
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAccelerating the electron-transfer of nitrogen electro-fixation through assembling Fe nanoparticles into Fe nanochains†
Rongkang
Wang‡
 *a,
Jingyu
Lu‡
*b,
Xu
Li
c and
Chunyu
Song
a
*a,
Jingyu
Lu‡
*b,
Xu
Li
c and
Chunyu
Song
a
aChongqing Chemical Industry Vocational College, Chongqing, 401228, China. E-mail: wrk1992@163.com
bSchool of Materials Science and Engineering, China University of Petroleum (East China), Qingdao 266580, China
cSouthwest Technology and Engineering Research Institute, Chongqing 401329, China
First published on 1st July 2024
Abstract
Electrochemically synthesizing NH3via N2 is a facile and sustainable approach that involves multistep electron and proton transfer processes. Thus, consecutive electron and proton transfer is necessary. Here, a universal method with the assistance of magnetic stirring that can assemble Fe, Co, and Ni nanoparticles into nanochains is developed. Notably, the Fe nanochain, composed of amorphous Fe nanoparticles, facilitates electron and proton transfer, resulting in an enhanced NH3 yield (92.42 μg h−1 mg−1) and faradaic efficiency (20.02%) at −0.4 V vs. RHE during the electrochemical reduction of N2. This work offers new insight into designing tandem electrocatalysts.
Introduction
Ammonia (NH3) is a vital component in many industrial applications, including fertilizers, pharmaceuticals, and refrigerants, underscoring its global significance.1–4 Currently, the industrial Haber–Bosch process, which is the primary method for producing NH3, typically operates at high temperatures (400–500 °C) and pressures (200–300 atm), leading to substantial energy consumption worldwide.5,6 In recent years, the electrochemical synthesis of NH3 from the N2 reduction reaction (NRR) has emerged as a promising and sustainable alternative, allowing for the hydrogenation of N2 molecules through multistep proton and electron transfers at ambient temperature and pressure.7–10 In this context, the role of electrocatalysts, particularly those based on transition metals, is of paramount importance. Inspired by natural nitrogenases such as FeMo-, FeV-, and FeFe-found in rhizobium plants, Fe-based electrocatalysts have been extensively studied.11–13 Various Fe compounds, including Fe oxides and Fe sulfides, have demonstrated excellent performance in the NRR.14–16Amorphous electrocatalysts, due to their high density of exposed active sites, have been extensively explored in the NRR.17,18 The concept of tandem active sites, which has been successfully applied in several multistep electrocatalytic processes, offers a promising approach.19–22 These active sites, each possessing distinct adsorption–desorption characteristics and electron transfer capabilities, can be sequentially arranged at the atomic level to enhance electrocatalytic performance.20 Additionally, the assembly of small particles into different morphologies or pore structures can further facilitate electron and proton transfer.23 Therefore, the strategic utilization of active sites in amorphous materials and the assembly of tandem electrocatalysts present a potential pathway for the design of electrocatalysts for nitrogen electro-fixation.
Herein, we assemble amorphous Fe nanoparticles into nanochains by a facile magnetic-assisted method. When the Fe nanochain was applied as the electrocatalyst for electrochemical reduction of N2, it takes full advantage of the active sites of amorphous Fe nanoparticles to promote the multistep electron and proton transfer and produce NH3.
Experimental section
Materials synthesis
The Fe nanochain was synthesized by the chemical reduction method. In detail, 50 mg FeCl2 was dissolved in 30 mL of deionized water. Then 500 mg NaBH4 was dosed into the above solution and magnetically stirred at 1600 rpm at room temperature for 2 h. The Fe nanochain precipitates were finally obtained by cleaning with deionized water three times. The Fe nanoparticles were obtained by a similar procedure to Fe nanochains but without magnetic stirring just standing for 2 h.Physical characterization
Morphology was observed on a field emission scanning electron microscope (FESEM, JEOL 7800F), transmission electron microscope (TEM), and high-resolution transmission electron microscope (HRTEM, Titan G2 60-300, a spherical and chromatic aberration imaging corrector). The crystal structure was examined by using an X-ray diffractometer (XRD, D8 Advance, Bruker, Karlsruhe, Germany 9 kw, 40 kV, 40 mA, λ = 1.5418 Å) with Cu-Kα radiation. The chemical state was analyzed by X-ray photoelectron spectroscopy (XPS, Thermo ESCALAB 250XI between 0 and 1400 eV). Ultraviolet-visible (UV-vis) absorption spectra were recorded on an Agilent Cary 60.Results and discussion
Synthesis and characterization
The Fe nanochain, assembled via magnetic stirring during the chemical reduction process, is composed of Fe nanoparticles. The morphology was visualized using scanning electron microscopy (SEM). As shown in Fig. 1a, the Fe nanochain extends to approximately 3.1 μm in length, and its interwoven structure forms conspicuous nanopores (Fig. 1b). Fig. 1c illustrates the orderly assembly of Fe nanoparticles into the nanochain. In contrast, in the absence of magnetic stirring, the Fe nanoparticles accumulate randomly into agglomerates (Fig. 1d and e). Upon magnification, the individual Fe nanoparticles become clearly visible (Fig. 1f). The STEM elemental mapping of Fe nanochains and Fe nanoparticles respectively demonstrates that Fe is uniformly dispersed in both the nanochains and nanoparticles (Fig. 1g–j). | ||
| Fig. 1 SEM images of (a–c) Fe nanochains and (d–f) Fe nanoparticles. (g and h) Element mapping of Fe nanochains. (i and j) Element mapping of Fe nanoparticles. | ||
Transmission Electron Microscopy (TEM) images further corroborate the nanochain morphology of Fe (Fig. 2a and b). Upon magnification of the edge of the Fe nanochain, no lattice fringe is observed. Examination of a single Fe nanoparticle within the nanochain reveals an edge approximately 4 nm thick and a core with a diameter of 23 nm. Within this structure, Fe atoms exhibit a gradual ordering from the edge (Fig. 2e) to the core (Fig. 2d). In addition, no diffraction dots and rings are observed in FFT from the corresponding TEM (Fig. 2f), suggesting its amorphous phase. In agreement with the TEM results, the XRD patterns also show the Fe nanoparticle and nanochain are amorphous (Fig. S1†). This amorphous material may expose a more active site to promote the electrocatalytic NRR. To explore the universality of the synthesized nanochain, magnetic element Co and Ni nano-chains were also prepared by the same method as the Fe nanochain. Fig. S2–S5† show that the Ni and Co nano-chains were successfully obtained which also exhibit their amorphous state.
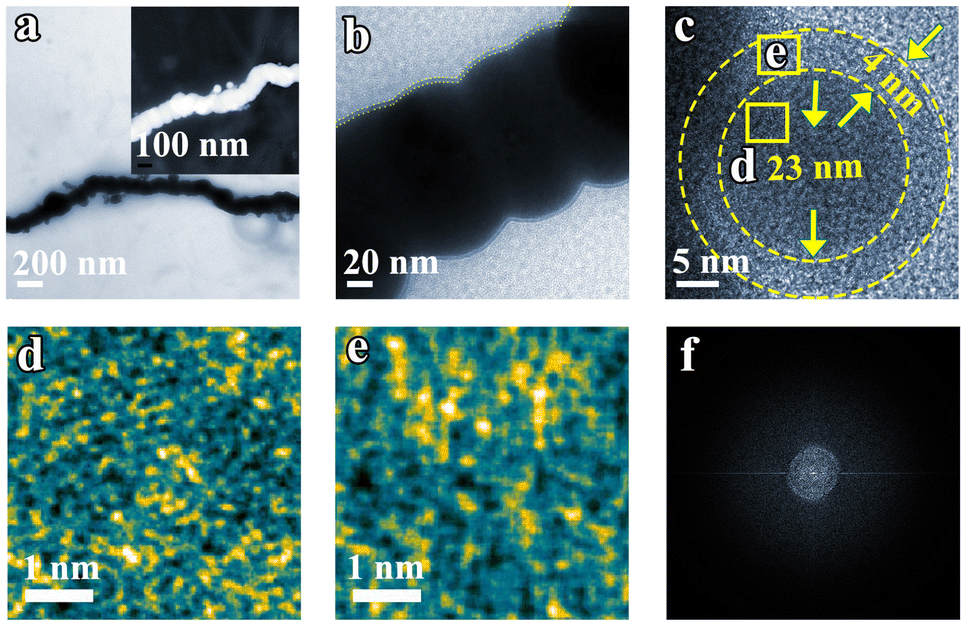 | ||
| Fig. 2 (a and b) TEM images of Fe nanochains. (c) HRTEM image of the Fe nanochain. (d and e) Selected area magnification HR-TEM from the edge (d) and interior (e). (f) Electron diffraction obtained by Fourier transform. | ||
Electrocatalytic nitrogen reduction performances
The electrocatalytic N2 fixation performances of the amorphous Fe nanochain and nanoparticle were evaluated. First, linear sweep voltammetry (LSV) was performed in N2 and Ar-saturated electrolytes (Fig. 3a) to show the response current of electrochemical N2 reduction. It shows that the current in N2-saturated electrolytes is higher than that in Ar-saturated electrolytes and suggests that N2 undergoes electrochemical reduction with the Fe nanochain electrode. Furthermore, the currents at different potentials were examined to show the electro-reduction of N2 (Fig. 3b). From −0.2 V vs. RHE to −0.6 V vs. RHE, the corresponding currents increase up to about 1.6 mA cm−1 at −0.6 V vs. RHE. Generally, N2 is reduced to NH3via the six-electron pathway or N2H4via the four-electron pathway through electrochemical fixation. Thus, the indophenol blue and Watt–Chrisp methods were applied to determine the finally generated products (Fig. S6–S10†). The resulting product NH3 was quantified. Corresponding with the increase of currents, from −0.2 to −0.4 V vs. RHE, the yield rate and faradaic efficiency of NH3 are gradually up to 92.42 μg h−1 mg−1 and 20.02% (Fig. 3c and d). Nevertheless, the potential at −0.4 V vs. RHE presents the highest yield rate and faradaic efficiency which is higher than the potential at −0.5 V vs. RHE (52.62 μg h−1 mg−1 and 6.02%) and −0.6 V vs. RHE (43.94 μg h−1 mg−1 and 2.61%). Therefore, compared to the state-of-the-art catalysts reported in the literature, our Fe nanowires are among the most outstanding NRR electrocatalysts disclosed to date (Table S1†). The stability of activity was examined using the current at −0.4 V vs. RHE. As shown in Fig. 3e, any decrease of current is presented for 60 h at −0.4 V vs. RHE and the LSV of the electrode after 60 h also suggests this result, which means the sustainable activity for electro reduction of N2 of this Fe nanochain. After the stability test, the XRD of the used Fe nanochain was used to characterize the crystal change (Fig. S11†). It presents a sharper peak ascribed to Fe of the used Fe nanochain than the fresh Fe nanochain. Additionally, the TEM images of the used Fe nanochains show that the nanochain structure remains intact after the stability test, with a more distinct interface between crystalline and amorphous regions. The observed lattice fringes can be attributed to the Fe (110) plane (Fig. S12†). This indicates that the amorphous Fe nanowires gradually crystallize during the electrocatalytic N2 reduction process. To trace the source of NH3, the yields of NH3 were measured at the open-circuit potential and applying the gas diffusion electrode at −0.4 V vs. RHE, suggesting almost no NH3 generation (0.64 μg h−1 mg−1 and 1.03 μg h−1 mg−1) (Fig. 3f). Besides, the yield of NH3 applying Ar-saturated electrolyte is 0.92 μg h−1 mg−1 but the N2-saturated electrolyte exhibits 92.42 μg h−1 mg−1 yield of NH3. It shows that the NH3 results from the electroreduction of N2 in electrolyte, suggesting the high activity of the Fe nanochain for N2 electro-fixation.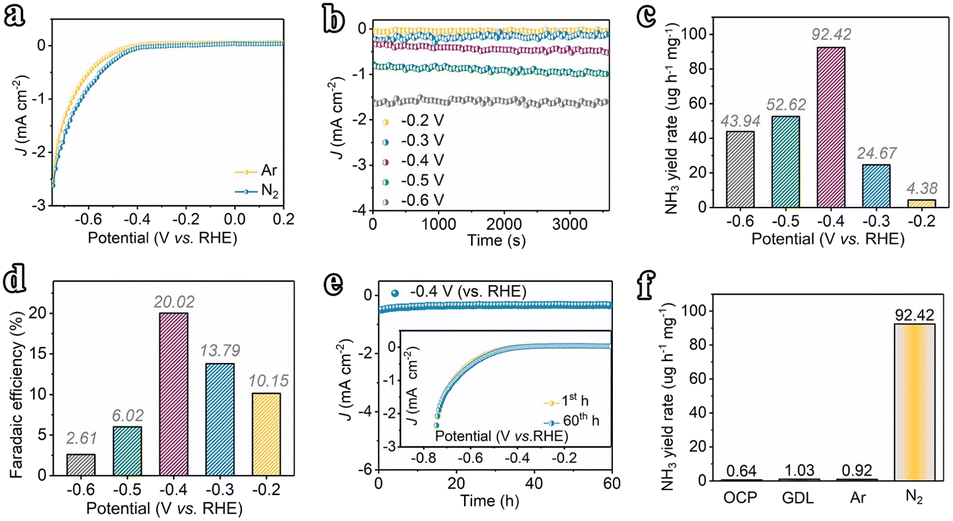 | ||
| Fig. 3 (a) LSV curves of the Fe nanochain electrode recorded for N2 electro-fixation in N2-saturated and Ar-saturated 0.1 M Na2SO4 solution (scan rate 5 mV s−1). (b) Chronoamperometry curves of the Fe nanochain at −0.2–0.6 V (vs. RHE). (c and d) NH3 yields and faradaic efficiencies of the Fe nanochain at −0.2–0.6 V (vs. RHE). (e) Stability test for 60 h at −0.4 V (vs. RHE); the inset of (e) shows LSVs of the electrode before and after the stability test. (f) NH3 yields of the Fe nanochain at −0.4 V (vs. RHE) under different testing conditions. | ||
Comparatively, the Fe nanoparticles are not assembled into nanochains and may not exhibit similar activity for N2 electro-fixation. The LSV of the Fe nanoparticle in Ar and N2 saturated electrolytes was measured (Fig. S13†). Almost few current responses are presented for electrocatalytic N2 reduction whereas high current densities are presented at different potentials (Fig. S14†). To accurately determine the generation of NH3 or not, the concentration of the produced NH3 was measured. As shown in Fig. 4a, the yield of NH3 of Fe nanoparticles is about 32.82 μg h−1 mg−1 accompanied by 6.01% faradaic efficiency, which is far below that of the Fe nanochain (Fig. S15†). The total current densities of Fe nanochains and Fe nanoparticles at different potentials were examined to show the electrochemical activity. The Fe nanoparticle presents a higher total current density at −0.4 V vs. RHE and −0.3 V vs. RHE than the Fe nanochain (Fig. 4b). However, the partial current densities for electrocatalytic N2 reduction are different between Fe nanochains and Fe nanoparticles (Fig. 4c), in which the difference at −0.4 V vs. RHE is the greatest, also indicating the higher activity of Fe nanochains than Fe nanoparticles. This superior activity may be ascribed to the continuous electron transfer process of Fe nanochains. Thus, the Tafel slopes are used to explain the electron transfer rate. As presented in Fig. 4d, the Fe nanochain exhibits a lower Tafel slope than the Fe nanoparticle, suggesting a faster electron transfer process for N2 reduction. Furthermore, the Fe nanochain exhibits a larger 0.85 mF cm−2 specific capacitance than the Fe nanoparticle (0.60 mF cm−2) (Fig. 4e and S16†), implying the larger electrochemical active surface,24–26 which could contribute to the electrochemical performance of N2 reduction. Therefore, for Fe nanochains, several single amorphous Fe nanoparticles were stringed into nanochains which present more consecutive electron transfer than a single nanoparticle. This plays an important role in the multistep electron transfers of electrochemical N2 reduction and mainly promotes the corresponding performance of Fe nanochains.
 | ||
| Fig. 4 (a) NH3 yields and faradaic efficiencies of Fe nanochains and Fe nanoparticles. (b) Current densities of Fe nanochains and Fe nanoparticles at −0.2–0.6 V (vs. RHE). (c) Partial current densities for N2 electroreduction at −0.2–0.6 V (vs. RHE). (d) Tafel slopes of Fe nanochains and Fe nanoparticles. (e) Capacitive plots of Fe nanochains and Fe nanoparticles at different scan rates (60–140 mV s−1). (f) Schematic mechanism for promoting the electroreduction of N2. | ||
Conclusions
In conclusion, amorphous Fe nanoparticles were assembled into Fe nanochains via the magnetic-assisted method. This magnetic-assisted method exhibits the universality that assembles the magnetic Co and Ni nanoparticles into nanochains. The assembled Fe nanochain exhibits more excellent performance for electrochemical N2 reduction than the amorphous Fe nanoparticle. The Fe nanochain as the electrode for electrochemical N2 reduction takes full advantage of the exposed active sites of the single amorphous Fe nanoparticle and facilitates the continuous electron transfer process, which is important to the multistep electron transfer for the reduction of N2 to NH3. This work adopts a novel method for assembling Fe nanoparticles into Fe nanochains and offers promising guidance for designing electrocatalysts.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
R. W. and J. L. conceived and designed the research; R. W. and J. L. synthesized the samples and performed the characterization; R. W., J. L., X. L. and C. S. carried out the electrochemical measurements. R. W. and J. L. discussed and all authors co-wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Research project of Chongqing Municipal Education Commission (KJQN202004501 and KJQN202304508).Notes and references
- Y. Sun, W. Fan, Y. Li, N. L. D. Sui, Z. Zhu, Y. Zhou and J. M. Lee, Adv. Mater., 2024, 36, e2306687 CrossRef PubMed.
- W. Qiu, X. Xie, J. Qiu, W. Fang, R. Liang, X. Ren, X. Ji, G. Cui, A. M. Asiri, G. Cui, B. Tang and X. Sun, Nat. Commun., 2018, 9, 3485 CrossRef.
- Y. Sun, L. Yu, S. Xu, S. Xie, L. Jiang, J. Duan, J. Zhu and S. Chen, Small, 2022, 18, 2106358 CrossRef CAS.
- C. Tang and S. Qiao, Chem. Soc. Rev., 2019, 48, 3166 RSC.
- Y. Sun, X. Li, Z. Wang, L. Jiang, B. Mei, W. Fan, J. Wang, J. Zhu and J. M. Lee, J. Am. Chem. Soc., 2024, 146, 7752 CrossRef CAS.
- X. Zhao, G. Hu, G. Chen, H. Zhang, S. Zhang and H. Wang, Adv. Mater., 2021, 33, 2007650 CrossRef CAS PubMed.
- Y. Sun, W. Wu, L. Yu, S. Xu, Y. Zhang, L. Yu, B. Xia, S. Ding, M. Li, L. Jiang, J. Duan, J. Zhu and S. Chen, Carbon Energy, 2023, 5, e263 CrossRef CAS.
- Y. Sun, S. Ding, B. Xia, J. Duan, M. Antonietti and S. Chen, Angew. Chem., Int. Ed., 2022, 61, e2021151 Search PubMed.
- Y. Sun, B. Xia and J. Duan, J. Mater. Chem. A, 2021, 9, 20040 RSC.
- X. Wu, L. Xia, Y. Wang, W. Lu, Q. Liu, X. Shi and X. Sun, Small, 2018, 14, 1803111 CrossRef.
- Y. Zhang, J. Zhao, D. Yang, B. Wang, Y. Zhou, J. Wang, H. Chen, T. Mei, S. Ye and J. Qu, Nat. Chem., 2022, 14, 46 CrossRef CAS PubMed.
- M. D. Walter, Nat. Chem., 2021, 14, 12 CrossRef.
- A. McSkimming and D. L. M. Suess, Nat. Chem., 2021, 13, 666 CrossRef CAS PubMed.
- T. Wu, X. Zhu, Z. Xing, S. Mou, C. Li, Y. Qiao, Q. Liu, Y. Luo, X. Shi, Y. Zhang and X. Sun, Angew. Chem., Int. Ed., 2019, 58, 18449 CrossRef CAS PubMed.
- J. Duan, Y. Sun, S. Chen and C. Zhao, J. Mater. Chem. A, 2020, 8, 18810 RSC.
- Y. Sun, T. Jiang, J. Duan, L. Jiang, X. Hu, H. Zhao, J. Zhu, S. Chen and X. Wang, ACS Catal., 2020, 10, 11371 CrossRef CAS.
- B. Jiang, H. Xue, P. Wang, H. Du, Y. Kang, J. Zhao, S. Wang, W. Zhou, Z. Bian, H. Li, J. Henzie and Y. Yamauchi, J. Am. Chem. Soc., 2023, 145, 6079 CrossRef CAS.
- J. Kang, X. Chen, R. Si, X. Gao, S. Zhang, G. Teobaldi, A. Selloni, L. Liu and L. Guo, Angew. Chem., Int. Ed., 2023, 62, e2022174 Search PubMed.
- H. Yan, K. He, I. A. Samek, D. Jing, M. G. Nanda, P. C. Stair and J. M. Notestein, Science, 2021, 371, 1257 CrossRef CAS.
- W. He, J. Zhang, S. Dieckhöfer, S. Varhade, A. C. Brix, A. Lielpetere, S. Seisel, J. R. C. Junqueira and W. Schuhmann, Nat. Commun., 2022, 13, 1129 CrossRef CAS PubMed.
- B. K. Mahato and A. Barman, J. Mater. Sci., 2021, 56, 19476 CrossRef CAS.
- Z. Wei, Y. Qiu, H. Chen, K. Yan, Z. Zhu, Q. Kuang and S. Yang, J. Mater. Chem. A, 2014, 2, 5508 RSC.
- S. Yu, D. Kim, Z. Qi, S. Louisia, Y. Li, G. A. Somorjai and P. Yang, J. Am. Chem. Soc., 2021, 143, 19919 CrossRef CAS PubMed.
- S. Cao, Y. Sun, S. Guo, Z. Guo, Y. Feng, S. Chen, H. Chen, S. Zhang and F. Jiang, ACS Appl. Mater. Interfaces, 2021, 13, 40618 CrossRef CAS.
- Y. Sun, S. Ding, C. Zhang, J. Duan and S. Chen, J. Mater. Chem. A, 2021, 9, 1603 RSC.
- Y. Sun, M. Li, J. Duan, M. Antonietti and S. Chen, Angew. Chem., Int. Ed., 2024, e202402678 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4na00281d |
| ‡ These authors equally contributed to the work. |
| This journal is © The Royal Society of Chemistry 2024 |