
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of solvothermal synthesis parameters on the crystallite size and atomic structure of cobalt iron oxide nanoparticles †
Olivia
Aalling-Frederiksen
a,
Rebecca K.
Pittkowski
 a,
Andy S.
Anker
a,
Jonathan
Quinson
ab,
Lars
Klemeyer
c,
Benjamin A.
Frandsen
d,
Dorota
Koziej
c and
Kirsten M. Ø.
Jensen
*a
a,
Andy S.
Anker
a,
Jonathan
Quinson
ab,
Lars
Klemeyer
c,
Benjamin A.
Frandsen
d,
Dorota
Koziej
c and
Kirsten M. Ø.
Jensen
*a
aUniversity of Copenhagen, Department of Chemistry, Nanoscience Center, 2100 Copenhagen Ø, Denmark. E-mail: kirsten@chem.ku.dk
bAarhus University, Department of Biological and Chemical Engineering, 8200 Aarhus, Denmark
cUniversity of Hamburg, Institute for Nanostructure and Solid-State Physics, Center for Hybrid Nanostructures, Luruper Chausse 149, 22761 Hamburg, Germany
dBrighham Young University, Department of Physics and Astronomy, Provo, Utah 84602, USA
First published on 16th September 2024
Abstract
We here investigate how the synthesis method affects the crystallite size and atomic structure of cobalt iron oxide nanoparticles. By using a simple solvothermal method, we first synthesized cobalt ferrite nanoparticles of ca. 2 and 7 nm, characterized by Transmission Electron Microscopy (TEM), Small Angle X-ray scattering (SAXS), X-ray and neutron total scattering. The smallest particle size corresponds to only a few spinel unit cells. Nevertheless, Pair Distribution Function (PDF) analysis of X-ray and neutron total scattering data shows that the atomic structure, even in the smallest nanoparticles, is well described by the spinel structure, although with significant disorder and a contraction of the unit cell parameter. These effects can be explained by the surface oxidation of the small nanoparticles, which is confirmed by X-ray near edge absorption spectroscopy (XANES). Neutron total scattering data and PDF analysis reveal a higher degree of inversion in the spinel structure of the smallest nanoparticles. Neutron total scattering data also allow magnetic PDF (mPDF) analysis, which shows that the ferrimagnetic domains correspond to ca. 80% of the crystallite size in the larger particles. A similar but less well-defined magnetic ordering was observed for the smallest nanoparticles. Finally, we used a co-precipitation synthesis method at room temperature to synthesize ferrite nanoparticles similar in size to the smallest crystallites synthesized by the solvothermal method. Structural analysis with PDF demonstrates that the ferrite nanoparticles synthesized via this method exhibit a significantly more defective structure compared to those synthesized via a solvothermal method.
Introduction
Ferrite nanoparticles with the spinel structure have been extensively studied over the past decades. Their magnetic properties are exploited for several applications including medical imaging,1–4 while their surface structure provides a high adsorption capacity, which can be used for water treatment.5,6 Moreover, the good electronic conductivity of some spinel metal oxides enables their use as electrocatalysts for the oxygen evolution reaction (OER) in alkaline media, and numerous studies have investigated the relationship between material composition, particle size and catalytic activity.7–9 Recently, increased interest has been directed towards exploring ‘amorphous’ and nanostructured ferrites, as improved activities can be achieved in amorphous materials compared to their crystalline counterparts, e.g., in OER electrocatalysis.10–13It is well established that in general, the properties of spinel-structured ferrites are size dependent.14–16 Such size-dependent changes in material properties are often attributed to the increased surface-to-volume ratio at the nanoscale. However, the small size may also impact the atomic structure of the material, as it has been observed for several other oxide materials.17–19 Nevertheless, the challenges of characterizing the atomic structure in nanostructured materials mean that these effects are not well understood.20,21 Here, we investigate how the solvent used in a solvothermal method influences the resulting crystallite size and the atomic structure in spinel-type CoFe2O4 and reveal local structural features. The solvothermal method, which has also been widely used for ferrites, is simple, low-cost, scalable and highly tuneable, allowing for the attainment of different particle characteristics.22,23
The spinel structure, with the general formula AB2O4 is presented in Fig. 1. The oxygens are arranged in a cubic closed packed structure, while 8 metal ions occupy tetrahedral sites, and 16 metal atoms occupy the octahedral sites.25 In the normal spinel structure, the available tetrahedral sites are occupied by A2+ ions, while the octahedral sites are occupied by B3+ ions, resulting in a [A2+]Td[B3+]2OhO4 structure. However, spinels can also be inverse or partially inverse. The inverse spinel has all A2+ ions in the octahedral sites, while B3+ are distributed between the tetrahedral and octahedral sites, resulting in the formula [B3+]Td[A2+B3+]2OhO4. The partially inverse spinel has an empirical formula of [A1−x2+Bx3+]Td[Ax2+B2−x3+]2OhO4, where x is the inversion degree and represents the fraction of A2+ occupying the octahedral sites.15,16,26–28 Cobalt iron oxide is known to crystallize as an inverse spinel in the bulk state. However, nanosized particles synthesized via solvothermal or microwave synthesis procedures can adopt a partially inverse spinel structure.15,26–28 Previous studies of spinel iron oxide nanoparticles have also shown a large effect of crystallite size on the atomic structure,29–31 where vacancies are observed in the tetrahedral sites in the atomic structure, which influences their surface structure and reactivity.29 It has not yet been uncovered whether similar effects are seen for other spinel ferrites including CoFe2O4. The inversion degree is well known to affect the magnetic properties of the material.15,32–34 It has also been suggested that the cation distribution affects the catalytic properties of cobalt ferrite,35–37 and structural characterization is thus essential for being able to elucidate structure/property relationships.
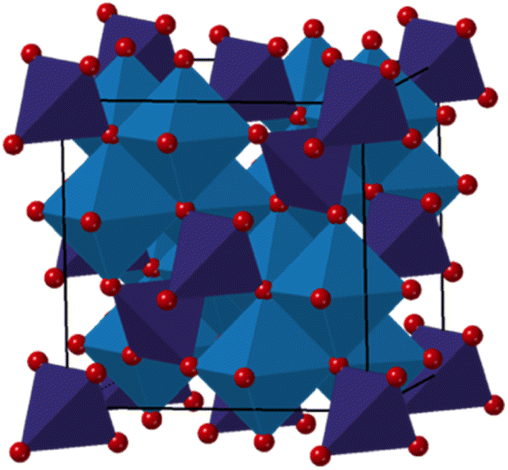 | ||
Fig. 1 Structural representation of a normal spinel in the space group Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m,24 where MOh cations are shown in blue, while MTd cations are shown in purple (M = Co/Fe). Oxygens are represented in red. m,24 where MOh cations are shown in blue, while MTd cations are shown in purple (M = Co/Fe). Oxygens are represented in red. | ||
Here, we synthesize spinel cobalt iron oxide nanocrystallites using a simple solvothermal synthesis approach and demonstrate that the spinel crystallite size can be tuned by changing the solvent from water to tert-butanol. We characterize the particles using small-angle X-ray scattering (SAXS), transmission electron microscopy (TEM), and Pair Distribution Function (PDF) analysis of X-ray and neutron total scattering data, which show that the atomic structure in even the smallest nanoparticles can be well described by the spinel structure, albeit with significant disorder. These effects can be explained by surface oxidation of the small nanoparticles, which is confirmed by X-ray near edge absorption spectroscopy (XANES). From the neutron total scattering data, we also use magnetic PDF (mPDF) to analyse the magnetic structure in the nanoparticles.38 We furthermore analyse the structure of cobalt ferrites synthesized by coprecipitation at room temperature. This synthesis leads to particles in the same size range as the small solvothermally synthesized particles; however their structure differs significantly due to the different synthesis method. Here, the PDF shows significant defects, including vacancies in the tetrahedral sites, as previously observed in pure iron oxide nanoparticles.29 The analysis using complementary structure characterization techniques thus provides insight into the structure–synthesis–size relationship of cobalt iron oxide nanocrystallites.
Methods
Solvothermal synthesis
Cobalt iron oxide nanoparticles were synthesized using a heterogenous solvothermal synthesis approach.10 The synthesis approach is illustrated in Fig. S1 in ESI.† 0.5 mmol of CoCl2·6H2O (ACS reagent, 98%) and 1.0 mmol of Fe(NO3)3·9H2O (ACS reagent, ≥98%) were added to 20 mL of solvent. Subsequently, 1.0 mmol of KOH (ACS reagent, ≥85%) was added. The solvents used herein are water and tert-butanol (ACS reagent, ≥99%). Prior to synthesis, the tert-butanol was dried over sodium sulfate (anhydrous, ACS, ISO, ≥99%). The solutions were sonicated for 30 min, and then heated for 2 hours in an autoclave at 60 °C, followed by 1 hour at 160 °C. The autoclave was cooled down, and the formed nanoparticles were washed, first in pure hexane and then in a mixture of hexane and ethanol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 volume ratio), and finally dried overnight under ambient conditions.
:3 volume ratio), and finally dried overnight under ambient conditions.
Room temperature synthesis
Besides the two samples prepared with a solvothermal approach, we investigated a sample prepared in water at room temperature (∼22 °C). In water, the addition of KOH to the clear solution of metal precursors resulted in the immediate precipitation of a black powder. Both metal salts and KOH remain insoluble in tert-butanol at room temperature.Characterization
The scattering data were processed using PDFgetX3 software to obtain the PDFs.39 Reciprocal-space Rietveld refinement of the total scattering data was performed using FullProf software.40 Real-space Rietveld refinements of the PDFs were performed using PDFgui.41 The cubic Fe3O4 structure in space group Fdm was used in the refinements for simplification, since Fe and Co scatter X-rays almost equally.42 γ-Fe2O3 in space-group P43212 was furthermore investigated.
Rietveld refinement was performed using FullProf software,43 including both an atomic structural model of CoFe2O4 in the Fdm space group and magnetic structure in the F![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group with antiparallel magnetic moment components on the tetrahedral and octahedral sites. The scattering patterns collected at each of the four detector banks were refined simultaneously.
space group with antiparallel magnetic moment components on the tetrahedral and octahedral sites. The scattering patterns collected at each of the four detector banks were refined simultaneously.
PDFgetN3 was used for data reduction to obtain the PDFs.44 The experimentally obtained PDFs contain information on both the atomic and magnetic configuration, as the neutrons interact with both nuclei and magnetic moments of the unpaired electrons. Therefore, the atomic PDFs were first modelled using PDFgui software,45 and the residual was treated as the mPDF, which was modelled with the diffpy.mpdf package in DiffPy-CMI.46
475). Both the Fe K-edge (7112 eV) and Co K-edge (7708 eV) were investigated using a Ge620 analyzing crystal with a Rowland diameter of 500 mm, and a Ge444 analyzing crystal with a Rowland diameter of 505 mm. We acquired Fe K-edge data in the scan range of 7.08 keV to 7.25 keV and Co K-edge data in the scan range of 7.65 keV to 7.95 keV. For both edges, a step size of 0.5 eV and an acquisition time of 2 s per point were used. While the Fe K-edge data were averaged over 15 individual scans, the Co K-edge data were averaged over 20 individual scans. The Demeter software package was used for XAS analysis.49
Results and discussion
Phase identification and data collection strategies
The solvothermal synthesis approach is adapted from a previously reported synthesis method, which showed that nanoparticle size can be tuned by selecting different solvents.10 Here, we applied water and tert-butanol as solvents for the synthesis. Fig. 2A and B show the diffraction patterns obtained for the two samples, using different data acquisition conditions. When compared with a spinel reference pattern (Fdm),24 the XRD patterns obtained with Ag Kα radiation, shown in black in Fig. 2A and B, show that spinel ferrite nanoparticles are formed in both cases. A broader peak profile is observed for the particles synthesized in tert-butanol, indicating smaller crystallite domain sizes.
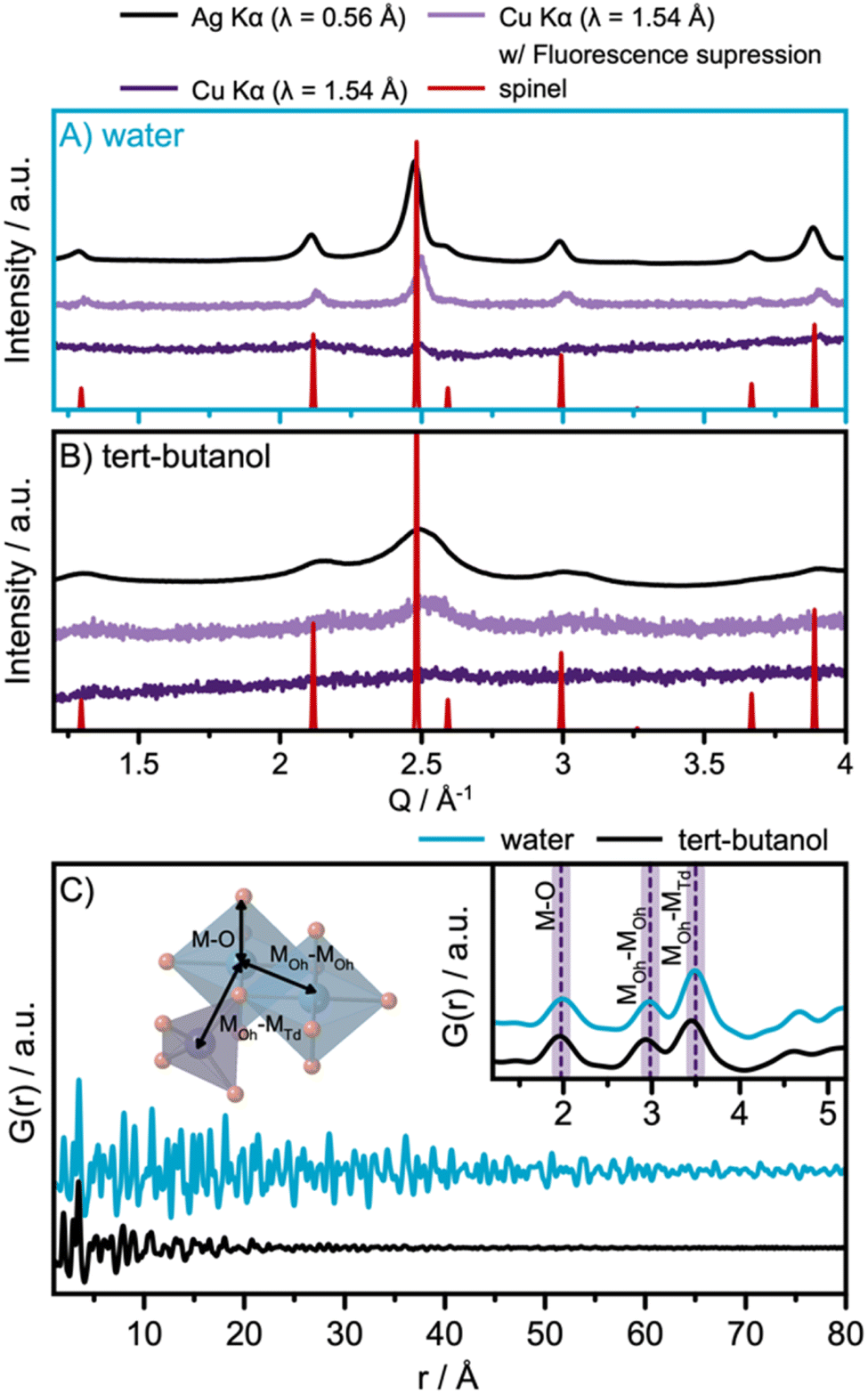 | ||
| Fig. 2 XRD patterns of the (A) water and (B) tert-butanol-synthesized nanoparticles obtained using three different data collection strategies: Cu Kα radiation (λ = 1.54 Å) as is used in most laboratory diffractometers, with Cu Kα radiation applying energy discrimination on the detector to suppress fluorescence, and with Ag Kα radiation (λ = 0.56 Å). (C) PDFs for the water- and tert-butanol-synthesized nanoparticles. The inset shows the local range of the PDF. The first three interatomic correlations, M–O (2.0 Å), MOh–MOh (3.0 Å), and MOh–MTd (3.5 Å), are highlighted. | ||
Previous studies of this synthesis approach have indicated that the product from the tert-butanol synthesis may be amorphous, which would manifest in the diffraction profile as very broad features.10 To further investigate this hypothesis, and to highlight the impact of different acquisition conditions on the quality of the collected diffraction patterns, both samples were investigated with X-rays under three different conditions: with Ag Kα radiation (λ = 0.56 Å), with Cu Kα radiation (λ = 1.54 Å) as is used in most laboratory diffractometers, and with Cu Kα radiation applying energy discrimination on the detector to suppress fluorescence. For diffraction using Cu Kα radiation, the standard voltage range of the detector discriminator is 0.110–0.250 V. To suppress fluorescence, the voltage range used is instead set to 0.190–0.270 V based on recommendations by the manufacturer.
The data obtained with standard Cu Kα settings, Fig. 2A and B, are dominated by a large background signal arising from fluorescence. Cu Kα radiation causes significant fluorescence from Co and Fe and is thus not well suited for the analysis of cobalt ferrites. Our data show almost flat diffraction profiles, which may lead to the wrong interpretation that completely disordered or amorphous nanoparticles have formed. A clear improvement in the data quality is observed when using energy discrimination on the detector. The best data are obtained using Ag Kα radiation, whose energy (λ = 0.56 Å) does not result in significant fluorescence from Co and Fe. The results emphasize the importance of selecting appropriate instruments for data collection from the samples in question. Special attention should be given when investigating disordered or nanocrystalline materials, as the Bragg peak profile may become smeared and fluorescence can dominate the patterns.
The use of Ag Kα radiation also allows data collection over a wide Q-range. Here, we acquired data up to a Qmax of 17 Å−1. PDFs from the samples are shown in Fig. 2C, which demonstrate that the water-synthesized nanoparticles exhibit extended structural order compared to the tert-butanol-synthesized nanoparticles. However, as visible in the inset of Fig. 2C, the two PDFs are locally quite similar, differing only in a slight shift of the PDF peaks towards lower r-values for the PDF of the tert-butanol-synthesized sample. In the inset of Fig. 2C, the first inter-atomic correlations are highlighted. The closest metal-to-oxygen distance (M–O, with M = Fe and Co) appears in the PDF at 2.0 Å. The edge-sharing octahedra give rise to a metal-to-metal (MOh–MOh) PDF-peak at 3.0 Å, while the corner-sharing octahedra–tetrahedra metal-to-metal distance (MOh–MTd) is evident in a peak at 3.5 Å.
PDF and XANES analysis of the atomic structure and oxidation state
Having identified the structural models for the two samples, we now move on to quantitative analysis. Real-space Rietveld refinements of the PDFs are shown in Fig. 3A and B. The spinel model24 can describe the PDFs of both the water- and the tert-butanol-synthesized nanoparticles, providing agreement factors (Rw-values) of 0.17 and 0.27, respectively. Refined parameters are listed in Table S1 in ESI.† A similar PDF analysis of synchrotron total scattering data was also conducted (Fig. S4 in ESI† and refined parameters can be found in Table S2†), providing similar results.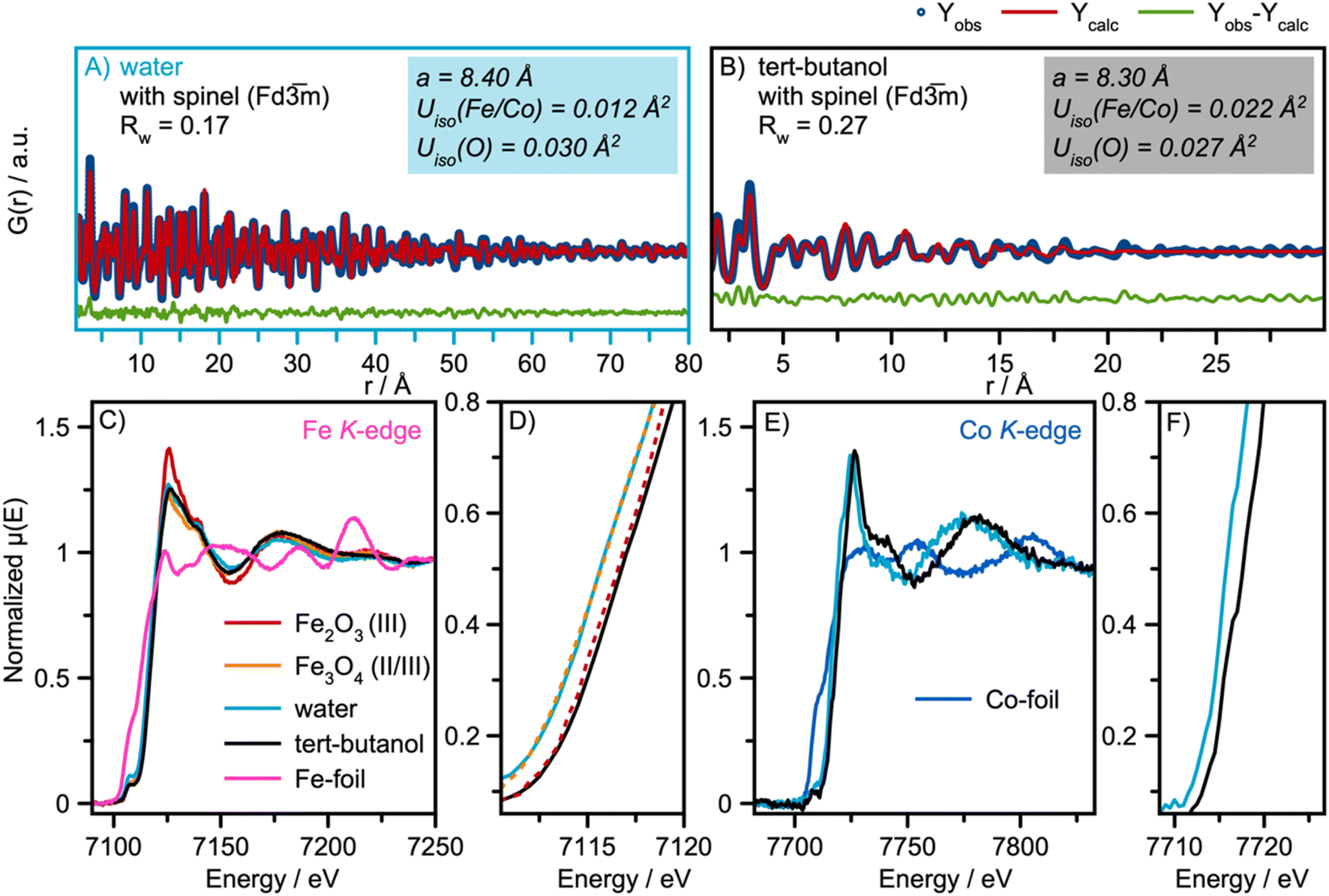 | ||
| Fig. 3 Real-space Rietveld refinement of the (A) water and (B) tert-butanol-synthesized nanoparticles. In both cases, the spinel structure in space-group Fdm was used as the structural starting model.24 (C) Fe K-edge XAS spectra of the water and tert-butanol synthesized nanoparticles with references γ-Fe2O3 (maghemite)50 and Fe3O4 (magnetite)24 reference spectra. (D) A Zoomed-in image of the absorption edge of the Fe K-edge XAS spectra shown in (C). (E) Co K-edge XAS spectra of the water and tert-butanol synthesized nanoparticles. (F) A Zoomed-in image of the absorption edge of the Co K-edge XAS spectra shown in (E). | ||
The data thus show that even the smallest particles retain the spinel structure. However, the refinement parameters presented in Fig. 3A and B show a contraction of the unit cell parameters, a, when exchanging water with tert-butanol. It is also observed that the atomic displacement parameters, (ADPs) Uiso, for the tetrahedral and octahedral metal sites are more than twice as large for the tert-butanol-synthesized nanoparticles compared with the water-synthesized nanoparticles. Similar results were obtained for Rietveld refinements of the XRD patterns in Q-space (Table S3 and Fig. S7 in ESI†). Besides describing the thermal vibrations of the atoms in the samples, the ADP-values also account for displacements resulting from static disorder.51,52 This points towards the formation of ultra-small spinel cobalt iron oxide nanoparticles with some structural disorder compared to the larger, more ordered nanoparticles formed in water. We note that the structural parameters of the particles formed in water are consistent with earlier studies on similar cobalt iron oxide materials.15,53
Previous in situ formation studies of CoFe2O4 and the closely related maghemite γ-Fe3O4 nanoparticles have shown an expansion of the unit cell for small nanoparticles compared to the bulk value.53–55 This effect has been explained by surface-restructuring in small nanoparticles, which results in an expansion of the crystallographic unit cell size. However, we observe the opposite trend here, as the refined unit cell parameter for the 2 nm particles is smaller than that of the 9 nm particles (8.30 Å vs. 8.40 Å). This effect could be related to the amount of cobalt in the sample. The smaller ionic radius of Co compared with Fe means that the unit cell parameter, a, of Co3O4 spinel is smaller than that for CoFe2O4.56 However, from SEM-EDS measurements, presented in Fig. S8–S9 and Tables S4 and S5,† we obtain a Co:Fe atomic ratio of 1:2 for both samples, ruling out the first possible explanation. Another explanation might be that the larger relative surface area of the smallest crystallites leads to more oxidized nanoparticles, which effectively decreases the ionic radii and therefore, results in a smaller refined unit cell for the smallest particles.
To further confirm this, we use X-ray absorption spectroscopy, XAS, to investigate the electronic state of the samples. Fig. 3C and D show the Fe K-edge XAS spectra from the samples, along with two reference spectra from γ-Fe2O3 (maghemite)50 and Fe3O4 (magnetite).24 Both iron oxide references have the spinel structure; however maghemite has vacancies in the octahedral sites to achieve charge balance.
The Fe K-edge XANES spectra of the nanoparticles synthesized in water align with the magnetite reference spectra. The XANES spectra of nanoparticles synthesized in tert-butanol exhibit a 1 eV shift of the white line to higher energies, indicating an increase in the Fe oxidation state of Fe3+, consistent with the reference spectra of maghemite. The derivatives of the normalized μ(E) are presented in Fig. S10 in the ESI.† The two samples exhibit a similar Co K-edge shape, as shown in their XAS spectra in Fig. 3E; however, an absorption edge shift for the tert-butanol-synthesized nanoparticles to higher energies also suggests a more oxidized state of Co, Fig. 3F. The observations from our XAS analysis thus indicate that more oxidized nanoparticles form in tert-butanol, which means that vacancies are most likely present in the small spinel nanoparticles to accommodate charge compensation. We investigate this explanation further below.
Crystallite size analysis
The PDF modelling results indicate a crystallite domain size of 2 nm for the particles synthesized in tert-butanol, while the crystalline domain size of the water-synthesized nanoparticles is determined to be ca. 9 nm. However, further inspection of the refinement of the water-synthesized particles (Fig. S3 in ESI†) shows that the single spinel nanoparticle model does not describe the PDF peaks in the high r-range (60–80 Å) well. Therefore, we introduced a second phase with the same spinel structure in the model, as shown in the fit presented in Fig. 3A. Only the scale factors are refined individually for the two phases, and the second phase was included as a bulk structure. This phase describes the presence of nanocrystallites of sizes larger than what can reliably be quantified from the total scattering measurements, which are optimized for r-resolution rather than Q-resolution. The crystallite size was refined to ca. 7 nm. The refined values are listed in Table S1 in the ESI.† A similar analysis approach was applied to the data collected for the nanoparticles synthesized in tert-butanol, as it is apparent that some structural peaks are not fully described in the r-range 15–25 Å using the one-phase model (Fig. 3B). Two two-phase models were tested: one where a bulk spinel was introduced, where only the scale factors are refined individually, and another where the lattice parameter for the bulk spinel is also refined, to account for potentially changes in lattice parameters with crystallite size. Both refinements are shown in Fig. S4† and parameters are listed in Table S1 in the ESI.† Although the fit quality is slightly improved with Rw-value decreasing from 0.27 to 0.24, the two-phase models do not account for the additional peaks at higher r-values, and thus the simple two-phase model does not account well for the size distribution present in the sample. Therefore, we choose to show the simplest one-phase model in Fig. 3B.We note here that data collection strategies should again be carefully considered regarding characterization of nanoscale materials and the information that can be extracted from PDF data. As mentioned above, X-ray total scattering data for PDF analysis were also collected using synchrotron radiation in the RA-PDF set-up,57 where a wide Q-range is obtained by using high energy X-rays (here λ = 0.1619 Å) and positioning the detector close to the sample (here 283 mm). This results in lower Q-resolution than in the Ag Kα diffractometer, and thus a lower limit for which crystallite sizes can reliably be quantified from PDF refinements. The refinement is shown in Fig. S6† and refined parameters are listed in Table S2 in the ESI.† PDF modelling of the synchrotron data shows an average crystallite size of only 4 nm for the water-synthesized nanoparticles (obtained using the one phase model). The crystallite size is thus clearly underestimated, again illustrating the need for a critical approach in data collection methods. The different Q-resolutions for the two data collection strategies are expressed in the Qdamp parameters, which describe instrumental broadening. While this parameter refines to 0.011 Å−1 for the Ag Kα diffractometer, it is 0.034 Å−1 for the synchrotron data.
TEM micrographs from the samples are shown in Fig. 4A and B. These confirm that close-to-spherical particles on the nanometer scale have been synthesized. A high level of polydispersity is observed for the water-synthesized nanoparticles, consistent with the findings of the PDF refinements. The high degree of agglomeration in the sample challenges the size determination from TEM; however, the nanoparticles synthesized in water have size in the range of 4–20 nm, comparable to the crystallite size of 7 nm obtained from PDF analysis. For the tert-butanol-synthesized sample, size estimation is difficult from the TEM images due to the agglomeration of the small nanoparticles. Additional TEM data are shown in Fig. S11 and S12 in the ESI.†
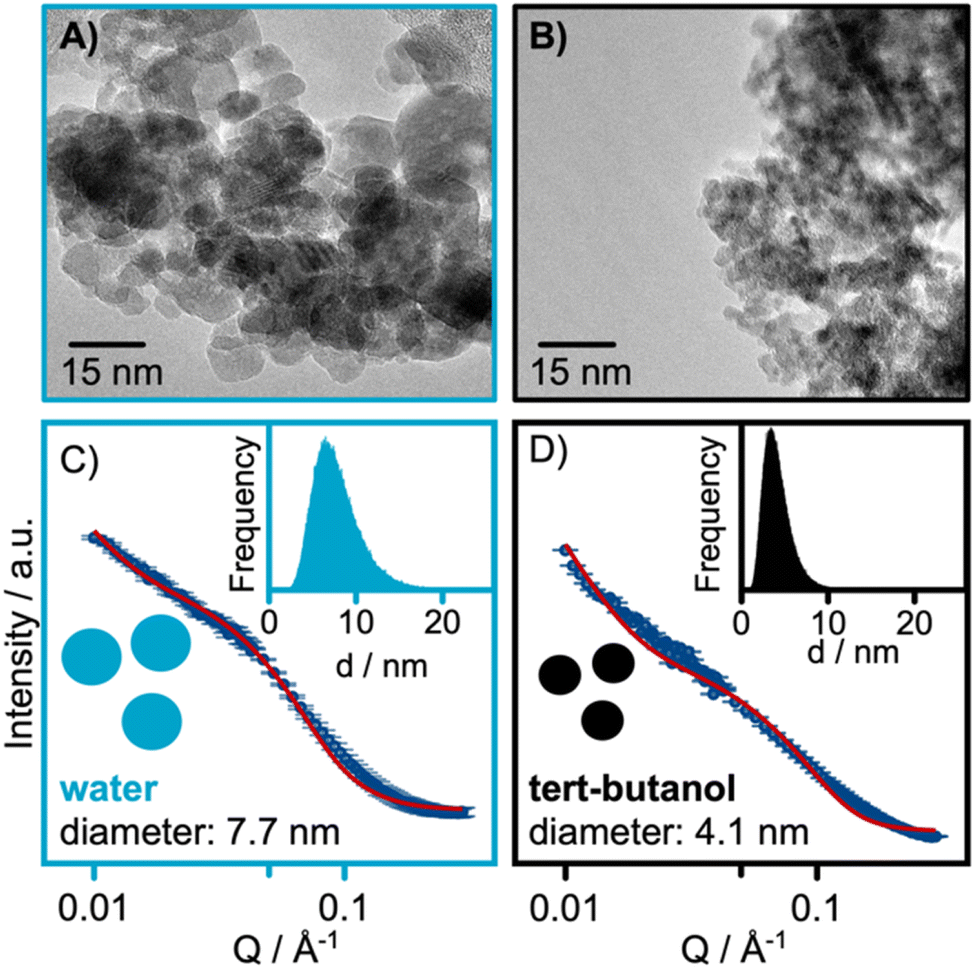 | ||
| Fig. 4 TEM micrographs of the (A) water-synthesized and (B) tert-butanol synthesized nanoparticles. SAXS refinements of the (C) water-synthesized and (D) tert-butanol synthesized nanoparticles. | ||
Complementary SAXS measurements were also conducted. The SAXS data were modelled with a polydisperse sphere model. The results from the SAXS refinements shown in Fig. 4C and D, reveal sizes of 7.7 and 4.1 nm for the water and tert-butanol synthesized nanoparticles, respectively. Our refinements reveal a wide size distribution for both samples as seen from the lognormal distribution presented in the insets of Fig. 4C and D. The refined SAXS parameters are listed in Table S6 in the ESI.† The full Q-range of the data collected is also presented in Fig. S13 in the ESI.†
Together, PDF, TEM and SAXS thus show that significantly larger particles are formed in water compared to tert-butanol. A broad size distribution is observed especially for the water synthesized samples, and particles in the size range of ca. 4 to 20 nm are obtained. The nanoparticles synthesized in tert-butanol falls in the size range of around 2 nm.
Cation site occupancies and magnetic structure – neutron scattering analysis
For a deeper understanding of the site occupancies in the spinel structure, neutron total scattering data for nPDF analysis were collected. The different neutron scattering lengths of iron (9.45 fm) and cobalt (2.49 fm) allow differentiation between these two elements, enabling us to investigate the distribution of cobalt and iron in the spinel structure of the nanocrystalline particles.58We use [Co1−xFex]tet[CoxFe2−x]octO4 in space group Fdm as the structural starting model for the refinements, constraining the Co:Fe atomic ratio to 1:2 as determined by SEM-EDS analysis (Fig. S7, S8, Tables S4 and S5 in ESI†). The nPDF fits are shown in Fig. 5, and refined parameters are given in Table S7 in ESI.† The full fitting range (1.5–60 Å) of the water synthesized sample is presented in Fig. S15 in the ESI.† From the nPDF refinements, we obtain inversion degrees of 0.57 and 0.46 for the spinel structures of the water- and tert-butanol-synthesized particles, respectively. This gives the formulas [Co0.43Fe0.57]Td[Co0.57Fe1.43]OhO4 for the water-synthesized nanoparticles, and [Co0.54Fe0.46]Td[Co0.46Fe1.54]OhO4 for the tert-butanol synthesized particles. Bulk CoFe2O4 has an inversion degree of 1, and our data thus confirm that nanoparticles have a more mixed character than bulk materials, as it has been demonstrated in related studies.28,53 Possibly, a decreasing size results in more mixing; however, analysis of data from a wider range of different sizes and data points would be needed to confirm this trend.
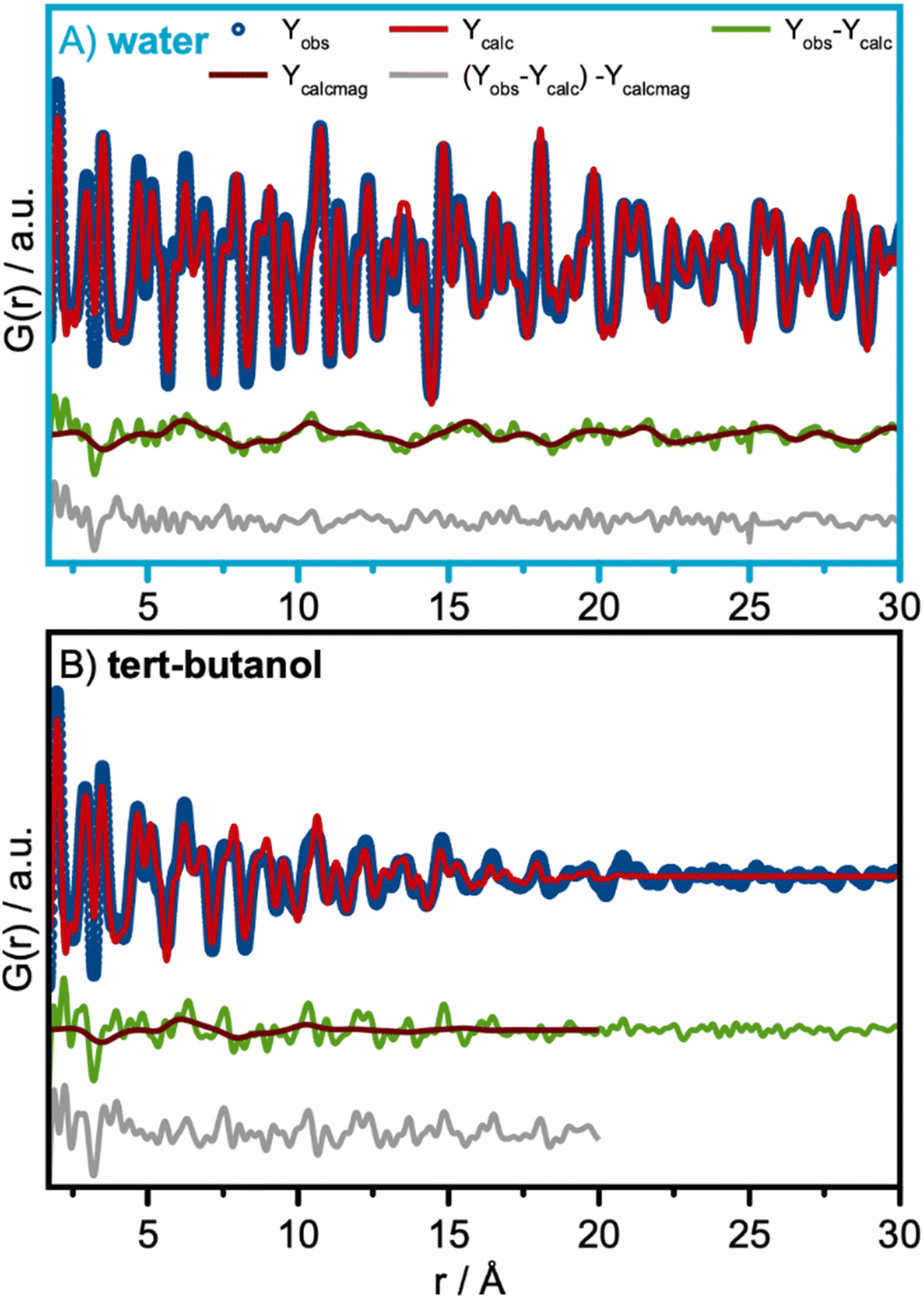 | ||
| Fig. 5 Neutron PDF (nPDF) and refinements using the CoFe2O4 spinel structure in space-group Fdm. (A) Refinement of the nPDF of the water-synthesized nanoparticles. Here the magnetic PDF is furthermore refined as the difference curve using a collinear model. (B) Refinement of the nPDF of the tert-butanol-synthesized nanoparticles. | ||
The nPDF refinements presented in Fig. 5A and B show a wave-like feature in the fit-residual shown in green. This underlying systematic feature in the fit-residual appears too regular to be mere noise and can be attributed to a contribution from the magnetic structure.38,59 Magnetic PDF (mPDF) analysis were therefore performed on the residuals of the nuclear PDF analysis and the fits are shown in brown. The refined parameters are listed in Table S8 in the ESI.†
The magnetic structure is described with a collinear model, where the magnetic moments on the tetrahedral and octahedral sites are oriented antiparallel. This has been described before for CoFe2O4 by Andersen et al.15,60 The magnetic model was constructed from the refined nuclear structure by adding magnetic species with antiparallel alignment on the tetrahedral and octahedral sites. The spin direction was 100 in the coordinate system of the cubic structure. The average magnetic moment size and the ratio of the moment size on the octahedral site to the moment on the tetrahedral site were refined, together with a parameter representing a spherical magnetic domain size. The average ordered moment for the water-synthesized sample was 3.83(13) μB, consistent with the expectations for mixed Co and Fe moments. The magnetic correlation length for the water-synthesized sample was determined to be 6.5 nm from refinements, which is 80% of the crystallite size refined from the neutron scattering data. The data thus reveal that the magnetic coherence is smaller than the crystallite size itself. This finding is consistent with a study of the magnetic structure of non-stoichiometric spinel iron oxide nanocrystallites, where the magnetic domain size was found to be 60–70% of the nanocrystallites.61Q-space Rietveld refinement is shown in Fig. S16† and refined parameters are listed in Table S9 in the ESI† for the water-synthesized nanoparticles. Here we obtain similar results, which are comparable to the refined values from the PDF-analysis. Hence, the lattice parameter was refined to 8.38 Å and an inversion degree of 0.63 was obtained.
We use the same approach to describe the contribution from a magnetic structure in the fit-residual of the nuclear nPDF refinement for tert-butanol synthesized nanoparticle, Fig. 5B. Here, the magnetic correlations length was determined to be 2.0 nm, which is close to the average crystallite domain size.
Synthesis of ultra-small defect-rich nanoparticles
Despite the small size achieved, the tert-butanol synthesis resulted in spinel-structured nanoparticles. While some structural disorder was observed, this is likely induced primarily by surface oxidation. Another approach for the synthesis of ultra-small cobalt iron oxide nanoparticles is to simply collect the precipitate that forms at room temperature (RT) when KOH is added to the metal precursor solution in water, described in detail in Fig. S1 in ESI.† Similar co-precipitation methods have previously been used to prepare iron-based nanoparticles for magnetic properties studies.62,63 To further explore size/structure relations for ferrite-based materials, we here perform structure analysis of the precipitate.The Q-space data and PDF for the nanoparticles synthesized at RT are plotted with the data from the particles of the solvothermal synthesis in Fig. S17 in the ESI† for comparison. At first glance, the Q-space data show features at positions expected for the spinel structure. However, the PDFs show significant differences. When performing a PDF refinement using the Fe3O4 spinel structural model in space-group Fdm (Fig. 6A), the spinel Fdm structure cannot fully describe the MOh–MOh to MOh–MTd ratio highlighted in purple in Fig. 6A. A similar effect has previously been observed in PDF analysis of small nanoparticles of iron oxide,29,55 where it was shown that vacancies in the tetrahedral sites appear with decreasing nanoparticle size. In these studies, the particles were found to adopt the maghemite structure, γ-Fe2O3, in space-group P43212.24,50 As described above, charge-balance in maghemite is achieved through ordered vacancies on the MOh-site. The MOh-sites (Fe4) which have occupancies of 0.33 are highlighted in orange in the structure presented in Fig. 6B. Here, we perform a similar analysis of the cobalt iron oxide nanoparticles at room temperature.
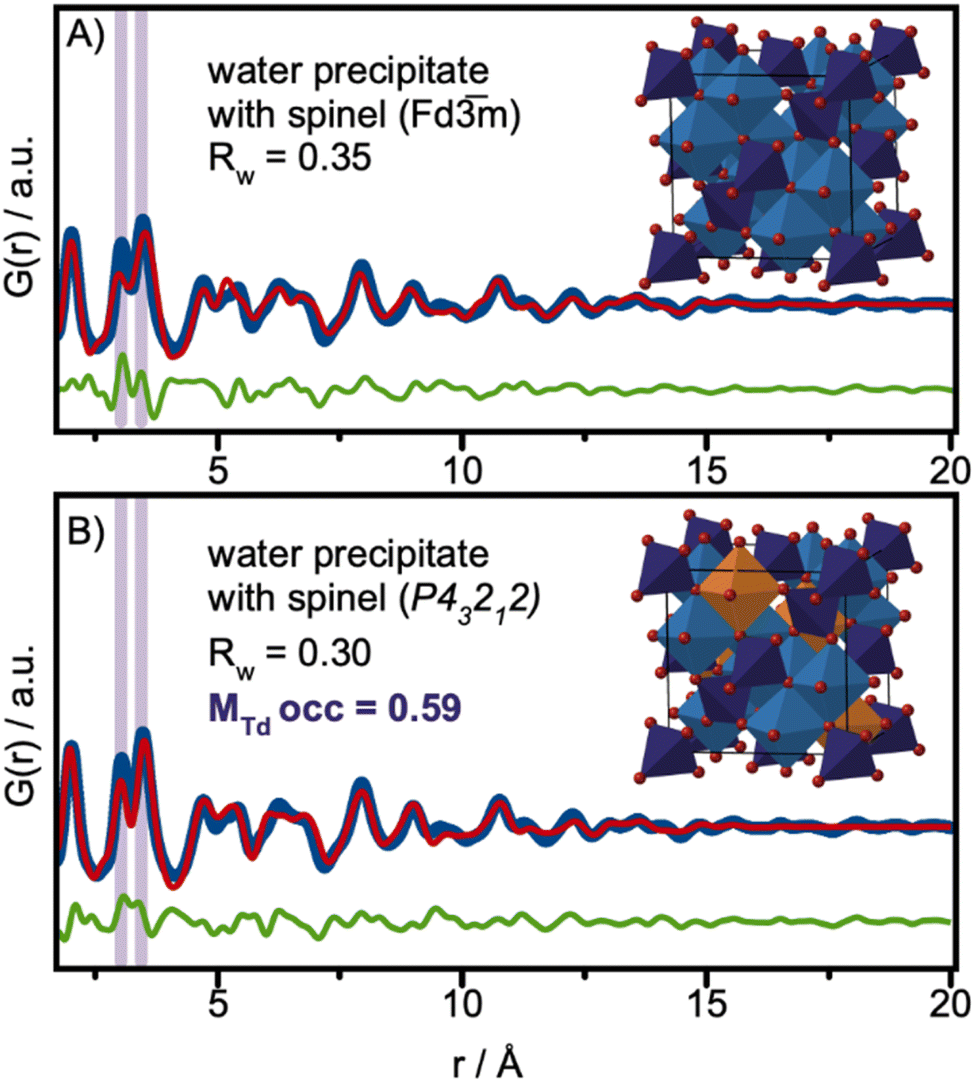 | ||
| Fig. 6 PDF analysis of the precipitate in water collected at RT. (A) PDF refinement using the magnetite model in space-group Fdm, (B) PDF refinement using the maghemite model in space-group P43212. | ||
Using maghemite γ-Fe2O3 as the structural model and refining the MTd site occupancies, we improve the description of the experimental PDF, as shown in Fig. 6B. The MTd site occupancy is refined to 0.59, and the spherical crystallite size is refined to 1.8 nm. All refined parameters are listed in Table S10 in the ESI.† The fit quality is further improved if we allow refinement of all metal positions. This fit is presented in Fig. S18 in the ESI.†
We note that when using the γ-Fe2O3 structural model to describe the PDF of the tert-butanol-synthesized nanoparticles discussed above, no significant improvement in the fit quality is achieved compared to the magnetite structure used in Fig. 3B. The fit is shown in Fig. S19 in the ESI.† The MTd site occupancy is refined to 0.81.
Our analysis thus shows that the ultra-small cobalt ferrite oxide nanoparticles that form at RT in water are significantly more defect-rich compared to the spinel-structured tert-butanol synthesized particles of similar size. When synthesized in water at RT, the particles show similarities with spinel nanoparticles of iron oxide, where a certain size is needed before the ideal spinel structure can describe the structure.29 The results thus illustrate a clear effect of the synthesis method and particle size on the atomic structure of the materials.
Conclusions
Here, a simple solvothermal synthesis that allows tuning the size of CoFe2O4 nanoparticles has been described. When the solvent is changed from water to tert-butanol, nanoparticles with an average size of ca. 2 nm are observed. Even though their small size corresponds to only 8–12 spinel unit cells, their atomic structure can be well-described by the spinel structure. Enlarged ADPs on the metal sites and lattice contraction are observed from X-ray PDF analysis. We explain these structural effects by oxidation of the nanoparticles, which is confirmed by XANES. We also use neutron total scattering to provide insight into the Co and Fe occupancies in the two distinct sites in the spinel structure. Our analysis shows that the smaller nanoparticles formed in tert-butanol have a higher Fe concentration in the tetrahedral sites. Neutron scattering experiments also provide information on the magnetic ordering in the nanoparticles. We found that the magnetic ordering in the largest particles correspond to ca. 80% of the crystallite size, and some ordering on the 2 nm-scale could be observed for the smallest nanoparticles.To further address size/structure/synthesis relationships, we synthesized ultra-small nanoparticles from co-precipitation at room temperature in water. Unlike the particles synthesized by a solvothermal method, we were not able to describe these with the simple spinel structure due to a high concentration of vacancies observed on the tetrahedral sites in the structure, altering the local atomic structure. The observation of vacancies aligns well with the size-dependent structure observed for iron oxide nanoparticles.29,55
Our study thus shows that size–structure relationships are not always specific for a given material, but are also highly influenced by the synthesis method. Apart from the structural analysis, we have provided data showing how appropriate data collection strategies are required, as the use of Cu Kα radiation can lead to misleading conclusions regarding the crystal structure of Co and Fe containing nanomaterials.
Data availability
Data for this article, including XRD, PDF, TEM and SAXS are available at https://sid.erda.dk/cgi-sid/ls.py?share_id=arkPjUfdTH.Author contributions
OA and KMØJ planned the project. OA synthesized the samples, collected X-ray total scattering data, analyzed the data, and wrote the original draft along with KMØJ. LK collected the XANES data and assisted in XANES data analysis under the supervision of DK. RP assisted in the analysis of XANES data. ASA collected the SAXS data and assisted in the SAXS data analysis. JQ performed the TEM analysis. BF assisted in and supervised the mPDF analysis. KMØJ supervised the project. All authors contributed to reviewing and editing of the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Villum Foundation for financial support through a Villum Young Investigator grant (VKR00015416). Funding from the Danish Ministry of Higher Education and Science through the SMART Lighthouse is gratefully acknowledged. We acknowledge support from the Danish National Research Foundation Center for High Entropy Alloy Catalysis (DNRF 149). This work is part of a project that has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 Research and Innovation Program (grant agreement No. 804066). We are grateful for funding from the Carlsberg Foundation (CF21-0278). LK and DK acknowledge support from the European Research Council (LINCHPIN project, grant no. 818941) and the Deutsche Forschungsgemeinschaft (DFG) through the Graduate School “Nanohybrid” (funding ID 408076438). This work was carried out with the support of the Diamond Light Source, instrument I15-I (proposal CY21791-4). We thank Phil Chater for X-ray total scattering data collection. A portion of this research used resources at the Spallation Neutron Source, a DOE Office of the Science User Facility operated by the Oak Ridge National Laboratory. The beam time was allocated to NOMAD under proposal number IPTS-25763. We thank Jue Liu for neutron total scattering data collection. We are grateful to Nicolas Schlegel and Beatrice Frey for SEM-EDS data collection and acknowledge access to the Microcopy Imaging Center (MIC) facilities at the University of Bern. The magnetic pair distribution function analysis performed by BAF was supported by the U.S. Department of Energy, Office of Science, Basic Energy Sciences through award no. DE-SC0021134.References
- J. t. Jang, H. Nah, J. H. Lee, S. H. Moon, M. G. Kim and J. Cheon, Angew. Chem., Int. Ed., 2009, 48, 1234–1238 CrossRef CAS PubMed
.
- J.-H. Lee, Y.-M. Huh, Y.-w. Jun, J.-w. Seo, J.-t. Jang, H.-T. Song, S. Kim, E.-J. Cho, H.-G. Yoon and J.-S. Suh, Nat. Med., 2007, 13, 95–99 CrossRef CAS PubMed
- H. B. Na, I. C. Song and T. Hyeon, Adv. Mater., 2009, 21, 2133–2148 CrossRef CAS
- Q. A. Pankhurst, J. Connolly, S. K. Jones and J. Dobson, J. Phys. D: Appl. Phys., 2003, 36, R167–R181 CrossRef CAS
- K. K. Kefeni, B. B. Mamba and T. A. M. Msagati, Sep. Purif. Technol., 2017, 188, 399–422 CrossRef CAS
- D. H. K. Reddy and Y. S. Yun, Coord. Chem. Rev., 2016, 315, 90–111 CrossRef CAS
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC
- F. Lu, M. Zhou, Y. Zhou and X. Zeng, Small, 2017, 13, 1701931 CrossRef PubMed
- M. Chatenet, B. G. Pollet, D. R. Dekel, F. Dionigi, J. Deseure, P. Millet, R. D. Braatz, M. Z. Bazant, M. Eikerling and I. Staffell, Chem. Soc. Rev., 2022, 51, 4583–4762 RSC
- A. Indra, P. W. Menezes, N. R. Sahraie, A. Bergmann, C. Das, M. Tallarida, D. Schmeißer, P. Strasser and M. Driess, J. Am. Chem. Soc., 2014, 136, 17530–17536 CrossRef CAS PubMed
- S. Anantharaj and S. Noda, Small, 2020, 16, 1905779 CrossRef CAS PubMed
- W. Z. Cai, R. Chen, H. B. Yang, H. B. Tao, H. Y. Wang, J. J. Gao, W. Liu, S. Liu, S. F. Hung and B. Liu, Nano Lett., 2020, 20, 4278–4285 CrossRef CAS PubMed
- R. D. Smith, M. S. Prévot, R. D. Fagan, S. Trudel and C. P. Berlinguette, J. Am. Chem. Soc., 2013, 135, 11580–11586 CrossRef CAS PubMed
- P. Guardia, A. Labarta and X. Batlle, J. Phys. Chem. C, 2011, 115, 390–396 CrossRef CAS
- H. L. Andersen, M. Saura-Múzquiz, C. Granados-Miralles, E. Canévet, N. Lock and M. Christensen, Nanoscale, 2018, 10, 14902–14914 RSC
- K. Chakrapani, G. Bendt, H. Hajiyani, I. Schwarzrock, T. Lunkenbein, S. Salamon, J. Landers, H. Wende, R. Schlögl and R. Pentcheva, ChemCatChem, 2017, 9, 2988–2995 CrossRef CAS
- M. Juelsholt, A. S. Anker, T. L. Christiansen, M. R. V. Jørgensen, I. Kantor, D. R. Sørensen and K. M. Ø. Jensen, Nanoscale, 2021, 13, 20144–20156 RSC
- T. L. Christiansen, E. D. Bøjesen, M. Juelsholt, J. Etheridge and K. M. Ø. Jensen, ACS Nano, 2019, 13, 8725–8735 CrossRef CAS PubMed
- T. L. Christiansen, S. R. Cooper and K. M. Jensen, Nanoscale Adv., 2020, 2, 2234–2254 RSC
- R. D. Smith, M. S. Prévot, R. D. Fagan, Z. Zhang, P. A. Sedach, M. K. J. Siu, S. Trudel and C. P. Berlinguette, Science, 2013, 340, 60–63 CrossRef CAS PubMed
- L. Liardet and X. Hu, ACS Catal., 2018, 8, 644–650 CrossRef CAS PubMed
- S. Yáñez-Vilar, M. Sánchez-Andújar, C. Gómez-Aguirre, J. Mira, M. A. Señarís-Rodríguez and S. Castro-García, J. Solid State Chem., 2009, 182, 2685–2690 CrossRef
- M. P. Reddy and A. M. A. Mohamed, Microporous Mesoporous Mater., 2015, 215, 37–45 CrossRef CAS
- M. Fleet, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1981, 37, 917–920 CrossRef
-
N. N. Greenwood and A. Earnshaw, Chemistry of the Elements, Elsevier, 2012 Search PubMed
- E. Solano, C. Frontera, T. Puig, X. Obradors, S. Ricart and J. Ros, J. Appl. Crystallogr., 2014, 47, 414–420 CrossRef CAS
- C. Liu, A. J. Rondinone and Z. J. Zhang, Pure Appl. Chem., 2000, 72, 37–45 CrossRef CAS
- A. L. Tiano, G. C. Papaefthymiou, C. S. Lewis, J. Han, C. Zhang, Q. Li, C. Shi, A. M. Abeykoon, S. J. Billinge and E. Stach, Chem. Mater., 2015, 27, 3572–3592 CrossRef CAS
- S. R. Cooper, R. O. Candler, A. G. Cosby, D. W. Johnson, K. M. Jensen and J. E. Hutchison, ACS Nano, 2020, 14, 5480–5490 CrossRef CAS PubMed
- K. M. Ø. Jensen, H. L. Andersen, C. Tyrsted, E. D. Bøjesen, A.-C. Dippel, N. Lock, S. J. L. Billinge, B. B. Iversen and M. Christensen, ACS Nano, 2014, 8, 10704–10714 CrossRef CAS PubMed
- M. Auffan, J. Rose, O. Proux, D. Borschneck, A. Masion, P. Chaurand, J.-L. Hazemann, C. Chaneac, J.-P. Jolivet and M. R. Wiesner, Langmuir, 2008, 24, 3215–3222 CrossRef CAS PubMed
- R. Roongtao and C. Ruttanapun, Jpn. J. Appl. Phys., 2019, 58, 101001 CrossRef CAS
- M. Fantauzzi, F. Secci, M. Sanna Angotzi, C. Passiu, C. Cannas and A. Rossi, RSC Adv., 2019, 9, 19171–19179 RSC
- K. R. Sanchez-Lievanos, J. L. Stair and K. E. Knowles, Inorg. Chem., 2021, 60, 4291–4305 CrossRef CAS PubMed
- A. K. Gomaa, M. G. Zonkol, G. E. Khedr and N. K. Allam, EES Catal., 2024 10.1039/D4EY00121D
- S. Jha, P. Jain, R. Palkovits and P. Popinand Ingole, J. Mater. Chem. A, 2023, 11, 23034–23047 RSC
- V. H. Ong, T. N. Pham, V. M. Tien, N. X. Dinh, N. Thi Lan, N. Van Quy, T. N. Bach, V. D. Lam, L. M. Tung and A.-T. Le, J. Alloys Compd., 2023, 949, 169880 CrossRef CAS
- B. A. Frandsen, X. Yang and S. J. Billinge, Acta Crystallogr., Sect. A: Found. Adv., 2014, 70, 3–11 CrossRef CAS PubMed
- P. Juhás, T. Davis, C. L. Farrow and S. J. Billinge, J. Appl. Crystallogr., 2013, 46, 560–566 CrossRef
- J. Rodríguez-Carvajal, Phys. B, 1993, 192, 55–69 CrossRef
- C. Farrow, P. Juhas, J. Liu, D. Bryndin, E. Božin, J. Bloch, T. Proffen and S. Billinge, J. Phys.: Condens. Matter, 2007, 19, 335219 CrossRef CAS PubMed
- F. S. Varley, J. Neutron Res., 1992, 3, 29–37 Search PubMed
- L. Yan, X. Rui, G. Chen, W. Xu, G. Zou and H. Luo, Nanoscale, 2016, 8, 8443–8465 RSC
- P. Juhás, J. N. Louwen, L. v. Eijck, E. T. Vogt and S. J. Billinge, J. Appl. Crystallogr., 2018, 51, 1492–1497 CrossRef
- C. Farrow, P. Juhás, J. Liu, D. Bryndin, E. Božin, J. Bloch, T. Proffen and S. Billinge, J. Phys.: Condens. Matter, 2007, 19, 335219 CrossRef CAS PubMed
- B. A. Frandsen, P. K. Hamilton, J. A. Christensen, E. Stubben and S. J. Billinge, J. Appl. Crystallogr., 2022, 55, 1377–1382 CrossRef CAS
- SasView, https://www.sasview.org.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld and B. Schmid, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed
- C. Greaves, J. Solid State Chem., 1983, 49, 325–333 CrossRef CAS
- B. Gilbert, F. Huang, H. Zhang, G. A. Waychunas and J. F. Banfield, Science, 2004, 305, 651–654 CrossRef CAS PubMed
- E. Božin, K. Knox, P. Juhás, Y. Hor, J. Mitchell and S. Billinge, Sci. Rep., 2014, 4, 4081 CrossRef PubMed
- J. V. Ahlburg, C. Granados-Miralles, F. H. Gjørup, H. L. Andersen and M. Christensen, Nanoscale, 2020, 12, 9440–9451 RSC
- H. L. Andersen and M. Christensen, Nanoscale, 2015, 7, 3481–3490 RSC
- K. M. Jensen, H. L. Andersen, C. Tyrsted, E. D. Bøjesen, A.-C. Dippel, N. Lock, S. J. Billinge, B. B. Iversen and M. Christensen, ACS Nano, 2014, 8, 10704–10714 CrossRef CAS PubMed
- W. Smith and A. Hobson, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1973, 29, 362–363 CrossRef CAS
- P. J. Chupas, X. Qiu, J. C. Hanson, P. L. Lee, C. P. Grey and S. J. Billinge, J. Appl. Crystallogr., 2003, 36, 1342–1347 CrossRef CAS
-
E. Prince, International Tables for Crystallography, Volume C: Mathematical, Physical and Chemical Tables, Springer Science & Business Media, 2004 Search PubMed
- B. A. Frandsen and S. J. Billinge, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 325–334 CrossRef CAS PubMed
- H. L. Andersen, C. Granados-Miralles, M. Saura-Múzquiz, M. Stingaciu, J. Larsen, F. Søndergaard-Pedersen, J. V. Ahlburg, L. Keller, C. Frandsen and M. Christensen, Mater. Chem. Front., 2019, 3, 668–679 RSC
- H. L. Andersen, B. A. Frandsen, H. P. Gunnlaugsson, M. R. Jørgensen, S. J. Billinge, K. Jensen and M. Christensen, IUCrJ, 2021, 8, 33–45 CrossRef CAS PubMed
- A. H. Lu, E. e. L. Salabas and F. Schüth, Angew. Chem., Int. Ed., 2007, 46, 1222–1244 CrossRef CAS PubMed
- C. Pereira, A. M. Pereira, C. Fernandes, M. Rocha, R. Mendes, M. P. Fernández-García, A. Guedes, P. B. Tavares, J.-M. Grenèche and J. o. P. Araújo, Chem. Mater., 2012, 24, 1496–1504 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4na00590b |
| This journal is © The Royal Society of Chemistry 2024 |