
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePtRu mesoporous nanospheres as electrocatalysts with enhanced performance for oxidation of methanol†
Jiangbin
Guo
 a,
Qiyu
Yan
a,
Man
Zhang
b,
Jun
Fang
a,
Shuiyuan
Luo
a and
Jing
Xu
*a
a,
Qiyu
Yan
a,
Man
Zhang
b,
Jun
Fang
a,
Shuiyuan
Luo
a and
Jing
Xu
*a
aCollege of Chemical Engineering and Materials, Quanzhou Normal University, Quanzhou, Fujian 362000, P. R. China. E-mail: jingxu@qztc.edu.cn
bState Key Laboratory of Molecular Engineering of Polymers, Department of Macromolecular Science, Fudan University, Shanghai 200433, P. R. China
First published on 8th August 2024
Abstract
Composition and morphology are crucial factors in the design of Pt-based catalysts with high performance, particularly in direct methanol fuel cells (DMFCs). Herein, PtRu mesoporous nanospheres (PtRu MNs) with tunable compositions were synthesized via a facile method and then deposited on a carbon support to act as electrocatalyst materials for the methanol oxidation reaction (MOR). Superior catalytic activity, better catalytic stability, and good tolerance to CO were achieved by the optimum PtRu (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) MNs/C catalyst compared with Pt MNs/C. The mass activity on PtRu (2:1) MNs/C reached 111.77 mA mgPt−1, which was approximately 6.45-fold higher than that of Pt MNs/C (17.33 mA mgPt−1). Meanwhile, PtRu (2:1) MNs/C retained much more current density (84.7%) than Pt MNs/C (17.7%) after 500 cycles. The improved catalytic performance is due to several factors, including the formation of a mesoporous nanostructure with abundant active sites and the favorable effects of the Ru species. This work provides guidance toward designing and fabricating effective Pt-based electrocatalysts for DMFC applications.
:1) MNs/C catalyst compared with Pt MNs/C. The mass activity on PtRu (2:1) MNs/C reached 111.77 mA mgPt−1, which was approximately 6.45-fold higher than that of Pt MNs/C (17.33 mA mgPt−1). Meanwhile, PtRu (2:1) MNs/C retained much more current density (84.7%) than Pt MNs/C (17.7%) after 500 cycles. The improved catalytic performance is due to several factors, including the formation of a mesoporous nanostructure with abundant active sites and the favorable effects of the Ru species. This work provides guidance toward designing and fabricating effective Pt-based electrocatalysts for DMFC applications.
Introduction
The consumption of fossil-based resources and subsequent pollution of the environment have led to growing research interest in sustainable and clean energy resources.1–5 Fuel cell technology, which directly transforms chemical energy into electrical energy, shows good potential as an alternative power source for a variety of applications.6–10 Direct methanol fuel cells (DMFCs) are particularly promising because they have excellent energy densities and low emissions. Moreover, methanol fuels are easy to handle.11–15 The anode in a DMFC is where the methanol oxidation reaction (MOR) happens, and platinum-based materials are the main MOR catalysts. However, the commercialization of DMFCs is hindered by several challenges, such as the scarcity of platinum and its easy CO poisoning, meaning that DMFCs with Pt-based MOR catalysts have high costs and poor fuel cell performance.16,17 Thus, developing low-cost Pt-based electrocatalysts that exhibit good catalytic properties would be highly significant for the use of DMFCs.The usage of Pt in MOR electrocatalysts can be reduced by the incorporation of Pt with another metal in an alloy. Moreover, this strategy can dramatically enhance the electrocatalytic performance toward the MOR.18–22 Among the various bimetallic alloys, PtRu nanocatalysts are regarded as superior DMFC anode catalysts, and the high catalytic activity of these catalysts is explained by the electronic effect and bifunctional mechanism.23–25 On one hand, Ru species facilitate the dissociative adsorption of H2O to provide abundant hydroxyl species at lower potentials. This mitigates the CO poisoning of Pt because oxidative CO removal is promoted. On the other hand, the Pt–CO bond strength is reduced by charge transfer from Ru to Pt, which alters the electronic properties of Pt.
Fabricating PtRu nanocatalysts with different morphologies to expose more active sites has been widely investigated to improve their electrocatalytic performance and utilization.26–30 For example, Huang et al.26 synthesized PtRu nanocubes, nanorods, and nanowires to study the effect of the exposed crystal face on electrocatalytic activity. The PtRu nanowires with {111}-terminated facets showed improved mass activity and better MOR durability than PtRu nanocubes terminated by {100} facets and commercial Pt/C. Dong et al.27 describe a strategy for synthesizing PtRu alloy nanodendrites with tunable compositions. For the electro-oxidation of methanol, the optimum Pt0.8Ru0.2 nanodendrite catalyst showed 6.6-fold improved mass activity compared with commercially available Pt/C. Yin and co-workers28 prepared a special core–shell nanostructured electrocatalyst that contained a bimetallic PtRu mesoporous shell surrounding a Pt core (Pt@mPtRu YSs). This core–shell material exhibited superb electrocatalytic performance, excellent stability, and satisfactory tolerance toward CO as a MOR electrocatalyst because the mesoporous PtRu nanocage shell and inner Pt nanoparticles had an abundance of active sites.
This study reports a simple approach for the hydrothermal fabrication of PtRu mesoporous nanospheres (PtRu MNs) with tunable compositions via RuCl3 and K2PtCl4 co-reduction using L-ascorbic acid. The crystal structure, morphology, and surface chemical states of the prepared catalysts were characterized. The optimum PtRu (2:1) MNs/C catalyst showed superb catalytic performance and stability for methanol electro-oxidation because its porous nanostructure and Ru species led to a synergistic improvement.
Experimental section
Chemicals and materials
Pluronic F127 (Mn = 13000), ruthenium chloride (RuCl3, 99%), potassium tetrachloroplatinate (K2PtCl4, 98%), L-ascorbic acid (99%), potassium bromide (KBr, AR), ethanol (C2H5OH, AR), sulfuric acid (H2SO4, 97%), methanol (CH3OH, AR), and formic acid (HCOOH, AR) were purchased from Sinopharm Chemical Reagent Co., Ltd (China). Vulcan XC-72 carbon black was obtained from Cabot Co. Perfluorinated Nafion ion-exchange resin solution (5 wt%, lower aliphatic alcohol solution with 15–20% water content) was provided by Sigma-Aldrich. No additional chemical purification was performed.
Catalyst preparation
000 rpm), and this solid was washed with distilled water three times to obtain PtRu MNs.
:2 v/v ratio of ethanol/water, 15 mL) via sonication for 30 min. Next, the PtRu MNs and Vulcan XC-72 suspensions were combined and homogeneously mixed by sonicating for an additional 1 h. This mixture was centrifuged to obtain PtRu MNs/C, which was then washed with distilled water three times. Finally, the product was dried for 24 h under vacuum at 60 °C.
Characterization of electrocatalysts
A Rigaku RINT Ultima X-ray diffractometer (XRD) was operated with Cu Kα X-rays (λ = 1.5418 Å) to study the crystallographic information. The spectra were acquired over the angular 2θ range 20–80° with a scanning rate of 10° min−1. A Phenom Pharos scanning electron microscope (SEM) was used to evaluate morphology. The samples were placed in the specimen holder, and scrutinized using a scanning electron detector at an accelerated voltage of 15 kV. An FEI Talos F200x G2 transmission electron microscope (TEM, 200 kV) was utilized to obtain TEM micrographs, high-angle annular dark-field scanning TEM (HAADF-STEM) micrographs, and elemental mappings. The samples were dispersed in ethanol, and small droplets of suspensions were placed on carbon-coated copper grids for observation. A PHI 5000 Versaprobe III X-ray photoelectron spectrometer (XPS) was operated with monochrome Al Kα radiation (1486.7 eV), and the analysis chamber was maintained at about 2 × 10−10 mbar. All of the binding energies were calibrated relative to the standard C 1s peak at 284.6 eV.Electrochemical testing
The working electrode had a 0.196 cm2 surface area and was prepared with a modified glassy carbon electrode (GCE), the counter electrode was a pure Pt wire, and the reference electrode was Ag/AgCl (saturated KCl). A Metrohm Autolab PGSTAT302N electrochemical analyzer was employed for measurements with a 3-electrode cell. Working electrodes were fabricated by thoroughly combining 2 mg of catalyst with 4.6 μL Nafion, 164 μL ethanol, and 164 μL distilled water. To prepare a homogeneously mixed ink, this mixture was ultrasonicated for 20 min. After that, 6 μL catalyst ink was used to coat the GCE, which was naturally dried under ambient conditions. Cyclic voltammetry (CV) curves were obtained in methanol or formic acid aqueous solutions. The methanol oxidation reaction (MOR) was performed in a solution containing 0.5 M CH3OH and 0.5 M H2SO4 within the potential range of 0–1.2 V at a 50 mV s−1 scan rate. Long-term catalytic stability was evaluated for 500 cycles under the same conditions. Chronoamperometry (CA) was performed for 3600 s in a solution containing 0.5 M CH3OH and 0.5 M H2SO4 at a potential of 0.85 V. Electrochemical impedance spectroscopy (EIS) was performed at the frequency range from 100 kHz to 0.1 Hz in 0.5 M CH3OH with 0.5 M H2SO4. CO stripping voltammetry was performed in 0.5 M H2SO4 electrolyte. CO was purged into the H2SO4 solution for fully adsorbing on the catalyst at 0 V, and the excess CO was purged out by N2. Electrocatalytic performance and long-term stability of the as-prepared nanocatalysts toward the formic acid oxidation reaction (FAOR) were studied in a solution containing 0.5 M HCOOH and 0.5 M H2SO4. Potentials were converted to values with respect to a reversible hydrogen electrode (RHE).Results and discussion
Synthesis
The PtRu MNs were prepared using a simple one-pot synthesis procedure with K2PtCl4 and RuCl3 as the metal precursors. These precursors were reduced using L-ascorbic acid, and the surfactant Pluronic F127 was employed as a structure-directing agent to provide porosity in the PtRu MNs. To study the effect of KBr in this synthesis, the use of KCl was also investigated in a control experiment. As shown in Fig. S1 (ESI),† almost no PtRu MNs are generated when KCl is used instead of KBr, indicating that the metal precursors are not reduced by L-ascorbic acid. This likely indicates that KBr causes the generation of [PtBr4]2− species with a lower redox potential (0.58 V) via ligand exchange between [PtCl4]2− and Br−, resulting in the co-reduction of the metal precursors to obtain PtRu MNs.31 By varying the amount of RuCl3, mesoporous nanospheres with different Pt/Ru ratios were fabricated (Table S1, ESI†). Each prepared PtRu MNs catalyst was deposited on a carbon support for further characterization and evaluation in the methanol oxidation reaction. For comparison, Pt MNs/C was also prepared using the same method.Morphology
SEM images of all PtRu MNs/C samples and Pt MNs/C are displayed in Fig. S2 (ESI),† revealing that the carbon support contains a uniform dispersion of the obtained mesoporous nanospheres. SEM and TEM images of a typical PtRu MNs sample show many surface protrusions, which indicate the formation of a mesoporous nanostructure (Fig. 1). The spherical bimetallic PtRu nanoparticles have diameters of approximately 180 nm. In addition, the composition of PtRu MNs was evaluated by studying the distribution of elements. The nanospheres contain a homogeneous distribution of the two metals, demonstrating the likely creation of a PtRu alloy (Fig. 2). | ||
| Fig. 1 Representative (a) SEM and (b) TEM micrographs of typical PtRu MNs. | ||
 | ||
| Fig. 2 (a) HAADF-STEM micrograph and elemental distributions of (b) Pt and (c) Ru, and (d) their overlap in PtRu MNs. | ||
Crystallography
PtRu MNs/C and Pt MNs/C were investigated by XRD, as depicted in Fig. 3. A peak at 28° is ascribed to the C (002) crystal plane of the carbon support. The presence of face-centered cubic (fcc) Pt is indicated by the diffraction peaks located at about 39°, 46°, and 67°, which correspond to the (111), (200), and (220) crystal facets, respectively. Compared with Pt MNs/C, the incorporation of Ru causes these peaks to slightly shift toward higher 2θ values. This indicates the generation of a PtRu bimetallic alloy via Ru atom incorporation into the Pt lattice.32 Moreover, no phase separation and the successful formation of the alloy nanostructure are also demonstrated by the absence of diffraction peaks originating from Ru and Ru oxides.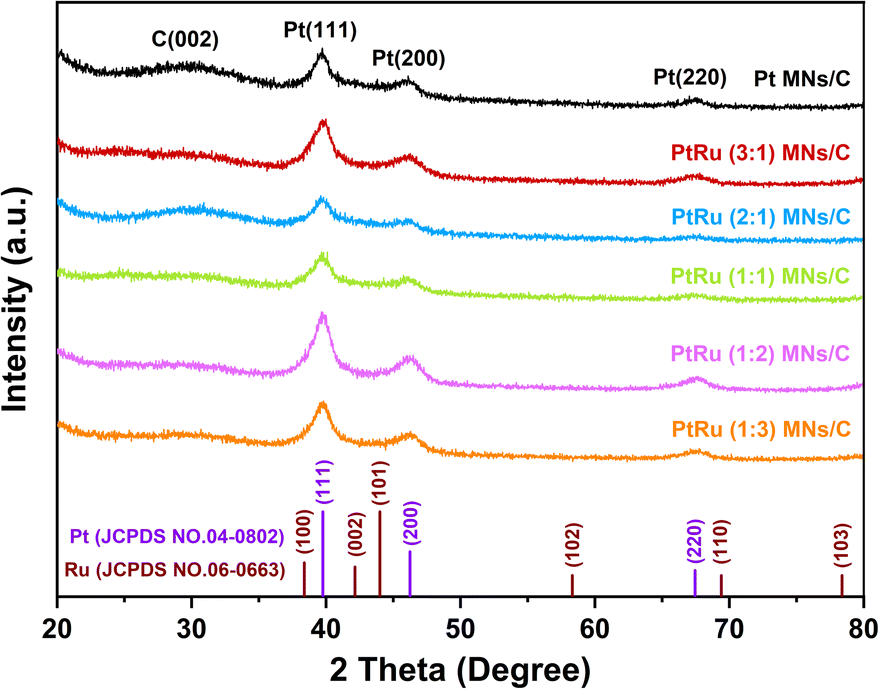 | ||
| Fig. 3 XRD patterns of PtRu MNs/C and Pt MNs/C. | ||
Surface chemical states
The electronic properties of PtRu MNs/C were explored by XPS. The Pt 4f region (Fig. 4a) displays a pair of deconvoluted Pt 4f7/2 and Pt 4f5/2 doublets, and the strong signals at 71.67 and 74.98 eV are ascribed to Pt metal. No peaks related to PtO, Pt(OH)2, or other Pt(II) species can be observed. The Ru 3d spectrum (Fig. 4b) shows Ru 3d5/2 and Ru 3d3/2 signals at 283.36 and 287.34 eV attributed to Ru(VI). Compared with pure Pt, the Pt 4f peaks of PtRu MNs/C show a slight peak shift, demonstrating that Pt may electronically interact with Ru.28 Ru is known to have a stronger affinity for oxygen than Pt.19 Therefore, the presence of Ru species in PtRu MNs/C prevents the oxidation of Pt. Moreover, the presence of Ru oxide species provides the catalyst with better electron-proton conductivity and facilitates the generation of Ru–OH species, resulting in much better catalytic performance.33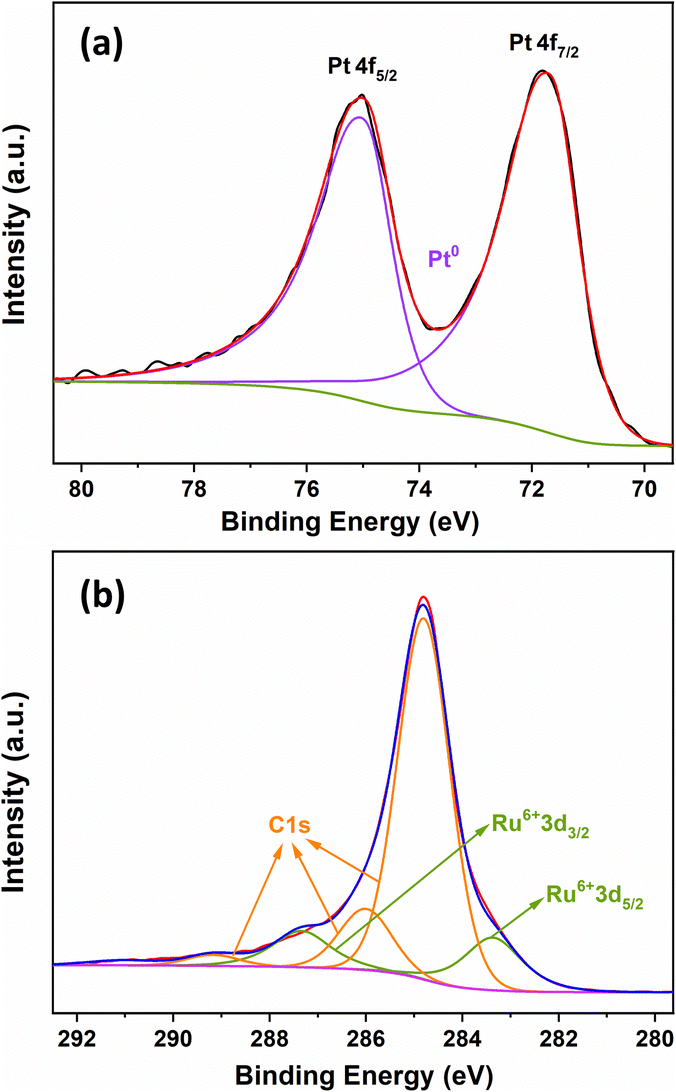 | ||
| Fig. 4 XPS analysis of PtRu MNs/C for (a) Pt 4f and (b) Ru 3d. | ||
Electrocatalytic activity and durability
Cyclic voltammetry (CV) was performed to evaluate PtRu MNs/C and Pt MNs/C electrocatalyst performance toward the MOR. Two irreversible oxidation peaks are observable in each CV curve (Fig. 5a). The oxidation of methanol on the Pt surface is responsible for the forward peak, while the reverse peak can potentially be attributed to either carbonaceous intermediate species oxidation or the MOR on regenerated Pt active sites after removing residual species.34,35 Electrocatalytic performance was evaluated by the forward peak current density and the ratio of If (forward scan peak current) to Ib (reverse scan peak current), as summarized in Table S2 (ESI).† Among the prepared PtRu-based catalysts, PtRu (2:1) MNs/C shows the best mass activity (111.77 mA mgPt−1). In contrast, the corresponding activity of Pt MNs/C (17.33 mA mgPt−1) is about 6.45 times lower than that of PtRu (2:1) MNs/C. Moreover, PtRu (2:1) MNs/C shows significantly lower methanol oxidation potentials than the other nanocatalysts. Specifically, the oxidation potential of PtRu (2:1) MNs/C is negatively shifted by ca. 0.26 V compared with Pt MNs/C at 10 mA mgPt−1 (dotted line in Fig. 5b). Electrochemical impedance spectroscopy (EIS) was performed to assess the charge transfer resistance of methanol oxidation for different catalysts (Fig. S3, ESI†). It is obvious that the main arc diameter of PtRu (2:1) MNs/C is smaller than that of Pt MNs/C, suggesting a faster charge transfer capacity toward the MOR.36,37 Besides that, PtRu (2:1) MNs/C also shows a larger If/Ib ratio (1.75) than the other prepared catalysts. A higher If/Ib ratio indicates that fewer residual carbonaceous species are accumulated on the Pt surface.38,39 Therefore, PtRu (2:1) MNs/C is more resistant to being poisoned by intermediates compared to the other catalysts. Moreover, the comparison of mass activities between PtRu (2:1) MNs/C and commercial Pt/C is illustrated in Fig. S4 (ESI).† It can be seen that the commercial one exhibits higher current density (208.86 mA mgPt−1) with a lower If/Ib ratio (0.91) than the as-prepared PtRu (2:1) MNs/C.
 | ||
| Fig. 5 (a) Cyclic voltammetry profiles of PtRu MNs/C and Pt MNs/C for the MOR. (b) Magnified view of CV curves between 0.2 and 0.85 V. | ||
The successful application of electrocatalysts in the MOR requires good long-term stability and durability. Therefore, CV curves of PtRu (2:1) MNs/C and Pt MNs/C were also obtained over 500 consecutive cycles. Long-term stability was estimated by comparing the If values of the initial cycle and the 500th cycle, as shown in Fig. 6a and b. PtRu (2:1) MNs/C retains 84.7% of its first-cycle If value after 500 cycles, while Pt MNs/C only retains 17.7% (Fig. 6c). Next, chronoamperometry (CA) measurements were performed to investigate the constant-potential durability of the catalysts (Fig. 6d). In the first few seconds, all catalysts show a rapid decline in MOR current density because intermediate carbonaceous species cause the active sites to be poisoned.40 However, PtRu (2:1) MNs/C exhibits an enhanced current density compared to Pt MNs/C across the entire testing period. To deeply investigate the CO tolerance of catalysts, CO stripping voltammograms were recorded in 0.5 M H2SO4 at a sweep rate of 20 mV s−1, as shown in Fig. S5 (ESI).† It is observed that PtRu (2:1) MNs/C and Pt MNs/C show a sharp peak in the first positive scan, which is due to oxidation of the adsorbed CO on the surface of the electrocatalyst. However, the CO oxidation peak of PtRu (2:1) MNs/C at 0.63 V is much more negative than that of Pt MNs/C (0.77 V), indicating a critical role of Ru species in enhancing the CO tolerance ability of the catalyst.41,42 These results clearly demonstrate that PtRu (2:1) MNs/C has excellent MOR activity and good catalytic stability/durability.
 | ||
| Fig. 6 CV curves of (a) PtRu (2:1) MNs/C and (b) Pt MNs/C before and after 500 potential cycles for the MOR. (c) Loss of mass activity of different electrocatalysts. (d) Chronoamperometry curves of PtRu (2:1) MNs/C and Pt MNs/C for the MOR. | ||
Another potential liquid fuel that can be used in fuel cells is formic acid. Platinum electrocatalysts have also been widely employed in the formic acid oxidation reaction (FAOR).43–45 To evaluate the applicability of PtRu MNs/C in the FAOR, CV curves of all as-synthesized samples were obtained. The forward scan has two oxidation peaks based on the reaction mechanism. The peak at ∼0.6 V (If1) is due to the direct pathway (direct formation of CO2), while that at ∼1.0 V (If2) is related to the indirect pathway (CO formed as an intermediate poison).46 As illustrated in Fig. S6 (ESI),† all the obtained CV curves show a higher If2 current density and negligible If1 current density. This suggests that the Ru in PtRu MNs/C does not promote the direct pathway, which can be ascribed to the presence of two oxygens in HCOOH.47 However, the If2 mass activity of PtRu (2:1) MNs/C is 218.90 mA mgPt−1, which exceeds those of the other PtRu MNs/C catalysts and is about 13.14 times higher than that of Pt MNs/C (16.66 mA mgPt−1). Electrocatalyst stability in the FAOR was tested by obtaining CV curves across 500 consecutive cycles (Fig. S7a and b, ESI†). The mass activity losses are summarized in Fig. S7c (ESI),† and the PtRu (2:1) MNs/C electrocatalyst shows a decline of 22.6% compared with its initial catalytic activity, while Pt MNs/C retains 79.9% of its initial activity.
The excellent electrocatalytic activity of PtRu (2:1) MNs/C can be attributed to two main reasons: (1) the mesoporosity of the PtRu (2:1) MNs/C structure provides an abundance of accessible active sites, leading to improved MOR activity; (2) the presence of the Ru element in PtRu MNs provides abundant oxygen-containing species during the MOR, which enhances catalyst durability by increasing the resistance to COads poisoning.
Conclusions
In summary, a series of electrocatalysts consisting of well-dispersed bimetallic PtRu MNs on carbon were successfully synthesized and employed as MOR catalysts. SEM and TEM images confirmed the presence of a mesoporous nanostructure, which effectively provided abundant active sites. XPS analysis indicated the existence of metallic Pt and oxidized Ru, which facilitated the removal of intermediate poisons adsorbed on the Pt surface. Benefiting from its nanostructure and composition, the optimum PtRu (2:1) MNs/C catalyst exhibited superb MOR catalytic activity. Electrochemical analysis showed that PtRu (2:1) MNs/C had a much larger current density compared to Pt MNs/C. Moreover, the larger If/Ib ratio of PtRu (2:1) MNs/C revealed that this catalyst had a stronger ability to resist the accumulation of carbonaceous substances. PtRu (2:1) MNs/C was also more durable (lower decline in mass activity and current density) than the other prepared catalysts, as demonstrated by CV curves and chronoamperometry. The results and analysis reported herein offer a compelling strategy for designing efficient Pt-based anode electrocatalysts for the MOR in DMFCs.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Jiangbin Guo: conceptualization, investigation, formal analysis, data curation, visualization, writing – original draft, writing – review & editing. Qiyu Yan: investigation, formal analysis, data curation. Man Zhang: investigation, formal analysis. Jun Fang: conceptualization, writing – review & editing, funding acquisition. Shuiyuan Luo: writing – review & editing, funding acquisition. Jing Xu: conceptualization, methodology, formal analysis, supervision, funding acquisition, writing – original draft, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Science Foundation of Fujian Province (grant no. 2022J011100 and 2020J05155), the National Natural Science Foundation of China (grant no. 21676222 and U1705252), the Science and Technology Major Special Project of Fujian Province (grant no. 2020HZ06019), the Quanzhou City Science and Technology Program of China (grant no. 2019C106), and the Scientific Research Foundation of Quanzhou Normal University (grant no. H18001 and H17015).References
- J. Li, L. Zhao, X. Li, S. Hao and Z. Wang, J. Energy Chem., 2020, 40, 7–14 CrossRef.
- B. Wu, C. Wu, J. Zhu, X. Li, J. Chu, X. Wang and S. Xiong, Ionics, 2020, 26, 4599–4608 CrossRef CAS.
- Y. V. Tolmachev and O. A. Petrii, J. Solid State Electrochem., 2017, 21, 613–639 CrossRef CAS.
- K. M. Yeo, S. Choi, R. M. Anisur, J. Kim and I. S. Lee, Angew. Chem., Int. Ed., 2011, 50, 745–748 CrossRef CAS PubMed.
- X. Liu, K. Wang, L. Zhou, H. Pu, T. Zhang, J. Jia and Y. Deng, ACS Sustainable Chem. Eng., 2020, 8, 6449–6457 CrossRef CAS.
- G. Montiel, E. Fuentes-Quezada, M. M. Bruno, H. R. Corti and F. A. Viva, RSC Adv., 2020, 10, 30631–30639 RSC.
- D. L. Quan and P. H. Le, Coatings, 2021, 11, 571 CrossRef CAS.
- J. Zhang, S. Lu, Y. Xiang and S. P. Jiang, ChemSusChem, 2020, 13, 2484–2502 CrossRef CAS PubMed.
- I. Staffell, D. Scamman, A. Velazquez Abad, P. Balcombe, P. E. Dodds, P. Ekins, N. Shah and K. R. Ward, Energy Environ. Sci., 2019, 12, 463–491 RSC.
- L. van Biert, M. Godjevac, K. Visser and P. V. Aravind, J. Power Sources, 2016, 327, 345–364 CrossRef CAS.
- B. G. Abraham, R. Bhaskaran and R. Chetty, J. Electrochem. Soc., 2020, 167, 024512 CrossRef CAS.
- Y. Feng, H. Liu and J. Yang, Sci. Adv., 2017, 3, e1700580 CrossRef PubMed.
- A. S. Moura, J. L. C. Fajín, M. Mandado and M. N. D. S. Cordeiro, Catalysts, 2017, 7, 47 CrossRef.
- Y. Zhao, J. Liu, C. Liu, F. Wang and Y. Song, ACS Catal., 2016, 6, 4127–4134 CrossRef CAS.
- Y. Wang, M. Zheng, H. Sun, X. Zhang, C. Luan, Y. Li, L. Zhao, H. Zhao, X. Dai, J.-Y. Ye, H. Wang and S.-G. Sun, Appl. Catal., B, 2019, 253, 11–20 CrossRef CAS.
- L. Bai, J. Sol–Gel Sci. Technol., 2022, 103, 118–124 CrossRef CAS.
- A. Garg, M. Milina, M. Ball, D. Zanchet, S. T. Hunt, J. A. Dumesic and Y. Román-Leshkov, Angew. Chem., Int. Ed., 2017, 56, 8828–8833 CrossRef CAS PubMed.
- A. Kaur, G. Kaur, P. P. Singh and S. Kaushal, Int. J. Hydrogen Energy, 2021, 46, 15820–15849 CrossRef CAS.
- J. Guo, M. Zhang, J. Xu, J. Fang, S. Luo and C. Yang, RSC Adv., 2022, 12, 2246–2252 RSC.
- E. Taylor, S. Chen, J. Tao, L. Wu, Y. Zhu and J. Chen, ChemSusChem, 2013, 6, 1863–1867 CrossRef CAS PubMed.
- Z. Qi, C. Xiao, C. Liu, T. W. Goh, L. Zhou, R. Maligal-Ganesh, Y. Pei, X. Li, L. A. Curtiss and W. Huang, J. Am. Chem. Soc., 2017, 139, 4762–4768 CrossRef CAS PubMed.
- Y. Lu, Y. Jiang and W. Chen, Nanoscale, 2014, 6, 3309–3315 RSC.
- M. Wakisaka, S. Mitsui, Y. Hirose, K. Kawashima, H. Uchida and M. Watanabe, J. Phys. Chem. B, 2006, 110, 23489–23496 CrossRef CAS PubMed.
- S. Lu, K. Eid, D. Ge, J. Guo, L. Wang, H. Wang and H. Gu, Nanoscale, 2017, 9, 1033–1039 RSC.
- Q. Wang, Y.-W. Zhou, Z. Jin, C. Chen, H. Li and W.-B. Cai, Catalysts, 2021, 11, 925 CrossRef CAS.
- L. Huang, X. Zhang, Q. Wang, Y. Han, Y. Fang and S. Dong, J. Am. Chem. Soc., 2018, 140, 1142–1147 CrossRef CAS PubMed.
- K. Dong, H. Pu, T. Zhang, H. Dai, X. Zhao, L. Zhou, K. Wang, Y. Wang and Y. Deng, J. Alloys Compd., 2022, 905, 164231 CrossRef CAS.
- S. Yin, R. D. Kumar, H. Yu, C. Li, Z. Wang, Y. Xu, X. Li, L. Wang and H. Wang, ACS Sustainable Chem. Eng., 2019, 7, 14867–14873 CrossRef CAS.
- H. Xu, H. Shang, C. Wang and Y. Du, Adv. Funct. Mater., 2020, 30, 2000793 CrossRef CAS.
- H. Tian, Y. Yu, Q. Wang, J. Li, P. Rao, R. Li, Y. Du, C. Jia, J. Luo, P. Deng, Y. Shen and X. Tian, Int. J. Hydrogen Energy, 2021, 46, 31202–31215 CrossRef CAS.
- H. Wang, H. Yu, S. Yin, Y. Li, H. Xue, X. Li, Y. Xu and L. Wang, Nanoscale, 2018, 10, 16087–16093 RSC.
- G. Garcia, V. Baglio, A. Stassi, E. Pastor, V. Antonucci and A. S. Aricò, J. Solid State Electrochem., 2007, 11, 1229–1238 CrossRef CAS.
- L. Bai, Appl. Surf. Sci., 2018, 433, 279–284 CrossRef CAS.
- A. Elangovan, J. Xu, A. Sekar, B. Liu and J. Li, ChemCatChem, 2020, 12, 6000–6012 CrossRef CAS.
- A. Sekar, N. Metzger, S. Rajendran, A. Elangovan, Y. Cao, F. Peng, X. Li and J. Li, ACS Appl. Nano Mater., 2022, 5, 3275–3288 CrossRef CAS.
- W. Qiao, L. Yu, J. Chang, F. Yang and L. Feng, Chin. J. Catal., 2023, 51, 113–123 CrossRef CAS.
- M. Li, F. Yang, J. Chang, A. Schechter and L. Feng, Acta Phys.-Chim. Sin., 2023, 39, 2301005 Search PubMed.
- Y. Shi, W. Zhu, H. Shi, F. Liao, Z. Fan and M. Shao, J. Colloid Interface Sci., 2019, 557, 729–736 CrossRef CAS PubMed.
- Y. Sang, R. Zhang, B. Xu, J. Yang, C. Zhao and H. Xu, ACS Appl. Energy Mater., 2022, 5, 4179–4190 CrossRef CAS.
- J. Zhang, X. Qu, Y. Han, L. Shen, S. Yin, G. Li, Y. Jiang and S. Sun, Appl. Catal., B, 2020, 263, 118345 CrossRef CAS.
- X. Ding, M. Li, J. Jin, X. Huang, X. Wu and L. Feng, Chin. Chem. Lett., 2022, 33, 2687–2691 CrossRef CAS.
- C. Liu and L. Feng, Chin. J. Struct. Chem., 2023, 42, 100136 CrossRef CAS.
- X. Yu and P. G. Pickup, J. Power Sources, 2008, 182, 124–132 CrossRef CAS.
- T. Chen, C. Ge, Y. Zhang, Q. Zhao, F. Hao and N. Bao, Int. J. Hydrogen Energy, 2015, 40, 4548–4557 CrossRef CAS.
- J. Xu, M. Zhao, S.-i. Yamaura, T. Jin and N. Asao, J. Appl. Electrochem., 2016, 46, 1109–1118 CrossRef CAS.
- K. Jiang, H.-X. Zhang, S. Zou and W.-B. Cai, Phys. Chem. Chem. Phys., 2014, 16, 20360–20376 RSC.
- M. S. Çögenli and A. B. Yurtcan, Int. J. Hydrogen Energy, 2018, 43, 10698–10709 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Optimization of synthetic conditions and characterization results. See DOI: https://doi.org/10.1039/d4na00210e |
| This journal is © The Royal Society of Chemistry 2024 |