
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNIR-triggered photooxygenation of α-terpinene with upconversion nanohybrids†
Laura
Francés-Soriano‡
 ,
Delia
Bellezza‡
,
Juan
Ferrera-González
,
María
González-Béjar
* and
Julia
Pérez-Prieto
*
,
Delia
Bellezza‡
,
Juan
Ferrera-González
,
María
González-Béjar
* and
Julia
Pérez-Prieto
*
Instituto de Ciencia Molecular (ICMol), Departamento de Química Orgánica, Universitat de València, Calle Catedrático José Beltrán 2, Paterna, Valencia 46980, Spain. E-mail: maria.gonzalez@uv.es; julia.perez@uv.es
First published on 11th September 2024
Abstract
Upconversion nanohybrids (UCNHs) consisting of rose bengal (RB) and upconversion nanoparticles (UCNPs) are able to promote terpinene oxidation upon near-infrared irradiation. The photophysical events occurring upon NIR-irradiation of the UCNH correlate well with the synthetic protocol used to prepare the UCNHs (RB loading and aggregation). These results highlight the importance of the optimization of UCNH composition for the photocatalysis outcome.
Introduction
Photocatalysts have been used in organic synthesis because of their inherent potential to implement organic transformations, which may be otherwise non-feasible under mild and green conditions.1 In this context, photocatalyzed oxidations involving molecular oxygen as a reactant are of great interest since they can form carbon–oxygen and heteroatom–oxygen bonds.2,3Near-Infrared (NIR) radiation accounts for ≈50% of the solar spectrum and offers fewer health risks and side reactions, as well as a deeper penetration than ultraviolet (UV) and visible (VIS) light through various reaction media.4 NIR-triggered photocatalysis has emerged as a greener alternative in organic synthesis over UV-VIS photocatalysis.5
Positive photocatalysis to accomplish organic transformations triggered by NIR light can be direct and indirect, involving single and dual photoactive catalysts, respectively.6 The direct one serves from molecular photocatalysts (PCs, e.g., cyanines,5,7 squaraines,8 and so on) to absorb NIR-light. The indirect one serves from an upconverting system to absorb NIR-light, e.g., upconversion nanoparticles (UCNPs) in which lower-energy photons are transformed into higher-energy photons. The upconverting system is combined with PCs (able to absorb the upconverted light). This combination is usually known as upconversion nanohybrid (UCNH). The major advantage of this strategy is that known PCs can be used to synthesize UCNHs.4,6 NIR-excitation of UCNHs has proven useful for photoinduced reactions,9 such as photopolymerization,10 photoisomerization, and photocoupling.11 For example, UCNHs containing [Ru(bpy)3]2+ as a PC have been used to accomplish the photoredox-catalysed aerobic oxidation of ethyl-2-bromo-2-phenylacetate.4
The attachment of a PC to a UCNP surface can confer upon the UCNH the ability to generate singlet oxygen (1O2) upon NIR excitation, since the PC can absorb the upconverted emission to generate singlet oxygen, a phenomenon previously demonstrated with numerous PCs.12–23
It would be expected that organic transformations carried out with the selected PC (absorption in the UV-VIS range) could easily be performed by using NIR light instead to activate the UCNHs containing the same PC.
In this context, we have recently prepared a colloidal UCNH made of a xanthenic dye, in particular rose bengal (RB), grafted to ytterbium and erbium co-doped upconversion nanoparticles (UCEr) to study the fundamental mechanisms which govern the dye photophysics upon interaction with the UCNPs.24 Moreover, among the previously reported UCNHs made up of UCNPs and RB,15,24–29 the generation of singlet oxygen (1O2) has been identified15,28 upon NIR-irradiation of UCNHs with a similar composition.
RB has been extensively used for visible light–driven reactions because the RB triplet excited state (3RB) transfers energy to ground state oxygen (3O2) to give singlet oxygen 1O2.30 The photooxygenation of α-terpinene has been extensively evaluated with heterogeneous systems containing RB as the PC, thereby facilitating the separation process and avoiding RB photodegradation.31–34 We envisaged that the synthesized UC@RBn NHs could serve as heterogeneous NIR-photocatalysts.
Up to now, the influence of photoactive lanthanides doping the UCNP and PC loading on the photocatalytic activity of UCNHs remains unexplored. The preparation of UCNHs is not a trivial task and we presumed that the photocatalytic performance of each UCNH would be strongly dependent on its photophysical properties. We have prepared and thoroughly characterized several UC@RB NHs with varying RB amounts.
To evaluate their capability to generate singlet oxygen, the photooxygenation of α-terpinene to produce ascaridole was tested as a benchmark reaction (Scheme 1). This work presents a comparative analysis between the photophysical properties of several UCEr@RB NHs and their photocatalytic outcome upon NIR-irradiation.
 | ||
| Scheme 1 NIR-triggered photooxidation of α-terpinene catalysed by UC@RB to give ascaridole and p-cymene (by-product). | ||
Results and discussion
Functionalization of UCNPs with RB and characterization
Oleate-capped β-NaYF4:Yb3+ (20%), Er3+ (2%) UCNPs (UC@OA) were synthesized by using a well-known thermal decomposition method with some modifications, as detailed in the ESI.†35 The transmission electron microscopy (TEM) image (Fig. S1†) showed homogeneous hexagonal prisms of UC@OA with a size of 21.0 ± 0.8 × 18.9 ± 0.7 nm.Functionalization with RB was achieved by following a two-step synthetic procedure (see the ESI†).24 First, the oleate was removed from the UC@OA surface by treatment with NOBF4. Then, oleate-free UCNPs were mixed with different RB concentrations ranging from 4.2 × 10−2 to 4.2 × 10−7 M (see the Experimental section for further details) in DMF for 24 hours, leading to the formation of different nanohybrids UC@RB1, UC@RB2, UC@RB3, UC@RB4, UC@RB5, and UC@RB6. The purification of the synthesized UC@RBn (n = 1–6) nanohybrids was based on cycles combining washing with DMF and centrifugation until the supernatant was colourless. Consequently, the number of cycles differed. Therefore, the lower the RB concentration, the lower the volume of DMF needed for washing UC@RBn. The dispersions of different UC@RBn nanohybrids in DMF resulted in pink dispersions with varying shades.
Among them, the most colourful was UC@RB2 (Fig. 1) thus indicating that the degree of RB aggregation in the starting stock solutions, aimed at preparing UC@RBn NHs, leads to less attachment.
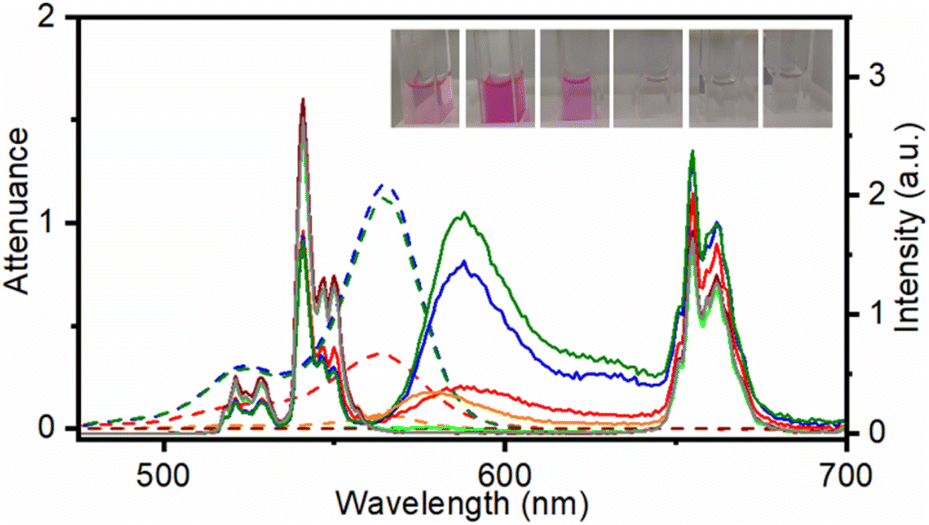 | ||
| Fig. 1 Attenuance (dashed line) and emission spectra (λexc = 980 nm) normalized at 843 nm of dispersions in DMF at 1 mg mL−1 of UC@RB1 (red), UC@RB2 (blue), UC@RB3 (green), UC@RB4 (orange), UC@RB5 (light green), UC@RB6 (brown), and UC@OA (grey). Inset: photographs of the different dispersions in DMF of the UC@RB nanohybrids. From right to left: UC@RB1, UC@RB2, UC@RB3, UC@RB4, UC@RB5, and UC@RB6. | ||
To quantify the amount of RB attached to the UCNP surface, the RB concentration was estimated from each UC@RBn NH absorption spectrum after scattering subtraction. Considering the number of UCNPs in 1 mg of UCNH36 and the RB moles derived from the absorbance, we initially estimated 34 ± 2, 104 ± 6, 98 ± 6, 6 ± 1, 1 ± 1, and 0.0 ± 0.1 RB molecules per UCNP for UC@RB1, UC@RB2, UC@RB3, UC@RB4, UC@RB5, and UC@RB6, respectively. It is noteworthy that these values are similar to those calculated in our previous work (20 RB/UCNP).24
Photophysical characterization of UC@RBn nanohybrids
To evaluate the influence of the RB concentration on the photophysical properties of the UC@RBn NHs, their absorption and emission spectra were recorded in dimethylformamide (DMF). The absorption spectra of the UC@RBn dispersions should exhibit a consistent trend of increasing absorption as the amount of RB on the NH increases. Notably, the absorption of UC@RB2 and UC@RB3, containing ∼104 RB/UCNP or with 98 RB/UCNP exceeded that of UC@RB1, with 34 RB/UCNP (Fig. 1). This is consistent with previous results, where RB aggregates were observed when preparing UCNHs with more than 100 RB molecules per UCNP. In these UCNHs, RB was covalently linked to a ligand coating the UCNPs.15 Absorption spectra revealed a band centered at 560 nm with a shoulder at shorter wavelengths (λmax: 523 nm), which enhances with an increasing RB concentration due to the presence of RB dimers or higher order aggregates. We previously hypothesized that the aggregation of RB molecules occurred on the surface of the NPs due to concentration-induced aggregation or by interaction with the positively charged surface of UCNPs.24 The fact that UC@RB1 presents the highest A560/A522 ratio can be attributed to a pre-aggregation of RB molecules in the stock solution. This pre-aggregation could lead to the direct attachment of RB dimers/aggregates to the UCNP surface. Moreover, the thermogravimetric analysis revealed a total mass loss of 26.1% that corresponds to 2286 ± 26 RB molecules attached to the surface of the UCNPs (Fig. S2†). Consequently, the number of RB molecules in UC@RB1 would be higher than 34, estimated from the absorption spectrum.Fluorescence quantum yield (ΦF) of RB in UC@RB dispersions supports this hypothesis (see Fig. 2).
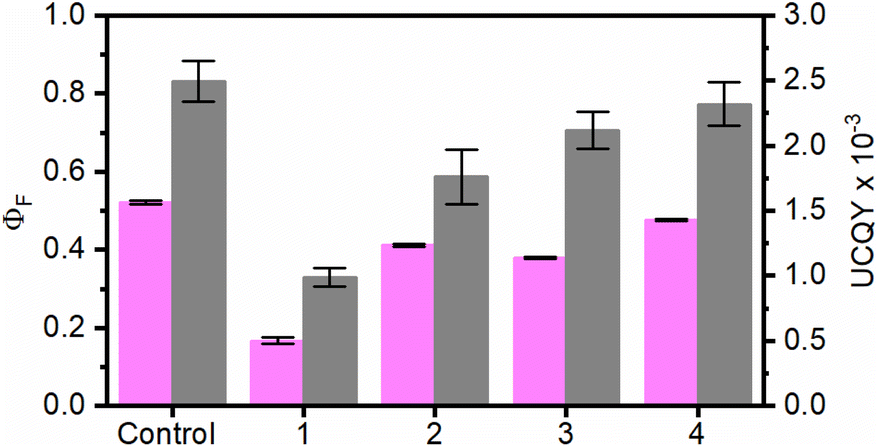 | ||
| Fig. 2 Fluorescence quantum yield of RB (ΦF; pink columns, λexc = 560 nm) and upconversion quantum yield (UCQY, grey columns, λexc = 980 nm) of UC@RBn NHs (n = 1–4). Control experiments correspond to RB for ΦF,RB and UC@OA for UCQY. | ||
All the UC@RBn NHs exhibited lower ΦF as compared to free RB, with UC@RB1 showing a particularly notable decrease of approximately 30% despite having the largest RB loading.
As previously reported, the reduced emission can be attributed to the self-deactivation effect due to π–π interactions in the aggregates of RB present in UC@RB1 NHs.24
The emission spectrum of the RB singlet excited state (1RB) in UC@RB2 is centered at 576 nm as reported for RB (Fig. S3†).
The lifetime of the 1RB was estimated to be between 2 and 2.3 ns in all the NHs and free RB. Neither 1RB emission nor the lifetime was sensitive to oxygen (see Fig. S3, 4 and Table S1†).
Under excitation at 980 nm, Yb3+ acted as a sensitizer, absorbing the incident light and subsequently transferring the energy to Er3+ cations. Accordingly, UC emission spectra of UC@RBn NHs displayed the characteristic emission bands of Er3+ ions in the UV, visible and NIR spectra (Fig. 1). The deactivation of Er3+ through emissive transitions resulted in six main bands: a very weak UV emission band centred at ∼410 nm was observed due to the 2H9/2 → 4I15/2 transition. Following this, two green bands were detected at ∼525 nm (2H11/2 → 4I15/2) and ∼540 nm (4S3/2 → 4I15/2), with the latter being the most intense in the original UC@OA. Additionally, a red emission band was observed at around 660 nm, corresponding to the transition 4F9/2 → 4I15/2. Finally, the 4S3/2 → 4I13/2 transition led to a broad emission band centred at ca. 842 nm (not shown).
Noteworthy quenching of the UC green emission by 36%, 42%, and 44% was observed for UC@RB1, UC@RB2, and UC@RB3, respectively (see Fig. S5†). The spectral overlap between the RB absorption and Er3+ green emission would explain the decrease of the UC emission in the NHs with a higher RB loading.
Interestingly, alongside the deactivation of UC emission, a new emission centered at 588 nm emerged, which would correspond to the sensitized RB emission. Accordingly, resonant energy transfer (RET) from the Er3+ of the UCNPs to RB resulted in a long-lived RB emission (∼50 μs; Table S2†); UC@RB3 exhibited the most intense emission followed by UC@RB2 and UC@RB1. These results were in accordance with the idea that higher RB concentrations during the synthesis led to the loading of non-fluorescent RB dimers/aggregates, consequently diminishing the fluorescence of RB on the UCNPs in the NH.
Time-resolved spectroscopy (λexc = 980 nm) was used to register the UC@RBs decays for all the emission bands (Fig. S6 and S7 and Table S2†). Non-significant differences were observed in lifetimes obtained in air or under N2. The emission lifetime at 540 nm was strongly shortened by the presence of RB in UC@RB1, UC@RB2, and UC@RB3, while in UC@RB4 the lifetime of the green emission remained practically unchanged, likely due to the low quantity of RB on the surface.
Notably, the amount of RB in UC@RB4, UC@RB5, and UC@RB6 was not enough to induce significant quenching of the UC green emission in comparison to the initial UC@OA. Nevertheless, while UC@RB5 and UC@RB6 exhibited no detectable RB emission, UC@RB4 demonstrated a notable emission band. Despite the low UC quenching efficiency (less than 1%), the RB emission peak was comparable to that of the UC@RB1 NH, evidencing an efficient energy transfer from UCNPs to RB. Furthermore, the peak maximum shifted to 577 nm, precisely matching that of free RB. This indicates that in UC@RB4, RB molecules are likely in a similar environment as they would be free in solution, allowing them to emit light at their characteristic wavelength.
In our prior investigation, we noted a red-shifting of the RB emission on UC@RB in 5 mg mL−1 dispersions in DMF due to concentration-induced aggregation.24 This effect was suppressed by using more dilute dispersions (1 mg mL−1). However, even when recording the UC emission spectra at this concentration, the increased RB loading amount could influence the aggregation degree of the UC@RB NHs within the dispersion, subsequently affecting the RB emissive properties.
Concerning the UCQY, a dependence on the RB concentration at the UCNP surface was observed with UC@RB1, the most affected NH with a decrease of more than 50% (Fig. 2 and S8†). This is consistent with an efficient deactivation of the UC luminescence in the presence of RB, due to RET processes from the UCNPs to RB upon NIR excitation.
Nanosecond-laser flash photolysis was used to record the transient absorption spectra of RB (Fig. 3) in the UCNHs and RB in DMF (absorbance was 0.24 at 560 nm). As we previously reported, RB triplet excited state (3RB) absorption spectra recorded for the UC@RB NHs exhibited three absorption bands at ca. 380, 470 and 610 nm.24 The lower the number of RB molecules in the NH, the higher the absorbance of 3RB in the spectra, which reveals better intersystem crossing (triplet formation). The generated 3RB subsequently evolved into a RB radical anion (RB˙−), which absorbs at ca. 440 nm (black trace in Fig. 3). However, when exciting RB in UC@RB, radical anion formation was clearly prevented for UC@RB2, UC@RB3 and UC@RB4, where 3RB was almost exclusively generated, as compared to RB in solution and UC@RB1. As expected, O2 quenched the 3RB (Fig. S9†) in the UCNH revealing a fast process, close to a diffusion-controlled mechanism (3τRB(O2) shorter than 1 μs) (Table S3†).
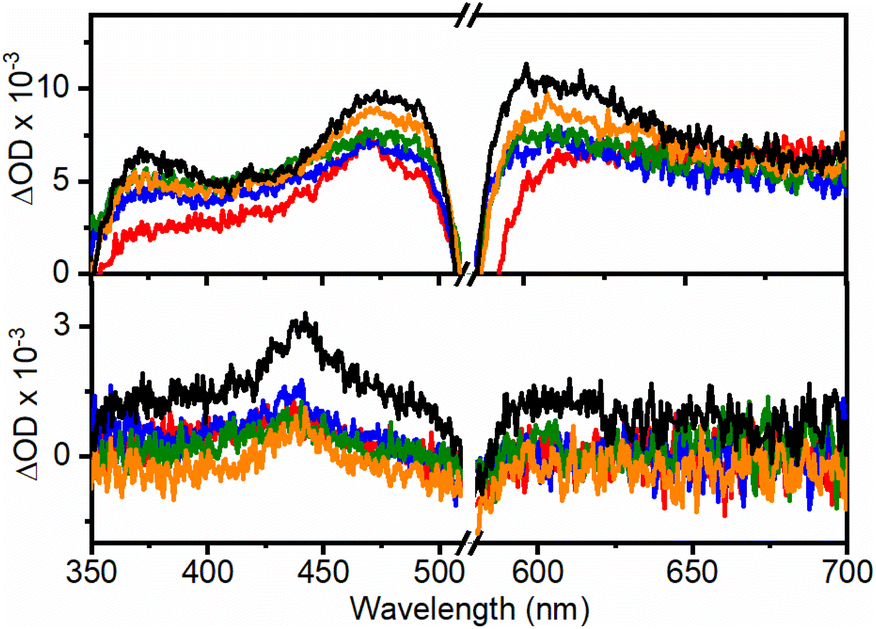 | ||
| Fig. 3 Transient absorption spectra recorded at 20 ns (top) and 550 μs (bottom) after the laser pulse under N2 (λexc = 560 nm, A = 0.24 at 560 nm) for UC@RB1 (red), UC@RB2 (blue), UC@RB3 (green), UC@RB4 (orange), and RB (black line) in DMF. | ||
As previously reported, room temperature phosphorescence of 3RB (670–1300 nm; maximum at ca. 750 nm) was also observed with a lifetime >150 μs and, as expected, was quenched by oxygen (Fig. S10, S11 and Table S4†).
Moreover, laser induced emission measurements revealed 3RB phosphorescence together with thermally activated delayed fluorescence under N2 (Fig. S12†).
Remarkably, when RB in the UCNH was selectively excited at 560 nm, an antenna effect from RB to the photoactive lanthanide ions in UC@RB was also observed. Fig. 4 shows two intense bands centered at 975 and 1550 nm attributed to the Yb3+·2F5/2 → 2F7/2 transition (the Er3+·4I11/2 → 4I15/2 transition could play a minor role due to lower doping) and the Er3+·4I13/2 → 4I15/2 transition, respectively. The emission intensities of these NIR bands at 975 nm (Yb3+ emission and 3RB phosphorescence) and at 1550 nm (Er3+ emission) were quenched by O2. Therefore, the emission bands registered in air were exclusively attributed to an energy transfer process from 1RB, since 3RB was efficiently deactivated in air (Fig. S13–S15 and Tables S5–S6†). Accordingly, the emission bands under an inert atmosphere are due to the energy transfer from 1RB (46%) and 3RB (54%) to the photoactive lanthanides.
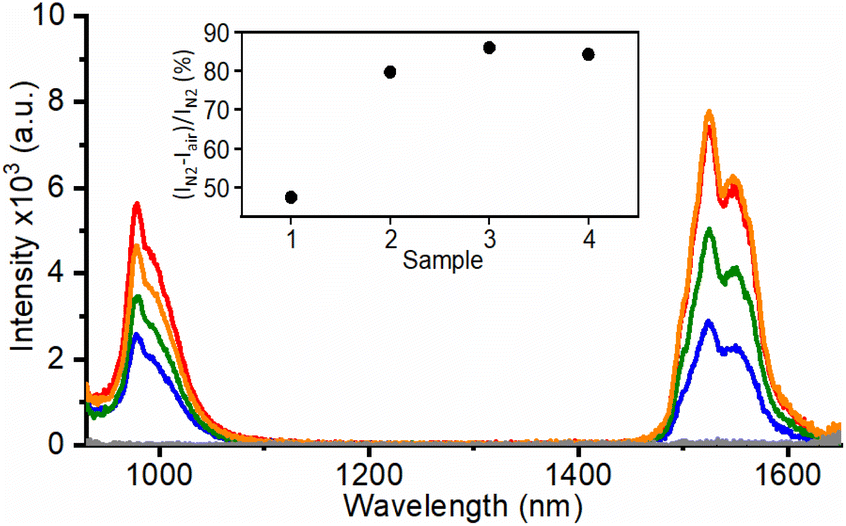 | ||
| Fig. 4 NIR emission spectra (λexc = 560 nm) of UC@RBn in DMF (5 mg mL−1) under N2 for UC@RB1 (red), UC@RB2 (blue), UC@RB3 (green), and UC@RB4 (orange). Inset: 3RB contribution in % for the different nanohybrids. | ||
Evaluation of 9,10-anthracenediyl-bis(methylene)dimalonic acid (ABDA) photoconsumption upon NIR-excitation of UC@RB nanohybrids
As stated above, upon NIR-irradiation of the UCNHs made up of UCNPs and RB, 1O2 production was detected (either by using 1,3-diphenylisobenzofuran (DPBF), as an optical probe28 or by direct detection of 1O2 phosphorescence at 1270 nm).15 Moreover, the optimal number of RB units for individual UCNPs has been determined to be ∼100.28 Even so, none of these UCNHs was used for synthetic purposes.Here, we initially tested the NIR-driven 1O2 generation of the UC@RB nanohybrids by using an anthracene-derivative, specifically 9,10-anthracenediyl-bis(methylene)dimalonic acid (ABDA), as a chemical 1O2-trapping agent, since 1O2 phosphorescence could not be detected spectroscopically under our experimental conditions.
The lifetime of singlet oxygen generation is solvent dependent.37 Therefore, the efficiency of its generation can vary for each UCNH according to the solvent and was estimated for three solvents capable of affording colloidal dispersibility of the UC@RB nanohybrids while dissolving ABDA in dimethylformamide (DMF), acetonitrile (ACN), and ethanol (EtOH).
Next, the decrease in ABDA fluorescence due to the formation of its nonfluorescent endoperoxide was followed.12 For this purpose, oxygen-saturated dispersions containing a mixture of UC@RB2 (with 100 RB/UCNP) and ABDA were irradiated with a 980 nm continuous wavelength laser diode to select the best solvent for the oxidation reaction. ABDA emission was recorded at different time intervals (Fig. 5). As shown in Fig. 5b and S16,† ABDA photoconsumption was observed in all solvents, indicating anthracene disappearance upon NIR-irradiation. The kinetic rates (k) of the process (monitored at 410 nm) are displayed in Table S7.† EtOH exhibited faster kinetics with a k value of 0.04, followed by ACN with a value of 0.024. However, ABDA photoconsumption did not progress after 2 hours of irradiation. Consequently, UC@RB2 dispersed in DMF was considered the most effective for ABDA photoconsumption, probably due to the superior dispersibility and stability of UC@RB in DMF.
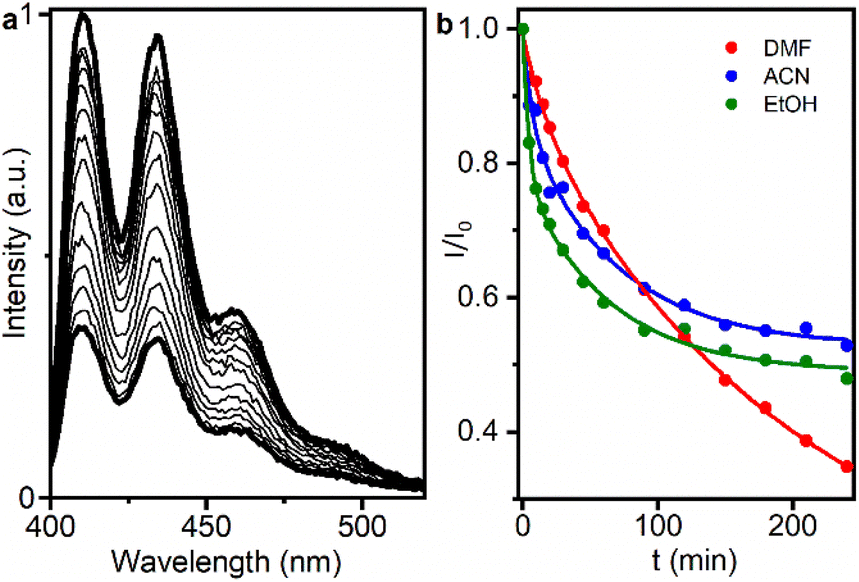 | ||
| Fig. 5 (a) Decrease in ABDA emission intensity generated upon NIR excitation of UC@RB2 in DMF (at different time intervals). (b) Decrease in the ABDA emission intensity recorded at 410 nm upon different NIR exposure times (up to 240 min) for UC@RB2 dispersions (1 mg mL−1) in DMF (red), ACN (blue), and EtOH (green). | ||
Evaluation of photocatalytic activity of UC@RB nanohybrids
The photooxygenation of α-terpinene involves the addition of 1O2 to the α-terpinene double bond, i.e., a [4 + 2] cycloaddition, resulting in the formation of ascaridole (Scheme 1) and autooxidation or dehydrogenation to p-cymene as a competitive reaction.31–33 The capability of UC@RBn NHs to photocatalyze this reaction and generate ascaridole upon NIR excitation was assessed in deuterated DMF (DMF-d7). For this purpose, an oxygen-saturated mixture of α-terpinene and the corresponding UC@RBn in DMF-d7 was irradiated with a 980 nm continuous wavelength laser diode for 8 h. The conditions and conversion efficiencies toward ascaridole and p-cymene are detailed in Table 1.| N | PC | [UC@RB] (mg mL−1) | Ascaridole (%) | p-Cymene (%) |
|---|---|---|---|---|
| a N2. b 24 h. c CD3CN. d Reaction performed with UC@RB recovered from entry 10. e Reaction performed with UC-RB recovered from entry 13. | ||||
| 1 | UC@RB1 | 1 | 12.4 | 4.1 |
| 2 | UC@RB2 | 1 | 17.0 | 5.3 |
| 3 | UC@RB3 | 1 | 16.5 | 4.7 |
| 4 | UC@RB4 | 1 | 8.3 | 4.1 |
| 5 | UC@BF4 | 5 | 4.1 | 5.2 |
| 6 | None | — | 4.1 | 6.2 |
| 7 | UC@RB3a | 1 | 2.7 | 12.4 |
| 8 | UC@RB2b | 1 | 20.8 | 4.0 |
| 9 | UC@RB3c | 1 | 12.4 | 7.2 |
| 10 | UC@RB2 | 5 | 41.3 | 3.1 |
| 11 | UC@RB3 | 10 | 53.8 | 6.2 |
| 12 | UC@RB2d | 5 | 12.4 | 2.6 |
| 13 | UC-RB | 20 | 25.7 | 17.6 |
| 14 | UC-RBe | 20 | 19.2 | 17.1 |
The photocatalyst activity of UC@RBn upon NIR irradiation was evidenced by the formation of ascaridole in all cases. At a concentration of 1 mg mL−1 of the UC@RBn photocatalyst, the reaction yield for ascaridole formation was calculated to be 12.4%, 17%, 16.5%, and 8.3% for UC@RB1, UC@RB2, UC@RB3, and UC@RB4, respectively (entries 1–4 in Table 1). These yields were calculated by using 1H-NMR spectroscopy after centrifugation to remove the NHs. UC@RB2 and UC@RB3 showed the best photocatalytic performance, which correlates well with their optical properties and corresponds to an RB/UCNP ratio of 100 (entries 2–3 in Table 1).
Control experiments with UCNPs without RB, in the absence of PC or oxygen (entries 5, 6 and 7, respectively), were carried out, thereby confirming that photooxidation was only possible in the presence of the UC@RBn NHs and O2. Although α-terpinene and reaction products are colourless with absorption bands in the UV region (no absorption at 980 nm), small amounts of the by-product, p-cymene, were detected in all cases even in the absence of PCs (entry 5).
Longer reaction times (24 h) gave rise to a marginal increase in the formation of ascaridole, reaching 20.8%. This result can be attributed to the combined effect of photo- and thermal-degradation of products (entry 8).38 Moreover, shorter reaction times of 1 and 4 h led to a lower ascaridole conversion, primarily due to the slow generation of singlet oxygen (Fig. S17†).
Alternatively, RB exhibits superior solubility and absorptivity in CD3CN. However, the photocatalytic activity to get ascaridole was slightly lower (12.4%) (entry 9). Therefore, DMF-d7 was selected as the solvent.
Then, experiments increasing the photocatalyst amount to 5 and 10 mg mL−1 were also conducted (entries 10 and 11). As a result, the yield of ascaridole reached 41.3% and 53.8%, respectively, after 8 hours of light exposure at 980 nm. Presumably, more 1O2 would be generated by increasing the quantity of RB, thus resulting in a two-fold and three-fold enhancement in the selective production of ascaridole.
To assess the photocatalyst recyclability, the reaction mixture from entry 10 ([NH] = 5 mg mL−1) was centrifuged (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g, 10 min), facilitating the precipitation of UC@RB2 NHs. Fresh α-terpinene and DMF-d7 were added to the recovered UC@RB2 NH, and after redispersion, the mixture was irradiated at 980 nm for 8 h (Table 1, entry 12) yielding only 12.4% of ascaridole. The reduction of the photocatalytic activity can primarily be attributed to two factors: RB photobleaching and detachment of RB from the surface. In fact, partial photodegradation of RB was observed (ca. 20%). This points out that strategies to avoid PC leaching, such as covalent linking of the PC, should also be evaluated.
000g, 10 min), facilitating the precipitation of UC@RB2 NHs. Fresh α-terpinene and DMF-d7 were added to the recovered UC@RB2 NH, and after redispersion, the mixture was irradiated at 980 nm for 8 h (Table 1, entry 12) yielding only 12.4% of ascaridole. The reduction of the photocatalytic activity can primarily be attributed to two factors: RB photobleaching and detachment of RB from the surface. In fact, partial photodegradation of RB was observed (ca. 20%). This points out that strategies to avoid PC leaching, such as covalent linking of the PC, should also be evaluated.
Consequently, we synthesized a previously reported NH in which RB is covalently linked to the UCNP surface, referred to as UC-RB,15 and further investigated its photocatalytic performance. Briefly, the surface of UC@BF4 was modified with 2-aminoethyl dihydrogen phosphate (AEP) and RB hexanoic ester (RB-HA) was linked via EDC/NHS chemistry.15 To ensure comparability with the RB-grafted NH assays, equivalent RB concentrations were desired. Therefore, the UC-RB concentration (20 mg mL−1) in the reaction mixture was adjusted to match the RB absorption observed in entry 10. Upon NIR irradiation, the total reaction yield was similar to that obtained with the non-covalent NH (ca. 43%). However, the reaction selectivity was significantly compromised, yielding 25.7% ascaridole and 17.6% p-cymene (Table 1, entry 13), which is almost half the ascaridole formation compared to the non-covalent NH. No RB leaching was observed after centrifugation of the sample (see Fig S18†). Notably, the recyclability experiment (Table 1, entry 14) evidenced a ca.25% reduction in the formation of ascaridole while keeping the yield towards p-cymene.
While UC-RB NHs exhibited superior resistance to PC leaching, the selectivity of the UC-RB NH was likely diminished due to interference caused by the organic moieties present in the UCNP environment. Hence, UC@RB NHs provide higher selectivity in product formation, specifically yielding a greater amount of ascaridole. In addition, the amount required to undergo the photocatalytic reaction was four times higher for UC-RB NHs than for UC@RB NHs, substantially increasing the resources needed. In conclusion, despite the limited recyclability of UC@RB NHs, their use in NIR-triggered photocatalytic reactions proved to be faster, more efficient, and more cost-effective.
When using UC@RB NHs as photocatalysts, RB is not directly excited using the light source; instead, RB is sensitized using the upconverted light from UCNPs, which have a low UCQY. In the photooxidation of α-terpinene, the limiting reactant is the singlet oxygen. Therefore, there are numerous steps required to achieve the formation of singlet oxygen: (i) excitation of UCNPs using NIR light; (ii) upconverting the light; (iii) resonant energy transfer from UCNPs to RB; (iii) formation of the RB triplet; (iv) energy transfer from the 3RB to O2; and (v) formation of 1O2 to finally achieve the photooxidation of α-terpinene. Given these factors, the effectivity of our system is comparable to that described in the most recent literature.39
Flow NIR-triggered photooxidations need to be evaluated in the near future for UC@RB NHs. In fact, 85% yield of ascaridole has been reported for RB-sensitized flow photooxidation of α-terpinene.35,40,41
Experimental
Synthesis of RB-capped UCNPs
Several UC@RB nanohybrids were synthesized following a previously described procedure.24 Briefly, six different vials were prepared containing 34.1 mg of UC@BF4 and increasing concentrations of RB: (1) 4.2 × 10−2, (2) 4.2 × 10−3, (3) 4.2 × 10−4, (4) 4.2 × 10−5, (5) 4.2 × 10−6, and (6) 4.2 × 10−7 M in DMF (1 mL total volume). The vials were kept in darkness and stirred at room temperature for 24 h. The corresponding UC@RB nanohybrids were recovered by centrifugation at 12000g for 30 min. Then, the precipitates were washed following cycles of redispersion in 5 mL of DMF and centrifugation at 10000g for 10 min, until the supernatants were transparent. The total volume used for the washing steps was 165 mL for UC@RB1, 270 mL for UC@RB2, 15 mL UC@RB3, and 5 mL for UC@RB4, UC@RB5, and UC@RB6. To eliminate large aggregates, 3 mL of DMF was added and a centrifugation step (1000 g, 2 min) was carried out, thus recovering the supernatant. The final concentrations of the six UC@RB dispersions were calculated by weighing the difference, leaving 100 μL of the dispersion to dry.
Chemical determination of singlet oxygen generation
The singlet oxygen generation upon NIR irradiation of different UC@RB nanohybrids was evaluated in DMF, ACN, and EtOH by using an anthracene derivative as an optical probe (ABDA).12 An AB2 series spectrofluorometer equipped with a NIR laser diode (980 nm CW, 308 W cm−2) was used for these experiments. In a quartz cuvette, 1 mL of solution containing ABDA (1.03 × 10−5 M) and the corresponding UC@RB nanohybrid (1 mg mL−1) was prepared and kept under stirring and oxygen-saturated conditions. Then, the solution was irradiated with an NIR laser. Subsequently, the ABDA emission spectrum from 398 nm to 550 nm was recorded at different time intervals (λexc = 378 nm, 750 V, excitation, and an emission bandwidth of 2 nm, 1 nm s−1). The 1O2 generation efficiency was evaluated from the decreases of the ABDA emission at 410 nm.In addition, a control sample containing UC@RB and ABDA was kept in the dark.
Evaluation of photocatalytic activity of UC@RB nanohybrids
200 μL of a mixture containing the corresponding synthesized UC@RB nanohybrids with different amounts of RB (i.e., UC@RB1, UC@RB2, UC@RB3, UC@RB4, UC@RB5, and UC@RB6) and 0.06 mM of α-terpinene in N,N′-dimethylformamide-d7 were placed in a 10 × 10 mm quartz cuvette. To ensure oxygen-saturated solutions, the mixtures were bubbled with oxygen for 15 minutes in an ice bath before irradiation. Then, the cuvette was irradiated with a CW 980 nm laser diode (308 W cm−2) for 8 hours under continuous stirring at 20 °C. After irradiation completion, the reaction mixture was centrifuged at 10000g for 10 min to separate the UC@RB nanohybrids from the reaction. Then, the supernatant was analyzed by 1H-NMR (see more details in ESI, Fig. S19†). Control experiments under identical conditions, but without nanohybrids and using BF4−-stabilized UCNPs (5 mg mL−1), were also carried out.
Conclusions
UC@RB nanohybrids have been used as near-infrared (NIR) light-harvesting systems for heterogeneous photocatalysis, specifically, for selective photooxidation of α-terpinene with moderate yields (∼50%).The photophysical events occurring upon NIR-irradiation proved to be strongly dependent on the number of RB molecules anchored to the UCNPs and their aggregation state (the monomer being preferred over the aggregates). The best photocatalytic performance corresponds to a ratio of ∼100 RB/UCNP for UC@RB2 and UC@RB3, which correlates well with their optical properties and the highest 3RB generation. To get the best photocatalytic performance, it is crucial to optimize the synthetic protocol used to prepare UCNHs, the PC concentration, solvent and irradiation time. This study highlights the potential of UC@RB NHs as an efficient and cost-effective NIR photocatalyst and compares it with the covalently linked analogue, UC-RB. The rapid preparation of UC@RB NHs, coupled with their higher selectivity towards the formation of ascaridole and the lower amount of NHs required (5 and 10 mg mL−1), results in a more selective photocatalytic reaction. This efficiency translates into lower material costs, minimized waste, and a smaller environmental footprint, thereby enhancing the sustainability of the process.
Next, the optimization of synthetic protocols for UCNHs comprising non-covalently and covalently linked PCs needs to be accomplished to simultaneously avoid PC leaching and improve recyclability together with flow photooxidation experiments.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Laura Francés-Soriano: investigation, methodology, data curation, visualization, formal analysis, writing-original draft; Delia Bellezza; investigation, methodology, data curation, visualization, writing – original draft; Juan Ferrera-González: investigation, methodology, data curation, formal analysis; María González-Béjar: conceptualization, funding acquisition, project administration, methodology, supervision, writing – original draft; writing – review & editing; Julia Pérez-Prieto: conceptualization, funding acquisition, project administration, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by Agencia Estatal de Investigación-AEI (grant number MMCIU Unit of Excellence “Maria de Maeztu” CEX2019-000919-M); Ministerio de Universidades (María Zambrano contract L. F. S. (ZA21-037)); and Generalitat Valenciana (IDIFEDER/2018/064 and IDIFEDER/2021/064), all of them partially cofinanced with FEDER funds. This study forms part of the Advanced Materials programme (MFA/2022/051) and was supported by MICIN with funding from European Union NextGenerationEU (PRTR-C17.I1) and by Generalitat Valenciana. We also thank Universitat de Valéncia (Programa Propi del Vicerectorat d'Investigació de la UV (UV-INV_AE-2653809).References
- M. González-Béjar, J. Pérez-Prieto and J. C. Scaiano, in Green Chem., Nova Science Publishers, Inc., 2014, pp. 71–105 Search PubMed.
- A. A. Ghogare and A. Greer, Chem. Rev., 2016, 116, 9994–10034 CrossRef CAS PubMed.
- M. Zamadar and A. Greer, in Handbook of Synthetic Photochemistry, 2009, pp. 353–386 Search PubMed.
- M. Freitag, N. Möller, A. Rühling, C. A. Strassert, B. J. Ravoo and F. Glorius, ChemPhotoChem, 2019, 3, 24–27 CrossRef CAS.
- A. R. Obah Kosso, N. Sellet, A. Baralle, M. Cormier and J. P. Goddard, Chem. Sci., 2021, 12, 6964–6968 RSC.
- N. Sellet, M. Cormier and J. P. Goddard, Org. Chem. Front., 2021, 8, 6783–6790 RSC.
- N. Sellet, L. Clement-Comoy, M. Elhabiri, M. Cormier and J. P. Goddard, Chem.–Eur. J., 2023, 29, 1–8 CrossRef PubMed.
- N. Sellet, M. Sebbat, M. Elhabiri, M. Cormier and J. P. Goddard, Chem. Commun., 2022, 58, 13759–13762 RSC.
- S. Wu, J. P. Blinco and C. Barner-Kowollik, Chem.–Eur. J., 2017, 23, 8325–8332 CrossRef CAS PubMed.
- P. Lederhose, Z. Chen, R. Müller, J. P. Blinco, S. Wu and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2016, 55, 12195–12199 CrossRef CAS PubMed.
- S. Wu and H. J. Butt, Phys. Chem. Chem. Phys., 2017, 19, 23585–23596 RSC.
- M. González-Béjar, M. Liras, L. Francés-Soriano, V. Voliani, V. Herranz-Pérez, M. Duran-Moreno, J. M. Garcia-Verdugo, E. I. Alarcon, J. C. Scaiano and J. Pérez-Prieto, J. Mater. Chem. B, 2014, 2, 4554–4563 RSC.
- X. Yang, Q. Xiao, C. Niu, N. Jin, J. Ouyang, X. Xiao and D. He, J. Mater. Chem. B, 2013, 1, 2757–2763 RSC.
- F. Chen, S. Zhang, W. Bu, Y. Chen, Q. Xiao, J. Liu, H. Xing, L. Zhou, W. Peng and J. Shi, Chem.–Eur. J., 2012, 18, 7082–7090 CrossRef CAS PubMed.
- K. Liu, X. Liu, Q. Zeng, Y. Zhang, L. Tu, T. Liu, X. Kong, Y. Wang, F. Cao, S. A. G. Lambrechts, M. C. G. Aalders and H. Zhang, ACS Nano, 2012, 6, 4054–4062 CrossRef CAS PubMed.
- Z. Zhang, J. N. Rahmat, R. Mahendran and Y. Zhang, Adv. Sci., 2020, 7, 2001831 CrossRef CAS PubMed.
- K. Lv, L. Yao, X. Fu, X. Gao, H. Wang, Y. Zhou, R. Zhang, Y. Lu, J. Feng and H. Zhang, Nano Today, 2023, 52, 101964 CrossRef CAS.
- S. Cui, H. Chen, H. Zhu, J. Tian, X. Chi, Z. Qian, S. Achilefu and Y. Gu, J. Mater. Chem., 2012, 22, 4861–4873 RSC.
- S. Cui, D. Yin, Y. Chen, Y. Di, H. Chen, Y. Ma, S. Achilefu and Y. Gu, ACS Nano, 2013, 7, 676–688 CrossRef CAS PubMed.
- Y. Il Park, H. M. Kim, J. H. Kim, K. C. Moon, B. Yoo, K. T. Lee, N. Lee, Y. Choi, W. Park, D. Ling, K. Na, W. K. Moon, S. H. Choi, H. S. Park, S.-Y. Yoon, Y. D. Suh, S. H. Lee and T. Hyeon, Adv. Mater., 2012, 24, 5755–5761 CrossRef PubMed.
- A. Nsubuga, K. Morice, N. Fayad, F. Pini, V. Josserand, X. Le Guével, A. Alhabi, M. Henry, D. Puchán Sánchez, N. Plassais, P. Josse, J. Boixel, P. Blanchard, C. Cabanetos and N. Hildebrandt, Adv. Funct. Mater., 2024, 2410077 CrossRef.
- L. Francés-Soriano, M. A. Zakharko, M. González-Béjar, P. A. Panchenko, V. Herranz-Pérez, D. A. Pritmov, M. A. Grin, A. F. Mironov, J. M. García-Verdugo, O. A. Fedorova and J. Pérez-Prieto, Chem. Mater., 2018, 30, 3677–3682 CrossRef.
- J. Wu, S. Du and Y. Wang, J. Mater. Chem. B, 2019, 7, 7306–7313 RSC.
- J. Ferrera-González, M. González-Béjar and J. Pérez-Prieto, Nanoscale, 2023, 15, 19792–19800 RSC.
- W. Liu, Y. Zhang, W. You, J. Su, S. Yu, T. Dai, Y. Huang, X. Chen, X. Song and Z. Chen, Nanoscale, 2020, 12, 13948–13957 RSC.
- F. Jin, J. Qi, D. Liu, Y. You, G. Shu, Y. Du, J. Wang, X. Xu, X. Ying, J. Ji and Y. Du, J. Controlled Release, 2021, 337, 90–104 CrossRef CAS PubMed.
- X. Chen, Y. Zhang, X. Zhang, Z. Zhang and Y. Zhang, Microchim. Acta, 2021, 188, 1–10 CrossRef PubMed.
- Y. Wang, K. Liu, X. Liu, K. Dohnalová, T. Gregorkiewicz, X. Kong, M. C. G. Aalders, W. J. Buma and H. Zhang, J. Phys. Chem. Lett., 2011, 2, 2083–2088 CrossRef CAS.
- V. Muhr, C. Würth, M. Kraft, M. Buchner, A. J. Baeumner, U. Resch-Genger and T. Hirsch, Anal. Chem., 2017, 89, 4868–4874 CrossRef CAS PubMed.
- S. Sharma and A. Sharma, Org. Biomol. Chem., 2019, 17, 4384–4405 RSC.
- F. Ronzani, N. Costarramone, S. Blanc, A. K. Benabbou, M. Le Bechec, T. Pigot, M. Oelgemöller and S. Lacombe, J. Catal., 2013, 303, 164–174 CrossRef CAS.
- L. Pessoni, S. Lacombe, L. Billon, R. Brown and M. Save, Langmuir, 2013, 29, 10264–10271 CrossRef CAS PubMed.
- C. Mendoza, N. Emmanuel, C. A. Páez, L. Dreesen, J. C. M. Monbaliu and B. Heinrichs, ChemPhotoChem, 2018, 2, 890–897 CrossRef CAS.
- G. O. Schenck, Angew. Chem., 1957, 69, 579–599 CrossRef CAS.
- Z. Li and Y. Zhang, Nanotechnology, 2008, 19, 345606 CrossRef PubMed.
- S. Wilhelm, M. Kaiser, C. Würth, J. Heiland, C. Carrillo-Carrion, V. Muhr, O. S. Wolfbeis, W. J. Parak, U. Resch-Genger and T. Hirsch, Nanoscale, 2015, 7, 1403–1410 RSC.
- M. Bregnhøj, M. Westberg, F. Jensen and P. R. Ogilby, Phys. Chem. Chem. Phys., 2016, 18, 22946–22961 RSC.
- G. W. Mcgraw, R. W. Hemingway, L. L. Ingram, C. S. Canady and W. B. Mcgraw, Environ. Sci. Technol., 1999, 33, 4029–4033 CrossRef CAS.
- J. Chen, R. Prinsloo and X. Ni, Technologies, 2024, 12, 29 CrossRef.
- N. P. Beard and A. J. De Mello, Electrophoresis, 2002, 23, 1722–1730 CrossRef CAS PubMed.
- M. C. Mitchell, V. Spikmans and A. J. De Mello, Analyst, 2001, 126, 24–27 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials, methods, TEM images, TGA, emission spectra, kinetic profiles, emission decay fittings, phosphorescence spectra, transient absorption spectra, laser-induced emission spectra, ABDA photoconsumption spectra, and 1H-NMR spectra. See DOI: https://doi.org/10.1039/d4na00528g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |