fig. 12. The energy densities of our cell ranges from 168.0 to 30.0 W h kg−1 for power densities ranging from 0.35 to 29.2 kW kg−1. It exhibits a high-power density of 2.27 kW kg−1 while maintaining an energy density of 127 W h kg−1. These characteristics make it suitable for developing efficient high-end energy devices for potential future use in automobiles and the electronic sector. The comparison between our cell and the reported LICs developed from different kinds of similar anodes and cathodes is shown in Fig. 12a. It is worth highlighting that the excellent energy density measured for our device in the high power region is higher than those of most of the previously reported LICs.44–49 Moreover, our LIHS device maintained a high energy density close to that of LIBs and higher power densities than the typical AC//AC supercapacitors (see Fig. 12b), which indicates its remarkable application prospects. We postulated that the porous hollow structures of the Ni-doped MnO2 nanocomposite can offer extraordinary surface properties with stable structures to sustain the strain generated by the MnO2 particles during cycling. Especially, Ni doping induces electronic conductivity and hampers the particle's aggregation during cycling, which in turn makes it a suitable electrode material for future energy storage device studies. In Fig. 12c, there is a diagram of our LIHS system in a LiPF6-based electrolyte. The anode side consists of Ni–MnO2 hollow material with Li-ion interactions, while the cathode side exhibits non-faradaic EDLC-like supercapacitor generated by the surface-adsorbed PF6− ions on the high-surface-area conductive AC carbon through electrostatic interactions.
 |
| | Fig. 12 (a) Ragone plots of the 20Ni-hMO//AC LiHSC cell compared with the literature on different Li-ion hybrid capacitors. (b) Ragone plot comparing the energy and power densities of LIBs and supercapacitors17 with our LIHS system. (c) Schematic illustration of the 20Ni-h-MO//AC LIHS device. | |
3. Conclusion
Ni-doped hollow structures of λ-MnO2 were synthesized using an easy procedure. The morphological analyses showed that the un-doped hollow MnO2 and 10 and 20 mol% doped Ni–MnO2 structures have tangled nano-sized hollow particles outfitted with 10 to 20 nm nanoparticles with walls that are nanometers-thick (∼50–100 nm). The 20% doped Ni–MnO2 material exhibits enhanced lithium insertion-extraction performance, especially at very high current rates, which is the best electrochemical performance noted among the reported MnO2-based anodes for LIBs and makes it a promising candidate for use as the negative electrode in lithium-ion hybrid capacitors. The Ni-doped MnO2 hollow particles can resist volume changes during repetitive lithiation–delithiation cycles, while the hierarchical porosity of the hollow particles provides very low resistance to ion diffusion, ensuring good specific capacities at high current rates. It was observed that the LIHS cell fabricated using a Ni-doped hollow MnO2 anode and an activated carbon cathode delivered a maximum gravimetric energy density of 168 W h kg−1 at 0.35 kW kg−1 and still 63 W h kg−1 at a prodigious power density of 24 kW kg−1. The robustness of our LIHS cell was endorsed by its signifying long-term stability over 5000 cycles at a high current of 10 A g−1 with a good capacity of about 15 mA h g−1. This study unlocks innovative research pathways for developing new nanostructured hollow materials from other metal oxides. This method is also easily extendable to doping metals into other metal oxides, making them an attractive choice for various energy storage applications. This exceptional performance makes our proposed LIHS a promising energy storage system standing out among its peers.
4. Experimental details
Mn(NO3)2·6H2O (0.069 M, Aldrich) and the appropriate stoichiometric amounts (10%, 20%, and 30%) of dopant precursor Ni(NO3)2·6H2O were dissolved separately in different vessels and added to isopropanol (100 mL). Then, Li2O2 (0.6 g, Aldrich) was added to the prepared solutions slowly while stirring at room temperature. Initially, a precipitate was formed, and after 1 h, the color of the solutions turned to light brown with a minor amount of pale green color. The reaction mixture was then kept under ambient conditions overnight with continuous stirring. The resultant dark brown precipitate was separated by centrifugation, washed with deionized water and ethanol, and dried at room temperature overnight. The obtained powders were then subjected to heat treatment at 500 °C for 1 h in air. For comparison, pure MnO2 was prepared by a similar approach without the addition of the dopant precursor solution. Bare hollow structured MnO2 and the samples with different molar amounts of Ni doping (10%, 20%, and 30%) were named as hMO, 10Ni-hMO, 20Ni-hMO, and 30Ni-hMO, respectively.
4.1. Material characterization
The crystal structure of the prepared materials was investigated using a Rigaku D/MAX-2200 X-ray diffractometer (XRD; Rigaku Corp., Tokyo, Japan). The surface morphology and microstructures of the prepared materials were analyzed by scanning electron microscopy SEM (JSM7000F, JEOL Ltd, Tokyo, Japan) and transmission electron microscopy TEM (JEM-2100F, JEOL Ltd, Tokyo, Japan). The Brunauer–Emmett–Teller (BET) method was applied to analyze the specific surface area and pore size distribution of the prepared composite materials. X-ray photoelectron spectroscopy (XPS) carried out using a PHI Quantera SXM (ULVAC-PHI Inc., Kanagawa, Japan) was employed to examine the surface chemical states of the prepared materials. The elemental composition of the Ni-doped MnO2 samples was examined by ICP-OES to confirm the amount of Mn and Ni loaded in the Ni-doped MnO2 nanoparticles. ICP-OES analysis was conducted by dissolving the powder samples in a concentrated acidic solution (HCl:HNO3, 3:1 v/v by volume).
4.2. Electrochemical measurements
All electrochemical measurements of the prepared materials were evaluated using CR2032R button-type coin cells. The working electrodes were prepared using 65% active material, 20% super-P conductive carbon, and 15% polyamide-imide (PAI-Typolymer. Co., Ltd Torlon (R) amide-imide, 4000 T-HV) binder in N-methyl 2-pyrrolidone (NMP) solvent. All electrode components were mixed well for 20 min using a mini-ball mill. Then the resultant slurry was coated on copper foil using the doctor blade technique. Later, the coated copper foil was dried at 80 °C for 3 h to evaporate excess solvent and then moved to a vacuum oven and heated at 200 °C for 3 h to activate the PAI binder. Small disks of about 14 mm in diameter were punched and used as anodes and the average active material mass loading on each electrode was about 1.8 mg cm−2 with an average thickness of 60 μm. In the preparation of the cells, metallic Li was used as the counter electrode with a polypropylene (PP Wellcos Corporation, Separator 2400) membrane as the separator and 1 M LiPF6 in a mixture of ethylene carbonate and diethyl carbonate (1:1, v/v) as the electrolyte. All coin cells were fabricated in a glove box filled with high-purity argon (H2O < 0.1 ppm and O2 < 1 ppm). The galvanostatic charge–discharge evaluation, cyclability, rate capability, and cyclic voltammetry experiments were carried out using a BioLogic battery testing system. Cyclability tests were carried out at 200 mA g−1 and the rate capability tests were carried out with the applied current in the range of 100–5000 mA g−1. All half-cell tests were conducted in the voltage range of 0.01–3.0 V. Cyclic voltammetry (CV) experiments were performed at the same cutoff voltage at a scan rate of 0.1 mV s−1. The electrochemical impedance spectroscopy measurements were carried out with the same battery testing system in the frequency range of 10 kHz to 10 MHz with an alternating current voltage of 5 mV.
For the LIHCs, the negative electrode with Ni-hMO was prepared by the same method as described for LIB. The positive electrode slurry was prepared by mixing 80 wt% activated carbon (PCT-AC-07, BET: 2000–2300 m2 g−1) with 10 wt% super-P and 10 wt% PAI in NMP. The slurry was mixed and coated on aluminum foil and then dried in an oven at 80 °C for 3 h. The measured thickness of the positive and negative electrodes was about 15 μm and 9 μm, respectively. The mass loading for the positive was 2–4 mg cm−2, and the negative electrode was within the range of 1–1.5 mg cm−2. The activated carbon cathode was tested at different potentials ranging from 1 to 4.5 V. LIHC (Ni-hMO//AC) full cells with a porous glass fiber separator (Whatman GFB) were assembled using 2032-type coin cells using a negative-to-positive electrode mass ratio of 1.20. All cells were fabricated using 1 M LiPF6 in EC:DEC (1:1) electrolyte. Before testing, the Ni-hMO anode electrode was pre-lithiated between 3 and 0.01 V vs. Li/Li+ to compensate for the initial capacity loss. The AC cathode was charged up to 4.2 V vs. Li/Li+. After this pre-lithiation process, Li-HSC full cells were built for further electrochemical studies. The applied current was calculated based on the total mass loading of the electrodes (5.2 mg); for example, 5.2 mA was applied to Li-HSC at a current density of 1 A g−1 and aged for 12 h at room temperature before measurement. The hybrid system delivers a non-linear galvanostatic charge–discharge curve, thus it is appropriate to determine the energy density by integrating the area under the discharge curve.
| | 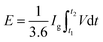 | (3) |
| | 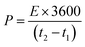 | (4) |
where
t1 and
t2 are the start and end times in the discharge process (s),
I is the current density (A g
−1), and
V is the potential (V). The energy density and power densities were calculated by using the reported equations.
50
Author contributions
Conceptualization, V. N.; methodology, V. N; measurement and analysis, V. N. and A. N.; measurement supervision, K. Y. S., and V. N.; writing – original draft preparation, V. N.; writing – review and editing, K. Y. S., A. N., and V. N.; project administration, K. Y. S.; funding acquisition, K. Y. S. All authors have read and agree to the published version of the manuscript.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
This work was supported by the 2023 Inje University research grant.
References
- D. Deng, Energy Sci. Eng., 2015, 3, 385–418 CrossRef.
- M. Li, X. H. Hou, Y. j. Sha, J. Wang, S. J. Hu, X. Liu and Z. P. Shao, J. Power Sources, 2014, 248, 721–728 CrossRef CAS.
- Y. K. Sun, S. T. Myung, B. C. Park, J. Prakash, I. Belharouak and K. Amine, Nat. Mater., 2009, 8, 320–324 CrossRef CAS PubMed.
- A. Nulu, V. Nulu and K. Y. Sohn, ChemElectroChem, 2020, 7, 4055–4065 CrossRef CAS.
- W. J. Zhang, J. Power Sources, 2011, 196, 13–24 CrossRef CAS.
- H. Kim, V. Nulu, J. Yoon and W. S. Yoon, J. Alloys Compd., 2018, 778, 37–46 CrossRef.
- S. Goriparti, E. Miele, F. D. Angelis, E. D. Fabrizio, R. P. Zaccaria and C. Capiglia, J. Power Sources, 2014, 257, 421–443 CrossRef CAS.
- X. H. Xia, J. P. Tu, Y. Q. Zhang, X. L. Wang, C. D. Gu, X. B. Zhao and H. J. Fan, ACS Nano, 2012, 6, 5531–5538 CrossRef CAS PubMed.
- D. Chen, Q. F. Wang, R. M. Wang and G. Z. Shen, J. Mater. Chem. A, 2015, 3, 10158–10173 RSC.
- S. H. Choi, J. K. Lee and Y. C. Kang, Nanoscale, 2014, 6, 12421–12425 RSC.
- S. Johnson, J. Power Sources, 2007, 165, 559–565 CrossRef.
- C. Zhang, M. Zeng, Y. Kong, Z. Pan and Z. Yang, Sens. Actuators, B, 2012, 162, 292–299 CrossRef.
- S. Ding, X. F. Shen, S. Sithambaram, S. Gomez, R. Kumar, V. M. B. Crisostomo, S. L. Suib and M. Aindow, Chem. Mater., 2005, 17, 5382–5389 CrossRef.
- Y. Tanaka, M. Tsuji and Y. Tamaura, Phys. Chem. Chem. Phys., 2000, 2, 1473–1479 RSC.
- V. Subramanian, H. Zhu, R. Vajtai, P. M. Ajayan and B. Wei, J. Phys. Chem. B, 2005, 109, 20207–20214 CrossRef CAS PubMed.
- J. B. Fei, Y. Cui, X. H. Yan, W. Qi, Y. Yang, K. W. Wang, Q. He and J. B. Li, Adv. Mater., 2008, 20, 452 CrossRef.
- J. S. Moon, A. Nulu, Y. G. Hwang, V. Nulu and K. Y. Sohn, ChemistrySelect, 2021, 6, 7012–7024 CrossRef CAS.
- X. Wang and Y. D. Li, Chem. Commun., 2002, 7, 764 RSC.
- T. Chen, Z. Wu, W. Xiang, E. Wang, T. Chen, X. Guo, Y. Chen and B. Zhong, Electrochim. Acta, 2017, 246, 931 CrossRef CAS.
- W. Luo, X. Hu, Y. Sun and Y. Huang, ACS Appl. Mater. Interfaces, 2013, 5, 1997 CrossRef CAS PubMed.
- Y. Chu, L. Guo, B. Xi, Z. Feng, F. Wu, Y. Lin, J. Liu, D. Sun, J. Feng, Y. Qian and S. Xiong, Adv. Mater., 2018, 30, 1704244 CrossRef PubMed.
- S. Yao, R. Zhao, S. Wang, Y. Zhou, R. Liu, L. Hu, A. Zhang, R. Yang, X. Liu, Z. Fu, D. Wang, Z. Yang and Y. M. Yan, J. Chem. Eng., 2022, 429, 132521 CrossRef CAS.
- Y. Sun, S. Wang, Y. Dai and X. Lei, Funct. Mater. Lett., 2016, 9, 1650005 CrossRef CAS.
- J. Dong, Z. Hou, Q. Zhao and Q. Yang, E3S Web Conf., 2019, 79, 03002 CrossRef CAS.
- M. Asif, M. Rashad, Z. Ali, H. Qiu, W. Li, L. Pan and Y. Hou, Mater. Today Energy, 2018, 10, 108–117 CrossRef.
- Y. G. Hwang, V. Nulu, A. Nulu and K. Y. Sohn, RSC Adv., 2023, 13, 22007 RSC.
- S. P. Takle, S. D. Naik, S. K. Khore, S. A. Ohwal, N. M. Bhujbal, S. L. Landge, B. B. Kale and R. S. Sonawane, RSC Adv., 2018, 8, 20394 RSC.
- L. Chen, J. Jia, R. Ran and X. Song, J. Chem. Eng., 2019, 369, 1129–1137 CrossRef CAS.
- M. Wang, K. Chen, J. Liu, Q. He, G. Li and F. Li, Catalysts, 2018, 8, 138 CrossRef.
- L. Huang, X. Luo, C. Chen and Q. Jiang, Ionics, 2021, 27(9), 3393–3941 Search PubMed.
- A. Nulu, V. Nulu and K. Y. Sohn, J. Alloys Compd., 2022, 911, 164976 CrossRef CAS.
- A. A. Amayaa, C. A. Gonzálezb, M. E. Niño-Gómeza and M. O. Fernando, J. Electron Spectrosc. Relat. Phenom., 2019, 233, 5 CrossRef.
- T. Zhang, F. Zhang, L. Zhang, Y. Lu, Y. Zhang, X. Yang, Y. Ma and Y. Huang, Carbon, 2015, 92, 106–118 CrossRef CAS.
- Y. Zhang, M. Xie, Y. He, Y. Zhang, L. Liu, T. Hao, Y. Ma, Y. Shi, Z. Sun, N. Liu and Z. J. Zhang, J. Chem. Eng., 2021, 420, 130469 CrossRef CAS.
- A. A. Kashale, A. S. Rasal, G. P. Kamble, V. H. Ingole, P. K. Dwivedi, S. J. Rajoba, L. D. Jadhav, Y. C. Ling, J. Y. Chang and A. V. Ghule, Composites, Part B, 2019, 167, 44–50 CrossRef CAS.
- Y. Zhao, G. Chen, D. Wang, C. Song, L. Xie, L. Chang, R. Wang and N. Zhong, Int. J. Electrochem. Sci., 2016, 11, 2525–2533 CrossRef CAS.
- K. Zhu, Y. Zhang, H. Qiu, Y. Meng, Y. Gao, X. Meng, Z. Gao, G. Chen and Y. Wei, J. Alloys Compd., 2016, 675, 399–406 CrossRef CAS.
- H. Sun, G. Xin, T. Hu, M. Yu, D. Shao, X. Sun and J. Lian, Nat. Commun., 2014, 5, 4526–4534 CrossRef CAS PubMed.
- Z. Li, J. Wang, Z. Wang, Y. Tang, C. S. Lee and S. Yang, RSC
Adv., 2014, 4, 54416–54421 RSC.
- J. Chen, Y. Wang, X. He, S. Xu, M. Fang, X. Zhao and Y. Shang, Electrochim. Acta, 2014, 142, 152–156 CrossRef CAS.
- Z. Hu, L. Wang, K. Zhang, J. Wang, F. Cheng, Z. Tao and J. Chen, Angew. Chem., 2014, 53, 12794–12798 CrossRef CAS PubMed.
- N. Ding, J. Xu, Y. X. Yao, G. Wegner, X. Fang, C. H. Chen and I. Lieberwirth, Solid State Ionics, 2009, 180, 222–225 CrossRef CAS.
- X. Yu, C. Zhan, R. Lv, Y. Bai, Y. Lin, Z. H. Huang, W. Shen, X. Qiu and F. Kang, Nano Energy, 2015, 15, 43–53 CrossRef CAS.
- X. Chen, Z. Cao, L. Xing, Y. Liao, Y. Qiu and W. Li, Nanoscale, 2017, 9, 18467–18473 RSC.
- D. Puthusseri, V. Aravindan, S. Madhavi and S. Ogale, Electrochim. Acta, 2014, 13, 766 CrossRef.
- W. J. Cao and J. P. Zheng, J. Power Sources, 2012, 213, 180–185 CrossRef CAS.
- Y. Zhao, Y. Cui, J. Shi, W. Liu, Z. Shi, S. Chen, X. Wang and H. Wang, J. Mater. Chem. A, 2017, 5, 15243–15252 RSC.
- J. H. Kim, J. S. Kim, Y. G. Lim, J. G. Lee and Y. J. Kim, J. Power Sources, 2011, 196, 10490–10495 CrossRef CAS.
- X. Sun, X. Zhang, W. Liu, K. Wang, C. Li, Z. Li and Y. Ma, Electrochim. Acta, 2017, 235, 158–166 CrossRef CAS.
- T. Panja, J. Ajuria, N. Díez, D. Bhattacharjya, E. Goikolea and D. Carriazo, Sci. Rep., 2020, 10, 10842 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: SEM and TEM analysis, EDAX, tabulated XPS results, EIS results, LIBs anode performance comparison graph with literature, CV studies, and AC cycling tests. See DOI: https://doi.org/10.1039/d4na00023d |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Arunakumari
Nulu
,
Arunakumari
Nulu













