
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tailoring surface morphology on anatase TiO2 supported Au nanoclusters: implications for O2 activation†
Muhammed Fasil
Puthiyaparambath
,
Julian Ezra
Samuel
and
Raghu
Chatanathodi
 *
*
Department of Physics, National Institute of Technology Calicut, Calicut, Kerala 673601, India. E-mail: raghuc@nitc.ac.in
First published on 18th September 2024
Abstract
Strong interaction between the support surface and metal clusters activates the adsorbed molecules at the metal cluster–support interface. Using plane-wave DFT calculations, we precisely model the interface between anatase TiO2 and small Au nanoclusters. Our study focusses on the adsorption and activation of oxygen molecules on anatase TiO2, considering the influence of oxygen vacancies and steps on the surface. We find that the plane (101) and the stepped (103) surfaces do not support O2 activation, but the presence of oxygen vacancies results in strong adsorption and O–O bond length elongation. Modifying the TiO2 surface with supported small Aun nanoclusters (n = 3–5) also significantly enhances O2 adsorption and stretches the O–O bond. We observe that manipulating the cluster orientation through discrete rotations results in improved O2 adsorption and promotes charge transfer from the surface to the molecule. We propose that the orientation of the supported cluster may be manipulated by making the cluster adsorb at the step-edge of (103) TiO2. This results in activated O2 at the cluster–support interface, with a peroxide-range bond length and a low barrier for dissociation. Our modeling demonstrates a straightforward means of exploiting the interface morphology for O2 activation under low precious metal loading, which has important implications for electrocatalytic oxidation reactions and the rational design of supported catalysts.
1. Introduction
Activation of molecular oxygen adsorbed over a catalyst surface is a critical step in several important processes such as catalytic oxidation reactions, photocatalysis, CO oxidation, propylene epoxidation, corrosion control, chemical sensing, etc.1–4 For example, the electrocatalytic oxygen reduction reaction (ORR) which occurs in hydrogen fuel cells is initiated by the adsorption and activation of an O2 molecule on a catalyst surface.5 The catalyst here may be a metal, non-metal or metal oxide. The progress of the ORR greatly depends on the bonding and activation of O2 on the active site on the catalyst surface. Understanding and controlling O2 activation provides a handle over phenomena like corrosion and oxide layer formation, which have immense practical implications.6 In general, the reactivity of a surface to O2 is indicated by the strength of the binding, measured in terms of the adsorption energy, the O–O bond length etc.7,8 Therefore, a study of how surface properties and their modification affect oxygen binding and activation can provide fruitful insights for desirable ends like controlling corrosion or designing efficient catalysts for oxidation reactions.The study of O2 binding and activation on metal and alloy surfaces has received a fair bit of attention over the years, both experimentally and theoretically. A comprehensive review of the area may be found in the work of Montemore et al.9 With reference to the ORR in particular and electrocatalysis in general, the search for efficient and earth abundant catalysts to replace expensive and kinetically sluggish Pt or Pt-based materials is an ongoing research endeavour. Development of such alternative catalysts can lead to the large scale deployment of fuel cells in place of fossil fuels, which would lead to the realization of some of our green energy aspirations. One class of alternate materials being pursued here are metal oxides, particularly transition metal oxides. Transition metal oxides (TMOs) have gained significant interest over time as applicable active materials in the fields of catalysis, energy storage and sensing, due to their low cost, variable oxidation state, ease of synthesis, stability, corrosion resistance and environmental friendliness.10–13
Titanium dioxide (TiO2) is a very well-known material amongst TMOs, due to its relatively large abundance, non-toxicity and several applications as a wide band gap electronic material and a photocatalyst.14 TiO2 occurs in nature in the form of rutile and anatase, of which rutile is thermodynamically more stable, but anatase is catalytically and technologically an important TMO.15 Over the past decade or so, the study of interaction between TiO2 and O2 molecules has attracted considerable interest. It happens that the anatase TiO2 surface is relatively inactive to the O2 molecule, and therefore not a suitable catalyst for the ORR. The surface becomes active when an excess of electrons is present.16 The adsorption of O2 on TiO2 is generally associated with the withdrawal of negative charge from the surface.17–19 Adsorbed O2 exists in either a peroxide (O22−), or superoxide (O2−) state, or may get dissociated into two O2− ions.9
Varied attempts have been made to enhance the activity of the TiO2 surface towards O2. Adsorbed metal single atoms or nanoscale clusters, surface defects, steps and dopant atoms can play a significant role in activating the surface.20 The interface formed between the metal oxide surface and the metal clusters dispersed over it plays an important role in the catalytic oxidation processes.21,22 The geometric structure and electronic properties of the metal clusters can get significantly modified by the supporting metal oxides, and thereby have a significant impact on their catalytic properties.23 Gold nanoparticles exhibit high catalytic activity for many reactions when dispersed on metal oxides. Interestingly, both Au and metal oxides individually were considered to be inactive for numerous catalytic reactions.24 However, they combine to form a very active interface. The pioneering work of Haruta25 has led to extensive experimental studies on Au/oxide systems, yielding significant results, and it is established that the catalytic activity of Au-based systems greatly depends on the choice of the underlying oxides.
Among all the catalytic reactions involving Au, the activation of O2 holds particular significance in the research field of Au/oxide systems. Strategies are currently being developed for the activation of O2 molecules using the Au-metal oxide interface. It is found that the catalytic activity of Au depends considerably on the nature of the support, the synthesis method and the size of Au nanoparticles. The most commonly used oxide supports for Au nanoparticles are TiO2, ZnO, Al2O3 and SiO2.26,27 The structural aspects of Au clusters supported on TiO2 were investigated by Hemmingson et al. using high-resolution electron microscopy (HREM).21 It was discovered that the catalytic activity can be tuned by controlling the size of the Au nanoparticles, and the oxygen molecule is activated at the interface of Au/TiO2.28–30 Green et al.31 experimentally and theoretically provided insights into the activation of Au clusters supported on rutile TiO2. The transmission IR spectroscopic data indicated the direct involvement of the Au–Ti4+ at the interface in the activation of O2. While most researchers agree that the activation of O2 is favoured at the perimeter site, the elementary process leading to O2 activation, whether solely on the Au clusters or at the cluster–support interface, is debatable.24,27,32,33 Moreover, a decrease in the size of the clusters resulted in a decrease in the activity of Au clusters.34 The interactions between Au and TiO2 support have been extensively studied using density functional theory (DFT).35,36 The results suggest that the adsorption strength of Au/TiO2 is sensitive to both the cluster size and facet. Additionally, the interface's geometric and electronic structure plays crucial roles in the catalytic activity.
Stepped edges are common defects found on the surface of crystalline materials. Despite being ubiquitous, they have been studied less extensively compared to other defects such as vacancies and interstitials. High-index surfaces exhibit unique properties compared to low-index surfaces because of their low coordination.37 Surface facets characterized by Miller indices {h, k, l} with at least one index greater than one, can be termed stepped surfaces. The step edges serve as active sites for the adsorption of molecules and also act as nucleation centres when metals are deposited on the oxide surface.38–40 The stepped surface of anatase TiO2 is of great interest and has been amply investigated by Gong et al.41,42 They observed that, depending on the terrace/step configuration, the steps can exhibit lower reactivity compared to the flat terraces. Rieboldt43 and co-workers experimentally studied vicinal rutile TiO2 surfaces and found that they have a strong influence on O2 due to the presence of step edges. Additionally, these surfaces are characterized by smaller gap states compared to flat surfaces. High-resolution TEM images show that the (103) surface of anatase nanoparticle can be exposed depending on the processing conditions.44 The work function of the TiO2 (441) surface was smaller by 0.7 eV compared to (110)45 due to Fermi-level pinning and downward band bending toward the surface. Du et al. recently reported that the larger Au nanoparticles exhibit a higher encapsulation tendency in the Au/TiO2 system due to size-dependent strong metal–support interaction.46
In the present work, we have looked at O2 adsorption on plane, stepped, reduced and Au cluster decorated surfaces of anatase TiO2. We have used plane-wave DFT calculations to model O2 adsorption, probing adsorption energy and the O–O bond length as descriptors of O2 activation over these surfaces. We attempt to demonstrate that alteration of surface morphology of anatase TiO2 through the presence of vacancies and steps, as well as supported Au clusters, at minimal Au loading, can lead to enhanced O2 activation. We find that the orientation of the Au nanoclusters plays an important role in enhancing O2 binding to the surface. We point out a simple way to realize such optimal orientation, viz., cluster adsorption at the step-edge in the stepped TiO2 surface. In the next section, we describe the computational methodology and in the subsequent section our results. Finally, we summarize and conclude our work.
2. Computational methods
All calculations in this work were performed using a spin-polarized density functional theory (DFT) approach with the Vienna Ab initio Simulation Package (VASP).47,48 The core electrons were treated using the projector augmented wave (PAW)49,50 method. The generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE)51 functional was employed to describe the exchange–correlation energy. The DFT-D2 method proposed by Grimme52,53 was used to model the dispersive interactions. The wave functions were expanded in a plane wave basis with an energy cut-off of 520 eV, and Gaussian smearing of 0.05 eV was applied. The Brillouin zone was sampled using 5 × 5 × 1 Monkhorst–Pack grid54 for the surface slab. A 15 Å vacuum was included to avoid the spurious periodic interaction along the z-axis. The structural relaxation was carried out until the force on each atom was less than 0.01 eV Å−1. It is well known that the standard DFT fails to model Ti atoms, primarily due to the self-interaction error inherent in the functionals used in the GGA. The GGA+U method was employed to calculate the electronic structure to overcome this limitation. The value of Hubbard parameter U was set to 4.2 eV, which is similar to the previous work 55,56The optimized lattice parameters for the bulk anatase TiO2 (a = 3.88 Å and c = 9.58 Å), and calculated bond lengths (Ti–O) in apical (2.01 Å) and basal (1.98 Å) directions agree with previous experimental values.57 In this work, we have considered the (101) plane and (103) stepped surfaces of anatase TiO2. The (101) and (103) surfaces were modelled using a 3 × 1 supercell consisting of 108 and 144 atoms, respectively. Au clusters were modelled in a unit cell of 15 × 15 × 15 Å, considering only the Γ-point. To identify the minimum energy path (MEP) for O2 dissociation on the TiO2 surface, the climbing image-nudged elastic band (CI-NEB)58 method was utilized to determine the transition state. The activation energy is defined as Ea = ETS − EIS, where ETS and EIS are the energies of the transition state and initial state, respectively. Bader charge59 analysis was employed to determine the local charge of the atom.
The surface energy (γ) is calculated using the expression:
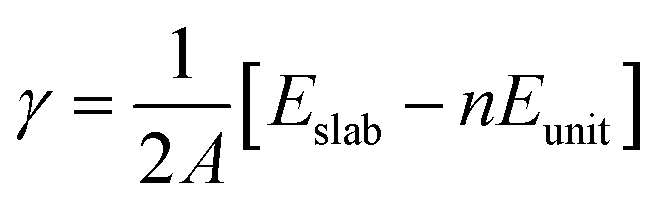 | (1) |
The adsorption energy of the molecular O2 is determined as:
| EO2 ad = EO2+surf − Esurf − EO2 | (2) |
The O vacancy is created in both (101) and (103) surfaces, with the formation energy given by the following equation:
| Ef = Edef − Epri − (1/2)EO2 | (3) |
The binding energy per Au for the Aun cluster on the TiO2 substrate is calculated using the formula
| EBE = (1/n)[Etot − Eslab − n × EAu] | (4) |
3. Results and discussion
3.1 O2 adsorption on (101) and (103) anatase TiO2
Bulk anatase TiO2 is cleaved into two different facets, the usual (101) and the stepped (103). The surface energy is calculated using the formula (1). The (101) facet has a surface energy of 0.46 J m−2 which is comparable to the previously calculated values.60 The optimized configuration of (101) is shown in Fig. 1(a) and (b). The (101) surface consists of fully saturated six-coordinated Ti and three-coordinated O atoms, as well as unsaturated five-coordinated Ti and two-coordinated O atoms on the surface. The (103) facet has two possible terminations that are commonly discussed: the “faceted” (103)f and “smooth” denoted as (103)s.60 The surface energy of (103)f is 1.12 J m−2, which is greater than 1.08 J m−2 of (103)s. This trend is similar to that observed in previous calculations,61 and this surface energy is lower than that of (001).62 Hence, the (103)s is stable, and we consider the (103)s facet throughout our calculation, denoted by (103). The optimized configuration is shown in Fig. 1(c) and (d). The hydration surface energy is also calculated for the (103) surface (for details, see S1†) at different coverage, and it is interesting to note that the surface energy of (103) is reduced for this coverage, when compared to those of (101). This indicates that hydration stabilizes the (103) surface over the (101). | ||
| Fig. 1 (101) TiO2 (a) side view (b) close view; (103) TiO2 (c) side view (d) close view; PDOS of (e) (101) and (f) (103) TiO2. | ||
The optimized configuration of the (103) surface consists of four-coordinated Ti (Ti4c) and three types of two-coordinated O (O2c), in addition to the usual coordination present in (101) anatase TiO2, and one three-coordinated (O3c1) (see Fig. 1(c) and (d)). The three types of O2c are: (a) linear along the b-axis making an angle of 152° (O2c1), (b) inclined at an angle of 161° (O2c2) and (c) bend at an angle of 101° (O2c3). Relaxation of the (103) stepped surface leads to satisfaction of unsaturated surface bonds and a 3% stabilization of the system. In the relaxed geometry, the angle between Ti4c–O–Ti4c along the b-axis is contracted by 4°, while the angle made in the b–c plane is increased by 5°.
To compare the electronic structure of two facets, the PDOS of (101) and (103) are calculated and plotted as Fig. 1(e) and (f), respectively. No significant change in the band gap is observable. In both cases, the top of the valence band (VB) is composed of O 2p states, and the bottom of the conduction band (CB) is composed of Ti 3d states, consistent with the previous literature.63 In (101), the valence band maximum (VBM) is dominated by px and pz, and the conduction band minimum (CBM) is composed of the dominant t2g states of Ti (dxy, dyz, and dxz). In case (103), all the states of O 2p are present in the VBM, and the CBM consists of Ti 3d. This may be due to the lowering of the symmetry following the formation of a surface. The Ti–O–Ti bond angles of (103) are larger compared to (101), resulting in better availability of oxygen for bonding with other species. All these contribute to making the (103) stepped surface a more active site for binding of adsorbates.
However, we find that the binding of the O2 molecule to the (103) facet improves only marginally, despite all considerations of the previous paragraph. The adsorption energy of the O2 molecule is calculated on (101) and (103) using formula (2). The O2 molecule gets physisorbed in both cases. There is a slight increase in the adsorption energy of O2 in the case of the stepped surface (−0.17 eV) compared to the plane (−0.13 eV) surface of TiO2. The gas phase O–O bond length of 1.23 Å remains unaltered after adsorption. Thus, O2 molecules desorb easily from (101) and (103) TiO2 surfaces.
3.2 O2 adsorption on reduced (101) and (103) TiO2 surfaces
We now discuss the case of oxygen defects on the TiO2 (101) and (103) surfaces. The formation energy of oxygen vacancies for both (101) and (103) surfaces is calculated using eqn (3) and is tabulated in Table 1. A large positive value indicates that more energy is needed to form the defect. The surface site defects V2c and V3c correspond to the vacancies created upon the removal of an O2c and O3c atom from the (101) surface (see Fig. 2(a)). Thus, it takes more energy to remove the O3c than the O2c atom. In the case of the (103) surface, V2c1, V2c2, V2c3, and V3c1 indicate the removal of O2c1, O2c2, O2c3 and O3c1 atoms, respectively, from the surface (see Fig. 2(b)). The formation energy is lowest for the removal of the O2c3 atom compared to all other sites.| Surface | Site | Formation energy (eV) |
|---|---|---|
| (101) | V2c | 4.23 |
| V3c | 5.02 | |
| (103) | V2c1 | 5.43 |
| V2c2 | 4.71 | |
| V2c3 | 3.22 | |
| V3c1 | 4.93 |
 | ||
| Fig. 2 The top view of the vacant surface of (a) (101) and (b) (103). | ||
We create oxygen vacancies both on the plane (101) and stepped (103) TiO2 (shown in Fig. 2) and compare both cases. After relaxation, in the case of V2c (removing the O2c atom from the (101) surface), the Ti atom moves downwards by a distance of 0.16 Å, while the bottom oxygen atom is shifted upward by a distance of 0.18 Å. For the V3c (removing the O3c atom from the (101) surface) vacancy, there was no change in the position of the Ti atom in the c-direction, but it is shifted by 0.18 Å in the lateral direction. On the vacant surface with V2c1, the uncoordinated Ti atom was shifted away from the vacant site by 0.42 Å. Consequently, the adjacent Ti–O bond distance was reduced by 0.20 Å. On the V2c2 vacant surface, the Ti has been displaced to the vacant site by a distance of 0.40 Å. At the V2c3 vacant site, the bottom oxygen atom shifted upwards by 1.08 Å, while the six-coordinated Ti atom moved along the surface by 0.50 Å. On the V3c1 vacant surface, the oxygen atom originally coordinated to O2c1 becomes three-coordinated, leading to a reduction in the Ti–O bond distance from 2.46 Å to 2.11 Å. These geometries are displayed in Fig. S2.†
The adsorption energy of an O2 molecule is calculated on the reduced surfaces of (101) and (103). The adsorbed O2 configuration is shown in Fig. S3.† In the case of V2c and V3c of (101), the O2 molecule adsorbs at the Ti site, with O–O stretching of 1.46 Å and 1.48 Å, respectively, bringing these to the peroxide regime. The adsorption energy, O–O bond length and charge transfer to the molecule are tabulated in Table 2. The capability to absorb more than one oxygen molecule and activate these is important to enhance the efficiency of processes like the ORR. The study of O2 coverage can provide insights into the correlation between excess electron charge and the reactivity of the TiO2 surface.16 For more than one O2 adsorbed, we report the average value of adsorption energy, bond length and charge transfer. As the adsorbed O2 acts as an electron scavenger, it inhibits further adsorption of O2 molecules. We observe that the average adsorption energy is lowered, in the case of two molecules, with reduced stretching of the O–O bond. The average charge transfer is also lower in the case of two O2. What happens is thus: the first O2 molecule becomes trapped at the lattice site upon adsorption, thereby withdrawing all available electron density and thus, the second molecule is only physisorbed on the nearest Ti site available. These optimized configurations for V2c and V3c are shown in Fig. S4.†
| No. of O2 molecules | Sites | ||
|---|---|---|---|
| V2c | V3c | ||
| E ads (eV) | 1 | −3.70 | −4.35 |
| 2 | −1.94 | −2.24 | |
| d O−O (Å) | 1 | 1.46 | 1.48 |
| 2 | 1.34 | 1.36 | |
| Q |e| | 1 | 0.92 | 1.08 |
| 2 | 0.46 | 0.54 | |
The average adsorption energy, bond length and charge transferred for adsorption of up to three O2 on the reduced (103) surface are tabulated in Table 3. The first O2 is adsorbed strongly, with a bond length in the peroxide regime. This is comparable with the case of the reduced (101) surface. In the case of V2c1, the adsorption of the second O2 molecule is much reduced, while there is practically no binding for the third. On the other hand, for V2c2, V2c3 and V3c1 sites, the first adsorbed O2 does not fill the vacant lattice site. The configuration of this adsorbed O2 is displayed in ESI Fig. S4.† Therefore, the second O2 has significant average adsorption energy, on the nearest Ti site available. This adsorbed O2 has a bond length in the superoxide regime. The third O2 is weakly bound, by physisorption since there is hardly any charge available on the surface, following the second binding. Thus, we see a graded lowering of the average adsorption energy and bond length with the number of O2 adsorbed in Table 3.
| No. of O2 molecules | Sites | ||||
|---|---|---|---|---|---|
| V2c1 | V2c2 | V2c3 | V3c1 | ||
| E ads (eV) | 1 | −5.09 | −3.76 | −3.02 | −2.73 |
| 2 | −2.68 | −2.47 | −1.97 | −2.35 | |
| 3 | — | −1.56 | −1.52 | −1.66 | |
| d O−O (Å) | 1 | 1.46 | 1.47 | 1.46 | 1.45 |
| 2 | 1.34 | 1.33 | 1.33 | 1.34 | |
| 3 | — | 1.30 | 1.29 | 1.30 | |
| Q |e| | 1 | 1.10 | 0.82 | 0.86 | 0.88 |
| 2 | 0.56 | 0.50 | 0.48 | 0.52 | |
| 3 | — | 0.34 | 0.32 | 0.35 | |
In order to understand further the capture of O2 by a vacant surface site, we performed CI-NEB calculations on reduced (101) and (103) TiO2 and the details of CI-NEB can be found in S2.† We found that an energy barrier exists for the adsorbed O2 to move to the vacant lattice site, in the case of V2c2, V2c3, and V3c1 on the reduced (103) surface. This barrier is estimated to be 1.15 eV, 1.98 eV, and 0.78 eV respectively (see Fig. S5†). This is not the case for V2c1 on reduced (103), or V2c and V3c on reduced (101), where the transfer is barrierless. It may hence be concluded that the advantage of vacancies on reduced (103) TiO2 is that these provide multiple sites for O2 activation, hosting more than one activated O2 (as seen from Fig. S4†), resulting in improved efficiency for catalytic reactions.
3.3 O2 adsorption over Au clusters supported on (101) and (103) TiO2
Next, we investigate a strategy to enhance O2 activation over low loading of small Au clusters supported on (101) and (103) TiO2 surfaces. Small Au clusters have been rigorously studied over several years, owing to a wide range of applications in catalysis, nanophotonics etc.64 The formation of energetically stable Aun on the (101) surface is discussed by Wan et al.35 and Vittadini et al.65 To find a stable structure for Aun, (n = 3–5), on stoichiometric (101) and (103), we grow the cluster over the surface step-by-step, adding one Au atom at a time and re-optimizing the geometry of the entire system. In this case, the binding energy per Au is calculated using eqn (4).The optimized configuration of an isolated Au3 cluster exhibits a planar, triangular geometry, which is more stable than the linear trimer. After adsorption and relaxation, the two Au atoms of the Au3 cluster bind to the O2c of the (101) TiO2 surface with a binding energy of −2.28 eV and an Au–O2c bond length of 2.04 Å. The Au3 cluster is inclined at an angle of 120°, to the a-axis. The optimized configuration is shown in Fig. 3(a) (i and ii). In Au4/TiO2, the fourth Au atom is attached to the optimized Au3/TiO2 structure and has a binding energy of −2.37 eV. Of the two Au atoms attached to the O2c in the case of Au3/TiO2, one Au gets detached in the case of Au4/TiO2 and is tilted at an angle of 148°. The optimized configuration is shown in Fig. 3(a) (iii and iv). The most stable Au5 on (101) TiO2 is in the form of a planar trapezoidal structure. The three Au atoms are bonded with O2c with an Au–O2c bond length of 2.13 Å with a binding energy of −2.50 eV. The Au5 cluster gets tilted at an angle of 154°. The stable structure is shown in Fig. 3(a) (v and vi).
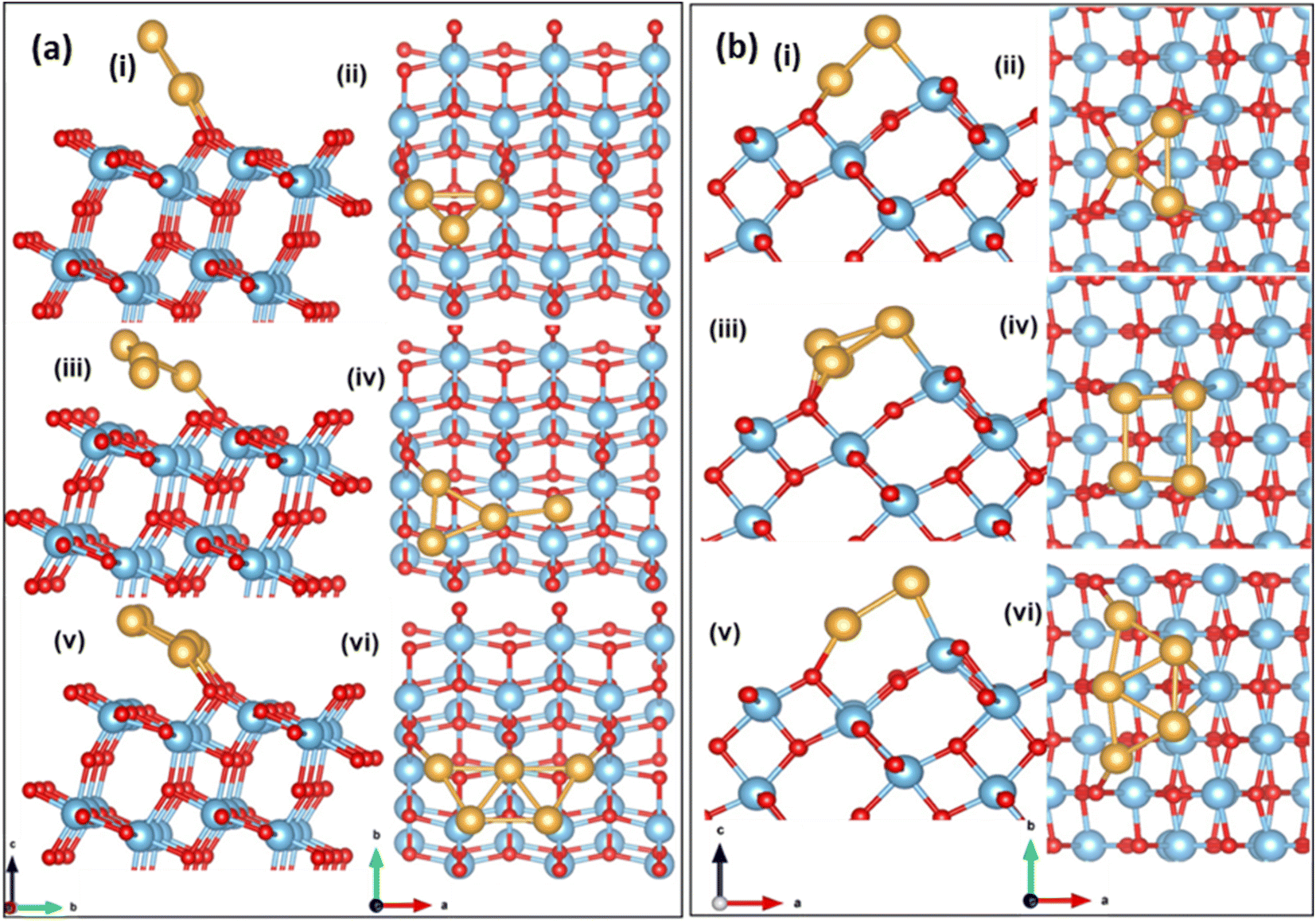 | ||
| Fig. 3 Side view and top view of the optimized configuration of Aun/TiO2 (a) (101); (b) (103) (i and ii) n = 3, (iii and iv) n = 4, (v and vi) n = 5. | ||
Aun (n = 3–5) has different possible binding sites on the (103) surface. Among these, three types of two-coordinated oxygen sites (O2c1, O2c2, and O2c3) are present. The planar Au3 cluster is grown atom by atom, and the binding energy is calculated using eqn (4). The favourable configuration of Au3 has a binding energy of −2.39 eV at the O2c2 site with an Au–O2c2 bond length of 2.24 Å. The optimized configuration is shown in Fig. 3(b) (i and ii). The Au3 cluster on (103) is tilted at an angle of 48° to the b-axis. The Au4 cluster binds to the (103) with a binding energy of −2.62 eV and adopts a square geometry after adsorption. The bond lengths Au–O2c2 and Au–Ti are 2.08 Å and 2.72 Å, respectively. The cluster is tilted at an angle of 38° (see Fig. 3(b) (iii and iv)). The Au5 cluster binds on the (103) surface in a trapezoidal geometry with a binding energy of −2.70 eV and has an Au–O2c2 bond length of 2.20 Å, with a tilting angle of 40° (see Fig. 3(b) (v and vi)). The binding energy of the cluster in case of stepped surfaces is higher due to the availability of more states at the step edge.
The adsorption of O2 molecules on the optimized configuration of Au supported (101) and (103) surfaces has been investigated, and the results are tabulated in Table 4. Here, we have considered O2 adsorption on Ti atoms at the interface and low-coordinated Au sites on the cluster. At the interface, the preferred adsorption sites are Ti5c and Ti4c, for (101) and (103) respectively. As Table 4 shows (see col. 4 and 8), there is weaker adsorption for O2 on the Au cluster itself, which is significant only in the case of the (103) surface.
| Aun (n) | (101) | (103) | ||||||
|---|---|---|---|---|---|---|---|---|
| E ads (eV) (Ti5c) | d O−O (Å) | E ads (eV) (on Au) | d O−O (Å) | E ads (eV) (Ti4c) | d O−O (Å) | E ads (eV) (on Au) | d O−O (Å) | |
| 3 | −0.73 | 1.32 | −0.39 | 1.27 | −1.66 | 1.43 | −0.78 | 1.30 |
| 4 | −0.44 | 1.32 | −0.38 | 1.24 | −1.88 | 1.44 | −0.90 | 1.36 |
| 5 | −0.99 | 1.34 | −0.85 | 1.32 | −1.84 | 1.44 | −1.10 | 1.35 |
On the Au3 supported (101) surface, the O2 molecule undergoes adsorption in a side-on configuration, with an adsorption energy of −0.73 eV. The O–O bond length is elongated to 1.32 Å from the gas-phase bond length of 1.23 Å, and the Ti–O bond length is 2.05 Å. The optimized configurations of O2 on Au3/TiO2 (101) and (103) are depicted in Fig. 4(a) and (b). This trend is similarly observed for Au4 and Au5, where O2 adsorbs on the Ti5c site with the elongation of the O–O bond (see Table 4). The corresponding configurations are not displayed here, for the sake of brevity. In all cases, the O–O bond exhibits an average length of 1.32 Å, which puts it in the superoxide regime. The adsorbed O2 has a magnetic moment of 1 μB, which indeed confirms the superoxide state.9
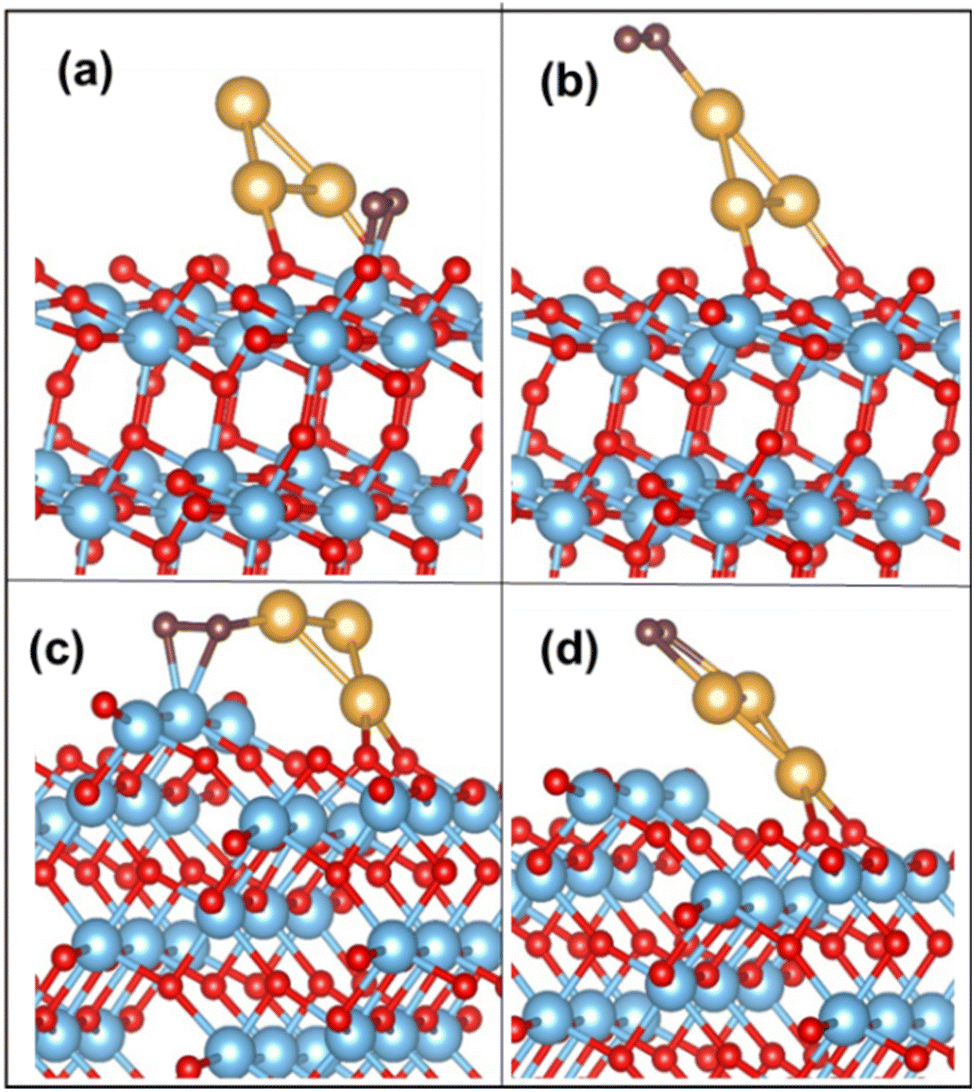 | ||
| Fig. 4 O2 adsorption on (a) Ti5c and (b) Au on the Au3/TiO2 (101) surface; (c) Ti4c and (d) Au on the Au3/TiO2 (103) surface. | ||
To explore further the interaction of the adsorbed O2 with the Au3 cluster, we first rotate the cluster from 0° to 180° along the a-axis, as illustrated in Fig. 5(a), on the (101) surface. As the cluster is rotated through discrete angles, the O–O bond length shrinks and goes through a minimum. Correspondingly, the Au–O distance undergoes a maximum. It is interesting to note that the maximal stretching of the O–O bond occurs at angles where the cluster Au atoms are closest to the adsorbed oxygen. From the plot in Fig. 5(a), these angles are 20° and 160° with the a-axis, and the O–O bond stretches by 0.04 Å, with an Au–O bond distance of 2.20 Å. For an angle of 60°, where the Au3 cluster is farthest away from the O2, the O–O stretching is minimized.
 | ||
| Fig. 5 Effect of Au3 clusters supported on TiO2 rotated along (a) a-axis; and (b) c-axis, on the O–O and Au–O bond lengths. | ||
We also rotate the Au3 cluster with respect to the c-axis (see Fig. 5(b)). The variation of the O–O bond length as a function of Au–O is plotted in Fig. 5(b). The O–O bonding exhibits maximum elongation at an angle of 60°, with the O–O bond length at 1.36 Å. At this point, the Au–O distance is 2.30 Å. The minimal stretching occurs at angles of 0° and 140°. The O–O bond undergoes maximum stretching when Au is closest to the O atom, forming a Ti–(O–O)–Au configuration. Thus, we understand from the above exercise that in terms of the O–O stretching, O2 activation may be achieved by re-orienting the Au3 cluster with respect to the adsorbed molecule, such that in some configurations, the Au atoms are close to the O2 molecule.
The above discussed rotation of the Au3 cluster (or Aun cluster) may be achieved through the application of a static electric field, lattice strain or steric effects due to bulky ligands.66,67 There is yet another simpler way to achieve this bending, using naturally occurring surface imperfections. A stepped surface may naturally tilt the adsorbed cluster in a manner mimicking the a-axis rotation of Fig. 5(a). Thus, we propose that surface morphology may be used to tune cluster orientation. If we look at Aun cluster adsorption on a (103) TiO2 surface, the cluster inclines itself with the “kinks” on the (103) surface. The O2 molecule adsorbs on the Ti4c site, with Aun at the perimeter, forming a Ti4c–(O–O)–Au bond. The calculated adsorption energy of O2 for the Au3 supported (103) surface is −1.66 eV, with the O–O distance stretched to 1.43 Å, corresponding to the peroxide bond length. The Au–O bond distance (i.e. O of O2) is measured at 2.07 Å. On the surface, we find that the Ti4c site is slightly lifted upward, by 0.20 Å, indicating a strong Ti–O interaction. The O–O activation also occurs on the Au cluster site exhibiting an adsorption energy of −0.78 eV. These optimized configurations of O2 adsorbed on Au3/TiO2 (103) are shown in Fig. 4(c) and (d). These findings hold true for Aun (n = 4, 5) clusters too. The corresponding binding energy values are tabulated in Table 4. The table reveals the O–O bond length in the peroxide regime. The calculated magnetic moment of the adsorbed O2 species is close to 0 μB. In the case of adsorption on the low coordinated Au on the cluster itself, Aun (n = 3–5) adsorb O2 as superoxide species, as can be seen from calculated bond lengths (compared to weak binding on the Au/(101) TiO2 surface) in Table 4. Thus, it appears that the interaction of the Au cluster with O2 has been modified by the support (103) surface, into strong chemisorption, leading to activated O2.
To address the question of O2 coverage, we calculate the adsorption energy for the second and third molecules, adsorbed on active sites on the surface. Table S1† displays the averages of adsorption energy per molecule, bond length and charge transfer (to O2 molecule) for both (101) and (103) surfaces. For Au3 supported (101), the adsorption energy of a single O2 molecule is −0.85 eV accompanied by an electron transfer of 0.45e. The second and third molecules bind within the range of physisorption. In the case of (103), the addition of a second O2 molecule results in an average adsorption energy of −1.10 eV, with a bond length of 1.31 Å falling within the superoxide range, and a charge transfer of 0.52e. The third O2 molecule does not undergo chemisorption and exhibits an average adsorption energy of −0.83 eV. Consequently, the Au3 supported (103) surface shows an optimal O2 binding compared to the (101) surface. The average adsorption energy per molecule for (103) is twice that of the (101) surface. Thus Au3/TiO2 (103) can adsorb more than one O2 molecule, in which two of them may be described as activated.
3.4 Stability and electronic structure considerations
To investigate the stability of the cluster configuration, molecular dynamics (MD) simulation for Au3 on (101) and (103) is performed. The temperature is maintained at 300 K by using the Nose thermostat68 for a duration of 6000 fs. Fig. S6(a)† depicts the time evolution for Au3/(101), along with the cluster configuration at 300 K. Monitoring the distance between Au and two-O2c (d1 and d2) of TiO2 reveals no observable fluctuations, with an average distance ranging from 2.02 to 2.39 Å, indicating dynamic stability. A similar situation (Fig. S6(b)†) is observed for (103), with an average distance of 2.05–2.57 Å. Thus, these structures are stable at room temperature.The electronic structures of O2 adsorbed Au3 supported (101) and (103) TiO2 are compared in Fig. 6(a) and (b). For the (101) surface we see localized states of Au 5d, below the Fermi energy, as well as O 2p from the adsorbed molecule. Additionally, we also notice Ti 3d states at the edge of the valence band. All these states are broadened out a little, indicating a weakly chemisorbed state for O2 on the Au/TiO2 (101) surface. In Fig. 6(b), over the (103) surface, we see a stronger hybridization between Au 5d, Ti 3d and O 2p states, whereby the cluster states now have a more delocalized character. Au 5d states have a larger contribution at the edge of the valence band, leading to the strengthening of O2 adsorption. Looking at the difference charge density (or deformation density) plots in Fig. 6(c) and (d), it is clearly seen that there is no electron density accumulation in the O2–Au for Au/TiO2 (101), while for Au/TiO2 (103), excess charge density is present on the Au cluster, leading to enhanced O2 adsorption. The charge accumulation on the O2 is also enhanced, leading to the stretching of the O–O bond.
 | ||
| Fig. 6 PDOS of (a) Au3/TiO2 (101), (b) Au3/TiO2 (103), after O2 adsorption; difference charge density plot of (c) O2/Au3/TiO2 (101), (d) O2/Au3/TiO2 (103). Yellow: charge accumulation; blue: charge depletion. Iso-surface level = 2 × 10−3 e Å−3. | ||
Calculating Bader charges in the ground state, it is observed that a total charge of 0.45e is transferred to the 2π* orbital of the O2 molecule in the Au3/TiO2 (101) configuration. This transfer can be partially from Ti on the surface, and also from Au3, through the surface (contributed during the relaxation of Au3 on the surface). With the Au–O distance of 3.70 Å, the probability of direct transfer between the Au cluster and O2 is very small. However, in the case of the Au3/TiO2 (103), a transfer of 0.78e is calculated to the O2 molecule. Now, the Au–O distance is 2.07 Å, and the cluster interacts directly with the O2 molecule. Thus, charge transfer to O2 is now enabled through an additional channel: transfer from Au3 directly. The elongation of the O–O bond length can be attributed to the stronger interaction of the Au cluster with the O2 molecule, enabled by the (103) step. We may also say that O2 is stabilized at the Au–Ti4+ interface through the formation of a di-σ bond involving Au–(O–O)–Ti, which is consistent with interpretations from previous work.24,69
The role played by the (103) stepped surface can be understood in terms of the relative shift of the 2π energy level of the O2 molecule and the d-band center70 of Au3. To obtain the energy of the 2π state, we start with the ground state configuration of (101) with the Au3 cluster and O2 molecule, remove the Au3 completely, and then obtain the energy for the 2π state of O2 through the energy calculations with the molecule placed 2.10 Å and 8.82 Å above (101). The difference in these energy values is a measure of the stabilization of the 2π state due to bonding with the surface. This calculation is repeated for the (103) surface. We find that the energy of the 2π level of the O2 molecule decreased by 0.61 eV for (101) and 1.03 eV for (103). In a similar manner, the d-band centre for Au3 over both (101) and (103) was calculated without the O2 molecule, with Au3 positioned 2.06 Å and 10.70 Å above TiO2. The d-band centre is lowered to 0.78 eV for (101) and 1.21 eV for (103). The shifts of these two energy levels are indicated in Fig. 7. These two effects, namely the shifts of the O2 2π state and the d-band centre are indicators of O2 activation which is enabled by (103) surface.
 | ||
| Fig. 7 Schematic illustration of the energy level shift for (101) and (103) Au3/TiO2. | ||
3.5 Activation barrier for O2 dissociation for (101) and (103)
Next, we turn our attention to the dissociation process of O2 on (101) and (103) Au3/TiO2. An estimate of the dissociation barrier is important information, in the context of CO oxidation as well as the ORR. For the CI-NEB pathway, we take the initial state (IS) as the ground state configuration of O2 adsorbed on (101) and (103) Au3/TiO2. During the dissociation of O2, the dissociated O atom can migrate to two configurations: the “uplayered” and the “interfacial”. In the uplayered state, the oxygen atom after dissociation, attaches to the low coordinated site of Au3. In the interfacial state, the oxygen atom attaches itself to the neighbouring Ti. These configurations are illustrated in Fig. 8. | ||
| Fig. 8 O2 dissociation barrier for (a) uplayered and (b) interfacial Au3/TiO2 (101); (c) uplayered and (d) interfacial Au3/TiO2 (103) (adsorbed O2 is in brown). | ||
In the case of (101), the barrier for O2 dissociation at the uplayered Au is 0.48 eV, which is favourable compared to the interfacial site with a barrier of 2.47 eV (refer to Fig. 8(a) and (b)). On the other hand, for (103) the barrier (0.41 eV) is notably lower for the uplayered site, making it kinetically favourable. The barrier for the interfacial site on the (103) surface is reduced by 1.15 eV in comparison to the (101) site, and the reaction is exothermic. Consequently, (103) Au3/TiO2 exhibits superior O2 dissociation compared to (101) Au3/TiO2. This conclusion is in line with the fact that the former activates adsorbed O2 better than the latter. In contrast, the (101) or (103) TiO2 without Au cluster has practically no stretching of the O–O bond, and hence very small probability for O2 dissociation. As for non-supported Au clusters, the dissociation barrier is greater than 2 eV, according to the previous DFT studies.24 Thus, we predict a lowering of the O2 dissociation barrier in the case of Au supported stepped TiO2 surfaces. This is comparable to Pt (111), where the dissociation barrier is 0.45 eV, as computed by DFT.71 Our results are also comparable with studies on larger sized Au clusters on rutile TiO2 (110), where barriers of the order of 0.4–0.6 eV are observed.72 In particular, for the ORR, the nature of the activated state can provide information about the pathway, i.e. whether a four-electron or a two-electron pathway is likely to be followed, and thus the overall efficiency of the process. This is also true for any oxidation process on the surface, which involves the scission of the O–O bond in an activated state.
4. Conclusions
We have studied the adsorption and activation of oxygen molecules on plane (101), stepped (103) and reduced anatase TiO2 surfaces in this paper using plane wave DFT-based modeling. While the (101) and (103) surfaces do not activate the adsorbed O2 molecule, the reduced surfaces of the same adsorb O2 strongly. If we consider multiple O2 adsorption, it is seen that the reduced (103) TiO2 surface binds more than one O2 strongly. Hence, adsorption over vacancies is advantageous in terms of greater coverage of activated O2 when compared to (101) or (103) surfaces. Small sized Au clusters supported on the TiO2 surface lead to strong adsorption and activation of O2. In particular, the orientation of these Au clusters can tune the activation of O2, which depends on the proximity of Au atoms to the molecule. One can manipulate the orientation of Au clusters on TiO2 by adsorbing these on a step-edge of the surface. In such a configuration, we find a large stretching of the molecular O–O bond, into the peroxide regime, and strong binding. The electronic structure of the cluster–TiO2 interface shows good hybridization between the Au d and O p states and considerable charge transfer to the O2 molecule. In such an activated state, we find that the barrier for O2 dissociation is lower compared to the case of free Au clusters or Au clusters supported on (101) TiO2. Thus, we have demonstrated a simple way to utilize the morphology of the surface to suitably orient the Au cluster for enhanced O2 activation. Additionally, activated O2 may also be contributed by the active sites on the gold cluster itself. Thus, there is an overall enhancement of O2 activation through the Au cluster loading of the TiO2 surface. Experimental verification of our proposal would open a new way to realize an active catalyst surface, supported by an inexpensive and earth-abundant material and realized at low Au loading. This has important implications for various catalytic oxidation reactions.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
M. F. P.: conceptualization, formal analysis, investigation, visualization, writing – original draft. J. E. S.: conceptualization, formal analysis, investigation. R. C.: conceptualization, formal analysis, writing – review and editing, supervision, project administration.Conflicts of interest
There are no conflicts for the authors to declare.Acknowledgements
We acknowledge the Science and Engineering Research Board (SERB), Department of Science and Technology (DST), Government of India, for project grant CRG/2021/002468 under its Core Research Grant (CRG) scheme, and also the Centre for Computational Modeling and Simulation (CCMS), NIT Calicut for the computational infrastructure.References
- C. Stampfl, A. Soon, S. Piccinin, H. Shi and H. Zhang, J. Phys.: Condens. Matter, 2008, 20, 184021 CrossRef.
- J. Song, L. Wang, A. Zibart and C. Koch, Metals, 2012, 2, 450–477 CrossRef CAS.
- C. Jia, X. Wang, W. Zhong, Z. Wang, O. V. Prezhdo, Y. Luo and J. Jiang, ACS Appl. Mater. Interfaces, 2019, 11, 9629–9640 CrossRef CAS PubMed.
- D. H. Wells Jr, W. N. Delgass Jr and K. T. Thomson, J. Am. Chem. Soc., 2004, 126, 2956–2962 CrossRef PubMed.
- W. Zhang, J. Chang and Y. Yang, SusMat, 2023, 3, 2–20 CrossRef CAS.
- D. Costa, T. Ribeiro, F. Mercuri, G. Pacchioni and P. Marcus, Adv. Mater. Interfaces, 2014, 1, 1300072 CrossRef.
- A. M. Joshi, W. N. Delgass and K. T. Thomson, J. Phys. Chem. B, 2006, 110, 23373–23387 CrossRef CAS PubMed.
- F. Allegretti, S. O'Brien, M. Polcik, D. I. Sayago and D. P. Woodruff, Phys. Rev. Lett., 2005, 95, 226104 CrossRef CAS PubMed.
- M. M. Montemore, M. A. Van Spronsen, R. J. Madix and C. M. Friend, Chem. Rev., 2018, 118, 2816–2862 CrossRef CAS PubMed.
- Y. Wang, X. Ding, F. Wang, J. Li, S. Song and H. Zhang, Chem. Sci., 2016, 7, 4284–4290 RSC.
- C. Yuan, H. B. Wu, Y. Xie and X. W. Lou, Angew. Chem., Int. Ed., 2014, 53, 1488–1504 CrossRef CAS PubMed.
- C. Das, N. Sinha and P. Roy, Small, 2022, 18, 2202033 CrossRef CAS PubMed.
- W. Xiong, H. Yin, T. Wu and H. Li, Chem.–Eur. J., 2023, 29, e202202872 CrossRef CAS PubMed.
- A. L. Linsebigler, G. Lu and J. T. Yates Jr, Chem. Rev., 1995, 95, 735–758 CrossRef CAS.
- D. A. H. Hanaor and C. C. Sorrell, J. Mater. Sci., 2011, 46, 855–874 CrossRef CAS.
- U. Aschauer, J. Chen and A. Selloni, Phys. Chem. Chem. Phys., 2010, 12, 12956 RSC.
- M. A. Henderson, M. Shen, Z.-T. Wang and I. Lyubinetsky, J. Phys. Chem. C, 2013, 117, 5774–5784 CrossRef CAS.
- T. L. Thompson and J. T. Yates, Chem. Rev., 2006, 106, 4428–4453 CrossRef CAS PubMed.
- C. L. Pang, R. Lindsay and G. Thornton, Chem. Rev., 2013, 113, 3887–3948 CrossRef CAS PubMed.
- M. F. Puthiyaparambath and R. Chatanathodi, Phys. Rev. Mater., 2023, 7, 115801 CrossRef CAS.
- S. L. Hemmingson and C. T. Campbell, ACS Nano, 2017, 11, 1196–1203 CrossRef CAS PubMed.
- X. Li, X. Yang, Y. Huang, T. Zhang and B. Liu, Adv. Mater., 2019, 31, 1902031 CrossRef CAS PubMed.
- C.-J. Pan, M.-C. Tsai, W.-N. Su, J. Rick, N. G. Akalework, A. K. Agegnehu, S.-Y. Cheng and B.-J. Hwang, J. Taiwan Inst. Chem. Eng., 2017, 74, 154–186 CrossRef CAS.
- Z.-P. Liu, X.-Q. Gong, J. Kohanoff, C. Sanchez and P. Hu, Phys. Rev. Lett., 2003, 91, 266102 CrossRef PubMed.
- M. Haruta, Catal. Today, 1997, 36, 153–166 CrossRef CAS.
- J. A. van Bokhoven, C. Louis, J. T. Miller, M. Tromp, O. V. Safonova and P. Glatzel, Angew. Chem., Int. Ed., 2006, 45, 4651–4654 CrossRef CAS PubMed.
- N. Weiher, A. M. Beesley, N. Tsapatsaris, L. Delannoy, C. Louis, J. A. van Bokhoven and S. L. M. Schroeder, J. Am. Chem. Soc., 2007, 129, 2240–2241 CrossRef CAS PubMed.
- T. Hayashi, K. Tanaka and M. Haruta, J. Catal., 1998, 178, 566–575 CrossRef CAS.
- T. Akita, K. Tanaka, S. Tsubota and M. Haruta, J. Electron Microsc., 2000, 49, 657–662 CrossRef CAS PubMed.
- Y. Zhang, J.-X. Liu, K. Qian, A. Jia, D. Li, L. Shi, J. Hu, J. Zhu and W. Huang, Angew. Chem., Int. Ed., 2021, 60, 12074–12081 CrossRef CAS PubMed.
- I. X. Green, W. Tang, M. Neurock and J. T. Yates, Acc. Chem. Res., 2014, 47, 805–815 CrossRef CAS PubMed.
- N. Siemer, A. Lüken, M. Zalibera, J. Frenzel, D. Muñoz-Santiburcio, A. Savitsky, W. Lubitz, M. Muhler, D. Marx and J. Strunk, J. Am. Chem. Soc., 2018, 140, 18082–18092 CrossRef CAS PubMed.
- I. N. Remediakis, N. Lopez and J. K. Nørskov, Appl. Catal., A, 2005, 291, 13–20 CrossRef CAS.
- M. Valden, X. Lai and D. W. Goodman, Science, 1998, 281, 1647–1650 CrossRef CAS PubMed.
- W. Wan, X. Nie, M. J. Janik, C. Song and X. Guo, J. Phys. Chem. C, 2018, 122, 17895–17916 CrossRef CAS.
- K. Tada, H. Koga, A. Hayashi, Y. Kondo, T. Kawakami, S. Yamanaka and M. Okumura, Bull. Chem. Soc. Jpn., 2017, 90, 506–519 CrossRef CAS.
- K. G. Lakshmikanth, M. F. Puthiyaparambath and R. Chatanathodi, Surf. Sci., 2022, 722, 122084 CrossRef CAS.
- S. C. Petitto, E. M. Marsh and M. A. Langell, J. Phys. Chem. B, 2006, 110, 1309–1318 CrossRef CAS PubMed.
- U. Diebold, Surf. Sci. Rep., 2003, 48, 53–229 CrossRef CAS.
- C. C. Chusuei, X. Lai, K. Luo and D. W. Goodman, Top. Catal., 2000, 14, 71–83 CrossRef CAS.
- X.-Q. Gong, A. Selloni, M. Batzill and U. Diebold, Nat. Mater., 2006, 5, 665–670 CrossRef CAS PubMed.
- X. Gong and A. Selloni, J. Catal., 2007, 249, 134–139 CrossRef CAS.
- F. Rieboldt, R. Bechstein, F. Besenbacher and S. Wendt, J. Phys. Chem. C, 2014, 118, 3620–3628 CrossRef CAS.
- Y. Gao and S. A. Elder, Mater. Lett., 2000, 44, 228–232 CrossRef CAS.
- H. Onishi, T. Aruga, C. Egawa and Y. Iwasawa, Surf. Sci., 1988, 193, 33–46 CrossRef CAS.
- X. Du, Y. Huang, X. Pan, B. Han, Y. Su, Q. Jiang, M. Li, H. Tang, G. Li and B. Qiao, Nat. Commun., 2020, 11, 5811 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- L. Yan and H. Chen, J. Chem. Theory Comput., 2014, 10, 4995–5001 CrossRef CAS PubMed.
- B. J. Morgan and G. W. Watson, Surf. Sci., 2007, 601, 5034–5041 CrossRef CAS.
- J. K. Burdett, T. Hughbanks, G. J. Miller, J. W. Richardson Jr and J. V. Smith, J. Am. Chem. Soc., 1987, 109, 3639–3646 CrossRef CAS.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- M. Lazzeri, A. Vittadini and A. Selloni, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 155409 CrossRef.
- Z. Zhou, Y. Yu, Z. Ding, M. Zuo and C. Jing, Eur. J. Inorg. Chem., 2018, 2018, 683–693 CrossRef CAS.
- G. S. Herman, M. R. Sievers and Y. Gao, Phys. Rev. Lett., 2000, 84, 3354–3357 CrossRef CAS PubMed.
- H. Peng, J. Li, S.-S. Li and J.-B. Xia, J. Phys.: Condens. Matter, 2008, 20, 125207 CrossRef.
- A. Gentile, F. Ruffino and M. G. Grimaldi, Nanomaterials, 2016, 6, 110 CrossRef PubMed.
- A. Vittadini and A. Selloni, J. Chem. Phys., 2002, 117, 353–361 CrossRef CAS.
- B. Richter, H. Kuhlenbeck, H.-J. Freund and P. S. Bagus, Phys. Rev. Lett., 2004, 93, 026805 CrossRef CAS PubMed.
- G. Deng, S. Malola, P. Yuan, X. Liu, B. K. Teo, H. Häkkinen and N. Zheng, Angew. Chem., Int. Ed., 2021, 60, 12897–12903 CrossRef CAS PubMed.
- S. Nosé, J. Chem. Phys., 1984, 81, 511–519 CrossRef.
- I. X. Green, W. Tang, M. Neurock and J. T. Yates Jr, Acc. Chem. Res., 2014, 47, 805–815 CrossRef CAS PubMed.
- B. Hammer and J. K. Nørskov, Adv. Catal., 2000, 45, 71–129 CAS.
- P. C. Jennings, H. A. Aleksandrov, K. M. Neyman and R. L. Johnston, Nanoscale, 2014, 6, 1153–1165 RSC.
- I. X. Green, W. Tang, M. McEntee, M. Neurock and J. T. Yates Jr, J. Am. Chem. Soc., 2012, 134, 12717–12723 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4na00744a |
| This journal is © The Royal Society of Chemistry 2024 |