
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAtomically dispersed Pt1Ir1 pair for synergetic hydrogenation of levulinic acid to γ-valerolactone†
Boyang
Liu
 a,
Lin-Wei
Chen
b and
Lei
Wang
*a
a,
Lin-Wei
Chen
b and
Lei
Wang
*a
aSchool of Chemistry and Chemical Engineering, Yangzhou University, Yangzhou, 225009, Jiangsu, China. E-mail: leiwang88@yzu.edu.cn
bSchool of Pharmacy & Institute of Pharmaceutics, Anhui University of Chinese Medicine, Hefei, 230012, China
First published on 19th July 2024
Abstract
Atomically dispersed metal catalysts have attracted considerable attention in various important reactions owing to their high atom utilization and specific coordination environment. However, monometallic single sites sometimes present undesirable catalytic performance, which usually need a synergistic effect with the neighboring metal atoms, such as dimers or trimers. Different metal pairs on various solid carriers have been reported; nonetheless, huge challenges remain to precisely prepare a metal pair-site. Herein, we present a versatile strategy to synthesize an atomically dispersed Pt1Ir1 pair via strong metal–sulfur interaction over porous sulfur-doped carbons. Pt1Ir1 pair sites presented high activity and stability for the hydrogenation of levulinic acid to γ-valerolactone.
1 Introduction
Noble metals (Pt, Au, Ir, Rh, etc.) present high catalytic activities in heterogeneous catalysis and energy conversion applications owing to their specific geometric/electronic structures.1–6 Nevertheless, the high cost of noble metals restricts their widespread applications. For supported nanoparticles, only the exposed metal atoms could directly access reactants, resulting in low metal utilization. In addition, irregular surface structures or different surface sites always afford undesired byproducts during the catalytic process.7,8 Atomically dispersed metal catalysts supported on solid carriers are of immense interest owing to their maximum metal utilization efficiency and specific electronic/geometric structures.9–14 Generally, the surface free energy of the particles increased sharply with a decreased particle size; thus, isolated metal atoms are prone to aggregate into particles.15 Single metal atoms are anchored onto solid carriers by coordinating with neighbouring atoms (S, O, N, P, etc.) to form different coordination environments.16–20 Homogeneous active sites and specific electronic structures endow this class of heterogeneous catalysts with the properties of homogeneous catalysts.21 More importantly, the catalytic performance could be versatilely regulated by tuning coordination ligands.Although single site catalysts presented distinguished performance in various important catalytic applications, the application of this type of catalyst is limited because the adsorption/activation of small molecules need the synergetic effect of neighbouring or adjacent metal atoms. For example, the splitting of H2 molecules usually occurs on the surface of metal particles or clusters with adjacent metal atoms.22 In addition, the optimum configuration of reaction intermediates might need more than one metal site; thus, single atom sites could not present desirable catalytic performance.23–25 Zeng successfully prepared high-density atomically dispersed Pt on MoS2 nanosheets.26 For the CO2 hydrogenation reaction, only methanol was formed on isolated Pt sites, while formic acid and methanol were detected with the help of adjacent Pt atoms, which could not only reduce the reaction barrier, but also change the reaction path. Wang successfully prepared metal dimers over mesoporous carbon nitride via a “precursor-preselected” wet-chemistry strategy.27 Fe2 dimers presented outstanding catalytic performance for the epoxidation of trans-stilbene to trans-stilbene oxide. In addition, Lu precisely synthesized NiCu2 trimers over graphitic carbon nitride through the atomic layer deposition (ALD) method.28 NiCu2 trimers displayed high activity and strong stability for the hydrogenation of acetylene. The past years have seen reports on numerous metal pairs with optimum catalytic structures;29 yet, these approaches still face practical implementation challenges, such as the expense of metal precursors employed in synthesis and the difficulty in obtaining large-scale catalysts.
In this work, we successfully prepared atomically-dispersed Pt1Ir1 bimetallic catalysts via the sulfur-fixing strategy. The strong chemical metal–S interaction could effectively prevent the agglomeration of the metal atoms. The fully dispersed Pt1Ir1 pair were characterized by aberration-corrected high-angle annular dark-field scanning transmission electron microscopy, X-ray photoelectron spectroscopy, and powder X-ray diffraction. The maximizing atom utilization of the atomically-dispersed Pt1Ir1 pairs displayed high catalytic performance with synergetic effect for the hydrogenation of levulinic acid to γ-valerolactone.
2 Experimental section
2.1 Synthesis of mesoporous S–C support
The S–C carrier was prepared via the transition metal-assisted carbonization of molecular precursor according to a previous report.30 Typically, 2,2-bithiophene (2 g), SiO2 sphere (∼7 nm, 2 g), and Co(NO3)2·6H2O (1 g) were added into 150 mL of tetrahydrofuran (THF) and stirred for 6 hours under room temperature. The THF solvent was then removed by rotary evaporation. The dried powder was ground in an agate mortar and then carbonized at 800 °C for 2 hours with a heating rate of 5 °C min−1 under flowing inert N2 atmosphere. The SiO2 template was removed by alkaline etching with 2.0 M NaOH for 3 days. Then, the Co particle was etched with 0.5 M H2SO4 for 6 hours at 80 °C successively. Finally, the S–C support was obtained after drying overnight at 100 °C.2.2 Synthesis of Pt1Ir1/S–C catalysts
The Pt1Ir1/S–C catalyst was prepared via the conventional wet-impregnation method. Typically, 50 mg of S–C was first dispersed into 30 mL of deionized water (DI) before a certain amount of H2PtCl6 (0.25 mg Pt) and IrCl3 (0.25 mg Ir) aqueous solution was added. After ultrasonic treatment for 30 min and stirring for 12 h, the DI was removed by rotary evaporation. The metal salts/S–C samples were reduced at 300 °C following 5% H2/Ar for 2 h with a heating rate of 5 °C min−1. The mono metallic Pt1/S–C and Ir1/S–C catalysts were also prepared by a similar process with 1 wt% content.2.3 Catalysts' characterization
Transmission electron microscopy (TEM) images were obtained using a JEOL-2010F transmission electron microscope with an acceleration voltage of 120 kV. High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images and energy dispersive X-ray spectroscopy (EDS) mappings were conducted on an FEI Talos F200X operated at 200 kV. Aberration-corrected HAADF-STEM was performed on a JEM-ARM200F with accelerating voltage of 200 kV. Powder X-ray diffraction (XRD) analysis was conducted on a Rigaku Smartlab equipped with Cu Kα radiation (λ = 1.54056 Å) in the 2θ range of 20°–80° at a scanning speed of 10° min−1. The chemical states of the bimetallic Pt1Ir1/S–C catalysts were analyzed by X-ray photoelectron spectroscopy (XPS) performed on a Thermo Fisher Scientific K-Alpha with an Al Kα source (1486.6 eV). The binding energy for all the elements were calibrated with the C 1s peak (284.8 eV) of adventitious carbon. The metal content was determined using an Optima 7300 DV (PerkinElmer) inductively coupled plasma atomic emission spectrometry (ICP-AES) instrument. The Pt metal was digested in aqua regia overnight at 373 K, while the digestion of Ir metal was done in a high-pressure reactor at 160 °C for 8 hours. X-ray absorption near edge structure (XANES) measurement of S L-edge was performed at the BL11U beamline of the National Synchrotron Radiation Laboratory (Hefei) operated at 0.8 GeV and 300 mA. The Brunner–Emmet–Teller (BET) surface area and pore size distribution of the support were measured by N2 sorption isotherms (77 K) and the Barrett–Joyner–Halenda model with an Autosorb-iQ (Quantachrome Instruments, USA).2.4 Catalysts performance evaluation
In a typical experiment, 0.5 mmol of levulinic acid, 5 mg of Pt1Ir1/S–C catalyst, and 2 mL of solvent were added into a glass reaction vessel. Next, the reaction vessel was first ultrasonicated for five minutes and loaded into an autoclave reactor (Parr reactor, 300 mL). The autoclave reactor was then purged with pure H2 for five times and pressurized to 4.0 MPa. The reactor was preheated to 180 °C and then stirred at 800 rpm to eliminate external mass transfer limitations. Orthoxylene was introduced into the reaction system as an internal standard after the reaction was finished and the autoclave was submerged in freezing water. Finally, the reaction products were analyzed using a Shimadzu gas chromatograph with a flame ionization detector with high-purity nitrogen as the carrier gas. The activity was calculated under the conversion of <25% based on the total amount of metal. For the recycling test, the catalyst was separated by centrifugation, washed with ethanol and ethyl acetate successively, and then reduced at 200 °C in H2/Ar for 2 h.3 Results and discussion
3.1. Synthesis and characterization of the catalysts
The S–C material showed a distinct hysteresis loop, as seen by the N2 adsorption–desorption isotherms, suggesting that the S–C material presented typical mesopores (Fig. 1a). The as-prepared mesoporous S–C carrier presented a large BET surface area of 1070 m2 g−1, and the main pore size diameter was about 7 nm (Fig. 1b), which is regulated by the SiO2 sphere template. The sulfur content of S–C was >11.3 wt%, as revealed by elemental analysis, and no obvious particles or clusters were observed after leaching by H2SO4 (Fig. 1c). A small amount of Co species (∼0.18 wt%) was left in the S–C carrier. EDS-mapping demonstrated the homogeneous distribution of sulfur in the mesoporous S–C support (Fig. 1d).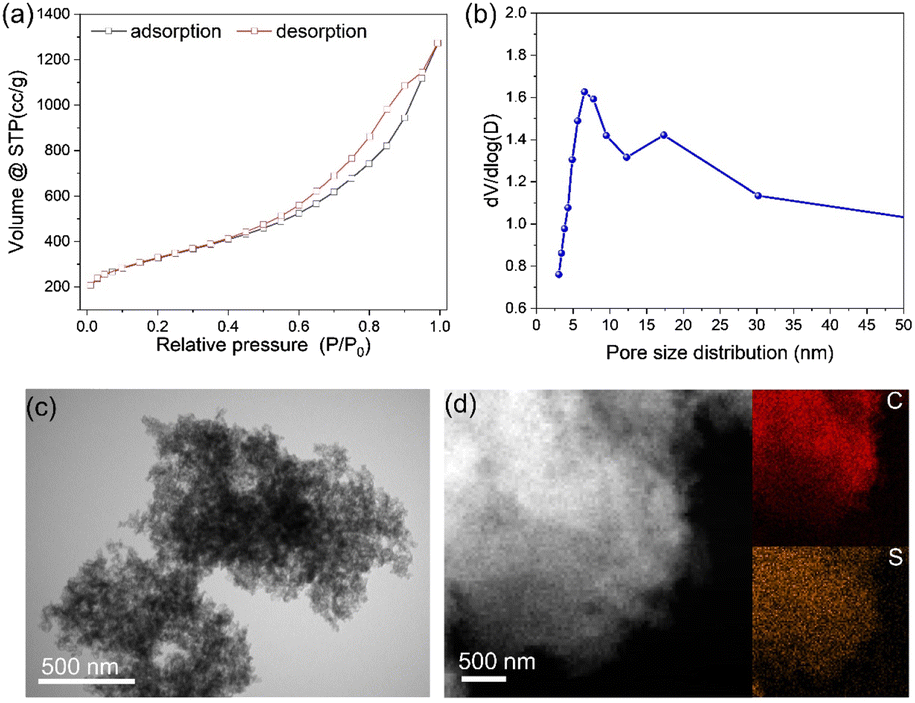 | ||
| Fig. 1 (a and b) N2 adsorption–desorption isotherms (a) and pore size distribution (b) of the S–C carrier. (c and d) TEM (c) and EDS-mappings (d) of the mesoporous S–C support. | ||
The Pt1Ir1/S–C catalysts were then synthesized via a traditional impregnation method that involved the wet impregnation of chloroplatinic acid and iridium chloride onto the S–C carrier and subsequent high-temperature reduction at 300 °C in H2 atmosphere. As revealed by the TEM and HAADF-STEM images (Fig. 2a and b), no obvious metal particles or tiny clusters were observed, demonstrating the high dispersion of the PtIr/S–C catalyst. The Pt and Ir metal atoms were homogeneously dispersed over the S–C carrier according to the EDS-mapping (Fig. 2b, S1 and S2†). No specific metal XRD peak was observed in the XRD pattern for PtIr/S–C sample according to the Pt (PDF#04-0802) and Ir (PDF#06-0598) PDF card (Fig. 2c), indicating the low crystallinity and absence of large metal particles. The broad peaks at 2θ ∼26° and 43° are attributed to the low graphitization degree of the S–C material.31 Aberration-corrected HAADF-STEM was taken to further identify the fine structure of the highly dispersed PtIr/S–C (Fig. 2d). Single atoms (white circles), dimers (yellow rectangles), and cluster with 4–6 atoms (red squares) were observed. Rough statistics based on the total atoms indicated that less than 20% of isolated single atoms were distributed over the S–C support. Most metal atoms were present in the form of dimers or clusters onto the S–C carrier (denoted Pt1Ir1/S–C), which is illustrated in Fig. 2e. The highly dispersed metals might be ascribed to the strong metal–S coordination.30,32,33 XPS results demonstrated that all the metal species for the atomically dispersed catalysts were in the same oxidation state (Fig. S3†), demonstrating charge transfer between the metal and S–C support via metal–S coordination, which is consistent with most of the reported single atom catalysts. The oxidized Pt site might provide a Lewis acid center during the synergetic hydrogenation process.34
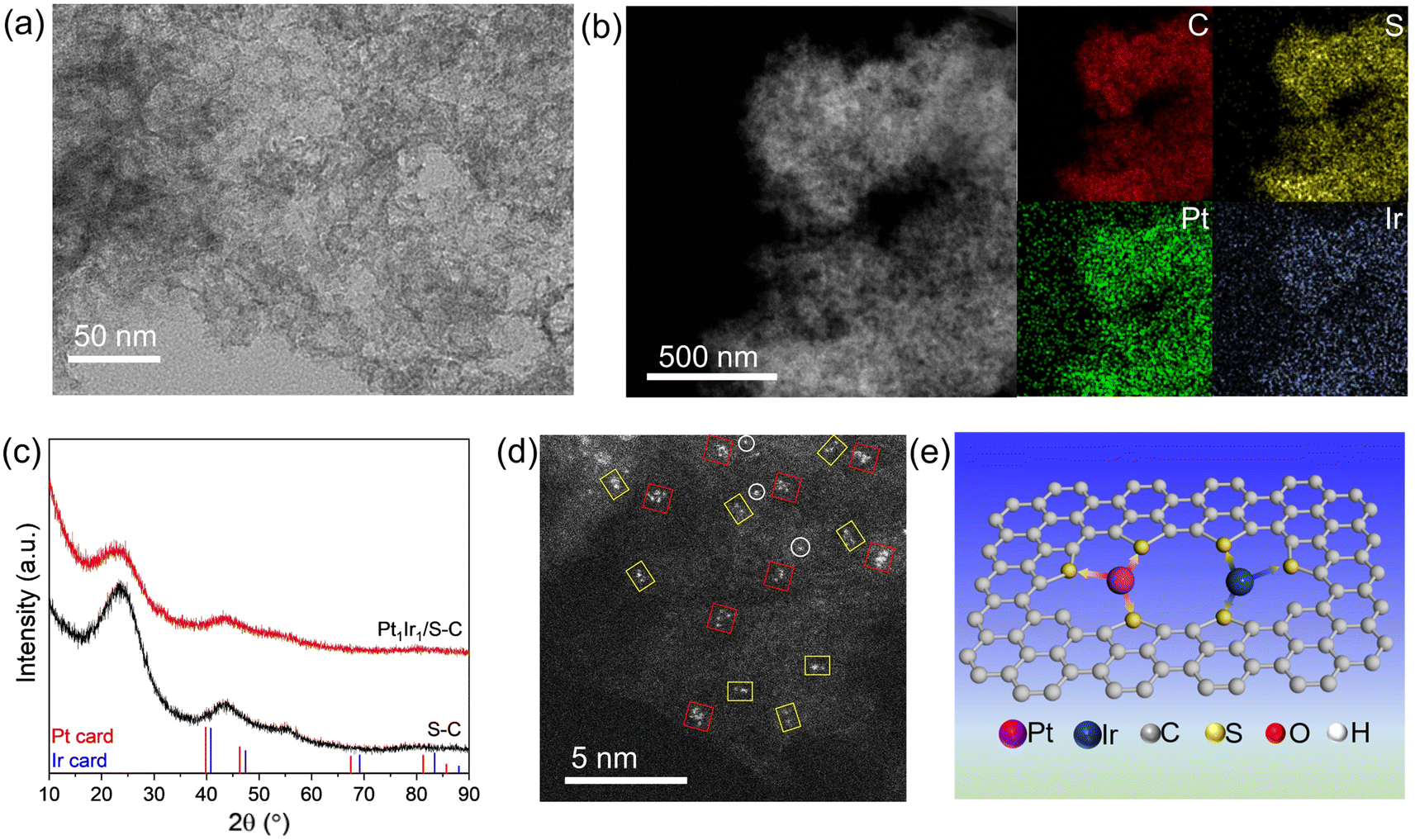 | ||
| Fig. 2 Characterization of the atomically dispersed Pt1Ir1/S–C catalyst. TEM (a) and (b) HAADF-STEM images of Pt1Ir1/S–C. (c) XRD curves of S–C and PtIr/S–C. (d) Atomic-resolution HAADF-STEM image of Pt1Ir1/S–C, where representative isolated atoms (Pt or Ir), dimers, and clusters are highlighted by white circles, yellow rectangles, and red squares, respectively. (e) Schematic of the Pt1Ir1 pair over the S–C support. | ||
As seen in Fig. 3a, two obvious new peaks at ∼174 and 183 eV were detected at the S L-edge XANES for Pt1/S–C, demonstrating that the metal–S bond was formed during the high temperature reduction process.33 Nevertheless, no obvious metal–S bond was found for the Ir1/S–C catalyst, indicating the relatively weak metal–sulfur interaction for the formation of the Ir1/S–C catalyst. Liang reported that the S 2p peak in the PtCo alloy shifted to a higher binding energy (0.1 eV) compared with the S–C carrier, demonstrating the electron transfer from S atoms to metal atoms.33 Nonetheless, we did not observe obvious changes among Pt1Ir1/S–C, Pt1/S–C, Ir1/S–C, and S–C carrier. This result might be ascribed to the low metal loading or less electron transfer from sulfur atoms to the metal atoms under relatively low reduction temperatures. In addition, the surface sulfur atoms decreased after all the samples underwent high temperature reduction treatment (Fig. S4 and Table S1†). The metal content of Pt and Ir was determined with ICP-AES (Table S2†), which was very close to the theoretical value.
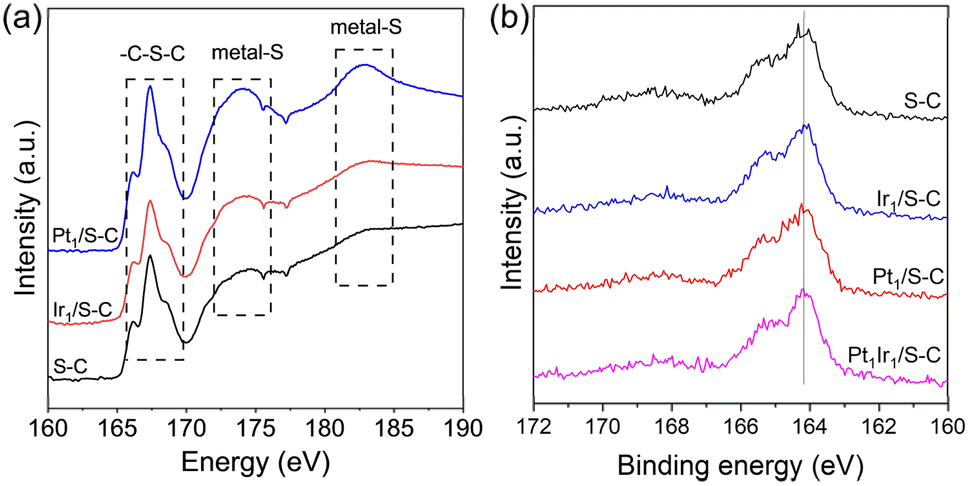 | ||
| Fig. 3 Characterization of metal–sulfur interaction. (a) XANES spectra at the S L-edge of the Pt1/S–C, Ir1/S–C, and S–C carrier. (b) XPS spectra (S 2p) of the Pt1Ir1/S–C, Pt1/S–C, Ir1/S–C, and S–C carrier. | ||
3.2 Catalytic performance for the hydrogenation of LA to GVL
The selective hydrogenation of biomass into renewable value-added products is a promising route to replace the traditional petroleum feedstock.35 For example, γ-valerolactone (GVL) can be used as a solvent, food and fuel additive, and intermediate for fine chemicals, which can be directly obtained from the selective hydrogenation of levulinic acid (LA) with H2 as the hydrogen donor (Scheme 1).36,37 GVL would undergo undesirable consecutive hydrodeoxygenation reactions to form 1,4-pentanediol and methyltetrahydrofuran (Scheme 1). Thus, we selected the hydrogenation of LA to GVL under different conditions as a model reaction to investigate the utility of this class of atomically-dispersed Pt1Ir1 pairs.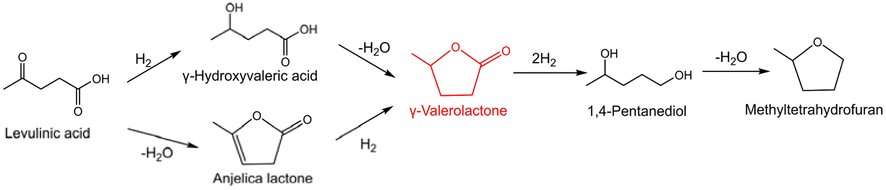 | ||
| Scheme 1 Possible reaction pathways of LA hydrogenation to GVL. | ||
The hydrogenation of LA was conducted in an autoclave reactor at high temperature. Monometallic Pt/S–C and Ir/S–C were also tested for comparison. The Pt/S–C and Ir/S–C in this work were atomically distributed on the S–C carrier, as we previously reported that the atomically-dispersed metal loading on S–C supports can reach up to 10 wt%. Given the importance of solvents in determining the reaction rate and product distribution, a variety of solvents has been explored in the hydrogenation of LA to GVL at 180 °C and 4 MPa H2. The experimental results demonstrated that the activity of Pt1Ir1/S–C in different solvents increase in the order dioxane < o-xylene < H2O < butanol (Fig. 4a). The Pt1Ir1/S–C catalyst presented >95% GVL selectivity in various solvents except H2O. The catalytic performance of the single site Pt1/S–C and Ir1/S–C were also tested for comparation. As shown in Fig. 4c, Pt1Ir1/S–C presented 64% LA conversion with ∼99% GVL selectivity after 4 h, while the conversion of single atom Pt1/S–C reached only ∼10% under the same reaction condition. Although the Ir1/S–C displayed high activity, the GVL selectivity <80%, which is far greater from most of the reported results. A certain amount of 1,4-pentanediol and methyltetrahydrofuran was detected for the single atom Ir1/S–C catalyst. To further evaluate the real activity of thePt1Ir1/S–C pairs, we calculated TOFs under the LA conversion below 25% for all the cases to ensure that the reaction remained under kinetic control. The Pt1Ir1/S–C pairs displayed a TOF value of 868 hour−1 for LA hydrogenation, giving about an 8-fold enhancement compared to single atom Pt1/S–C catalyst (104 hour−1). As demonstrated by the XANES (Fig. 3a), the low activity of the Pt1/S–C might be ascribed to the strong chemical sulfur–Pt interaction, providing weak adsorption ability towards the reaction molecules. The high catalytic performance of Pt1Ir1 pairs might come from the heterolytic dissociation route for the H2 activation process, which could robustly enhance the hydrogenation reactions. In addition, the Ir/S–C displayed much higher activity than Pt1/S–C, demonstrating that the cleavage of H2 is more energetically favourable on single site Ir1/S–C than Pt1/S–C. The hydrogenation of LA to GVL might react via the following mechanism. First, H2 splits on the isolated sites with the heterolytic dissociation process at the single Ir site.12,38 Then, the Hδ+/Hδ− pair transfers to the carbonyl group (Fig. S5†).39 The LA molecule is adsorbed by the hydroxyl oxygen of the carboxyl group on coordinatively oxidized Pt site (Lewis acid center).34 Then, the Pt atom attaches two hydrogen atoms to form 4-hydroxypentanoic acid (4-HPA, intermediate). 4-HPA suffers from further dehydration process to form GVL. Then, the adjacent Pt atom might provide Lewis acid site to accelerate the reaction activity. Subsequently, the activation energy experiment was performed (Fig. S6†), in which Pt1Ir1/S–C and Ir1/S–C present similar activation energy but higher than that of Pt1/S–C, demonstrating that high temperature is more suitable for Pt1Ir1/S–C.
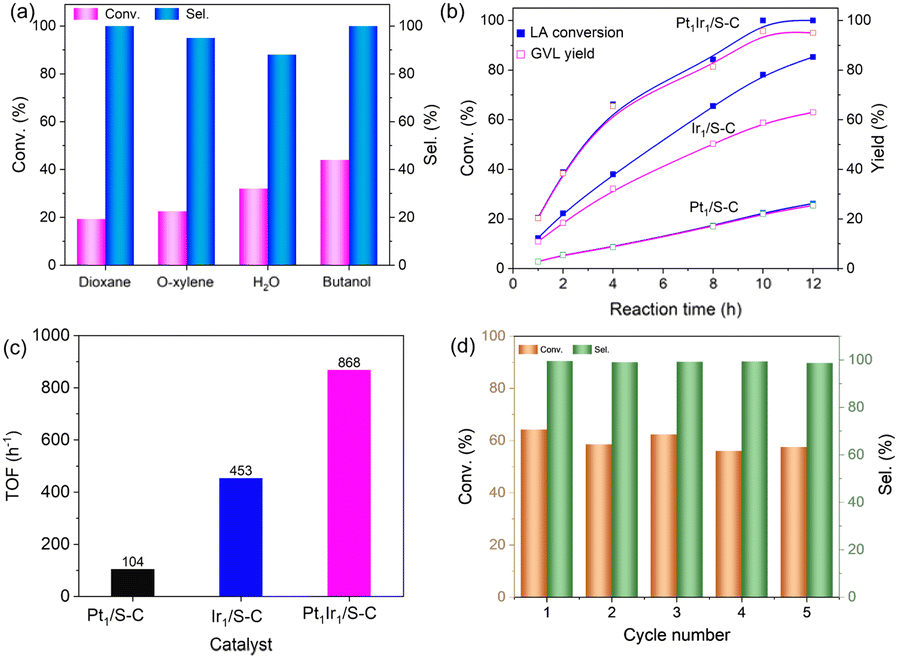 | ||
| Fig. 4 Catalytic performance for the hydrogenation of LA. (a) Hydrogenation of LA over the Pt1Ir1/S–C catalyst in different solvents. Reaction condition: 5 mg PtIr/S–C catalyst, 1 mmol LA, 3 mL solvent, 180 °C, 4 MPa H2, and 4 h. (b) Reaction kinetics over Pt1Ir1/S–C, Pt1/S–C, and Ir1/S–C catalysts. (c) Activity of different catalysts for the hydrogenation of LA. (d) Recycling of Pt1Ir1/S–C for phenylacetylene semihydrogenation within 4 h. Reaction conditions: 1 mmol LA, 3 mL butanol, 180 °C, 4 MPa H2, and 4 h. | ||
The reusability of the Pt1Ir1/S–C catalyst could be separated from the reaction system by filtration. After five recycling runs, the Pt1Ir1/S–C catalyst could nearly maintain its original activity and selectivity (Fig. 4d). No obvious agglomeration of metal atoms was observed by HAADF-STEM after recycling tests (Fig. S7†), indicating the high stability of Pt1Ir1 pairs on S–C when catalyzing LA hydrogenation.
4 Conclusions
In summary, we successfully synthesized atomically-dispersed Pt1Ir1 pairs over the sulfur-doped carbon via the sulfur-tethering strategy. The atomically-dispersed Pt1Ir1 pair presented desirable catalytic performance for the hydrogenation of LA to GVL. In contrast, the single site monometallic Pt1 presented a very low activity. The single Ir atoms split H2 through the heterolytic dissociation process, and the oxidized Pt synergistically accelerated the reaction activity as the Lewis acid center. The Pt1Ir1 pairs displayed high stability after five cycles under harsh reaction conditions. The synthesis method and synergetic effect of the atomically-dispersed metal pairs may open a new gate for fabricating dimers or trimers with an easy scale-up route for various important reactions.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
L. W. and B. L. contributed to the idea and experimental design and wrote the manuscript. B. L. and L.-W. C. collected data and analysed the results. All authors contributed to data analysis, interpreted the data, and approved the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (22301267), Natural Science Foundation of the Jiangsu Higher Education Institutions of China (23KJD150013), and Yangzhou University Start-up Foundation.Notes and references
- A. H. Motagamwala, R. Almallahi, J. Wortman, V. O. Igenegbai and S. Linic, Science, 2021, 373, 217 CrossRef CAS PubMed.
- H. Yan, K. He, I. A. Samek, D. Jing, M. G. Nanda, P. C. Stair and J. M. Notestein, Science, 2021, 371, 1257 CrossRef CAS PubMed.
- M. Escudero-Escribano, P. Malacrida, M. H. Hansen, U. G. Vej-Hansen, A. Velázquez-Palenzuela, V. Tripkovic, J. Schiøtz, J. Rossmeisl, I. E. L. Stephens and I. Chorkendorff, Science, 2016, 352, 73–76 CrossRef CAS PubMed.
- J. C. Matsubu, S. Zhang, L. DeRita, N. S. Marinkovic, J. G. Chen, G. W. Graham, X. Pan and P. Christopher, Nat. Chem., 2017, 9, 120–127 CrossRef CAS PubMed.
- S. Liu, W. Xu, Y. Niu, B. Zhang, L. Zheng, W. Liu, L. Li and J. Wang, Nat. Commun., 2019, 10, 5790 CrossRef CAS PubMed.
- L. Wang, Z. Ma, J. Xue, Z. Yuan, L.-W. Chen and S. Li, Inorg. Chem., 2024, 63, 3452–3459 CrossRef CAS PubMed.
- F. F. Tao and P. A. Crozier, Chem. Rev., 2016, 116, 3487–3539 CrossRef CAS PubMed.
- Z. Li, S. Ji, Y. Liu, X. Cao, S. Tian, Y. Chen, Z. Niu and Y. Li, Chem. Rev., 2020, 120, 623–682 CrossRef CAS PubMed.
- S. Yao, X. Zhang, W. Zhou, R. Gao, W. Xu, Y. Ye, L. Lin, X. Wen, P. Liu, B. Chen, E. Crumlin, J. Guo, Z. Zuo, W. Li, J. Xie, L. Lu, C. J. Kiely, L. Gu, C. Shi, J. A. Rodriguez and D. Ma, Science, 2017, 357, 389–393 CrossRef CAS PubMed.
- Y. Zhai, D. Pierre, R. Si, W. Deng, P. Ferrin, A. U. Nilekar, G. Peng, J. A. Herron, D. C. Bell, H. Saltsburg, M. Mavrikakis and M. Flytzani-Stephanopoulos, Science, 2010, 329, 1633–1636 CrossRef CAS PubMed.
- Y. Xiong, J. Dong, Z.-Q. Huang, P. Xin, W. Chen, Y. Wang, Z. Li, Z. Jin, W. Xing, Z. Zhuang, J. Ye, X. Wei, R. Cao, L. Gu, S. Sun, L. Zhuang, X. Chen, H. Yang, C. Chen, Q. Peng, C.-R. Chang, D. Wang and Y. Li, Nat. Nanotechnol., 2020, 15, 390–397 CrossRef CAS PubMed.
- P. Liu, Y. Zhao, R. Qin, S. Mo, G. Chen, L. Gu, D. M. Chevrier, P. Zhang, Q. Guo, D. Zang, B. Wu, G. Fu and N. Zheng, Science, 2016, 352, 797–801 CrossRef CAS PubMed.
- S. Qian, F. Xu, Y. Fan, N. Cheng, H. Xue, Y. Yuan, R. Gautier, T. Jiang and J. Tian, Nat. Commun., 2024, 15, 2774 CrossRef CAS PubMed.
- L. Wang, S. Lyu and S. Li, ChemPhysMater, 2024, 3, 24–35 CrossRef.
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed.
- C. Xia, Y. Qiu, Y. Xia, P. Zhu, G. King, X. Zhang, Z. Wu, J. Y. Kim, D. A. Cullen, D. Zheng, P. Li, M. Shakouri, E. Heredia, P. Cui, H. N. Alshareef, Y. Hu and H. Wang, Nat. Chem., 2021, 13, 887–894 CrossRef CAS PubMed.
- L. Jiao, J. Li, L. L. Richard, Q. Sun, T. Stracensky, E. Liu, M. T. Sougrati, Z. Zhao, F. Yang, S. Zhong, H. Xu, S. Mukerjee, Y. Huang, D. A. Cullen, J. H. Park, M. Ferrandon, D. J. Myers, F. Jaouen and Q. Jia, Nat. Mater., 2021, 20, 1385–1391 CrossRef CAS PubMed.
- H. W. Liang, S. Bruller, R. Dong, J. Zhang, X. Feng and K. Mullen, Nat. Commun., 2015, 6, 7992 CrossRef CAS PubMed.
- C. H. Choi, M. Kim, H. C. Kwon, S. J. Cho, S. Yun, H.-T. Kim, K. J. J. Mayrhofer, H. Kim and M. Choi, Nat. Commun., 2016, 7, 10922 CrossRef CAS PubMed.
- H. Yan, H. Cheng, H. Yi, Y. Lin, T. Yao, C. Wang, J. Li, S. Wei and J. Lu, J. Am. Chem. Soc., 2015, 137, 10484–10487 CrossRef CAS PubMed.
- X. Cui, W. Li, P. Ryabchuk, K. Junge and M. Beller, Nat. Catal., 2018, 1, 385–397 CrossRef CAS.
- D. R. Aireddy and K. Ding, ACS Catal., 2022, 12, 4707–4723 CrossRef CAS.
- H. Yan, Y. Lin, H. Wu, W. Zhang, Z. Sun, H. Cheng, W. Liu, C. Wang, J. Li, X. Huang, T. Yao, J. Yang, S. Wei and J. Lu, Nat. Commun., 2017, 8, 1070 CrossRef PubMed.
- Q. Wang, C. M. De Brito Mendes, O. V. Safonova, W. Baaziz, C. A. Urbina-Blanco, D. Wu, M. Marinova, O. Ersen, M. Capron, A. Y. Khodakov, M. Saeys and V. V. Ordomsky, J. Catal., 2023, 426, 336–344 CrossRef CAS.
- X. Long, J. Wang, C. Nie, Y. Xi and F. Li, Chem Catal., 2024, 4, 100810 CrossRef CAS.
- H. Li, L. Wang, Y. Dai, Z. Pu, Z. Lao, Y. Chen, M. Wang, X. Zheng, J. Zhu, W. Zhang, R. Si, C. Ma and J. Zeng, Nat. Nanotechnol., 2018, 13, 411–417 CrossRef CAS PubMed.
- S. Tian, Q. Fu, W. Chen, Q. Feng, Z. Chen, J. Zhang, W.-C. Cheong, R. Yu, L. Gu, J. Dong, J. Luo, C. Chen, Q. Peng, C. Draxl, D. Wang and Y. Li, Nat. Commun., 2018, 9(1), 2353 CrossRef PubMed.
- J. Gu, M. Jian, L. Huang, Z. Sun, A. Li, Y. Pan, J. Yang, W. Wen, W. Zhou, Y. Lin, H.-J. Wang, X. Liu, L. Wang, X. Shi, X. Huang, L. Cao, S. Chen, X. Zheng, H. Pan, J. Zhu, S. Wei, W.-X. Li and J. Lu, Nat. Nanotechnol., 2021, 16, 1141–1149 CrossRef CAS PubMed.
- E. Guan, J. Ciston, S. R. Bare, R. C. Runnebaum, A. Katz, A. Kulkarni, C. X. Kronawitter and B. C. Gates, ACS Catal., 2020, 10, 9065–9085 CrossRef CAS.
- L. Wang, M.-X. Chen, Q.-Q. Yan, S.-L. Xu, S.-Q. Chu, P. Chen, Y. Lin and H.-W. Liang, Sci. Adv., 2019, 5, eaax6322 CrossRef CAS PubMed.
- L. Wang, P. Yin, L.-L. Zhang, S.-C. Shen, S.-L. Xu, P. Chen and H.-W. Liang, J. Catal., 2020, 389, 297–304 CrossRef CAS.
- L. Z. Li, S. L. Xu, S. C. Shen, L. Wang, M. Zuo, P. Chen and H. W. Liang, ChemNanoMat, 2020, 6, 969–975 CrossRef CAS.
- C.-L. Yang, L.-N. Wang, P. Yin, J. Liu, M.-X. Chen, Q.-Q. Yan, Z.-S. Wang, S.-L. Xu, S.-Q. Chu, C. Cui, H. Ju, J. Zhu, Y. Lin, J. Shui and H.-W. Liang, Science, 2021, 374, 459–464 CrossRef CAS PubMed.
- I. I. Protsenko, L. Z. Nikoshvili, V. G. Matveeva and E. M. Sulman, Top. Catal., 2020, 63, 243–253 CrossRef CAS.
- Y. Gu, L. Wang, B.-Q. Xu and H. Shi, Chin. J. Catal., 2023, 54, 1–55 CrossRef CAS.
- W. Luo, M. Sankar, A. M. Beale, Q. He, C. J. Kiely, P. C. A. Bruijnincx and B. M. Weckhuysen, Nat. Commun., 2015, 6, 6540 CrossRef CAS PubMed.
- X. Gong, X. Feng, J. Cao, Y. Wang, X. Zheng, W. Yu, X. Wang and S. Shi, Chem. Commun., 2023, 59, 14717–14720 RSC.
- A. Comas-Vives, C. González-Arellano, A. Corma, M. Iglesias, F. Sánchez and G. Ujaque, J. Am. Chem. Soc., 2006, 128, 4756–4765 CrossRef CAS PubMed.
- D. Ren, L. He, L. Yu, R.-S. Ding, Y.-M. Liu, Y. Cao, H.-Y. He and K.-N. Fan, J. Am. Chem. Soc., 2012, 134, 17592–17598 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4na00419a |
| This journal is © The Royal Society of Chemistry 2024 |