
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFano enhancement of SERS for rapid early diagnosis of colorectal cancer†
Tianxun
Gong
 a,
Zhenjiang
Wei
a,
Libin
Huang
b,
Yan
Hong
c,
Yuan
Li
b,
Ke-ling
Chen
b,
Wen
Huang
a,
Xiaojing
Zhong
d,
Jinzhao
He
d,
Ming-Yi
Lee
e,
En-Chi
Chang
e,
Kien Voon
Kong
*e,
Xiaosheng
Zhang
*a and
Zongguang
Zhou
*b
a,
Zhenjiang
Wei
a,
Libin
Huang
b,
Yan
Hong
c,
Yuan
Li
b,
Ke-ling
Chen
b,
Wen
Huang
a,
Xiaojing
Zhong
d,
Jinzhao
He
d,
Ming-Yi
Lee
e,
En-Chi
Chang
e,
Kien Voon
Kong
*e,
Xiaosheng
Zhang
*a and
Zongguang
Zhou
*b
aState Key Laboratory of Electronic Thin Films and Integrated Devices, School of Electronic Science and Engineering (National Exemplary School of Microelectronics), University of Electronic Science and Technology of China, Chengdu, China. E-mail: zhangxs@uestc.edu.cn
bLaboratory of Digestive Surgery, State Key Laboratory of Biotherapy and Cancer Center, Department of Gastro-intestinal Surgery, West China Hospital, West China School of Medicine, Sichuan University, China. E-mail: zhou767@scu.edu.cn
cSchool of Materials and Energy, University of Electronic Science and Technology of China, Chengdu, China
dDepartment of Internal Medicine-Neurology, Heyuan People's Hospital, China
eDepartment of Chemistry, National Taiwan University, Taipei, 10617, Taiwan. E-mail: kvkong@ntu.edu.tw
First published on 30th September 2024
Abstract
Patients benefit greatly from early detection of colorectal cancer, but present diagnostic procedures have high costs, low sensitivity, and low specificity. However, it is still difficult to develop a strategy that can effectively detect cancer early using high-throughput blood analysis. Fano resonance-boosted SERS platform label-free serum creates an effective diagnostic system at the point of care. We obtained 220 high-quality SERS serum spectral datasets from 88 healthy volunteers and 132 patients with colorectal cancer. The biomarker detected in serum was further evaluated using 100 colorectal cancer tissues and adjacent normal intestinal tissues collected from West China Biobanks, West China Hospital, Sichuan University. The results showed that in 97 out of 100 paired samples, the biomarkers were successfully detected using the SERS platform. This demonstrates that Fano resonance-based SERS is highly effective for diagnosing colorectal cancer.
1. Introduction
Hybridized collective plasmonic modes, such as Fano resonance, can generate peculiar spectral shapes due to near-field coupling between a bright super-radiant mode (bright mode) and a narrow sub-radiant mode (dark mode). Fano resonance has recently demonstrated excellent performance in biophotonic sensing, such as surface-enhanced Raman scattering (SERS).1–4 Fano resonance exhibits outstanding properties over conventional plasmon resonance in that it significantly reduces or even eliminates the radiation loss of the system, thereby enhancing local electromagnetic fields.5Another feature of Fano resonance is its asymmetric non-Lorentzian spectral profile. It shows a distinct dip shape.6–9 Such symmetry breaking has been observed in a variety of metal nanostructures, such as the asymmetric structure of silicon–gold core–shell,10–12 non-concentric ring-disk cavities,13,14 metal nanoparticle heptamers and quadrumers,15–17 Au dolmen-like structures,3,13,18 and various heterodimers.19–23
The use of simple dielectric materials to induce Fano resonance has been actively investigated. This is due to the fact that Fano resonance can be created by coupling between plasmonic modes of a dielectric substrate. Zhongqun Tian et al. developed a self-assembling structure of a gold nanosphere-dielectric-gold substrate to produce magnetic Fano resonance in the visible wavelength range that can detect catalytic events ultra-sensitively through Raman scattering.24
The remarkable property of Raman detection using Fano resonance is that the Fano resonance generated by the magnetic dipole mode can be coupled with the electric and magnetic components of the incident light that leads to a significant increase in optical coupling efficiency. Clearly, this is superior to the conventional Fano resonance dominated by the electric dipole mode, which is mainly influenced by the electric field component of incident light. Additionally, the smaller electric dipole moment in the resonance can reduce the radiation loss. Therefore, the Fano resonance based on magnetic dipole mode produced a stronger near-field Raman enhancement effect.
Herein, we demonstrated that SERS substrates can be prepared in-house without the need for complicated instruments and chemistry. Our group intends to create an easy-access technology without sacrificing its accuracy and quality as a fundamental need for accelerating technological progress and benefits to society. In order to form the dielectric layer, polyethylene glycol (PEG)–biotin and avidin were used as linking molecules. By adjusting the length of PEG, we can in-house tune the Fano resonance dip according to experiment requirements of incident laser wavelength that can in-turn lead to maximum SERS enhancement and high sensitivity for the detection of tumor markers of colorectal cancer (CRC) in blood.
CRC is among the most common cancers and is the second leading cause of cancer-related deaths worldwide. Approximately 10% of stage IV CRC patients survive for five years.25 The two most common screening procedures for CRC are endoscopic examination and fecal immunochemical testing.26 SERS-based noninvasive blood biopsies can be used to improve the current state of the art to detect CRC and to provide rapid diagnosis and rapid mass deployment.
In this paper, we report that a Fano resonance system combined with a Raman spectrometer can achieve highly sensitive detection for small molecules. The blood serum sample was dropped onto the platform without the need for further processing. The SERS data were then measured by Raman spectrometry using sample detection within 5 seconds. The data agreed with the mass spectroscopy data. In order to explore the blood serum diagnostic information, we further analyzed 100 paired colorectal cancer tissues and adjacent normal intestinal tissues provided by West China Biobanks. To the best of our knowledge, this is the first label-free blood serum SERS detection system for CRC using a Fano resonance platform. The results suggested that Fano resonance-enhanced SERS could be a technology for rapid early diagnosis of CRC.
2. Experimental section
2.1. Fabrication of the Au-NS substrate
Clean silicon wafers were cut into 10 mm × 10 mm squares. The wafers were then ultrasonically treated with acetone, ethanol, and deionized water for 300 s, respectively. A 300 nm gold film was coated on a silicon wafer at 1 × 10−5 Pa (Shenyang Scientific Instrument Co., Ltd). The gold film coated wafer was then incubated with biotin–PEG–SH (Shanghai Aladdin Biochemical Technology Co., Ltd, 95%, MW 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da, 3400 Da, and 1000 Da) solution (50 μM) for 30 minutes and rinsed with water. A 50 μL (5 mg mL−1) of 200 nm avidin linked silica nanosphere (Microspheres-Nanospheres, a Corpuscular Company, 2.5% (w/v)) was then dropped on the surface of the substrate and spin coated at 100 rpm for 10 minutes. Finally, 15 nm gold was sputtered on the substrate.
000 Da, 3400 Da, and 1000 Da) solution (50 μM) for 30 minutes and rinsed with water. A 50 μL (5 mg mL−1) of 200 nm avidin linked silica nanosphere (Microspheres-Nanospheres, a Corpuscular Company, 2.5% (w/v)) was then dropped on the surface of the substrate and spin coated at 100 rpm for 10 minutes. Finally, 15 nm gold was sputtered on the substrate.
2.2. Surface characterization
The scanning electron microscopy (SEM) pictures of the NPGF substrate and F-NP substrates were obtained using a Gemini SEM 500 (ZEISS) at 5.00 kV, standard detector SE2 and secondary electron detector InLens.2.3. Scattering spectrum of the SERS substrate
All optical measurements were performed before SEM characterization to avoid possible carbon contamination and molecular fragmentation. A HORIBA iHR 550 spectrometer was used to collect scattering spectra of Au-NS substrates. A non-polarized white light source was used to illuminate the sample vertically. The scattered light is collected through a 100×, 0.55 NA objective lens. All scattering spectra are subtracted from the dark background and the wavelength response is normalized by using the scattering spectrum obtained from the silver mirror.2.4. Sample collection
The human subject study was approved by the Ethics Committee on Biomedical Research at West China Hospital of Sichuan University (approval no. 2020(374)). All experiments were performed in accordance with the Guidelines of the Declaration of Helsinki. Experiments were approved by the Ethics Committee on Biomedical Research at West China Hospital of Sichuan University. Informed consents were obtained from human participants in this study. Fresh blood and tissue samples were collected from the Department of Gastrointestinal Surgery, West China Hospital, Sichuan University. All serum samples were collected from subjects who had fasted for more than 10 hours and then used anticoagulant-free vacuum blood collection tubes to draw 2 mL of peripheral venous blood in the early morning. The blood samples were allowed to stand at 4 °C for 2 hours (to precipitate red blood cells) and centrifuged at 4000 rpm for 10 minutes at 4 °C. After centrifugation, 1 mL of the supernatant was pipetted into a cryotube, divided into 3 portions. All the collected serum was aliquoted and stored in a refrigerator at −80 °C for later use. At the same time, the patient's tumor resection tissue and normal samples from the distant cancer intestine as the control were collected. The tumor samples were collected by taking 0.5 × 0.5 cm size on the surface of tumor tissue after resection, and the control samples were collected by marking the intestinal mucosa farthest from the tumor and removing the same size tissue. The tissue was immediately placed in the cryotube and stored in a liquid nitrogen tank for later use.2.5. Treatment of serum samples
100 μL of blood serum was taken from the same patient to extract hydrophilic metabolites/hydrophobic metabolites/proteins respectively.000 g for 10 min. The hydrophilic metabolite was obtained by concentrating and drying the supernatant.
000 g for 5 min. After the sample was stratified, the upper layer was discarded and the lower chloroform layer was taken through the intermediate protein layer using a pipette tip. The hydrophobic metabolite was prepared by concentrating and drying the chloroform layer solution.
000 g for 5 min. After the sample was stratified, the upper layer was discarded, and 400 μL methanol was added to the solution. The mixture was repeatedly vortexed and centrifuged at 4 °C at 20000 g for 15 min. The supernatant was discarded, and the protein sample was prepared by precipitation of the solution at room temperature.
2.6. Tissue samples
The frozen tissue samples were taken out and thawed. 100 mg of tissue was cut into pieces and placed into a 5 mL centrifuge tube. Normal saline was added at a ratio of 1:5 and homogenized in a homogenizer at 4 °C at 15000 rpm for 2 min. The homogenate was centrifuged at 4 °C at 20000 g for 15 min. Small molecule compounds and proteins were extracted from the supernatant by the methods described in 2.3 and transferred into a sample tube, respectively. All collected samples were stored at −80 °C for later use.
2.7. Chromatographic analysis
400 μL of precooled methanol was added into samples prepared in 2.3, respectively. The mixtures were repeatedly vortexed, and then placed at −80 °C for 2 h. The samples were then centrifuged at 4 °C at 12000 g for 10 min. The supernatants were taken out and dried. 100 μL of 70% acetonitrile/30% methanol solution was used for re-dissolution and separation on a high-performance liquid phase chromatography (HPLC) instrument and separation conditions are as follows: instrument: Agilent-1260, mobile phase buffer A: 100% water, mobile phase buffer B: 100% acetonitrile, normal phase column: Kromail, 60-5-HILIC-D, 4.6 × 250 mm, ultraviolet detection: 240 nm wavelength, flow rate: 1 mL min−1, column temperature: 40 °C. The flow gradient is shown in Table S3.† HPLC was performed for 60 min. According to the running time, 1 tube of sample was collected every minute and marked with a number according to the collection time and stored at −80 °C for later use.
2.8. SERS detection of samples
20 μL of each sample was dropped onto the surface of the SERS substrate and vacuum dried in an oven at 37 °C for 2 hours. Raman spectra of the blood serum samples were collected using a Horiba iHR550 spectrometer using an excitation laser wavelength of 785 nm under a ×100 objective lens. The laser power is ∼2.7 mW, and the accumulation time is 5 s. The Raman detection range is from 400 to 1800 cm−1. Five detection points are randomly selected for each sample. The characteristic spectrum of each sample was obtained using the average spectrum collected from five points.2.9. Mass spectrometry detection
The retention time of the potential normal phase column was determined according to the spectral signal results. The pre-packed products were analyzed by non-targeted mass spectrometry. The mass spectrometry conditions were as follows: instrument: Q-extractive, mobile phase buffer A: 10 mM ammonium formate/0.1% formic acid/acetonitrile solution, mobile phase buffer B: 10 mM ammonium formate/0.1% formic acid/water solution, reverse column: Waters, Atlantis HILIC Silica column, 2.1 × 150 mm, flow rate: 300 μL min−1, column temperature: 35 °C. The flow rate gradient and mass spectrum parameters are shown in Tables S2–S4.† Non-targeted mass spectrometry analysis cooperated with Tsinghua University's metabolomics platform. The mass spectrometry raw data were processed using Tracefinder v3.1 (Thermo Fisher Scientific, USA) software through the local database and Tsinghua University's metabolomics database.3. Results and discussion
3.1. Simulation of the gold nanosphere (Au-NS) substrate
In the simulation model, we started with gold (Au) coated silica with a 1 nm gap distance between Au film to account for the presence of the PEG layer, with a refractive index of 1.4. The dielectric constant of Au was obtained from Rakic et al.27 (Fig. 1a). The absorption, scattering and extinction spectra and the electric field and magnetic field distributions are calculated by the finite element method (simulation details can be found in the ESI†).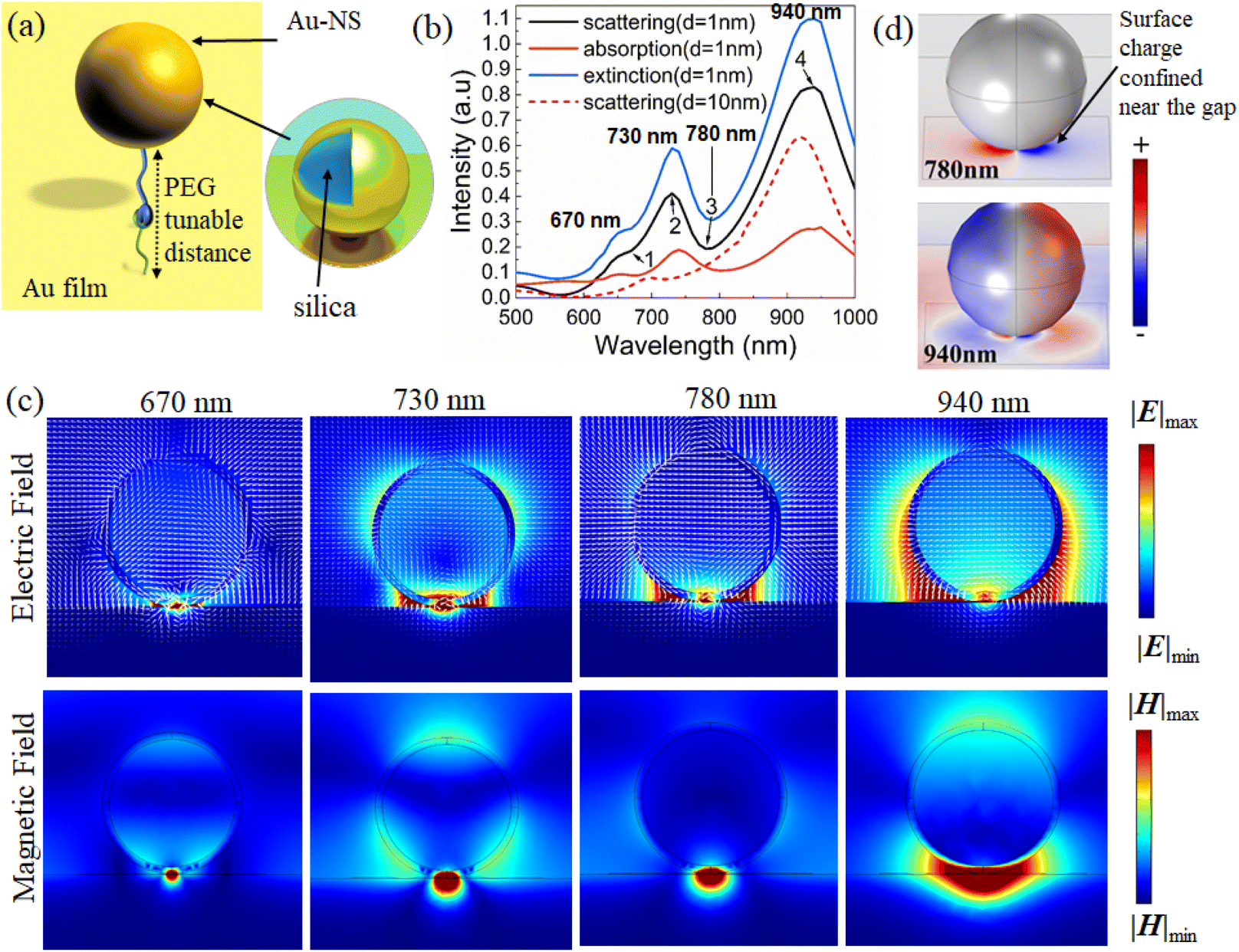 | ||
| Fig. 1 (a) The nano-heterostructure simulation model of Au-NSs with 200 nm size on a Au film. (b) The scattering and absorption curves for the Au-NS substrate with a diameter of 200 nm (d is the dielectric gap distance). (c) The electric displacement vector and the electric/magnetic field distribution of the Au-NS substrate. The electric field and magnetic field intensities have been normalized to the same scale, respectively. (d) The surface charge distribution of the Au-NS substrate at 780 nm and 940 nm. | ||
Au nanospheres (Au-NSs) and the Au film will generate electric dipoles when normal incident light is irradiated on the Au substrate because of the strong resonance between the localized surface plasmon and the propagating surface plasmon. According to the mirror imaging field effect, the imaging sphere also experiences similar electric modes with antiphase distributions.28 Due to the antiparallel distributions on the real and imaging spheres, a current loop may be formed between the two particles due to plasmon hybridization in the Au-NS substrate. As a result, it is possible to simulate the Au-NS substrate using an inductor capacitor (LC) resonator circuit.29
In principle, nanospheres and imaging spheres can be viewed as two resistors, whose resistance depends on their radius. The PEG layer and the surrounding air are considered capacitors. An LC resonator is sensitive to external parameters. The possibility of excitation and the spectral position of magnetic mode are dependent on the diameter of the nanosphere and the gap distance of the PEG layer. According to Fig. 1b, the absorption, scattering, and extinction spectra of the Au-NS substrate with a diameter of 200 nm and an Au thickness of 15 nm were calculated. As shown in Fig. 1b, there is a distinct valley in the scattering spectrum at 780 nm (mark 3). Fano resonance is characterized by an asymmetric curve, which occurs when super-radiation (bright mode) and sub-radiation (dark mode) combine. Furthermore, the wavelength of the average electric field at the nanogap in Fig. S1† reaches its maximum at around 780 nm, indicating that the plasmon coupling of Au-NSs not only generates electric dipoles, but also generates new plasma modes.
3.2. Fano resonance mechanism of Au-NSs
Au-NS near-field distributions corresponding to four different wavelengths (670 nm, 730 nm, 780 nm and 940 nm) were calculated to better understand the Fano resonance mechanism of the Au-NSs.In the gap between the Au-NS and Au film, there is a very concentrated localized “hot spot” of the electric (Fig. 1c (780 nm)) and magnetic fields (Fig. 1b; mark 3). As shown in Fig. 1c, the electric displacement vector circulates at the gap, with the field distribution resulting in a magnetic dipole mode perpendicular to the cross section.
At 670 nm (Fig. 1b; mark 1), the near-field magnetic and electric fields are distributed differently than those at 780 nm when a smaller “hot spot” appears at the gap. Fig. 1c (670 nm) shows a double-circle displacement vector ring and demonstrates the presence of “hot spots” at the gap, which are indicative of higher order magnetic modes.
For the electric field at 940 nm wavelength (Fig. 1b; mark 4) it can be seen that “hot spots” are widely distributed on the nanosphere's surfaces, especially on the left and right sides, and the “hot spots” do not dominate in the gap (Fig. 1c (940 nm)). It can be seen that the distribution of the magnetic field also extends from the gap to the left and right sides of the nanosphere. Therefore, the electrical response dominates, and this mode represents the electric dipole mode.
The electric field distribution at the second scattering peak at 730 nm (Fig. 1b; mark 2) extends toward both sides of the nanosphere, but it is prominently displayed in the gap. Similarly, the magnetic field distribution (Fig. 1c (730 nm)) represents an intermediate state between the electric dipole mode and the magnetic dipole mode. Here, the surface charge behaves as an LC resonator because of the reverse polar surface charge on the nanospheres' surface and on the gold film's surface.
In comparison with the charge distribution at 780 nm and 940 nm, it was found that the electromagnetic field is concentrated at 780 nm and distributed on the surface of the nanosphere at 940 nm, which confirms the previous explanation.
Hence, the Fano resonance results from the coupling of the electric dipole and magnetic dipole modes. The electric dipole mode is considered a super-radiation mode (bright mode), since the total electric dipole moment is proportional to the volume of the nanospheres, as shown in the surface charge distribution (Fig. 1d, 940 nm).30 The electric dipole moment on the nanosphere surface is weakened because of the presence of the inverted electric dipole on the Au film surface. However, the total electric dipole moment is still very large. The surface charge of most dipoles (Fig. 1d, 780 nm) is confined near the gap between the nanospheres and the Au film, resulting in a significant reduction in the total magnetic dipole moment. As for the magnetic dipole mode, it serves as the sub-radiation mode (dark mode).
The second scattering valley at 670 nm denotes the coupling of electric dipoles and higher-order magnetic modes, indicating subradiation modes (Fig. 1b). The resonant positions of plasmon modes are generally regarded as the optimal locations of experimental laser lines to obtain optimally enhanced near fields for most plasmonic systems. To understand the potential mechanism of plasmon-enhanced Raman spectroscopy, it will be essential to examine the relationship between near-field enhanced and scattering spectra of Au-NSs.
Fano resonance can be controlled using geometric parameters (Au thickness, the diameter of Au-NSs, and the gap between Au-NSs and the Au plate).
The Fano resonance attenuated with the decrease of the Au thickness, and a significant red shift can be observed in the magnetic dipole mode (Fig. 2). The electric field distribution of Au nanospheres with a thickness of 5 nm at 785 nm wavelength is shown in Fig. 2a; the gap is nearly free of “hot spots”. There was no Fano resonance observed for an Au thickness of 5 nm (Fig. 2b) because the influence of the dielectric environment inside the silica nanosphere becomes more significant, resulting in a great attenuation of the magnetic resonance.
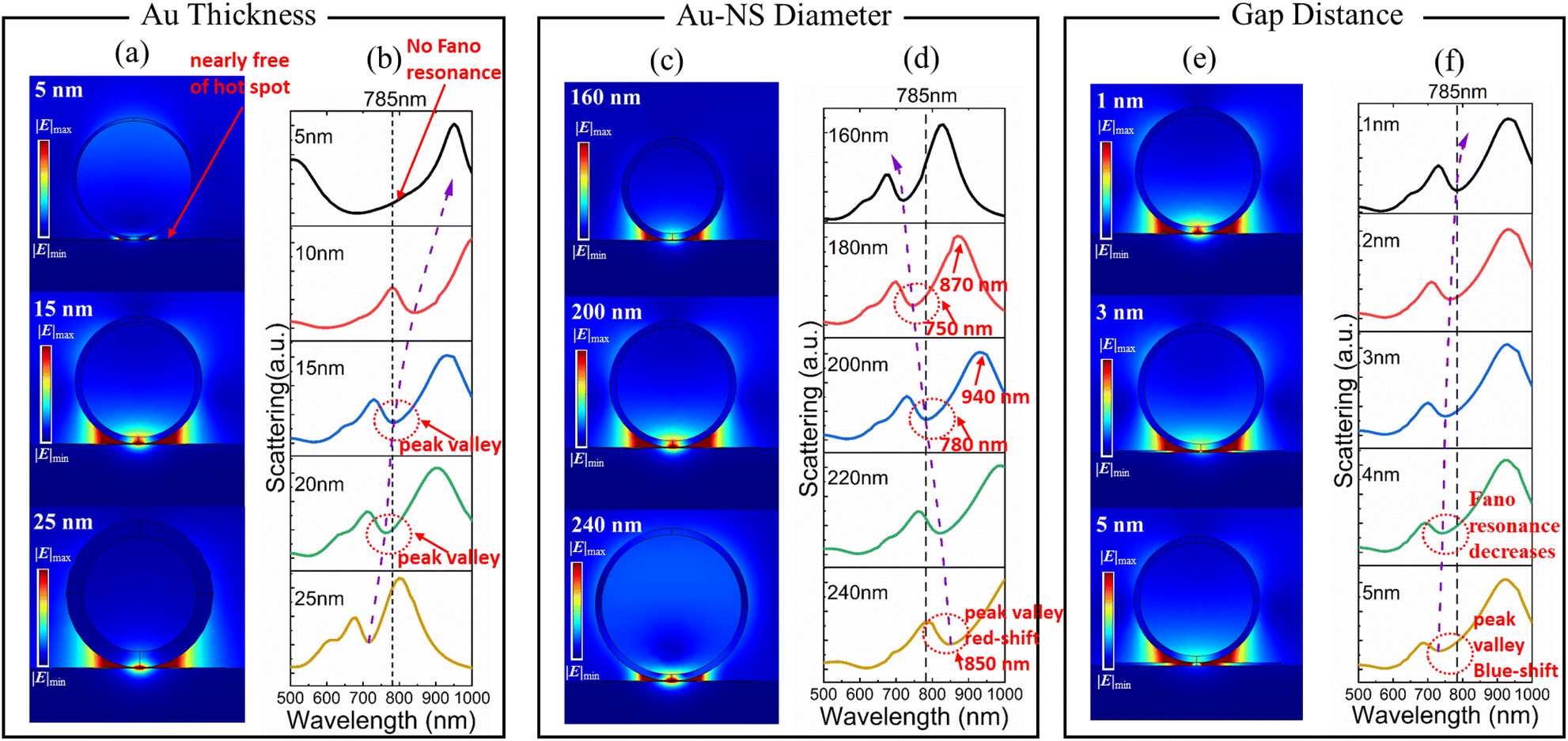 | ||
| Fig. 2 Fano resonance regulated using geometric parameters. (a) Electric field distribution at 785 nm wavelength corresponding to different Au thicknesses. (b) Calculated scattering spectra of Au thickness. Note: the diameter of Au-NSs is fixed at 200 nm and the gap distance is fixed at 1 nm. (c) Electric field distribution at 785 nm wavelength corresponding to different Au-NS diameters. (d) Calculated scattering spectra of Au-NSs with diameters ranging of 160–240 nm. Note: the Au thickness is fixed at 15 nm and the gap distance is fixed at 1 nm. (e) Electric field distribution at 785 nm wavelength corresponding to different dielectric gap distances ranging from 1–5 nm. (f) Calculated scattering spectrum of the dielectric gap distance ranging from 1 nm to 5 nm. Note: the Au thickness is fixed at 15 nm and the diameter of Au-NSs is fixed at 200 nm. | ||
When the Au thickness is 15 nm, the magnetic dipole matches the laser excitation at 785 nm. Compared with the Au thicknesses of 10 nm and 15 nm, the scattering intensity of Fano resonance is reduced from 22% to 16% according to the peak valley ratio. The electric field distribution with an Au thickness of 15 nm is shown in Fig. 2a, that has stronger “hot spots” at the gap compared to an Au thickness of 25 nm (Fig. 2a).
The effect of the diameter of the Au-NSs and gap distance on the plasmon coupling of the Au-NS substrate was also studied (Fig. 2c–f). We set the thickness of Au to 15 nm. Observed Fano valleys are red-shifted from 720 nm to 850 nm for diameters of Au-NSs between 160 nm and 240 nm (Fig. 2d). Additionally, the peak and valley of the scattering spectrum shifted with the increasing diameter (Fig. 2d); from 180 to 200 nm, the peak is blue-shifted by 60 nm (940 nm to 870 nm), while the valley is shifted by 30 nm (780 nm to 750 nm).
The electric field distribution at 785 nm wavelength is shown in Fig. 2c. It can be found that in the electric field distribution with a diameter of 160 nm (Fig. 2c) and 240 nm (Fig. 2c), the “hot spots” at the gap are not as strong as those at a diameter of 200 nm (Fig. 2c). Magnetic and electric dipolar modes are found to be different in their sensitivity to the diameter and the former is more sensitive to the sphere diameter than the latter, because of the dipolar moment of the magnetic resonance being intimately related to the equivalent resistance, which varies minutely with the diameter. In contrast, electric dipolar modes largely depend on the diameter.31 Thus, magnetic modes cannot be observed in our interested wavelength regions (785 nm) when the diameter in the Au-NS substrate is small, as the equivalent resistance is too small to excite the magnetic mode.32 Because of such non-synchronization, Fano resonance diminishes as the diameter increases, and the valley widens as the electrical dipolar resonance's radiative damping becomes increasingly dominant.
For the gap distance ranges from 1 nm to 5 nm, their calculated scattering spectra are shown in Fig. 2e and f. With the gap distance increasing, the scattering valley was blue shifted, while the Fano linear asymmetry became less pronounced (Fig. 2f). The position of the blue-shifted peak and valley can be seen in Fig. S3.† The peaks' position changed insignificantly, which is around 940 nm, but the valley's position shows a clear blue shift. As a result, we can conclude that the change in gap distance has little influence on the electric dipole mode, but is very significant for the magnetic dipole mode. It is because of the direct relationship between the magnetic dipole mode and the equivalent capacitance of the structure. With different gap distances, Fano resonance wavelength (valley) changed almost linearly (Fig. 2f). The electric field distribution at 785 nm wavelength is shown in Fig. 2e. The “hot spots” at the gap may not be as strong at dielectric gap distances of 3 and 5 nm as those at 1 nm (Fig. 2e). As the dielectric gap distance increases, the Fano resonance intensity gradually decreases.
3.3. Performance of SERS on the Au-NS substrate
The simulation results show that an Au-NS with 15 nm Au thickness, 200 nm diameter, and a 1 nm gap distance between the Au-NS and the Au film is optimal for SERS enhancement at 785 nm. To immobilize the Au-NS on the Au film, we used the avidin–biotin interfacial technique. Our group has previously demonstrated the application of avidin–biotin complexes in SERS,33 and it is the strongest non-covalent interaction known.The Au film was first coated using commercial biotin–PEG–SH. Au-NSs coated with avidin–SH were deposited onto the Au film to form a hetero-nanostructure. The Au-NS is firmly bound on the Au film. A sub-nano gap is formed with the presence of the PEG layer (Fig. 3a).
 | ||
| Fig. 3 Preparation and characterization of the Au-NSs. (a) Au-NS was immobilized via biotin–avidin complexation. (b) SEM image of the Au-NS substrate. (c) Calculated and experimentally measured scattering spectrum of the Au-NS substrate. Raman spectra and intensity of 4-aminothiophenol obtained from the Au-NS substrate with (d) different Au thicknesses of Au-NSs, (e) different diameters of Au-NSs, and (f) different dielectric gap distances between Au-NSs and the Au film. | ||
As shown in Fig. 3b, the surface of the substrate was observed using a scanning electron microscope (SEM). Due to the dielectric properties of PEG, it creates a dielectric layer sandwiched between the gold film and gold nanospheres. The Au-NSs are evenly distributed with separation distance to avoid mutual coupling of the electric and magnetic fields. From the enlarged picture of a single nanosphere on the substrate, the diameter of the sphere is ∼200 nm.
In addition, the scattering spectrum of Au-NS substrates was obtained, as shown in Fig. 3c. It can be observed that the Fano resonance valley of the scattering spectrum obtained from the experiment is located at around 785 nm, but broadened compared to the calculated spectrum. This discrepancy may be attributed to several factors: the spectrum was collected from scattered light originating from multiple nanospheres on the substrate; the nanosphere varied in shape and size; the uneven thickness of the gold coating and gold film; the refractive index and thickness changing of the PEG layer; and the collective effects of these variations resulted in a broadened Fano resonance valley. In contrast, the simulation was run on an idealized single nanosphere on a flat gold film with a gap fixed at 1 nm.
The coherent excitation of the magnetic mode in the Au-NS structure captures the incident light at the near field and enhances the electromagnetic field at the gap. Thus, it is to be expected that the enhanced electromagnetic field greatly amplifies the Raman scattering signal. The Raman enhancement of the Au-NS substrate was compared using 5 μL of 1 mM of 4-aminothiophenol as a marker. Raman spectra marker's 1080 cm−1 peak intensities obtained from Au-NSs with different Au thicknesses, different Au-NS diameters, and different dielectric gap distances between Au-NSs and the Au film have been analyzed.
It can be observed that when the Au thickness is 15 nm, the Raman signal has the strongest enhancement (Fig. 3d). When the Au thickness is 5 nm and 10 nm, Raman signal enhancement is not evident, indicating that the thickness cannot excite Fano resonance. There is a relatively larger Raman enhancement with Au thicknesses of 20 nm and 25 nm. The greatest enhancement can be observed with an Au thickness of 15 nm, as the Fano resonance valley matches with the wavelength of the 785 nm laser. Fig. 3e shows the spectra corresponding to different nanosphere diameters. The strongest Raman signal occurs when the diameter is 200 nm, followed by 160 nm, 300 nm, and 400 nm. Fig. 3f shows the spectra corresponding to different PEG lengths (gap distances). It can be found that the enhancement of the Raman signal with a dielectric gap at 0 nm (without PEG) is the weakest. As the molecular weight of PEG becomes larger and larger, the Raman signal becomes weaker and weaker. On the other hand, the best enhancement effect is achieved with PEG with 1000 Da (detailed comparison of PEG with different molecular weights can be seen in Fig. S4†). These results are similar to simulations. Based on the Raman signal comparison, the Au-NS substrate has the strongest enhancement due to its unique magnetic Fano resonance. The enhancement factor of the prepared Au-NS substrate is calculated to be ∼1 × 1015 (see detailed calculation in Fig. S5†).
3.4. SERS screening of colorectal cancer biomarkers
Through the use of the Fano resonance-assisted sensing platform, we have developed a novel method for the rapid diagnosis of CRC through the measurement of SERS in blood serum. SERS spectra from collected blood serum samples show prominent Raman enhancement (Fig. 4a). Clearly, the spectra from cancer and healthy groups share many features in common, but there are also some differences. | ||
| Fig. 4 Characteristic Raman peaks of blood serum. (a) Averaged Raman spectra of blood serum from colorectal cancer patients and normal controls. (b) Raman spectra of blood serum from cancer patients and healthy controls. (c) Heat map of the Raman spectra of hydrophilic fractions (1–20 fractions) from cancer patient's blood serum. (d) Mass spectrometry analysis of cancer serum hydrophilic compounds. | ||
To assess the reliability of the SERS measurements, a more detailed analysis of these spectral assignments is performed. The healthy group showed a higher concentration of tyrosine (648, 838, and 1165 cm−1) and phenylalanine (622, 1029, and 1573 cm−1).34,35 The amino acid tyrosine is an aromatic amino acid that is necessary for the synthesis of thyroid hormones, catecholamines, and melanin. A low level of tyrosine may be due to the metabolic disturbance that can result from the development of colorectal tumors in serum samples from patients with CRC. Several studies have also indicated that levels of tyrosine and glutamine–leucine in serum can be used to diagnose CRC.36
Hypoxanthine's main characteristic is its SERS band at 726 cm−1.37 SERS spectra of incubated blood samples usually exhibit this characteristic due to purine degradation.38 In CRC samples, levels of hypoxanthine in the SERS spectrum increased compared with healthy controls, which is in line with the results of metabolomic profiling.39 It was also found that the thymine signal at 741 cm−1 was generally stronger in the cancer group because CRC cells require large quantities of these molecules for adenosine triphosphate (ATP) transmission and synthesis of nucleic acids. These strong bands at 1141, 1263, and 1484 cm−1 are caused by lipids in the plasma or extracellular vesicles.40 A blood sample may contain total cholesterol, high-density lipoprotein cholesterol (HDL), low-density lipoprotein cholesterol (LDL) or triglycerides. Dyslipidemia, caused by high triglyceride levels and low HDL cholesterol levels, is linked to a higher incidence of CRC.41 CRC is associated with both blood lipids and lipids from extracellular vesicles, making the relative lipid intensity an important factor.
The hydrophilic and hydrophobic fractions were collected from blood serum. The Raman spectrum of the hydrophilic fraction exhibits greater consistency, especially the characteristic peak at 726 cm−1. While hydrophobic compounds also exhibit a characteristic peak at 726 cm−1, the overall average spectral signal is more variable. Furthermore, there is no significant 726 cm−1 peak for the protein fraction (Fig. S6†).
3.5. Metabolomics analysis
The combination of metabolomics methods that also study changes in the level of small molecule metabolites may be able to further hypoxanthine which is a tumor marker associated with colorectal cancer. Therefore, the hydrophilic fraction was further analyzed. The hydrophilic fraction was first separated by high-performance liquid chromatography (HPLC). Total 60 fractions were separated according to the separation time, and two fractions show a strong peak at 726 cm−1. The results suggested that the molecule corresponding to the characteristic peak at 726 cm−1 is retained in the specific fractions (Fig. 4c and S6b†). These two fractions were then analyzed by mass spectrometry. Among all 185 metabolites detected by mass spectrometry, the contents were analyzed according to the database results, and the substances with more than 85 credible scores were selected. It was found that the content of hypoxanthine was the highest, as shown in Fig. 4d. The content of hypoxanthine in cancer patients' serum was significantly higher than that in healthy controls, which is consistent with the Raman intensity of the 726 cm−1 peak. For further verification, pure hypoxanthine was mixed with cancer patient's blood serum with hypoxanthine concentrations of 50 μM, 100 μM, 200 μM, 500 μM, and 1 mM for Raman detection (Fig. S7†). We successfully detected 50 μM hypoxanthine in blood samples, despite the presence of millions of interfering molecules. This level of sensitivity is sufficient and appropriate for clinical applications. It can be seen that the characteristic peak from hypoxanthine is located at 726 cm−1, which is consistent with the previous experiment. Furthermore, the concentration of hypoxanthine is positively correlated with its Raman peak intensity.Hypoxanthine is an important intermediate product in purine metabolism. There are two main pathways for purine synthesis in the body. In the process of tumorigenesis and development, the rapid proliferation and replication of tumor cells may lead to abnormal levels of metabolites in tissues, while the higher concentration of hypoxanthine in colorectal cancer patients' serum indicates the possible abnormal metabolism of hypoxanthine in tumor patients.
For verification, we measured the concentration of hypoxanthine in tumor tissue samples and the adjacent normal tissue samples collected during the serum sample collection. Through hypoxanthine targeted mass spectrometry detection, we found that the hypoxanthine concentration in the tumor tissue was significantly higher than that in the normal tissue (p < 0.05, t-test), as shown in Fig. 5a. This suggests that the method has good specificity for colorectal cancer.
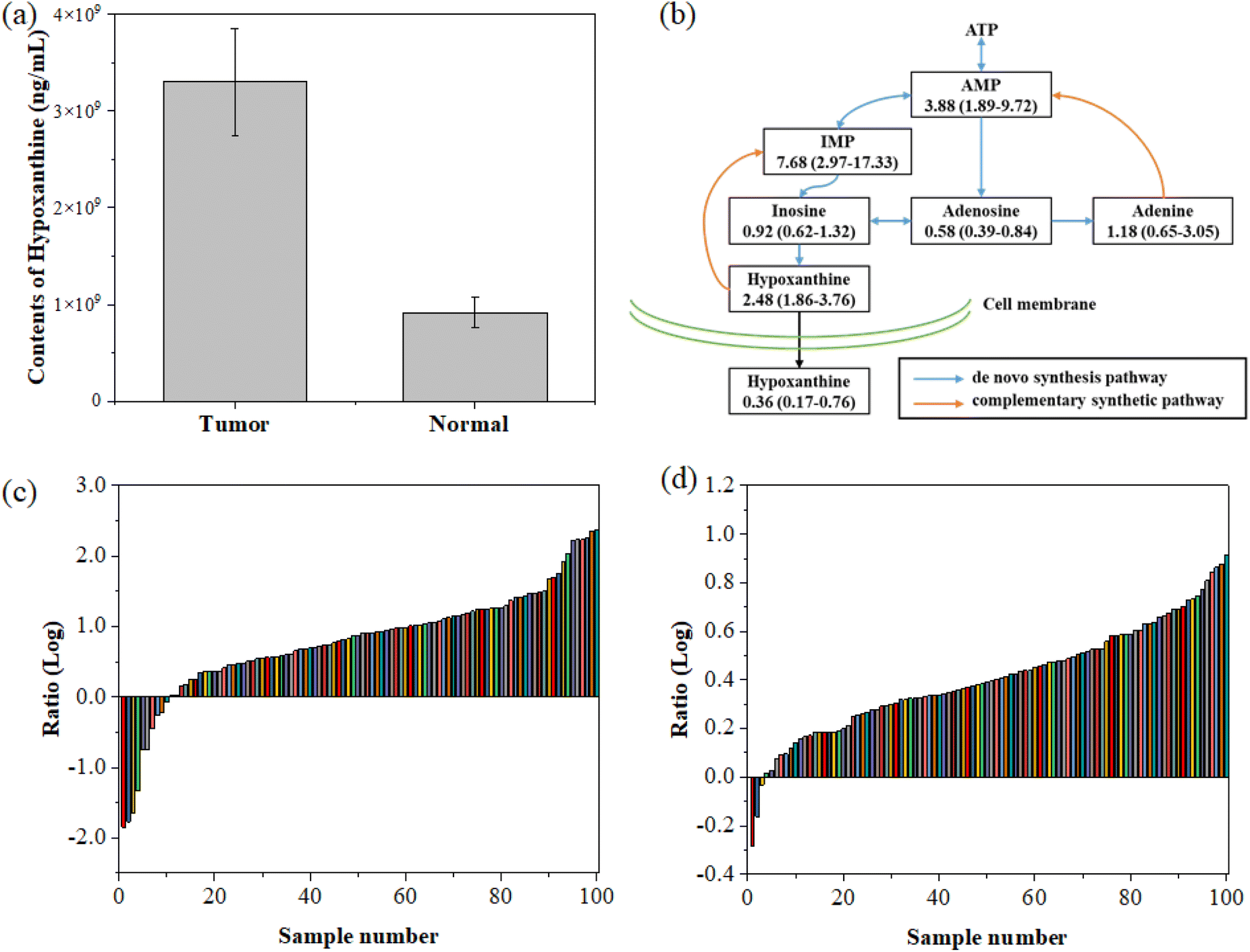 | ||
| Fig. 5 Hypoxanthine concentration in tissues. (a) Comparison of the hypoxanthine concentration between tumor tissue and normal tissue. (b) The key metabolites in the purine metabolism pathway (ATP: adenosine triphosphate, AMP: adenine ribonucleotide, and IMP: hypoxanthine nucleotide). (c) Paired detection of the hypoxanthine concentration ratio in colorectal cancer tissue and adjacent normal tissue. (d) Paired detection of the hypoxanthine nucleotide (IMP) concentration ratio in colorectal cancer tissues and adjacent normal tissue. The numbers in the figure are the median ratio of tumor tissue and normal tissue and the corresponding interquartile range. | ||
We further detected the concentration of hypoxanthine in 100 colorectal cancer tissues and adjacent normal intestinal tissues collected from West China Biobanks, West China Hospital, Sichuan University. The results showed that in 97 out of 100 paired samples, the concentration of hypoxanthine in tumor tissue was significantly higher than that in the adjacent normal tissues (Fig. 5c), highlighting the accuracy of this diagnostic approach. Furthermore, the concentration of IMP in tumor tissue is also higher than that of its paired normal tissues (Fig. 5d). By comparison, we found that in the tissue purine metabolism pathway, the concentrations of metabolites that are closely related to the complementary synthetic pathway, such as hypoxanthine, IMP, and AMP, are significantly higher than those of normal tissues, while the proportions of metabolites related to de novo metabolic pathways such as inosine, adenosine, and adenine are equivalent or down-regulated in tumor tissues compared to normal tissues.
To establish the ground-truth for differentiating between normal and tumor tissues, hypoxanthine concentrations were measured in paired colorectal cancer tissue samples and adjacent normal tissues. As illustrated in Fig. 5a, the tumor tissues consistently exhibited significantly higher levels of hypoxanthine compared to normal tissues. To further differentiate between the two tissue types, we analyzed the log-transformed ratios of hypoxanthine concentrations in tumor versus normal tissues across all samples, as shown in Fig. 5c. The data revealed a distinct inflection point where the log ratio transitions from predominantly negative values in normal tissues to positive values in tumor tissues. Based on this observation, we established a cut-off threshold at a log ratio of approximately 0.5. This threshold effectively separates the majority of tumor samples from normal samples, ensuring accurate classification. The validity of this cut-off was further corroborated by analyzing the paired concentration ratios of hypoxanthine nucleotides (IMP) in tumor and normal tissues, as presented in Fig. 5d. The consistent differentiation provided by this cut-off underscores its reliability for distinguishing between normal and tumor tissues in the context of our study.
Our study underscores the potential of hypoxanthine as a biomarker for the early diagnosis of colorectal cancer (CRC). The concentration of hypoxanthine was significantly elevated in the serum of patients diagnosed with CRC, as well as in tumor tissues when compared to adjacent normal tissues (Fig. 5). This elevation is particularly notable in early-stage CRC, where metabolic changes are less pronounced and more difficult to detect using conventional diagnostic methods. By utilizing the Fano resonance-enhanced SERS platform, we achieved highly sensitive detection of hypoxanthine, even in the early stages of the disease. The increased hypoxanthine levels in both serum and tissue samples from early-stage CRC patients support the reliability of our method for early diagnosis. This is further validated by the significant difference in the hypoxanthine concentration between tumor and normal tissues, as shown in 97 out of 100 paired samples analyzed. The early detection capability of our method highlights its potential utility in clinical settings, where early diagnosis is critical for improving patient outcomes.
4. Conclusions
Based on the above analysis, we conclude that there is possible a disorder of the purine metabolism pathway in tumor tissues, and the complementary synthetic pathway replaces the de novo metabolic synthesis pathway. It has become the main pathway of purine metabolism in tumor tissue, and this change is closely related to the proliferation of tumor cells and nucleic acid synthesis. Under normal circumstances, purine metabolism is regulated by the feedback of related nucleotides in order to maintain the balance of the purine concentration. However, various mutations in tumor cells can disrupt this balance, resulting in an abnormal increase in the concentration of nucleotides and other metabolites, which leads to the alteration of hypoxanthine content in peripheral blood.42 This explains the increased hypoxanthine concentration in peripheral blood from colorectal cancer. We demonstrated a self-assembled magnetic Fano SERS substrate based on the heterodimer of a nanosphere–EPG–gold film. The Au-NS substrate generates sub-radiate magnetic dipole mode which is coupled with the electric dipole super-radiation mode, resulting in higher optical coupling efficiency and lower radiation loss. Furthermore, the self-assembled Au-NS substrate can tune its Fano resonance wavelength by changing the dielectric layer thickness, nanosphere diameter, and Au thickness to match with the incident laser, thus obtaining strong SERS enhancement for sensitive detection of trace amounts of biomolecules in samples. The Au-NS substrates were adopted for screening of the biomarker for colorectal cancer in blood serum. It is found that the increased 726 cm−1 Raman peak intensity in blood serum of tumor patients is due to the increase in hypoxanthine content. With the disorder of the purine metabolism pathway in patients with colorectal cancer, the content of hypoxanthine is increased by tumor tissue metabolism to the peripheral blood circulation, which increases the intensity of its 726 cm−1 characteristic peak in blood serum. The results suggest that hypoxanthine in peripheral blood can be used as a reliable biomarker for rapid SERS screening of colorectal cancer in early stages.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Tianxun Gong, Zhenjiang Wei, Wen Huang, Ming-Yi Lee, En-Chi Chang and Kien Voon Kong were responsible for the SERS testing section. Libin Huang, Yan Hong, Yuan Li, Ke-ling Chen, Xiaojing Zhong, Jinzhao He and Zongguang Zhou were responsible for sample collection and clinical analysis. Tianxun Gong, Libin Huang and Xiaosheng Zhang performed substrate fabrication and simulation experiments. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank West China Biobanks, Department of Clinical Research Management, West China Hospital, Sichuan University, for their assistance in providing tissue samples. We also thank Lun-Zhi Dai and Yan-Qiu Gong and acknowledge the support from the metabolomics facility at the Technology Center for Protein Sciences of Tsinghua University. The authors acknowledge the financial support from the National Natural Science Foundation of China (61905035 and 62074029), the Natural Science Foundation of Sichuan Province (No. 2022NSFSC0652), the 1·3·5 Project for Disciplines of Excellence, West China Hospital, Sichuan University (ZY2016105), and the Open Fund of Radiation Oncology Key Laboratory of Sichuan Province (2020FSZLX-03). K. V. Kong acknowledges the funding support from the Ministry of Science and Technology (MOST), Taiwan (Grant No. 110-2628-M-002-003-MY3) and National Taiwan University (Grant No. 111L7805, 111PNTUS04, 112-2124-M-002-014 and 111L7760).Notes and references
- E. Prodan, C. Radloff, N. J. Halas and P. Nordlander, Science, 2003, 302, 419–422 CrossRef CAS PubMed.
- B. Luk'yanchuk, N. I. Zheludev, S. A. Maier, N. J. Halas, P. Nordlander, H. Giessen and C. T. Chong, Nat. Mater., 2010, 9, 707–715 CrossRef PubMed.
- Y. Francescato, V. Giannini and S. A. Maier, ACS Nano, 2012, 6, 1830–1838 CrossRef CAS PubMed.
- B. Gallinet and O. J. F. Martin, ACS Nano, 2011, 5, 8999–9008 CrossRef CAS PubMed.
- J. B. Lassiter, H. Sobhani, J. A. Fan, J. Kundu, F. Capasso, P. Nordlander and N. J. Halas, Nano Lett., 2010, 10, 3184–3189 CrossRef CAS PubMed.
- F. Hao, Y. Sonnefraud, P. Van Dorpe, S. A. Maier, N. J. Halas and P. Nordlander, Nano Lett., 2008, 8, 3983–3988 CrossRef CAS PubMed.
- L. Shao, C. Fang, H. Chen, Y. C. Man, J. Wang and H.-Q. Lin, Nano Lett., 2012, 12, 1424–1430 CrossRef CAS PubMed.
- W.-S. Chang, J. B. Lassiter, P. Swanglap, H. Sobhani, S. Khatua, P. Nordlander, N. J. Halas and S. Link, Nano Lett., 2012, 12, 4977–4982 CrossRef CAS PubMed.
- H. Chen, L. Shao, T. Ming, K. C. Woo, Y. C. Man, J. Wang and H.-Q. Lin, ACS Nano, 2011, 5, 6754–6763 CrossRef CAS PubMed.
- S. Zhang, G. C. Li, Y. Q. Chen, X. P. Zhu, S. D. Liu, D. Y. Lei and H. G. Duan, ACS Nano, 2016, 10, 11105–11114 CrossRef CAS PubMed.
- Z. Y. Fang, J. Y. Cai, Z. B. Yan, P. Nordlander, N. J. Halas and X. Zhu, Nano Lett., 2011, 11, 4475–4479 CrossRef CAS PubMed.
- Y. Hu, S. J. Noelck and R. A. Drezek, ACS Nano, 2010, 4, 1521–1528 CrossRef CAS PubMed.
- N. Verellen, Y. Sonnefraud, H. Sobhani, F. Hao, V. V. Moshchalkov, P. Van Dorpe, P. Nordlander and S. A. Maier, Nano Lett., 2009, 9, 1663–1667 CrossRef CAS PubMed.
- S. Mukherjee, H. Sobhani, J. B. Lassiter, R. Bardhan, P. Nordlander and N. J. Halas, Nano Lett., 2010, 10, 2694–2701 CrossRef CAS PubMed.
- J. A. Fan, K. Bao, C. H. Wu, J. M. Bao, R. Bardhan, N. J. Halas, V. N. Manoharan, G. Shvets, P. Nordlander and F. Capasso, Nano Lett., 2010, 10, 4680–4685 CrossRef CAS PubMed.
- J. A. Fan, C. H. Wu, K. Bao, J. M. Bao, R. Bardhan, N. J. Halas, V. N. Manoharan, P. Nordlander, G. Shvets and F. Capasso, Science, 2010, 328, 1135–1138 CrossRef CAS PubMed.
- M. Hentschel, M. Saliba, R. Vogelgesang, H. Giessen, A. P. Alivisatos and N. Liu, Nano Lett., 2010, 10, 2721–2726 CrossRef CAS PubMed.
- S. Zhang, D. A. Genov, Y. Wang, M. Liu and X. Zhang, Phys. Rev. Lett., 2008, 101, 047401 CrossRef PubMed.
- T. Shegai, S. Chen, V. D. Miljkovic, G. Zengin, P. Johansson and M. Kall, Nat. Commun., 2011, 2, 481 CrossRef PubMed.
- L. Shao, C. H. Fang, H. J. Chen, Y. C. Man, J. F. Wang and H. Q. Lin, Nano Lett., 2012, 12, 1424–1430 CrossRef CAS PubMed.
- G. Bachelier, I. Russier-Antoine, E. Benichou, C. Jonin, N. Del Fatti, F. Vallee and P. F. Brevet, Phys. Rev. Lett., 2008, 101, 197401 CrossRef CAS PubMed.
- A. Q. Fang, S. White, P. K. Jain and F. P. Zamborini, Nano Lett., 2015, 15, 542–548 CrossRef CAS PubMed.
- T. Pakizeh and M. Kall, Nano Lett., 2009, 9, 2343–2349 CrossRef CAS PubMed.
- S. Chen, Y. Zhang, T. M. Shih, W. Yang, S. Hu, X. Hu, J. Li, B. Ren, B. Mao, Z. Yang and Z. Tian, Nano Lett., 2018, 18, 2209–2216 CrossRef CAS PubMed.
- G. B. Jung, S. W. Nam, S. Choi, G.-J. Lee and H.-K. Park, Biomed. Opt. Express, 2014, 5, 3238–3251 CrossRef PubMed.
- F. Bray, J. Ferlay, I. Soerjomataram, R. L. Siegel, L. A. Torre and A. Jemal, Ca-Cancer J. Clin., 2018, 68, 394–424 CrossRef PubMed.
- A. D. Rakic, A. B. Djurisic, J. M. Elazar and M. L. Majewski, Appl. Opt., 1998, 37, 5271–5283 CrossRef CAS PubMed.
- J. J. Mock, R. T. Hill, A. Degiron, S. Zauscher, A. Chilkoti and D. R. Smith, Nano Lett., 2008, 8, 2245–2252 CrossRef CAS PubMed.
- S. Chen, Y. Zhang, T.-M. Shih, W. Yang, S. Hu, X. Hu, J. Li, B. Ren, B. Mao, Z. Yang and Z. Tian, Nano Lett., 2018, 18, 2209–2216 CrossRef CAS PubMed.
- P. C. Ray, Z. Fan, R. A. Crouch, S. S. Sinha and A. Pramanik, Chem. Soc. Rev., 2014, 43, 6370–6404 RSC.
- N. J. Halas, S. Lal, W. S. Chang, S. Link and P. Nordlander, Chem. Rev., 2011, 111, 3913–3961 CrossRef CAS PubMed.
- S. Chen, Z. L. Yang, L. Y. Meng, J. F. Li, C. T. Williams and Z. Q. Tian, J. Phys. Chem., 2015, 119, 5246–5251 CAS.
- T. Gong, K. V. Kong, D. Goh, M. Olivo and K.-T. Yong, Biomed. Opt. Express, 2015, 6, 2076–2087 CrossRef CAS PubMed.
- N. Stone, P. Stavroulaki, C. Kendall, M. Birchall and H. Barr, Laryngoscope, 2000, 110, 1756–1763 CrossRef CAS PubMed.
- J. Wang, D. Lin, J. Lin, Y. Yu, Z. Huang, Y. Chen, J. Lin, S. Feng, B. Li, N. Liu and R. Chen, J. Biomed. Opt., 2014, 19, 087003 CrossRef PubMed.
- J. Li, J. Li, H. Wang, L.-W. Qi, Y. Zhu and M. Lai, Gastroenterology, 2019, 157, 257–259 CrossRef CAS PubMed.
- A. Bonifacio, S. D. Marta, R. Spizzo, S. Cervo, A. Steffan, A. Colombatti and V. Sergo, Anal. Bioanal. Chem., 2014, 406, 2355–2365 CrossRef CAS PubMed.
- W. R. Premasiri, J. C. Lee and L. D. Ziegler, J. Phys. Chem., 2012, 116, 9376–9386 CrossRef CAS PubMed.
- Y. Long, B. Sanchez-Espiridion, M. Lin, L. White, L. Mishra, G. S. Raju, S. Kopetz, C. Eng, M. A. T. Hildebrandt, D. W. Chang, Y. Ye, D. Liang and X. Wu, Cancer, 2017, 123, 4066–4074 CrossRef CAS PubMed.
- R. Böhme, M. Richter, D. Cialla, P. Rösch, V. Deckert and J. Popp, J. Raman Spectrosc., 2009, 40, 1452–1457 CrossRef.
- Y. Shen, M. Sun, J. Zhu, M. Wei, H. Li, P. Zhao, J. Wang, R. Li, L. Tian, Y. Tao, P. Shen and J. Zhang, Mol. Omics, 2021, 17, 464–471 RSC.
- C. Y. Chan, H. Zhao, R. J. Pugh, A. M. Pedley, J. French, S. A. Jones, X. Zhuang, H. Jinnah, T. J. Huang and S. J. Benkovic, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 1368–1373 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The Raman spectra of Au-NS substrates, the Raman spectra of blood serum from cancer patients, and the liquid chromatography conditions. See DOI: https://doi.org/10.1039/d4na00543k |
| This journal is © The Royal Society of Chemistry 2024 |