
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh yield seedless synthesis of mini gold nanorods: partial silver decoupling allows effective nanorod elongation with tunable surface plasmon resonance beyond 1000 nm and CTAB-free functional coating for mTHPC conjugation†
Mike
Rozenberg
 a,
Matěj
Bárta
b,
Anya
Muzikansky
a,
Melina
Zysler
a,
Karolína
Šišková
b,
Yitzhak
Mastai
a and
David
Zitoun
*a
a,
Matěj
Bárta
b,
Anya
Muzikansky
a,
Melina
Zysler
a,
Karolína
Šišková
b,
Yitzhak
Mastai
a and
David
Zitoun
*a
aDepartment of Chemistry, Bar-Ilan Institute of Nanotechnology and Advanced Materials (BINA), Bar-Ilan University, Ramat Gan 5290002, Israel. E-mail: David.Zitoun@biu.ac.il
bDepartment of Experimental Physics, Faculty of Science, Palacký University Olomouc, Tř. 17. Listopadu 12, 77900 Olomouc, Czech Republic
First published on 18th July 2024
Abstract
Gold nanorods with small dimensions demonstrate better cellular uptake and absorption efficiency. The ability to synthesize gold nanorods while maintaining a tunable high aspect ratio is challenging as it requires careful control of reaction conditions, often employing additional steps such as pH modification or the use of polymeric additives. We demonstrate a seedless approach for the synthesis of mini (width < 10 nm) gold nanorods with tunable longitudinal surface plasmon resonance from ∼700 nm to >1000 nm and aspect ratios ranging from ∼3 to ∼7 without the use of any polymeric additives or pH modification. A single mild reducing agent, hydroquinone, allowed for up to ∼98% reaction yield from a gold precursor. A mechanism for elongation is proposed based on partial silver decoupling from the reaction. Finally, the particles were coated with various capping agents to allow functionalization and conjugation of mTHPC drug molecules, which are used in photodynamic treatments, and cytotoxic CTAB was removed to increase their biocompatibility.
1. Introduction
Metallic nanoparticles (NPs) have gained increasing interest in different fields as their unique chemical and physical properties allow for their integration into various applications, such as catalysis,1,2 biomedicine,3,4 sensing,5,6 imaging,7,8 and nanophotonics.9,10Gold nanoparticles, in particular, are one of the most interesting and useful nanoparticles owing to their ability to interact with light irradiation, which induces the collective oscillation of their conduction band electrons. This phenomenon is known as localized surface plasmon resonance (LSPR) and is sensitive to the composition, size, shape, and dielectric environment surrounding the nanostructure.11 Because of their intrinsic bio-compatibility,12 gold nanoparticles show potential in many applications ranging from surface-enhanced Raman spectroscopy (SERS),13,14 optoelectronics,15,16 photocatalysis,17,18 drug delivery,19,20 photothermal and dynamic treatments,21,22 and nanomedicine.23,24
Spectral properties of gold nanorods (GNRs) can be tuned by changing their aspect ratio (AR, length/width), which offers a particular advantage as they can be modified via small variations in their synthetic conditions, thus opening up a realm of particle design possibilities to match particular requirements based on their application.25 For these reasons, they are used as and in SERS substrates,26 photothermal therapy in vitro and in vivo,27,28 and unique catalytic structures.29
Since the discovery of pure wet-chemistry, seed-mediated, template-assisted synthesis of GNRs, the synthesis process has gone through a multitude of modifications in order to improve GNR physical properties, reproducibility of synthesis, and the novelty of preparation methods and conditions.30–34 Many methods for creating higher AR nanorods, such as using a binary surfactant template,31 lowering the cetyltrimethylammonium bromide (CTAB) content due to its influence on the nanorod quality and shape-yield,34 exchange of reduction agents for improved reduction yield,35 pH manipulation for AR and synthetic tunability,36 and seedless procedures for streamlined preparation, have been demonstrated.37
The most widely used synthetic protocol31 produces gold nanorods with widths between 10 and 25 nm and lengths between 30 and 80 nm using CTAB alone or in a binary surfactant mixture, depending on the desired AR. Smaller gold nanorods exhibit increased cellular uptake, better photothermal conversion efficiency,38 and generally, faster organ clearance and lower toxicity.39 For these reasons, numerous methods have been developed for the preparation of a variety of narrow gold nanorods with widths smaller than 10 nm, denoted as mini gold nanorods (mGNR).40 The dependency of the spectral properties of gold nanorods on AR allows the preservation of optical tunability while decreasing the size of the nanoparticles, thus making them suitable for therapeutic applications.33
Some of these protocols include the seedless preparation of GNRs with widths smaller than 5.5 nm and longitudinal LSPR ranging from 700 to 810 nm41 and the poly(vinylpyrrolidone) (PVP)-assisted method, in which a trace amount of PVP was used to elongate the nanorods and produce GNRs with increased ARs while maintaining the widths below 10 nm.42 Multiple seeded growth methods have exhibited similar results,43–48 while the most noteworthy approach40 produced AR-controlled mGNRs using very high volumes of seed solution such that the longitudinal LSPR peak appeared beyond 1000 nm, surpassing previous works.
In this work, we present a streamlined and facile synthesis procedure of mini gold nanorods, for the first time, based on a seedless, one-pot approach that produces high yields and allows tuning of the longitudinal LSPR of the mini GNRs beyond 1000 nm. We present a unique observation of the effect of high silver quantity on particles prepared using the seedless mediated approach, suggest the possible mechanism, and further apply and leverage this principle to elongate the mGNRs while maintaining their widths below 10 nm to achieve an AR of ∼7 without pH modification or polymer additives, unlike previous works. Additionally, we present a protocol for the surface functionalization of mGNRs via multi-step ligand exchange, and surface binding of the mTHPC drug used in photodynamic therapy is demonstrated as an example to highlight their potential application as drug carriers. This approach maintains the colloidal dispersion and removes CTAB from the surface, thus offering an alternative to coating the mGNRs with polymers for mitigating (without removal) the cytotoxicity of CTAB.
2. Materials and methods
2.1 Chemicals used
Sodium chloride (>99%, Sigma-Aldrich), hexadecyltrimethylammonium bromide (CTAB, 99%, Sigma-Aldrich), silver nitrate (99.9%, STREM Chemicals), hydrogen tetrachloroaurate(III) hydrate (99.9% Au, System Chemicals), meso-tetrahydroxyphenylchlorin (mTHPC, Frontier Scientific, Logan, UT, USA), 3-mercaptopropionic acid (MPA, ACROS ORGANICS), double-distilled water (ddH2O, 18 MΩ cm−1, Millipore), hydroquinone (99%+, Sigma-Aldrich), poly(sodium 4-styrene sulfonate) 70k (PSS, Sigma-Aldrich), NaOH 0.1 N (CARLO ERBA Reagents), and sodium borohydride (99%, Sigma-Aldrich) were obtained from commercial sources.2.2 Structural and spectroscopic characterization
The morphology, size, and distribution of the NPs were examined using a JEOL JEM 1400 transmission electron microscope (TEM) operated at 120 kV and a JEOL JEM 2100 high-resolution TEM (HR-TEM) operated at 200 kV. The presence of bromine in the samples was checked by EDAX (HR-TEM). Particle size measurement was done using ImageJ software by observing at least 150 particles for each sample.To infer the gold reduction yield, inductively coupled plasma optical emission spectroscopy (ICP-OES) was performed using a Multiview FHX22 Spectro Arcos instrument. The samples were prepared by performing dialysis of the as-prepared particles at 30 °C against 20 L of singly deionized water for 24–72 hours.
X-ray photoelectron spectroscopy (XPS) was used to follow and ensure the CTAB removal alongside the mTHPC conjugation using a Nexsa X-ray photoelectron spectrometer system (Thermo Scientific) with a monochromated Al Kα X-ray source. Measurements were taken at room temperature under high vacuum (<3.0 × 10−9 torr). A spot size of 400 μm was used with a pass energy of 40 eV. The samples were prepared by drop-casting on Al foil for the survey spectrum and on Cu tape for the survey and high-resolution data. The mTHPC-only sample was prepared by drop-casting 200 μL of 1 mg per mL mTHPC in ethanol on Cu tape to obtain both survey and high-resolution information.
Optical characterization of the gold nanostructures was performed using a Shimadzu UV-1280 UV-vis-NIR spectrophotometer.
3. Experimental methods
3.1 Regular and standard seedless synthesis of mGNRs
To a vial containing 4.725 mL of 0.2 M CTAB solution, 4.725 mL of ddH2O was added. Under stirring at 800 rpm, either 165 μL (regular) or 200 μL (standard) of a 25 mM HAuCl4 solution, 16 μL of 40 mM AgNO3 and 500 μL of 0.1 M HQ were added in sequence, followed by 30 seconds of monitoring at the end of which the solution turned completely transparent. 50 μL of a 0.01 M NaBH4 solution was immediately injected in one shot, with continuous stirring for 60 seconds and an open cap.The vial was closed and placed overnight (16–20 hours) without any further agitation or stirring in an incubator pre-heated to 30 °C.
The particles were washed by centrifugation at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 30 minutes at 25 °C three times and re-dispersed in 10 mL ddH2O each time.
000 rpm for 30 minutes at 25 °C three times and re-dispersed in 10 mL ddH2O each time.
3.2 Partially silver decoupled seedless synthesis of elongated mGNRs
To a vial containing 4.725 mL of a 0.2 M CTAB solution, 4.725 mL of ddH2O was added. Under stirring at 800 rpm, either 165 μL (regular) or 200 μL (standard) of a 25 mM HAuCl4 solution, 16 μL of 40 mM AgNO3 and 500 μL of 0.1 M HQ were added in sequence, followed by 30 seconds of monitoring at the end of which the solution turned completely transparent. 47 μL of a 0.01 M NaBH4 solution was immediately injected in one shot, with continuous stirring for 60 seconds and an open cap. After 120 more seconds without stirring or any movement of the vial, 13–30 μL of 40 mM AgNO3 was rapidly injected, followed by gentle hand stirring for 3 seconds.The vial was closed and placed overnight (16–20 hours) without any further movement or modification in an incubator pre-heated to 30 °C.
The particles are washed by centrifugation at 13000 rpm for 30 minutes at 25 °C three times and re-dispersed in 10 mL ddH2O each time.
3.3 Seeded synthesis of 80 μL silver stock mini gold nanorods
The seed solution was prepared according to the standard protocol.31 In brief, to a 9.9 mL solution of 0.1 M CTAB, 100 μL of a 25 mM HAuCl4 solution was added and mixed together. 600 μL of freshly prepared 0.01 M NaBH4 was added in one shot under stirring at 1200 rpm for 120 seconds. The vial was then placed with an open cap inside a pre-heated incubator at 30 °C for 1 hour prior to use.The growth solution was prepared by mixing 7.8 mL of a 0.1 M CTAB solution with 200 μL of a 25 mM HAuCl4 solution, followed by 80 μL of a 40 mM AgNO3 solution and 500 μL of a 0.1 M HQ solution. Under vigorous stirring, 2 mL of the seed solution was added to the growth solution; stirring was continued for 30 seconds and the vial was then sealed and placed overnight (16–20 hours) in a pre-heated incubator at 30 °C.
The particles were washed by centrifugation at 13000 rpm for 30 minutes at 25 °C three times and re-dispersed in 10 mL ddH2O each time.
3.4 1 L standard seedless synthesis of mGNRs
To a glass container, 472.5 mL of a 0.2 M CTAB solution and 472.5 mL of ddH2O were added. Under stirring at 800 rpm, 20 mL of a 25 mM HAuCl4 solution, 1.6 mL of 40 mM AgNO3 and 25 mL of 0.2 M HQ were added in sequence, followed by 30 seconds of monitoring at the end of which the solution turned completely transparent (it appeared slightly yellowish at times due to a change in the optical path). 5 mL of a 0.01 M NaBH4 solution was immediately injected in one shot, with continuous stirring for 60 seconds and an open cap.The vial was closed and placed overnight (16–20 hours) without any further agitation or stirring in an incubator pre-heated to 30 °C.
The particles were washed by centrifugation at 13000 rpm for 30 minutes at 25 °C three times and re-dispersed in 10 mL ddH2O each time.
3.5 PSS coating of mGNRs
PSS coating of the nanorods was carried out based on a previously reported procedure.49 In brief, to 10 mL of the mini gold nanorod solution, 200 μL of 10 mg per mL PSS prepared in 0.01 M NaCl and 10 μL of 0.1 M NaCl were added in sequence and stirred at 800 rpm for 1 hour.The particles were washed by centrifugation three times at 12000 rpm for 15 minutes and re-dispersed each time in 10 mL of ddH2O.
3.6 MPA coating of mGNR@PSS
To 1 mL of PSS-coated mini nanorods, 10 μL of 10% v/v% MPA dissolved in 0.1 N NaOH was added. The solution was hand-stirred mildly and left to incubate undisturbed at room temperature for 24 hours.The particles were washed by centrifugation twice at 12000 rpm for 15 minutes and re-dispersed each time in 1 mL of ddH2O.
3.7 Binding of mTHPC to mGNR@MPA
To 1 mL of MPA-coated mini nanorods kept in a vial covered by aluminum foil, 10 μL of 1 mg per mL mTHPC in ethanol and 10 μL of 0.1 N NaOH were added sequentially under stirring on a hotplate. The temperature was increased to 80 °C, and the reaction was continued for 2.5 hours.The particles were cleaned by dialysis (cellulose, cutoff 12–14 kDa) for 24 hours against at least 20 L of singly deionized water under stirring, and the setup was covered by aluminum foil as soon as the dialysis bag containing the solution was placed in it.
4. Results and discussion
Mini gold nanorods (mGNR) with different spectral properties and aspect ratios were synthesized via a seedless approach. The growth solution containing hydroquinone (HQ) initiated the reduction of Au(III) to Au(I), which was followed by the initiation of the nanorod growth with the addition of sodium borohydride (NBH), which created nucleation sites directly inside the growth solution. Unlike previous works36,37 the quantity of NBH was significantly increased in this study to elevate the number of nucleation sites formed, thereby limiting the possible thickening of the growing gold nanorods beyond 10 nm. These mGNRs were tuned to have longitudinal LSPR beyond 1000 nm (Fig. 1A and B).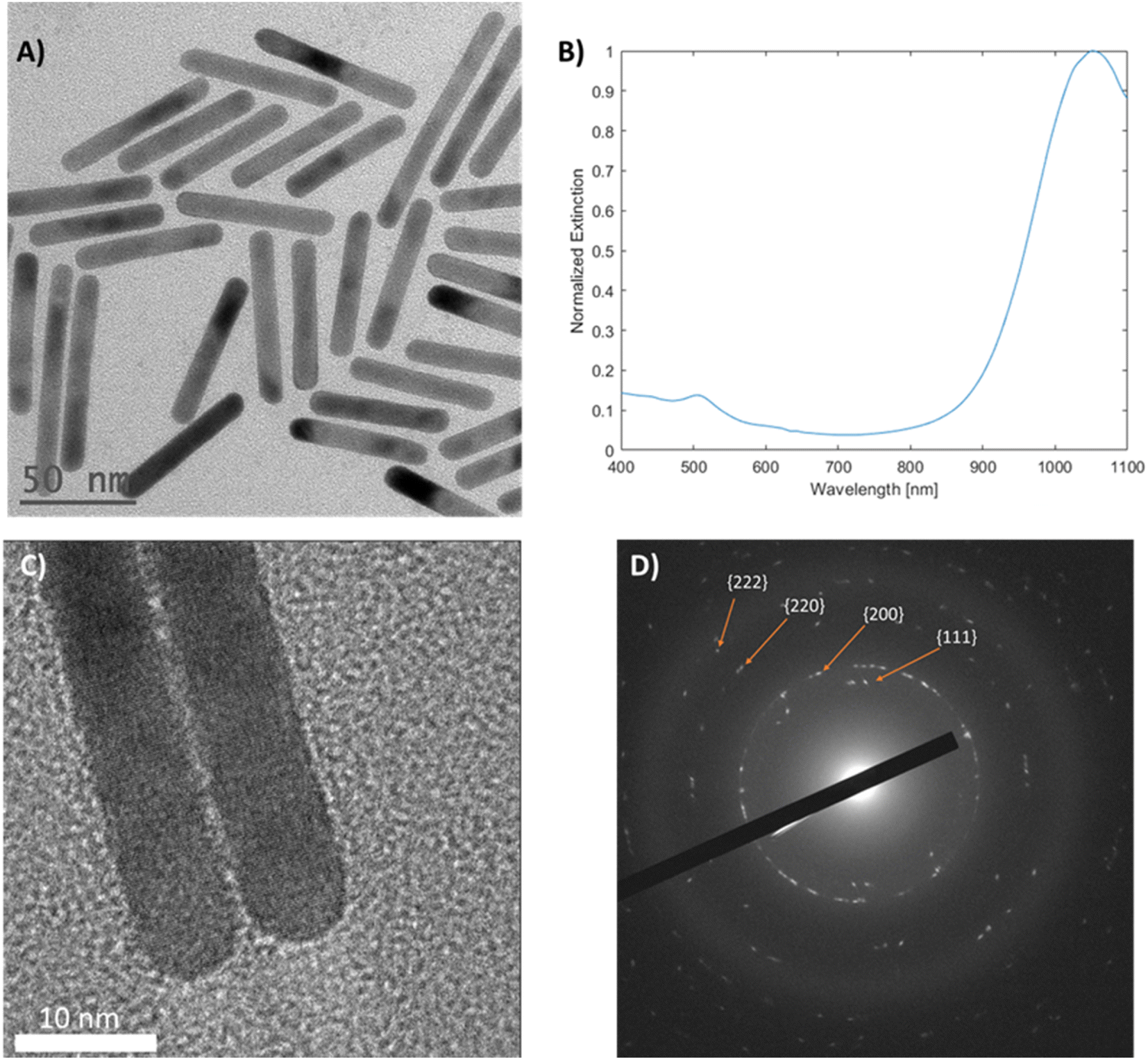 | ||
| Fig. 1 (A) TEM image of ∼7 AR mGNRs, (B) UV-vis-NIR spectrum of the corresponding mGNRs with longitudinal LSPR beyond 1000 nm and (C) high-magnification HR-TEM image and (D) diffraction pattern of random mGNRs prepared using our method. | ||
The mGNRs produced by this method were optically tunable, uniform in size and crystalline, as indicated by the HR-TEM images and electron diffraction pattern in Fig. 1. Synthetic parameters, size data and more information regarding all the samples synthesized in this work can be found in Tables 1S and 2S.†
4.1 Spectral reproducibility and parametric variations
Reproducibility and tunability were evaluated by varying the reaction conditions (UV-vis-NIR spectra and corresponding TEM images in Fig. 2–5). Initially, the particles were prepared by adding 200 μL of 25 mM gold complex stock to the growth solution, and these particles are denoted as ‘standard’ as this is the commonly used concentration in the literature for the synthesis of gold nanorods.31,33 An up-scaled to 1 L reaction is also demonstrated in Fig. 1S,† with more shape impurities, as evidenced by the decrease in the ratio50 between the longitudinal and transverse LSPRs from 3.03 to 2.03 in comparison with the standard mGNRs. Notably, hydroquinone was not required for this reaction as ascorbic acid could be effectively used to produce mGNRs (Fig. 2S†), although with potentially decreased reductive capability.31,40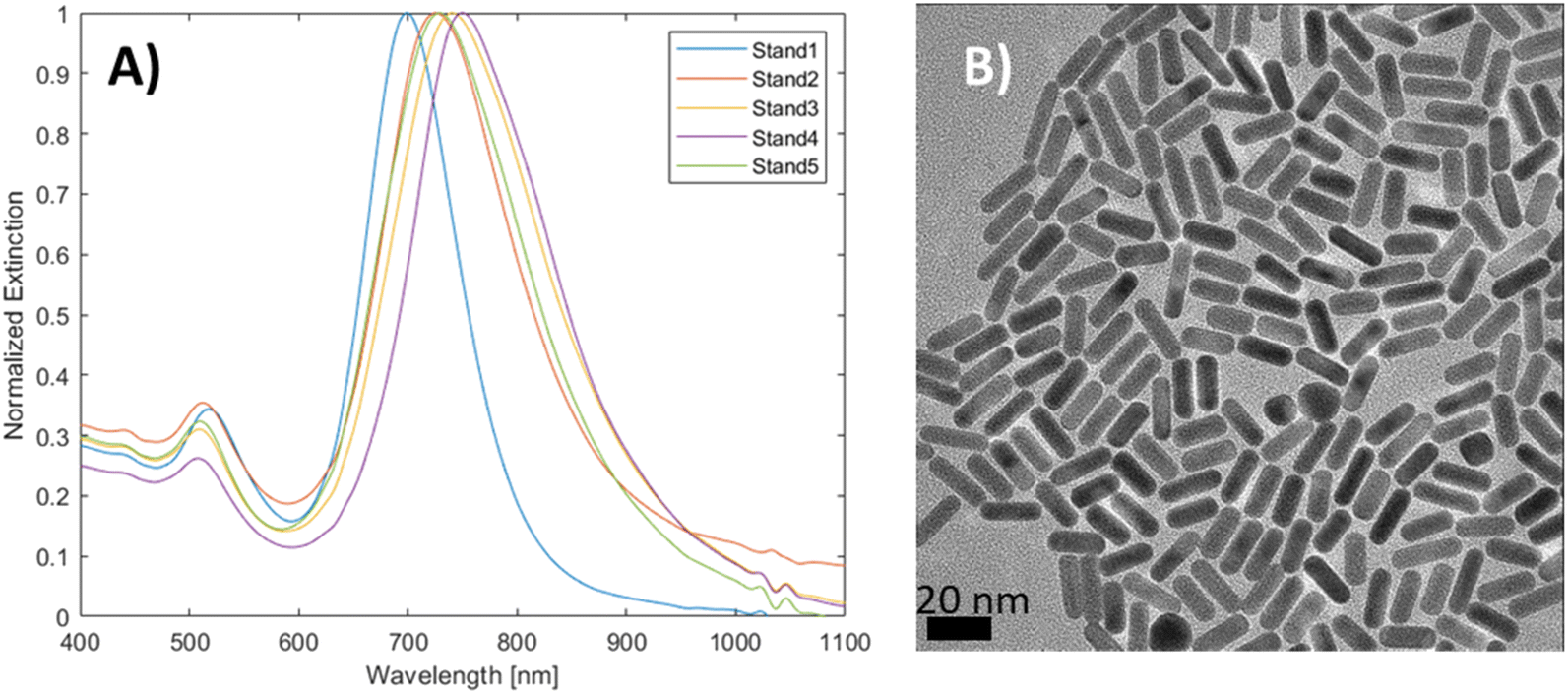 | ||
| Fig. 2 (A) UV-vis-NIR spectra of mGNRs prepared via the standard approach at different times with different stock solutions and (B) the corresponding HR-TEM image. | ||
The spectral reproducibility of mGNRs produced via this standard method was high (Fig. 2), with the longitudinal LSPR position at around 729 ± 17 nm, the samples were prepared using different gold, silver and HQ stock solutions at different times over the course of several months.
Variation in gold concentration in the nanorods was found to induce spectral changes (Fig. 3A). When 250 μL of the gold stock solution was added, the particles showed a single peak at 563 nm, potentially indicating that GNRs were not formed. At 225 μL, the longitudinal peak was blue-shifted (relative to the standard method) towards 670 nm, while at 165 μL (denoted as regular) the longitudinal peak was observed at around 718 nm, which is within the range of the standard; finally, at 103 μL of Au stock, the longitudinal peak was red-shifted towards 797 nm.
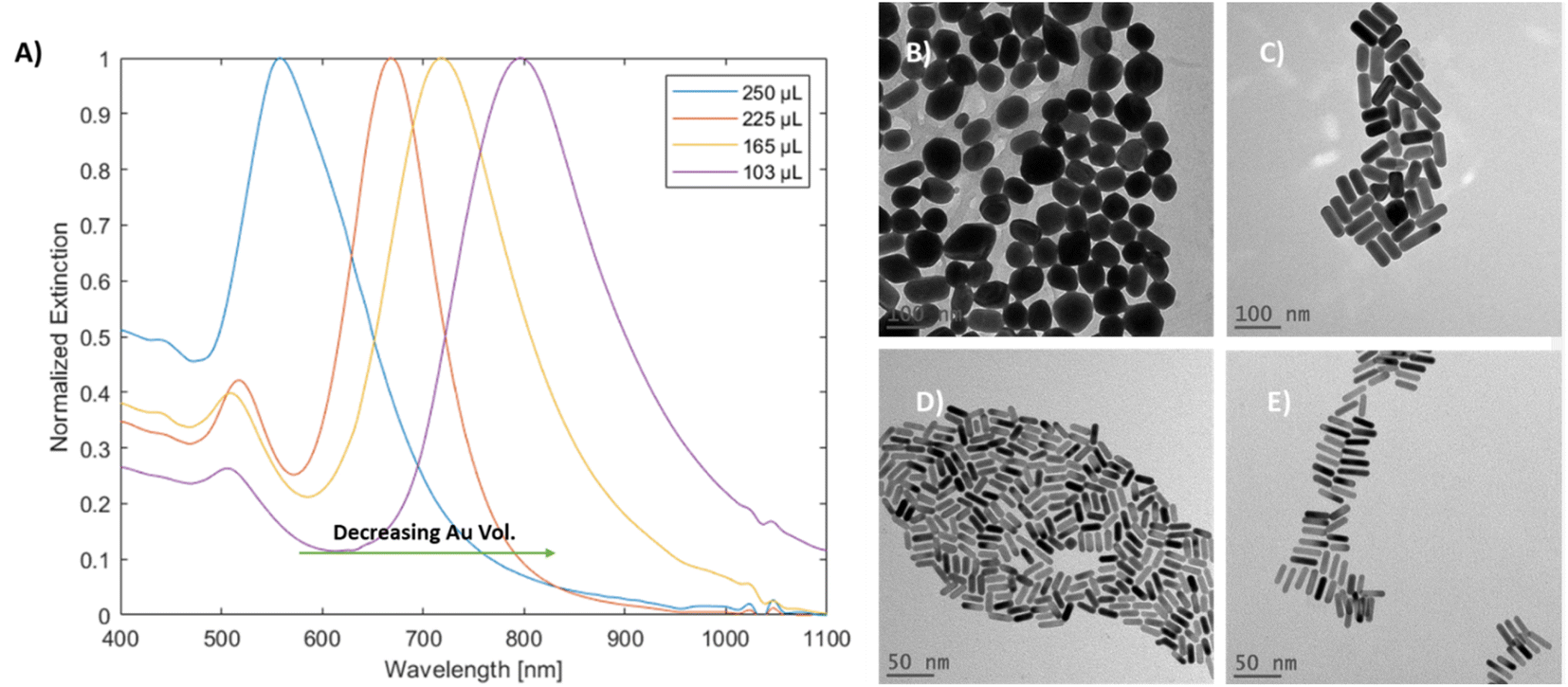 | ||
| Fig. 3 (A) UV-vis-NIR spectra of mGNRs prepared with different Au stock volumes. TEM images of (B) 250 μL of Au stock, (C) 225 μL of Au stock, (D) 165 μL of Au stock and (E) 103 μL of Au stock. | ||
The TEM images and width histograms (Fig. 3B–E and 3S,† respectively) exemplify that the GNRs produced with 225 μL stock had widths beyond the 10 nm threshold for the ‘mini’ designation. To ensure that the width of the mGNRs produced is below the threshold throughout the synthetic work, the volume of gold stock added to the growth solution was decreased to 165 μL (regular) unless stated otherwise; while the standard conditions also formed mGNRs, the conditions were quite close to the “edge” of the synthetic limit in terms of gold concentration.
Variations in the silver concentration added to the growth solution were monitored (Fig. 4), and as expected,31,33 an increase in the amount of silver resulted in a red-shifted longitudinal peak due to the increase in the aspect ratio of the gold nanorods. Notably, in concordance with previous observations,40 the mGNR produced through the seeded growth method showed both a blue-shifted longitudinal peak and widening of the mGNRs beyond the 10 nm threshold with a decrease in the amount of silver. In our case, as seen in Fig. 4B–F, in the samples prepared with 200 μL of Au stock, both 10 μL and 13 μL silver stock volumes produced GNRs with widths beyond the threshold, and this behavior was observed for the sample prepared using 10 μL of silver stock in the regular conditions as well. The longitudinal peak of the mGNRs can, therefore, be tuned from ∼700 nm to ∼850 nm by varying the silver volume during the synthesis.
 | ||
| Fig. 4 (A) UV-vis-NIR spectra of mGNRs prepared with different Au stock volumes and Ag stock volumes. TEM images for (B) 200 μL of Au stock and 10 μL Ag stock, (C) 200 μL of Au stock and 13 μL Ag stock, (D) 10 μL of Ag stock, (E) 19 μL of Ag stock and (F) 23 μL of Ag stock. | ||
The effect of sodium borohydride volume variations on mGNR production was examined (Fig. 5A and D). Initially, an unexpected36 redshift of the longitudinal peak was observed; the TEM images of the samples at the edges of Fig. 5A (Fig. 5B and C, from left to right) the aspect ratio of the produced particles is increasing.
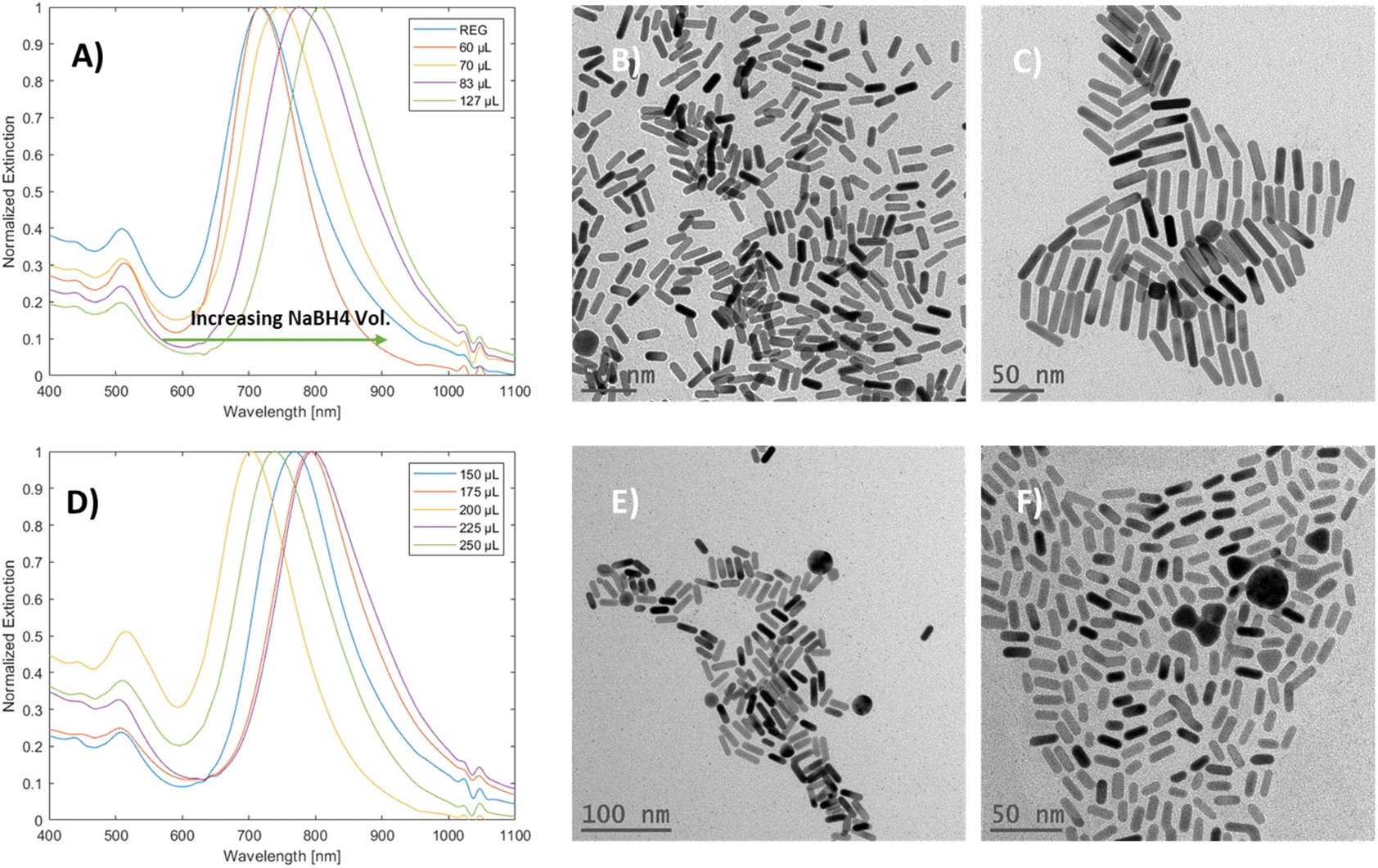 | ||
| Fig. 5 (A) Spectra of mGNRs prepared with increasing NBH volume from 50 to 127 μL with corresponding TEM images of: (B) regular – 50 μL NBH solution and (C) 127 μL NBH solution. (D) Spectra of mGNRs prepared with increasing NBH volume from 150 to 250 μL; TEM images of mGNRs prepared using (E) 150 μL NBH solution and (F) 250 μL NBH solution. | ||
Interestingly, once the NBH volume reached 150 μL, this trend was broken up to 250 μL, and there was no clear relationship between the volume of NBH and the longitudinal LSPR peak position in this range (Fig. 5D and the corresponding TEM images in Fig. 5E and F). It is worth mentioning that particles with irregular shapes were produced alongside nanorods as the volume of NBH was increased, while the width of the particles decreased (Fig. 3S(L) and (M)†) in that range.
4.2 High silver volume and partial silver decoupling
When a high volume of silver (80 μL) was added to the growth solution, an unexpected product was observed. The particles no longer exhibited dual mode LSPR; instead, they showed a singular peak at 531 nm, and they appeared spherical, as corroborated by the UV-vis-NIR and TEM results, respectively (Fig. 6A and B). Such changes were not observed for the samples prepared by the seeded method with the same volume of silver used (Fig. 6A and C). These results indicate a direct relationship between the seedless approach and silver concentration in the solution.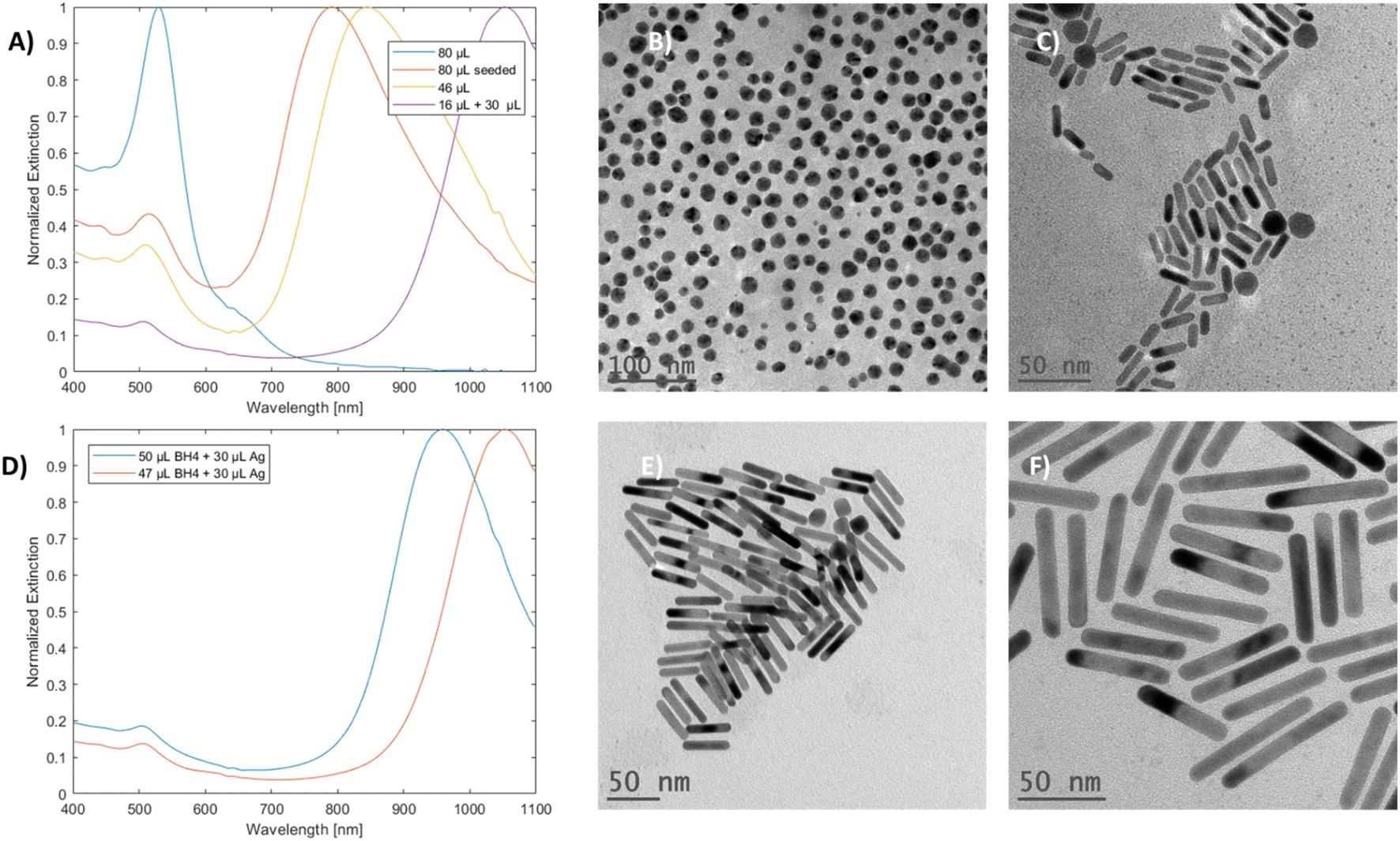 | ||
| Fig. 6 (A) Spectra of particles produced by the seedless and seeded approach with a silver stock volume of 80 μL used alongside mGNRs prepared using 46 μL of silver stock and the partially decoupled reaction of optimized regular synthesis with 30 μL silver stock added after 180 seconds. (B) and (C) are the TEM images of the particles obtained by the seedless and seeded approach when a silver stock volume of 80 μL was used, respectively. (D) Spectra of the non-optimized (50 μL NBH) and optimized (47 μL NBH) partially silver-decoupled reaction with 30 μL of silver stock solution added after 180 seconds. (E) and (F) are the TEM images of the non-optimized and optimized reactions, respectively. | ||
Partial silver decoupling was done by performing the reaction under regular conditions and waiting for NBH to decompose over a period of 180 seconds with an open cap; finally, more silver stock solution was added to the vial with quick and mild rotational shaking.
The resulting mGNRs with the optimized and non-optimized NBH volumes (Table 2S†) exhibited higher AR compared with mGNRs prepared using a high but not deleterious volume of silver stock (46 μL). When the NBH volume was slightly lowered to allow for slightly increased gold availability, it was possible to elongate the mGNRs to have longitudinal LSPRs beyond 1000 nm (Fig. 6D–F).
mGNRs prepared using partially decoupling silver could be tuned between ∼700 nm to beyond 1000 nm by adjusting the volume of silver stock added after the waiting period (Fig. 7) while maintaining widths below 10 nm (Fig. 4S†).
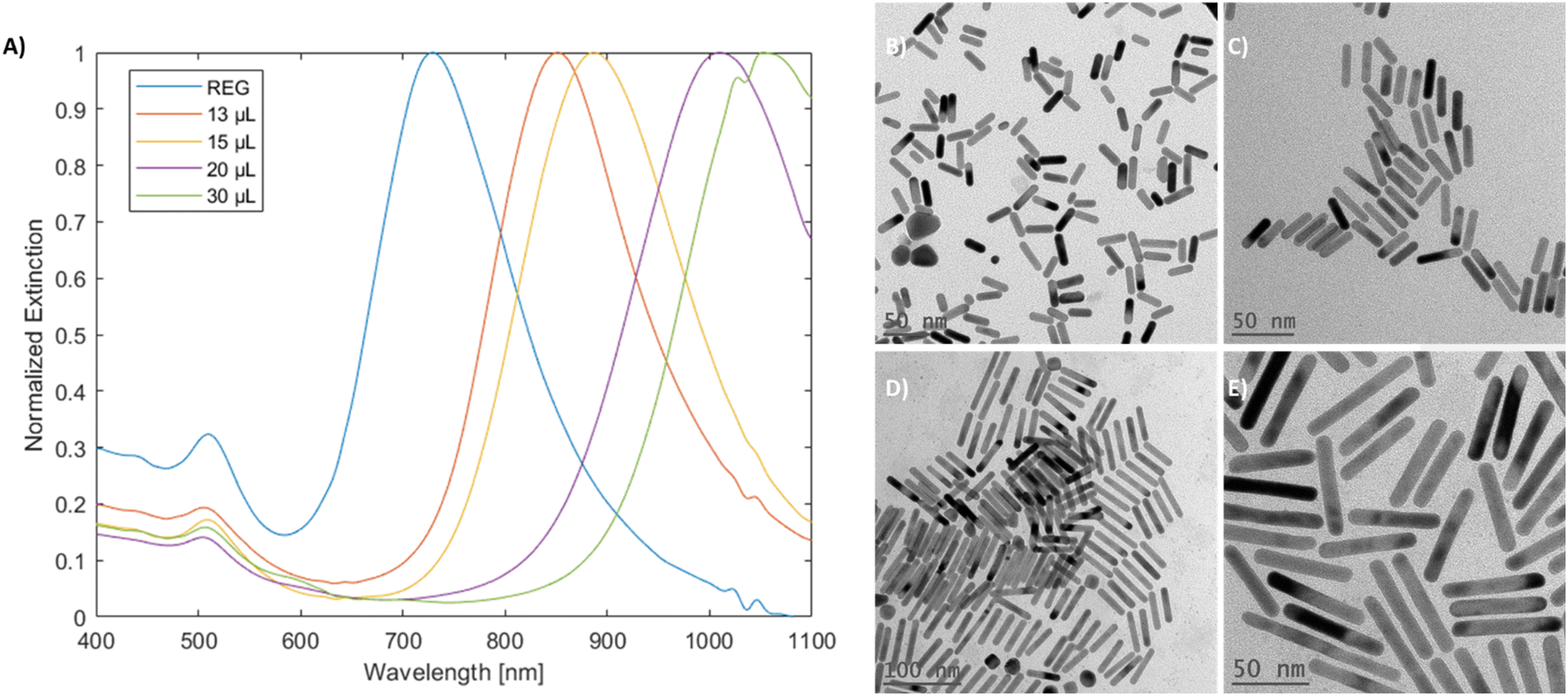 | ||
| Fig. 7 (A) Spectra of mGNRs prepared via the optimized partially silver-decoupled reaction by adding varying silver stock volumes after a 180 second waiting period and the corresponding TEM images for (B) 13 μL, (C) 15 μL, (D) 20 μL and (E) 30 μL of silver stock solution added. | ||
4.3 Ligand exchange and mTHPC conjugation
Coating the nanorods and removal of cytotoxic CTAB51 were performed as the main application of the as-produced mGNRs is in biological settings.40,43,52,53 This was achieved via a multi-step process. Initially, the nanorods possessed a positive charge due to the CTAB bilayer.54 As such, PSS was added to stabilize and inverse the charge through a well-known process.49,55,56 Then, the PSS-coated mGNRs were functionalized with mercaptopropionic acid (MPA) to form a monolayer of carboxylate functional groups facing outward, thus facilitating the conjugation of different species necessary for drug conjugation.57 Finally, a model drug (mTHPC) was conjugated to the mGNRs considering its increased performance in photodynamic and photothermal treatment when conjugated with gold nanoparticles, as previously demonstrated by our group.58Monitoring the presence of CTAB and its removal was achieved by XPS. The disappearance of the ∼Br 3d peak at 68 eV, Br 3p peak at ∼181/189 eV (181 eV marked) and N 1s peak at ∼402.5 eV54 along with the corresponding TEM images at each step showed that the mGNRs remained stable and dispersed (Fig. 8), which was verified by Br presence in EDAX (Fig. 5S–8S†).
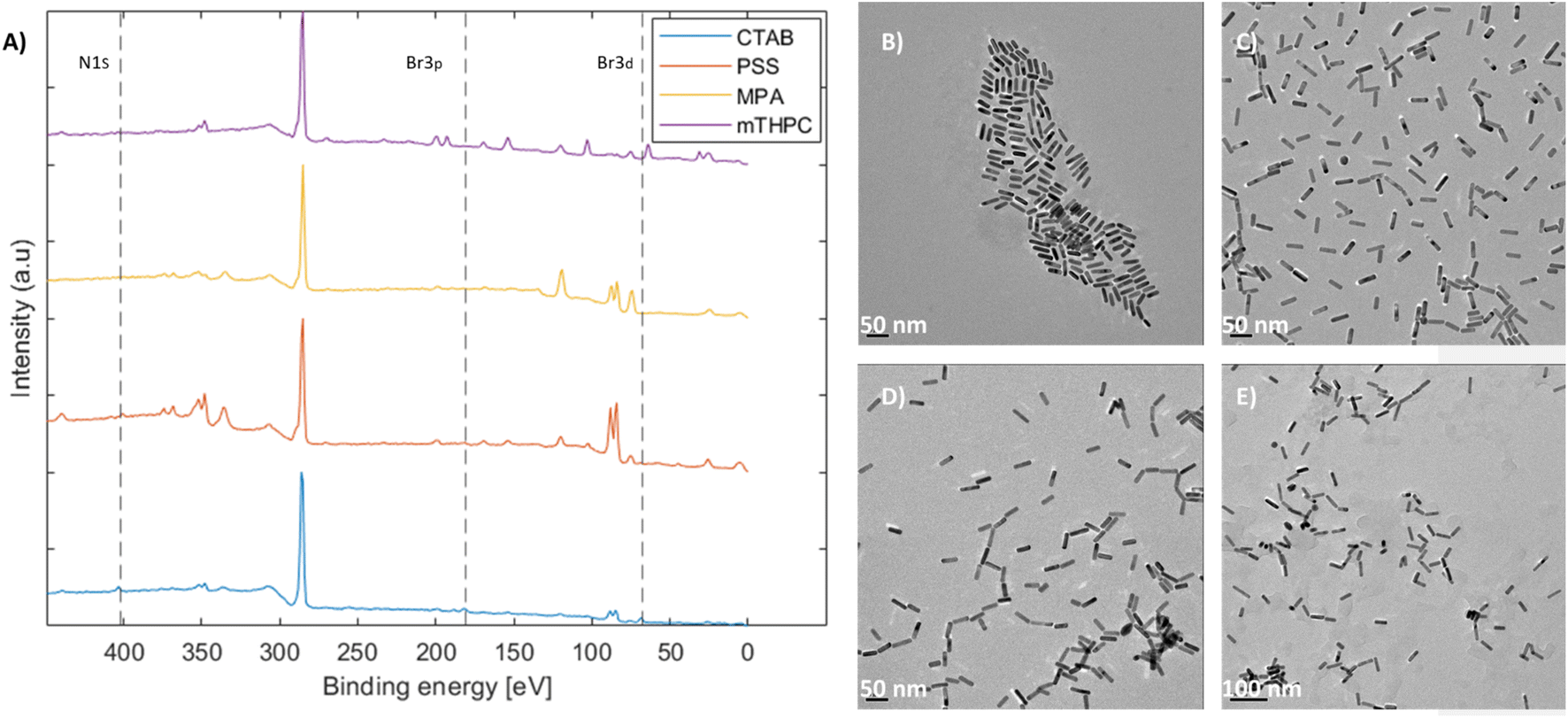 | ||
| Fig. 8 (A) XPS survey spectra showing the N 1s, Br 3p and Br 3d orbital peak positions for each step of the ligand exchange process; the corresponding HR-TEM images of (B) CTAB-capped mGNRs, (C) PSS-capped mGNRs, (D) MPA-capped mGNRs and (E) mTHPC-capped mGNRs. | ||
Complete XPS survey spectra and the detailed high-resolution data of the coatings with reference samples can be found in Fig. 9S–15S,† respectively. All XPS peaks were attributed according to previous works.59,60 The conjugation of mTHPC was confirmed by the high-resolution N 1s data, which showed the characteristic doublet (Fig. 14S(B)†) corresponding to the N–H bond (∼400 eV) and the R![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–R (aromatic) bond (∼398.5 eV) belonging to mTHPC, as confirmed by a standalone measurement (Fig. 15S(A)†). Additionally, the O 1s peak at ∼533–534 eV attributed to C–OH also corresponded well with the standalone mTHPC measurement (Fig. 15S(B)†). EDAX indicated the complete disappearance of CTAB only after dialysis was performed at the last step; although the values of Br were within the margin of error, Br had to be specifically selected for the MPA and mTHPC samples to appear in the EDAX analysis software. By comparison, Fig. 8A and high-resolution XPS data confirm that Br was indeed removed from the MPA samples, but not N 1s, which still featured an ammonium peak at ∼402.5 eV (Fig. 13S(B)†), corroborating the EDAX results and indicating that more than one elemental peak and analytical method are required to properly assess the removal of CTAB.
N–R (aromatic) bond (∼398.5 eV) belonging to mTHPC, as confirmed by a standalone measurement (Fig. 15S(A)†). Additionally, the O 1s peak at ∼533–534 eV attributed to C–OH also corresponded well with the standalone mTHPC measurement (Fig. 15S(B)†). EDAX indicated the complete disappearance of CTAB only after dialysis was performed at the last step; although the values of Br were within the margin of error, Br had to be specifically selected for the MPA and mTHPC samples to appear in the EDAX analysis software. By comparison, Fig. 8A and high-resolution XPS data confirm that Br was indeed removed from the MPA samples, but not N 1s, which still featured an ammonium peak at ∼402.5 eV (Fig. 13S(B)†), corroborating the EDAX results and indicating that more than one elemental peak and analytical method are required to properly assess the removal of CTAB.
Fig. 16S† shows the UV-vis-NIR spectra of the PSS-coated, mTHPC-capped and mTHPC reference samples, indicating the presence of mTHPC on the mGNRs and also showing a noticeable red-shift of the longitudinal peak probably due to a change in the dielectric environment.61 Additionally, more off-resonance peaks appeared, which could be associated with neither plasmon resonance nor mTHPC itself, and are considered beyond the scope of this work.
4.4 Elucidation of the reaction mechanism and mGNRs elongation via partial silver decoupling
Mechanisms underlying processes occurring both under NBH volume variations, leading to increased ARs and red-shifted longitudinal LSPR peak, and the high silver content resulting in spherical particles, can be traced to the direct addition of a high volume of NBH to the solution. In the case of seeded growth, the addition of more seeds will lower the aspect ratio,62 and a similar trend appears at lower volumes of NBH in seedless synthesis.36Initially, at high NBH volumes up to 127 μL (Fig. 5E), the increase in AR can be explained by both the decrease in available AuBr2− due to its reduction to Au0 and continued nucleation, in addition to the increased ratio between silver and remaining AuBr2−. Both less availability of gold for growth and the decreased ratio of AuBr2−:Ag+ become dominant features due to the high NBH volume used in this work in comparison to previous works,36 thus resulting in higher ARs as symmetry breaking of the nuclei occurs and the available Au+ required for growth.63 Notably, the difference in AR was not large (Table 2S,† ΔAR = 1.03), which can be due to the competition with the increased nuclei in the solution, effectively dampening the extension of the rods.
This mechanism is in line with the observations under varying gold complex concentrations, that is, less gold stock volume produced higher ARs and vice versa.
As for the formation of spherical particles when a high content of silver is added (Fig. 6B), supposedly this mechanism falls short as the ratio of AuBr2−:Ag+ decreases substantially, resulting in the symmetry breaking of the nuclei at a smaller size, which would induce the formation of higher AR rods. However, the observed AR was significantly lower (spheres, AR ∼ 1). This can be explained by an additional process occurring when a very high content of silver is present in the seedless approach, which is the co-reduction of silver by NBH. This leads to the formation of far more Ag0 atoms in solution compared with the standard conditions.
Based on that premise, we suggest a possible route that governs the oxidation of Ag0 and results in kinetically unfavorable conditions for the formation of gold nanorods through galvanic-process-enhanced reduction kinetics. In this pathway, Ag0 can reduce the Au+ present in the solution, and Au3+ is formed due to the comproportionation reaction (eqn (1)) due to its lower reduction potential (eqn (2)–(4)), as described in general in eqn (5) and (6).33,64,65
| Au3+ + 2Au0 → 3Au+ | (1) |
| Ag0 → Ag1+ + e−, E0 = 0.799 vs. NHE | (2) |
| AuBr2− + e− → Au0 + 2Br−, E0 = 0.959 vs. NHE | (3) |
| AuBr4− + 3e− → Au0 + 2Br−, E0 = 0.854 vs. NHE | (4) |
| AuBr2− + Ag0 → Ag1+ + Au0 + 2Br− | (5) |
| AuBr4− + 3Ag0 → 3Ag1+ + Au0 + 4Br− | (6) |
| Hydroquinone + Au3+ → Au+ + quinone | (7) |
These processes open a secondary avenue for the reduction of Au+ and modify the kinetics of the growth process (Fig. 9), which is usually dominated by eqn (1) and (7), especially in the seed-mediated growth process.
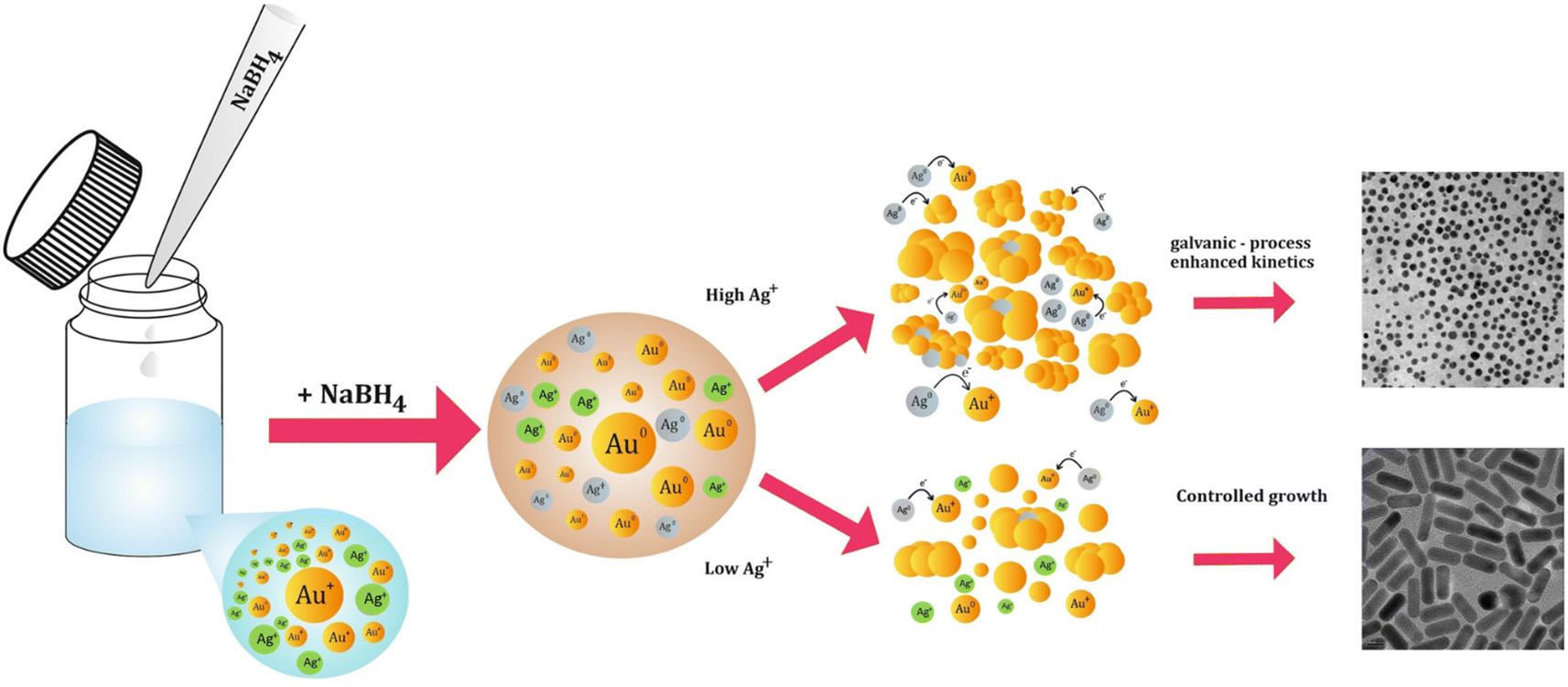 | ||
| Fig. 9 Illustration of the proposed galvanic-process-enhanced kinetics in the presence of high silver content in the seedless reaction and the outcome. | ||
Assuming that this mechanism indeed modifies the growth kinetics, it can be concluded that partially decoupling silver from the growth process under seedless conditions can potentially result in the effective elongation of the mGNRs when the silver content in the reaction is increased after borohydride is partially consumed, which is exactly the behavior observed in Fig. 6 and 7.
Using this method, we have demonstrated the possibility of effectively elongating mGNRs by partially decoupling silver from the reaction in a seedless approach without the use of pH modification or polymeric additives whilst maintaining the width below 10 nm and tuning the longitudinal LSPR of the GNRs beyond 1000 nm by increasing the AR in a one-pot process.
5. Conclusions
We demonstrate a facile seedless synthesis method of mGNRs with tunable aspect ratios from ∼3 to ∼7 and optical properties, with longitudinal LSPR ranging from ∼700 to >1000 nm, by the variation of silver stock volume, gold stock volume, NBH volume and partial silver decoupling. The reaction yields could be increased from 15% for the standard gold nanorod synthesis process to 98% by the as-developed strategy. Considering multiple synthetic variations, clear synthetic limits to achieving widths below 10 nm in the formed GNRs were observed, and the synthetic protocol was optimized around them. Unusual observations with regard to the presence of high silver content and the unexpected trend observed with the use of different NBH volumes are explained, and a mechanism of galvanic-enhanced reduction kinetics for the formation of the nanorods is suggested. This is further leveraged to achieve elongation in seedless synthesis by employing partial silver decoupling. This work bridges the gap in the seedless method to produce nanorods with longitudinal LSPR beyond 1000 nm without the use of acids or polymers while also conserving the width below 10 nm. Scale-up of the reaction to 1 L volume is demonstrated, thus offering a streamlined approach in contrast to the seeded-growth approach that poses the caveat of increased shape impurities. Finally, the mGNRs were functionalized, and the CTAB bilayer could be removed while maintaining the colloidal dispersion, as demonstrated by the XPS, EDAX and HR-TEM imaging data. Furthermore, the analytical requirement of more than one characterization technique for the confirmation of CTAB removal is presented and discussed. Conjugation of the model drug mTHPC illustrates the potential of these mGNRs as functional drug carriers for future applications in PDT and PTT.Data availability
All the data are available in the ESI.†Conflicts of interest
All authors declare that they have no conflicts of interest.Acknowledgements
We would like to thank the Erasmus+ Programme of the European Union, the U.S.-Israel Binational Science Foundation (BSF) and the Israel Science Foundation (ISF) for their financial support and contribution to this research. Their support allowed us to advance our understanding of the seedless synthesis of mini gold nanorods, their elongation and the realization of our surface modification goals presented in this work.References
- N. Narayan, A. Meiyazhagan and R. Vajtai, Materials, 2019, 12, 1–12 CrossRef PubMed.
- C. Gao, F. Lyu and Y. Yin, Chem. Rev., 2021, 121, 834–881 CrossRef CAS PubMed.
- K. McNamara and S. A. M. Tofail, Adv. Phys.: X, 2017, 2, 54–88 CAS.
- M. Nikzamir, A. Akbarzadeh and Y. Panahi, J. Drug Deliv. Sci. Technol., 2021, 61, 102316 CrossRef CAS.
- K. Saha, S. S. Agasti, C. Kim, X. Li and V. M. Rotello, Chem. Rev., 2012, 112, 2739–2779 CrossRef CAS PubMed.
- H. Kumar, K. Kuča, S. K. Bhatia, K. Saini, A. Kaushal, R. Verma, T. C. Bhalla and D. Kumar, Sensors, 2020, 20, 1–19 Search PubMed.
- L. K. Bogart, G. Pourroy, C. J. Murphy, V. Puntes, T. Pellegrino, D. Rosenblum, D. Peer and R. Lévy, ACS Nano, 2014, 8, 3107–3122 CrossRef CAS PubMed.
- T. T. V. Phan, T. C. Huynh, P. Manivasagan, S. Mondal and J. Oh, Nanomaterials, 2020, 10(1), 66 CrossRef CAS PubMed.
- A. Samanta, S. Banerjee and Y. Liu, Nanoscale, 2015, 7, 2210–2220 RSC.
- H. Altug, S. H. Oh, S. A. Maier and J. Homola, Nat. Nanotechnol., 2022, 17, 5–16 CrossRef CAS PubMed.
- E. C. Dreaden, A. M. Alkilany, X. Huang, C. J. Murphy and M. A. El-Sayed, Chem. Soc. Rev., 2012, 41, 2740–2779 RSC.
- M. Kus-liśkiewicz, P. Fickers and I. B. Tahar, Int. J. Mol. Sci., 2021, 22(20), 10952 CrossRef PubMed.
- G. P. Szekeres and J. Kneipp, Front. Chem., 2019, 7, 1–10 CrossRef PubMed.
- F. Tian, F. Bonnier, A. Casey, A. E. Shanahan and H. J. Byrne, Anal. Methods, 2014, 6, 9116–9123 RSC.
- G. M. A. Gad and M. A. Hegazy, Mater. Res. Express, 2019, 6, 085024 CrossRef CAS.
- S. Gravelsins, M. J. Park, M. Niewczas, S. K. Hyeong, S. K. Lee, A. Ahmed and A. A. Dhirani, Commun. Chem., 2022, 5, 103 CrossRef CAS PubMed.
- R. K. Singh, S. S. Behera, K. R. Singh, S. Mishra, B. Panigrahi, T. R. Sahoo, P. K. Parhi and D. Mandal, J. Photochem. Photobiol., A, 2020, 400, 112704 CrossRef CAS.
- M. Luna, Á. Cruceira, A. Díaz, J. M. Gatica and M. J. Mosquera, Environ. Technol. Innov., 2023, 30, 103070 CrossRef CAS.
- M. Yafout, A. Ousaid, Y. Khayati and I. S. El Otmani, Sci. Afr., 2021, 11, e00685 CAS.
- F. Y. Kong, J. W. Zhang, R. F. Li, Z. X. Wang, W. J. Wang and W. Wang, Molecules, 2017, 22(9), 1445 CrossRef PubMed.
- L. Pan, J. Liu and J. Shi, ACS Appl. Mater. Interfaces, 2017, 9, 15952–15961 CrossRef CAS PubMed.
- J. B. Vines, J. H. Yoon, N. E. Ryu, D. J. Lim and H. Park, Front. Chem., 2019, 7, 1–16 CrossRef PubMed.
- S. A. C. Carabineiro, Molecules, 2017, 22(5), 857 CrossRef PubMed.
- X. Hu, Y. Zhang, T. Ding, J. Liu and H. Zhao, Front. Bioeng. Biotechnol., 2020, 8, 1–17 CrossRef PubMed.
- J. Zheng, X. Cheng, H. Zhang, X. Bai, R. Ai, L. Shao and J. Wang, Chem. Rev., 2021, 121, 13342–13453 CrossRef CAS PubMed.
- V. D'Elia, J. Rubio-Retama, F. E. Ortega-Ojeda, C. García-Ruiz and G. Montalvo, Colloids Surf., A, 2018, 557, 43–50 CrossRef.
- M. Moros, A. Lewinska, F. Merola, P. Ferraro, M. Wnuk, A. Tino and C. Tortiglione, ACS Appl. Mater. Interfaces, 2020, 12, 13718–13730 CrossRef CAS PubMed.
- S. Liao, W. Yue, S. Cai, Q. Tang, W. Lu, L. Huang, T. Qi and J. Liao, Front. Pharmacol., 2021, 12, 664123 CrossRef CAS PubMed.
- T. C. Lebepe, S. Parani and O. S. Oluwafemi, Nanomaterials, 2020, 10, 1–24 CrossRef PubMed.
- N. R. Jana, L. Gearheart and C. J. Murphy, J. Phys. Chem. B, 2001, 105, 4065–4067 CrossRef CAS.
- B. Nikoobakht and M. A. El-Sayed, Chem. Mater., 2003, 15, 1957–1962 CrossRef CAS.
- A. Sánchez-Iglesias, K. Jenkinson, S. Bals and L. M. Liz-Marzán, J. Phys. Chem. C, 2021, 125, 23937–23944 CrossRef PubMed.
- L. Scarabelli, A. Sánchez-Iglesias, J. Pérez-Juste and L. M. Liz-Marzán, J. Phys. Chem. Lett., 2015, 6, 4270–4279 CrossRef CAS PubMed.
- X. Ye, L. Jin, H. Caglayan, J. Chen, G. Xing, C. Zheng, V. Doan-Nguyen, Y. Kang, N. Engheta, C. R. Kagan and C. B. Murray, ACS Nano, 2012, 6, 2804–2817 CrossRef CAS PubMed.
- L. Vigderman and E. R. Zubarev, Chem. Mater., 2013, 25, 1450–1457 CrossRef CAS.
- X. Xu, Y. Zhao, X. Xue, S. Huo, F. Chen, G. Zou and X. J. Liang, J. Mater. Chem. A, 2014, 2, 3528–3535 RSC.
- K. Liu, Y. Bu, Y. Zheng, X. Jiang, A. Yu and H. Wang, Chem.–Eur. J., 2017, 23, 3291–3299 CrossRef CAS PubMed.
- A. Malik, J. M. Khan, A. S. Alhomida, M. S. Ola, M. A. Alshehri and A. Ahmad, Chem. Pap., 2022, 76, 6073–6095 CrossRef CAS.
- J. Song, X. Yang, O. Jacobson, P. Huang, X. Sun, L. Lin, X. Yan, G. Niu, Q. Ma and X. Chen, Adv. Mater., 2015, 27, 4910–4917 CrossRef CAS PubMed.
- H. H. Chang and C. J. Murphy, Chem. Mater., 2018, 30, 1427–1435 CrossRef CAS PubMed.
- M. R. K. Ali, B. Snyder and M. A. El-Sayed, Langmuir, 2012, 28, 9807–9815 CrossRef CAS PubMed.
- K. I. Requejo, A. V. Liopo, P. J. Derry and E. R. Zubarev, Langmuir, 2017, 33, 12681–12688 CrossRef CAS PubMed.
- H. Jia, C. Fang, X. M. Zhu, Q. Ruan, Y. X. J. Wang and J. Wang, Langmuir, 2015, 31, 7418–7426 CrossRef CAS PubMed.
- Z. Li, S. Tang, B. Wang, Y. Li, H. Huang, H. Wang, P. Li, C. Li, P. K. Chu and X. F. Yu, ACS Biomater. Sci. Eng., 2016, 2, 789–797 CrossRef CAS PubMed.
- D. Xu, J. Mao, Y. He and E. S. Yeung, J. Mater. Chem. C, 2014, 2, 4989–4996 RSC.
- S. Seibt, H. Zhang, S. Mudie, S. Förster and P. Mulvaney, J. Phys. Chem. C, 2021, 125, 19947–19960 CrossRef CAS.
- Y. Xiong and Y. Xia, Adv. Mater., 2007, 19, 3385–3391 CrossRef CAS.
- G. González-Rubio, P. Llombart, J. Zhou, H. Geiss, O. Peña-Rodríguez, H. Gai, B. Ni, R. Rosenberg and H. Cölfen, Chem. Mater., 2024, 36, 1982–1997 CrossRef.
- A. Gole, J. W. Stone, W. R. Gemmill, H. C. Z. Loye and C. J. Murphy, Langmuir, 2008, 24, 6232–6237 CrossRef CAS PubMed.
- B. N. Khlebtsov, V. A. Khanadeev and N. G. Khlebtsov, J. Phys. Chem. C, 2008, 112, 12760–12768 CrossRef CAS.
- Z. Singh and I. Singh, Sci. Rep., 2019, 9, 1–13 CrossRef CAS PubMed.
- Á. M. Nunes, P. Falagan-Lotsch, A. Roslend, M. R. Meneghetti and C. J. Murphy, Nanoscale Adv., 2022, 5, 733–741 RSC.
- C. J. Murphy, H. H. Chang, P. Falagan-Lotsch, M. T. Gole, D. M. Hofmann, K. N. L. Hoang, S. M. McClain, S. M. Meyer, J. G. Turner, M. Unnikrishnan, M. Wu, X. Zhang and Y. Zhang, Acc. Chem. Res., 2019, 52, 2124–2135 CrossRef CAS PubMed.
- R. Del Caño, J. M. Gisbert-González, J. González-Rodríguez, G. Sánchez-Obrero, R. Madueño, M. Blázquez and T. Pineda, Nanoscale, 2020, 12, 658–668 RSC.
- A. Gole and C. J. Murphy, Chem. Mater., 2005, 17, 1325–1330 CrossRef CAS.
- J. G. Mehtala, D. Y. Zemlyanov, J. P. Max, N. Kadasala, S. Zhao and A. Wei, Langmuir, 2014, 30, 13727–13730 CrossRef CAS PubMed.
- L. Papaioannou, A. Angelopoulou, S. Hatziantoniou, M. Papadimitriou, P. Apostolou, I. Papasotiriou and K. Avgoustakis, AAPS PharmSciTech, 2019, 20 DOI:10.1208/s12249-018-1226-6.
- E. Varon, G. Blumrosen, M. Sinvani, E. Haimov, S. Polani, M. Natan, I. Shoval, A. Jacob, A. Atkins, D. Zitoun and O. Shefi, Int. J. Mol. Sci., 2022, 23(4), 2286 CrossRef CAS PubMed.
- H. Hantsche, Adv. Mater., 1993, 5, 778 CrossRef.
- C. D. Wagner, W. M. Riggs, L. E. Davis, J. F. Moulder and G. E. Muilenberg, Handbook of X-ray Photoelectron Spectroscopy, PerkinElmer Corp., 1979, vol. 192 Search PubMed.
- M. Tebbe, C. Kuttner, M. Männel, A. Fery and M. Chanana, ACS Appl. Mater. Interfaces, 2015, 7, 5984–5991 CrossRef CAS PubMed.
- M. Z. Wei, T. S. Deng, Q. Zhang, Z. Cheng and S. Li, ACS Omega, 2021, 6, 9188–9195 CrossRef CAS PubMed.
- W. Tong, M. J. Walsh, P. Mulvaney, J. Etheridge and A. M. Funston, J. Phys. Chem. C, 2017, 121, 3549–3559 CrossRef CAS.
- J. Rodríguez-Fernández, J. Pérez-Juste, P. Mulvaney and L. M. Liz-Marzán, J. Phys. Chem. B, 2005, 109, 14257–14261 CrossRef PubMed.
- A. Ott, S. K. Bhargava and A. P. O'Mullane, Surf. Sci., 2012, 606, L5–L9 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Tables of sizes, synthetic parameters and notes for their respective samples, additional histograms, TEM images, XPS and UV-vis-NIR spectra, reduction yields and synthesis information. See DOI: https://doi.org/10.1039/d4na00507d |
| This journal is © The Royal Society of Chemistry 2024 |