
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImpacts of heteroatom substitution on the excited state dynamics of π-extended helicenes†
Yuto
Kondo
a,
Yusuke
Tsutsui
 *ab,
Yusuke
Matsuo
a,
Takayuki
Tanaka
*a and
Shu
Seki
*a
*ab,
Yusuke
Matsuo
a,
Takayuki
Tanaka
*a and
Shu
Seki
*a
aDepartment of Molecular Engineering, Graduate School of Engineering, Kyoto University, Nishikyo-ku, Kyoto 615-8510, Japan. E-mail: tsutsui@moleng.kyoto-u.ac.jp; tanaka@moleng.kyoto-u.ac.jp; seki@moleng.kyoto-u.ac.jp
bJST-PRESTO, Honcho 4-1-8, Kawaguchi, Saitama 332-0012, Japan
First published on 1st August 2024
Abstract
Benzo-annulated aza[9]helicene ([9]AH) and thia[9]helicene ([9]TH) were prepared as novel π-extended heterohelicenes. [9]TH showed a quite short fluorescence lifetime of ∼0.3 ns and intense phosphorescence at low temperature that were attributed to its larger spin–orbit coupling and faster intersystem crossing between pseudo-degenerate S1/2 and triplet states.
π-Extended aromatic molecules are promising candidates for optical applications since they effectively delocalize π electrons on their molecular skeleton giving rise to unique optical properties in the UV-Vis-NIR regime. Most of the organic materials are designed as planar to enhance intramolecular π-conjugation. On the other hand, considering the unique electronic properties of curved π-systems such as carbon nanotubes and fullerene, the modulation in planarity and/or twisting in molecular design induces symmetry breaking in sp2-based orbitals and unique electronic structures compared to the planar molecules.1–8 Such symmetry breaking sometimes induces chirality to the original molecule; in other words, spatially delocalized chirality could be tailored by molecular design. Direct implementation of chirality based on the π-orbital enables the emergence of fascinating chiroptical properties in the Vis-NIR range. Recently, researchers have extended their efforts on such curved π-conjugated systems to unveil their potential and applicability in materials science.9–12
Helicene is one of the classical families of non-planar π-conjugated systems with a helical structure, mirror axis symmetry and spring-like molecular skeleton (Fig. 1(a)). Its unique properties have been widely demonstrated in a variety of fields such as asymmetric catalysis, molecular switches, electron spin filters and chiroptical applications including circular dichroism (CD) and circularly polarized luminescence (CPL) devices.13–18 On the other hand, since helicenes are also known for their low fluorescence quantum yield, it is highly demanded to design a new helicene motif with high fluorescence quantum yield.
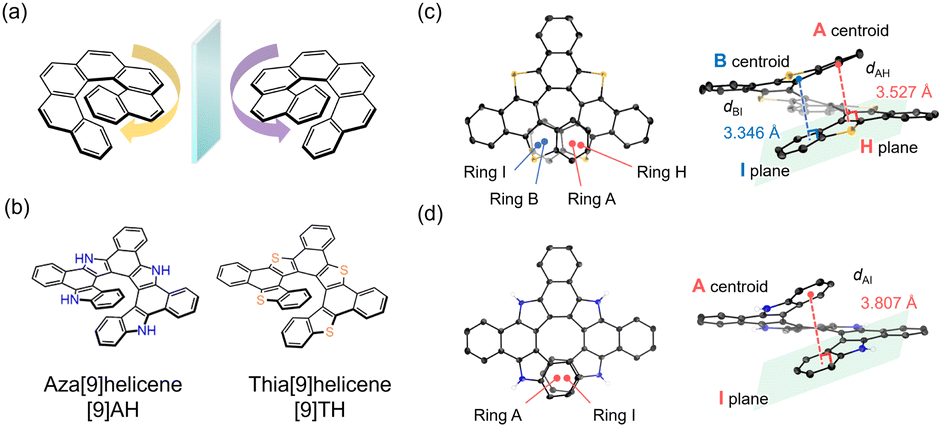 | ||
| Fig. 1 (a) Chemical structures of a carbo[7]helicene showing its chirality. (b) Chemical structures of newly designed hetero[n]helicenes (aza[9]helicene: [9]AH and thia[9]helicene: [9]TH). Molecular configurations of (c) [9]TH and (d) [9]AH extracted from their single crystal structures. Hydrogen atoms are partly omitted for clarity. | ||
One of the approaches is the introduction of heteroatoms in helicenes to drastically change their electronic structure.19–27 For example, a facile synthetic method for polyaza[7]helicene, a helicene with nitrogen atoms, was proposed in 2017 with higher fluorescence quantum yield and CPL activity than the original carbohelicenes.28 Fascinating phenomena of heterohelicenes were also reported such as thermally activated delayed fluorescence in B/N embedded derivatives,29,30 room temperature circularly polarized phosphorescence31 and electrocatalytic CO2 reduction.32 Recently, we have reported the facile synthesis of benzo-annulated aza[n]helicenes.33,34 Their high fluorescence quantum yield and great stability under ambient conditions are suited to the study of their excited state dynamics. It is also beneficial to derivatise this synthetic method to other heterohelicenes such as thiahelicenes by simply changing the starting materials. In this paper, aza[9]helicene ([9]AH) and newly designed thia[9]helicene ([9]TH) were spectroscopically characterized and their excited state dynamics were analyzed in detail (Fig. 1(b)).
[9]AH was synthesized based on a previously described procedure using an ortho-phenylene bridged tetrapyrrole as a precursor.33 By replacing the pyrrole to thiophene, [9]TH was newly synthesized and characterized with a similar procedure (see the ESI† for details). Single crystals of [9]TH were successfully obtained, from which the structure of [9]TH was revealed to include no solvent molecules in the lattice (Fig. S1–S7†). As shown in Fig. 1(c) and (d), due to the larger orbital of a sulfur atom, two aromatic rings (A–B and H–I) were almost completely overlapping each other to form a longer helix in [9]TH while only the terminal rings (A and I) were overlapped in [9]AH. The interplanar (centroid-to-plane) distances of [9]TH were estimated to be 3.346 and 3.527 Å which were shorter than that in [9]AH (3.807 Å). [9]TH exhibited a reversible first oxidation cycle from cyclic voltammetry with its half-wave oxidation potential of +0.63 V against Fc/Fc+ (Fig. S2†). The positive shift in oxidation potential compared to [9]AH (+0.089 V against Fc/Fc+) is in accordance with the deeper level in the HOMO, which is quasi-degenerate with the HOMO−1 and distributed to the whole molecular skeleton (Fig. S16†). The generation of the oxidized molecule was also confirmed by electrochemically induced absorption spectra (Fig. S3†).
The absorption and emission spectra of hetero[9]helicenes in dichloromethane are shown in Fig. 2. Both showed complex absorption spectra due to multiple contributions of optically allowed transitions to Sn states. Emission spectra are well-separated with significant vibronic progressions. The highest energy vibronic peak maxima were observed at 438 and 426 nm for [9]AH and [9]TH, respectively. A tiny Stokes shift and well resolved vibronic feature reflects the rigid structures of these hetero[9]helicenes. Further analysis of the excitation spectra of heterohelicenes (Fig. S13 and S14†) revealed that [9]AH showed emission sorely from S1 longer than 350 nm, but in [9]TH, S1 and S2 states are very close (∼60 meV) and the peaks at 400 and 426 nm are contributed also from the higher excited state (anti-Kasha behaviour). In our previous report, the enantiomers of [9]AH were successfully separated and their CD and CPL spectra were analyzed.33 Similarly, chiral HPLC allowed us to separate [9]TH with different retention times for (M)- and (P)-enantiomers (Fig. S4†). The observed electronic circular dichroism (ECD) and theoretically simulated ECD spectra suggested that the second fraction corresponded to the (P)-enantiomer (Fig. S5†). Circularly polarized luminescence (CPL) spectra were also observed with the opposite sign from [9]TH, where the g-value was estimated to be |gCPL| ∼2.6 × 10−3 (Fig. S6†).
 | ||
| Fig. 2 Absorption and emission spectra of (a) [9]AH and (b) [9]TH in dichloromethane. The excitation wavelength for emission measurement was set to 380 (350) nm for [9]AH ([9]TH). | ||
The absolute fluorescence quantum yield ΦF recorded was 0.20 and 0.01 for [9]AH and [9]TH, respectively (Table 1). The radiative and non-radiative rate constants (kf and knr) were estimated from the relationships ΦF = kf/(kf + knr) and τF = (kf + knr)−1. The fluorescence lifetime τF of [9]TH was 0.3 ns, one order of magnitude shorter compared to the 5.0 ns for [9]AH (Fig. S10†). Compared to carbo[9]helicene (ΦF = 0.014 and τF = 9.6 ns),35,36 the fluorescence quantum yield is enhanced especially for [9]AH, while lifetime is shorter in [9]AH, implying a larger radiative constant due to an enhanced transition dipole moment. It is suggested that the presence of partial n–π* character in the excited states using a lone pair of nitrogen atoms enhances the fluorescence quantum yield.37 On the other hand, fluorescence lifetime is significantly shorter in [9]TH. Since simple modification with heteroatoms drastically changed their photophysical properties, we decided to investigate these derivatives with more detailed analysis, especially for their excited states.
| Φ F | τ F/ns | k f/s−1 | k nr/s−1 | |
|---|---|---|---|---|
| [9]AH | 0.20 | 5.0 | 4.0 × 107 | 1.6 × 108 |
| [9]TH | 0.01 | 0.3 | 3.2 × 107 | 3.2 × 109 |
First, the temperature dependence of the emission spectra was evaluated as shown in Fig. 3. When the [9]TH solution was cooled down, the enhancement of the emission and sharpening of the peaks were observed due to the restricted molecular motion (Fig. 3(b)). Upon further cooling below 93 K, a new emission band emerged at 510–650 nm. As for the [9]AH solution, increases in the emission and sharpening of peaks at lower temperature were similarly observed. Tiny new bands also appeared at 520 and 560 nm, although the intensity was much weaker than that of [9]TH.
 | ||
| Fig. 3 Temperature dependent emission from (a) [9]AH and (b) [9]TH in 2-methyltetrahydrofuran with concentration of 1 μM. The excitation wavelength was set to 310 nm. | ||
To investigate the origin of the strong new band from [9]TH in more detail, we carried out several time-domain measurements. Time-resolved photoluminescence lifetime measurement revealed that the emission of [9]TH at 550 nm showed basically two components at 83 K, a fast (∼0.3 ns) and a very slow component, as shown in Fig. S11.† The former is the contribution of the tail from the fluorescence band suggested in steady state emission spectra (Fig. 3(b)), while the latter long lifetime component is indicative of the phosphorescence. A yellow afterglow was indeed readily confirmed by the naked eye. The phosphorescence lifetime of [9]TH was estimated to be ∼230 ms for 83 K and strongly depends on the temperature (Fig. S12†). The origin of phosphorescence at low temperature could be attributed to the reduction in non-radiative relaxation rate from the T1 state.
In order to observe the generation of the triplet state, transient absorption spectroscopy was carried out. Transient absorption spectra for [9]TH showed two significant bands: one was the (spontaneous and/or stimulated) emission at ∼420 nm to the negative direction and the other was the positive absorption band around 490 nm (Fig. 4(a)). The latter absorption band lasted for 0.74 μs at room temperature, but treatment with oxygen bubbling resulted in a decreased lifetime of 0.24 μs as shown in Fig. 4(c), suggesting the existence of the triplet excited state or related process. This process can be considered as an inherent nature of [9]TH since this kinetics and behaviour remained basically unchanged for various solvents and concentration (toluene, dichloromethane, 2-methyltetrahydrofuran, and benzonitrile as shown in Fig. S8 and Table S2†). On the other hand, [9]AH did not exhibit prolonged transient absorption spectra (Fig. S8(b)†) showing only the prompt contribution from fluorescence. Picosecond transient absorption spectra of [9]TH showed a prompt increase in absorption with a long-lasting feature. After 200 ps, the 500 nm peak slightly shifted to a shorter wavelength with a slight increase in intensity, implying the increase of the T1 state (Fig. S9†).
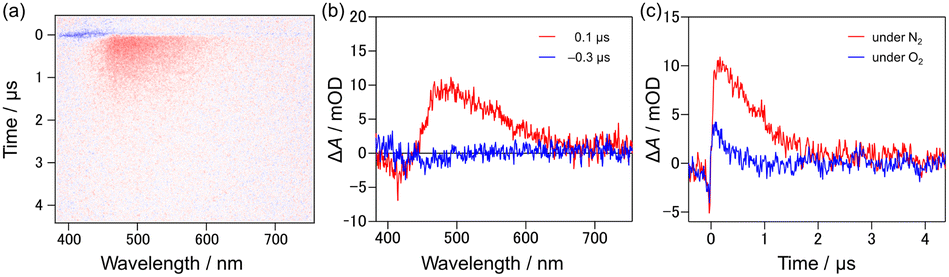 | ||
| Fig. 4 Transient absorption spectra of [9]TH in dichloromethane with the concentration of 0.1 mM. (a) Two-dimensional map of transient absorption. (b) Transient absorption spectra at 0.1 μs (red) and −0.3 μs (blue). (c) Kinetic traces at 490 nm under N2 saturated (red) and O2 bubbled (blue) conditions. | ||
To obtain further information on the difference between [9]AH and [9]TH, DFT-calculation was carried out. As shown in Fig. 5, six triplet states (Tn) lie at lower energy levels than their S1 states both in [9]AH and [9]TH. In [9]TH, besides the proximity of S1 and S2, T7 and T8 are also very close to them. Although intersystem crossing from S2 to T7/8 is energetically unfavourable by ∼50 meV at S1 configuration (Fig. 5), the S2 level is higher than T7/8 at the Franck–Condon state (S0 configuration, Fig. S17†), meaning energetically favourable intersystem crossings and level crossing could be expected during geometrical relaxation (Fig. S17†). Since the intersystem crossing rate is correlated with the square of spin–orbit matrix elements and exponential of the energy difference of the related states,38 fast intersystem crossing is expected for S2 and T7/8. In a ps-transient absorption experiment, it is considered that S1 and S2 states are simultaneously excited by the broadband (∼7 nm FWHM) pulse. Then, the S2 state can generate Tn and S1 in the picosecond time scale, followed by the decay of the S1 state with ∼0.3 ns. Indeed, longer excitation wavelengths seemed to delay the generation of the triplet state (Fig. S15†). However, we cannot rule out the contribution from other species at ∼500 nm in the early stage. To confirm the whole scenario, it would be required to carry out an excitation wavelength dependence test with higher resolution and longer pulse width (∼1 ps).
 | ||
| Fig. 5 Summarized energy landscape for [9]AH and [9]TH. Most of the excited states are calculated S1 optimized structures (@S1) unless otherwise noted. Excited states at S0 and T1 optimized structures are denoted by dashed and dotted lines. SOC constants (S1/Tn for S1 configuration, T1/S0 for T1 configuration) are shown in parentheses in cm−1. Calculated at the CAM-B3LYP/6-31G(d) level. | ||
The spin–orbit coupling (SOC) matrix elements between each excited state were also theoretically estimated. [9]AH showed a maximum coupling amplitude of only 0.28 cm−1 between the S1 and T1 states at the S1 configuration (denoted as S1@S1 and T2@S1). The small SOC suggests less contribution of intersystem crossing for its excited dynamics. On the other hand, much higher couplings were estimated for [9]TH, especially for S1/T3 (1.8 cm−1), S1/T1 (1.8 cm−1) and S1/T6 (1.4 cm−1). This larger coupling partly contributes to the faster intersystem crossing in [9]TH, hence suppressed fluorescence quantum yield and short fluorescence lifetime in [9]TH (Table 1). As for the relaxation from the T1 to S0 state, much larger coupling amplitudes from T1@T1 to S0@T1 were estimated in [9]TH (7.1 cm−1) than [9]AH (1.16 cm−1), supporting the strong phosphorescence observed from [9]TH. To conclude, the emergence of the stronger phosphorescence in [9]TH originated from the enhanced SOC and swift intersystem crossing from the S1/2 to the triplet state. On the other hand, [9]AH showed higher fluorescence quantum yield due to its enhanced transition dipole moment compared to carbo[9]helicene. Triplet states cannot readily be populated by intersystem crossing in [9]AH, resulting in the suppression of phosphorescence and absence of transient absorption from triplet species.
Conclusions
Benzo-annulated [9]AH and [9]TH, a new family of heterohelicenes, were newly designed and spectroscopically investigated in detail. Introduction of nitrogen atoms improved the fluorescence quantum yield ΦF to 0.20 compared to its carbon-analogue (ΦF = 0.014) at room temperature. On the other hand, introduction of sulfur atoms resulted in a much shorter fluorescence lifetime of 0.3 ns with small quantum yield. The suppressed fluorescence quantum yield and shorter lifetime of [9]TH was attributed to the efficient intersystem crossing due to large spin–orbit coupling and energetically close S1/2 and T7/8 states in this system. [9]TH showed enhanced phosphorescence at low temperature and a long-lifetime component in transient absorption, supporting the generation of triplet species. As shown in these results, the photophysical dynamics and their outcomes can be tailored through proper design of hetero[n]helicenes. Introduction of heteroatoms in π-extended helicenes will be a fascinating method for broadening the scope of future research, potentially leading to new discoveries and advancements in chiroptic properties from curved π-systems.Experimental
Materials and synthesis
All the details are provided in the ESI.†Absorption and emission spectra
Absorption spectra were recorded on a JASCO V-770 spectrometer. The concentration of [9]TH ([9]AH) was 1 × 10−4 M (1.3 × 10−4 M), and a 1 mm quartz cell was used for the measurements. For electrochemical absorption spectra, the voltage was modulated using a BAS ALS612E electrochemical analyzer. Tetrabutylammonium tetrafluoroborate was dissolved as an electrolyte at a concentration of 0.1 M. Emission spectra were measured on a SHIMADZU RF-6000 fluorescence spectrophotometer. The concentration of each sample was 1 × 10−6 M, and a 1 cm quartz cell was used for the measurements. Bandwidth was typically set at 3–5 nm for emission spectra. Excitation spectra of [9]TH ([9]AH) were measured with a concentration of 1 × 10−6 M (1.3 × 10−6 M) with an excitation/emission bandwidth of 1.5/1.0 nm. Temperature was controlled using a UNISOKU CoolSpek UV USP-203. Fluorescence quantum yield measurement was carried out with a C9920-02S (Hamamatsu Photonics) at room temperature. Circular dichroism spectroscopy was carried out with J-1700 JASCO under the same condition for absorption measurements. Circularly polarized photoluminescence spectra of [9]TH were measured with CPL-200S, JASCO from its dichloromethane solution.Transient absorption measurements
The sample cell was excited with a Nd:YAG nanosecond laser (10 Hz, EKSPLA NT341A-OPO). The diameter of the excitation beam was approximately 1 cm and the pump intensity was 0.5 mJ (for OPO experiments, it was adjusted to 1.2, 0.8, 1.5, 1.9, and 1.8 mJ for 355, 410, 420, 430, and 440 nm, respectively). A continuous Xe-lamp (Hamamtsu Photonics, L14972-01) was used as a probe light and orthogonally guided to the sample cuvette (1 cm, quartz). The transmitted probe light was dispersed both in wavelength and time to afford a two-dimensional image using a spectrograph and a streak scope (Hamamatsu Photonics).Lifetime measurements
Photoluminescence lifetime measurements were carried out using a Quantaurus-tau (Hamamatsu Photonics) at room temperature. The hetero[9]helicenes were dissolved in dichloromethane with a concentration of 10−6 M and excited at 365 nm. Temperature dependence was separately measured by a home-built set-up as follows. The ps-pulsed laser diode (405 nm, LDB-160C-405, Tama-denshi) excited the sample loaded in a liq-N2 cryostat (CoolSpek, Unisoku). Fluorescence was collected with a 90 degree configuration through a band-pass filter (FBH450-10 or FBH550-40, Thorlabs). It was then detected using an Si-APD (C16533, Hamamatsu Photonics) and counted using a universal frequency counter/timer (53230A, Keysight).Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Author contributions
Y. K. and Y. T. carried out most of the optical experiments, theoretical calculations, and wrote the manuscript. Y. M. synthesized and characterized the helicene derivatives, and measured the fluorescence quantum yield and lifetime at room temperature. T. T. and S. S. guided, reviewed, and edited the manuscript. T. T. also carried out the circularly polarized photoluminescence measurements.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JST, PRESTO Grant Number JPMJPR21Q5 and a Grant-in-Aid for Scientific Research (no. 22H00314, 20H05862, 20H05867, 22H00314 and 23K17942). We also acknowledge Dr Shohei Saito for photoluminescence lifetime measurements at room temperature, and Dr Masayuki Gon and Prof. Dr Kazuo Tanaka for circularly polarized photoluminescence measurements.Notes and references
- X. Lu and Z. Chen, Chem. Rev., 2005, 105, 3643–3696 CrossRef CAS PubMed.
- M. A. Majewski and M. Stępień, Angew. Chem., Int. Ed., 2019, 58, 86–116 CrossRef CAS PubMed.
- W. E. Barth and R. G. Lawton, J. Am. Chem. Soc., 1966, 88, 380–381 CrossRef CAS.
- L. T. Scott, M. M. Hashemi, D. T. Meyer and H. B. Warren, J. Am. Chem. Soc., 1991, 113, 7082–7084 CrossRef CAS.
- G. Povie, Y. Segawa, T. Nishihara, Y. Miyauchi and K. Itami, Science, 2017, 356, 172–175 CrossRef CAS PubMed.
- H. Sakurai, T. Daiko and T. Hirao, Science, 2003, 301, 1878 CrossRef CAS PubMed.
- M. Baba, J. Phys. Chem. A, 2011, 115, 9514–9519 CrossRef CAS PubMed.
- F. Guinea, A. K. Geim, M. I. Katsnelson and K. S. Novoselov, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 035408 CrossRef.
- T. Matsuno, S. Sato, R. Iizuka and H. Isobe, Chem. Sci., 2015, 6, 909–916 RSC.
- M. M. Chaolumen, Y. Sugano, A. Wakamiya and Y. Murata, Angew. Chem., Int. Ed., 2015, 54, 9308–9312 CrossRef CAS PubMed.
- T. Hosokawa, Y. Takahashi, T. Matsushima, S. Watanabe, S. Kikkawa, I. Azumaya, A. Tsurusaki and K. Kamikawa, J. Am. Chem. Soc., 2017, 139, 18512–18521 CrossRef CAS PubMed.
- H. Kawashima, N. Fukui, Q. M. Phung, T. Yanai and H. Shinokubo, Cell Rep. Phys. Sci., 2022, 3, 101045 CrossRef CAS.
- M. Gingras, Chem. Soc. Rev., 2013, 42, 968–1006 RSC.
- Y. Shen and C.-F. Chen, Chem. Rev., 2012, 112, 1463–1535 CrossRef CAS PubMed.
- W. Liu, T. Qin, W. Xie and X. Yang, Chem.–Eur. J., 2022, 28, e202202369 CrossRef CAS PubMed.
- W.-L. Zhao, M. Li, H.-Y. Lu and C.-F. Chen, Chem. Commun., 2019, 55, 13793–13803 RSC.
- L.-J. Peng, X.-Y. Wang, Z.-A. Li and H.-Y. Gong, Asian J. Org. Chem., 2023, e202300543 CrossRef CAS.
- Y. Nakakuki, T. Hirose, H. Sotome, M. Gao, D. Shimizu, R. Li, J. Hasegawa, H. Miyasaka and K. Matsuda, Nat. Commun., 2022, 13, 1475 CrossRef CAS PubMed.
- M. Miyasaka, M. Pink, S. Rajca and A. Rajca, Angew. Chem., Int. Ed., 2009, 48, 5954–5957 CrossRef CAS PubMed.
- K. Nakano, Y. Hidehira, K. Takahashi, T. Hiyama and K. Nozaki, Angew. Chem., Int. Ed., 2005, 117, 7298–7300 CrossRef.
- S. K. Pedersen, K. Eriksen and M. Pittelkow, Angew. Chem., Int. Ed., 2019, 58, 18419–18423 CrossRef CAS PubMed.
- A. Rajca, M. Pink, S. Xiao, M. Miyasaka, S. Rajca, K. Das and K. Plessel, J. Org. Chem., 2009, 74, 7504–7513 CrossRef CAS PubMed.
- K. Tanaka, N. Fukawa, T. Suda and K. Noguchi, Angew. Chem., Int. Ed., 2009, 48, 5470–5473 CrossRef CAS PubMed.
- S. K. Collins and M. P. Vachon, Org. Biomol. Chem., 2006, 4, 2518–2524 RSC.
- S. Maiorana, A. Papagni, E. Licandro, R. Annunziata, P. Paravidino, D. Perdicchia, C. Giannini, M. Bencini, K. Clays and A. Persoons, Tetrahedron, 2003, 59, 6481–6488 CrossRef CAS.
- S. Graule, M. Rudolph, N. Vanthuyne, J. Autschbach, C. Roussel, J. Crassous and R. Réau, J. Am. Chem. Soc., 2009, 131, 3183–3185 CrossRef CAS PubMed.
- Z. Sun, W. Xu, S. Qiu, Z. Ma, C. Li, S. Zhang and H. Wang, Chem. Sci., 2023, 15, 1077–1087 RSC.
- T. Otani, A. Tsuyuki, T. Iwachi, S. Someya, K. Tateno, H. Kawai, T. Saito, K. S. Kanyiva and T. Shibata, Angew. Chem., Int. Ed., 2017, 56, 3906–3910 CrossRef CAS PubMed.
- S. Oda, B. Kawakami, Y. Yamasaki, R. Matsumoto, M. Yoshioka, D. Fukushima, S. Nakatsuka and T. Hatakeyama, J. Am. Chem. Soc., 2022, 144, 106–112 CrossRef CAS PubMed.
- S.-Y. Yang, S.-N. Zou, F.-C. Kong, X.-J. Liao, Y.-K. Qu, Z.-Q. Feng, Y.-X. Zheng, Z.-Q. Jiang and L.-S. Liao, Chem. Commun., 2021, 57, 11041–11044 RSC.
- S. Tanaka, D. Sakamaki, N. Haruta, T. Sato, M. Gon, K. Tanaka and H. Fujiwara, J. Mater. Chem. C, 2023, 11, 4846–4854 RSC.
- P. Karak, S. K. Mandal and J. Choudhury, J. Am. Chem. Soc., 2023, 145, 7230–7241 CrossRef CAS PubMed.
- Y. Matsuo, M. Gon, K. Tanaka, S. Seki and T. Tanaka, J. Am. Chem. Soc., 2024, 146, 17428–17437 CrossRef CAS PubMed.
- F. Chen, T. Tanaka, T. Mori and A. Osuka, Chem.–Eur. J., 2018, 24, 7489–7497 CrossRef CAS PubMed.
- J. B. Birks, D. J. S. Birch, E. Cordemans and E. Vander Donckt, Chem. Phys. Lett., 1976, 43, 33–36 CrossRef CAS.
- E. V. Donckt, J. Nasielski, J. R. Greenleaf and J. B. Birks, Chem. Phys. Lett., 1968, 2, 409–410 CrossRef.
- K. Schmidt, S. Brovelli, V. Coropceanu, D. Beljonne, J. Cornil, C. Bazzini, T. Caronna, R. Tubino, F. Meinardi, Z. Shuai and J.-L. Brédas, J. Phys. Chem. A, 2007, 111, 10490–10499 CrossRef CAS PubMed.
- D. Beljonne, Z. Shuai, G. Pourtois and J. L. Bredas, J. Phys. Chem. A, 2001, 105, 3899–3907 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4na00516c |
| This journal is © The Royal Society of Chemistry 2024 |