
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTitanium dioxide functionalized silicon carbide phases as heterogeneous epoxidation catalysts†
Léa
Gonçalves
,
Olinda
Gimello
,
Karim
Bouchmella
,
Peter
Hesemann
 and
Johan G.
Alauzun
*
and
Johan G.
Alauzun
*
ICGM, Univ Montpellier-CNRS-ENSCM, 1919, Route de Mende, Montpellier Cedex 05 34293, France. E-mail: johan.alauzun@umontpellier.fr
First published on 7th October 2024
Abstract
We report silicon carbide (SiC) based epoxidation catalysts constituted of a silicon carbide core and a silica/titania (SiO2/TiO2) shell. The catalysts were obtained via surface modification of SiC microparticles and were used as heterogeneous catalysts for the epoxidation of cyclohexene using tert-butyl hydroperoxide or cumyl hydroperoxide as the oxidant. Conversions up to 83% and selectivities of more than 90% were obtained.
1. Introduction
Alkene epoxidation is a chemical process of considerable academic and industrial relevance because epoxides are valuable intermediates in the production of various important pharmaceuticals, agrochemicals, and industrial chemicals.1 Alkene epoxidation is therefore a key process in synthetic organic chemistry, in the chemical industry, and in process engineering.2Epoxidation reactions can be promoted by different types of either homogeneous or heterogeneous catalysts.3 As heterogeneous catalysis benefits from easy catalyst recovery and product separation together with limited metal leaching,4 huge attention has been paid to the development of heterogeneous epoxidation catalysts. Among the various heterogeneous catalyst systems that have been reported,4–6 Ti-based formulations represent the most important class. Since the discovery of the TS-1 zeolite by researchers of EniChem in the early 1980s,7 considerable effort has been made to develop efficient and selective titanosilicate based heterogeneous epoxidation catalyst systems.8,9 A variety of titanosilicates featuring different characteristics in terms of porosity, architecture or morphology have been reported, in particular Ti-containing zeolites such as Ti-ZSM-5,10 Ti-UTL,11 Ti-MWW12 and mesoporous materials such as Ti-MCM-41 (ref. 13 and 14) or Ti-SBA-15.15,16
Our paper describes titanium dioxide (TiO2) grafted silicon carbide (SiC) as novel heterogeneous epoxidation catalysts. Due to its physicochemical, chemical, and morphological properties, silicon carbide recently emerged as an interesting support material for heterogeneous catalysis.17,18 SiC displays high chemical inertness, a relatively high specific surface area, good thermal conductivity, and high oxidation resistance. SiC therefore combines unique physicochemical properties for the elaboration of advanced heterogeneous catalysts. As the surface of SiC is covered by a nanometric layer of oxidized silicon species such as silicon oxycarbide (SiOxCy) and/or silica (SiO2),19 it offers sufficient anchorage sites for the dispersion and stabilization of deposited active phases, in particular allowing the surface grafting of titanium alkoxides and the formation of SiC-supported TiO2 phases.20–23
Here, we report the synthesis and detailed characterization of TiO2-functionalized SiC microparticles via surface grafting of titanium tetraisopropoxide (Ti[OiPr]4) onto SiC surfaces. The resulting TiO2/SiO2@SiC phases were then used as heterogeneous catalysts for the epoxidation reaction of cyclohexene.
2. Experimental section
2.1 Materials
The catalytic carrier (powder, SiC4-P80/125-HP, polytype 3C-SiC with phase β-SiC) was purchased from SiCAT (France).The surface of SiC-based supports was modified using titanium tetraisopropoxide Ti[OiPr]4 (97%, Sigma-Aldrich) and hexamethyldisilazane (99%, Sigma-Aldrich). The reagents for the cyclohexene epoxidation tests, such as tert-butyl hydroperoxide (5–6 M in nonane), cumyl hydroperoxide (80% in cumene), n-decane (99%, anhydrous) and cyclohexene (99%) were also supplied by Sigma-Aldrich.
The solvents propan-2-ol (99.7%) and toluene (99.8%) were supplied by VWR and Fisher-Scientific, respectively. These solvents were dried by using molecular sieves (rods; 1.6 mm; 0.3 nm pore size; Supelco®, Sigma-Aldrich). These were activated at 200 °C under reduced pressure for 15 h and introduced into hermetically sealed flat-bottomed flasks. The solvents were then added and stored under argon. After at least 48 h, the water content of the solvents was measured by Karl-Fischer titration (TitroLine KF trace, SI Analytics) and showed values below 15 ppm.
The catalytic reactions were monitored by gas chromatography (GC) with a flame ionisation detector (FID), with an Agilent 6850 Network GC System Gas Chromatograph. The column used for the chromatography is a (5%-phenyl)-methylpolysiloxane nonpolar column (Agilent), of 30 m length and 0.25 mm internal diameter, with a film thickness of 0.25 μm. The injections (1 μl) are performed in a split mode of 65 with an air flow of 400 ml min−1 and a dihydrogen flow of 40 ml min−1. The gas carrier was hydrogen at a flow of 0.8 ml min−1. The temperature program was set to an initial temperature of 50 °C and kept for 5 min then it was increased to a temperature of 150 °C at a rate of 10 °C min−1 and held for 5 min. The final temperature reached 250 °C at 10 °C min−1 with a hold of 20 min. The injector was held at 250 °C.
2.2 Syntheses of materials
The preparation of the materials consists of three steps: the oxidation/activation of the silicon carbide surface, the titanium grafting onto the surface of the carrier and the surface hydrophobisation, also called passivation. 5 g of SiC carrier is first calcined in a muffle furnace at 1000 °C for 2 h to form a silica layer, necessary for the grafting of metal oxides.1 The SiC support is then activated by stirring the solid in 150 ml of deionised water for 3 h at room temperature. The solid is recovered by water evaporation (40 °C, 72 mbar) and dried at 77 °C under low pressure for 12 h.To remove all traces of air and water, the pre-treated silicon carbide carrier is dried and heated at 200 °C (low pressure overnight then Ar for 6 h). The dried silicon carbide is suspended in 25 ml of anhydrous isopropanol and 0.5 g (1.7 mmol) of titanium isoproxide is added. The suspension is heated under reflux at 110 °C and stirred for 22 h with an argon atmosphere. The solid is finally recovered by filtration, washed with isopropanol and dried under low pressure at 200 °C overnight. It is finally calcined at 600 °C for 5 h with a muffle furnace (5 °C min−1).
A passivation treatment was finally performed to hydrophobize the titanium-grafted solid surface. This treatment consists of replacing the surface hydroxyl groups with SiMe3 groups. The passivation is a really important step for several reasons. It makes the solid surface hydrophobic which will avoid the adsorption of water and make easier the adsorption of the reactants. By doing the passivation, we also decrease the acidity of the catalyst surface to avoid the epoxide cycle opening. 1 g of TiO2-grafted silicon carbide is heated at 150 °C for 1 h under argon and dried under low pressures at 150 °C for 2 h with a thermostatically controlled silicon oil bath. The solid is next suspended in anhydrous toluene. 1 ml (mol) of hexamethyldisilazane (HMDS) is added and the suspension is heated under reflux at 130 °C for 2 h. Then, the solid is recovered by filtration and washed 3 times with 20 ml of toluene, acetone and ethanol until it has a filtrate with a neutral pH. The solid is finally dried under low pressures at 200 °C for 12 h.
2.3 Catalytic tests – general procedure
The cyclohexene epoxidation tests were performed in batch mode with two different oxidising agents: tert-butyl hydroperoxide (TBHP) and cumene hydroperoxide (CHP).The catalytic tests were carried out in a 25 ml two-neck round-bottom flask, equipped with a condenser and a cryostat (15 °C) and thermostatically controlled using a silicon oil bath. The catalyst (100 mg) is first dried at 60 °C overnight under low pressures directly in the round-bottom flask. Then, cyclohexene (0.821 g, 10 mmol) is added with 5 ml of anhydrous toluene and the suspension is heated with stirring to a defined temperature that depends on the used peroxide (65 °C for CHP/90 °C for TBHP). Once this temperature is reached, 10 mmol of peroxide is added. Aliquots of the reaction mixture were collected after defined times and analysed by GC-FID.
3. Results and discussion
The synthesis of the SiC-supported catalyst was achieved in a three-step sequence starting from pure SiC (SiCAT, France) including (i) the activation of the SiC-surface by oxidation, (ii) the surface grafting of titanium dioxide, and finally (iii) the passivation/hydrophobization of the surface via trimethylsilylation of silanol groups. The SiC surface was activated via calcination of the parent SiC at 1000 °C during 2 h, followed by dispersion of the material in deionized water and finally drying under reduced pressure, yielding a material labelled SiC/SiO2. In the second step, titanium dioxide was grafted via impregnation of the activated surface of the SiC/SiO2 material with titanium tetraisopropoxide (Ti[OiPr]4) followed by calcination of the material at 600 °C for 5 h (Scheme 1A – Ti-grafting). This material was directly used for passivation. Silanol groups located on the surface of the material were passivated using hexamethyldisilazane (HMDS) (Scheme 1A – passivation), yielding the material labelled TiO2@SiC/SiO2-p. For detailed information about the synthesis of the materials, please refer to the ESI.†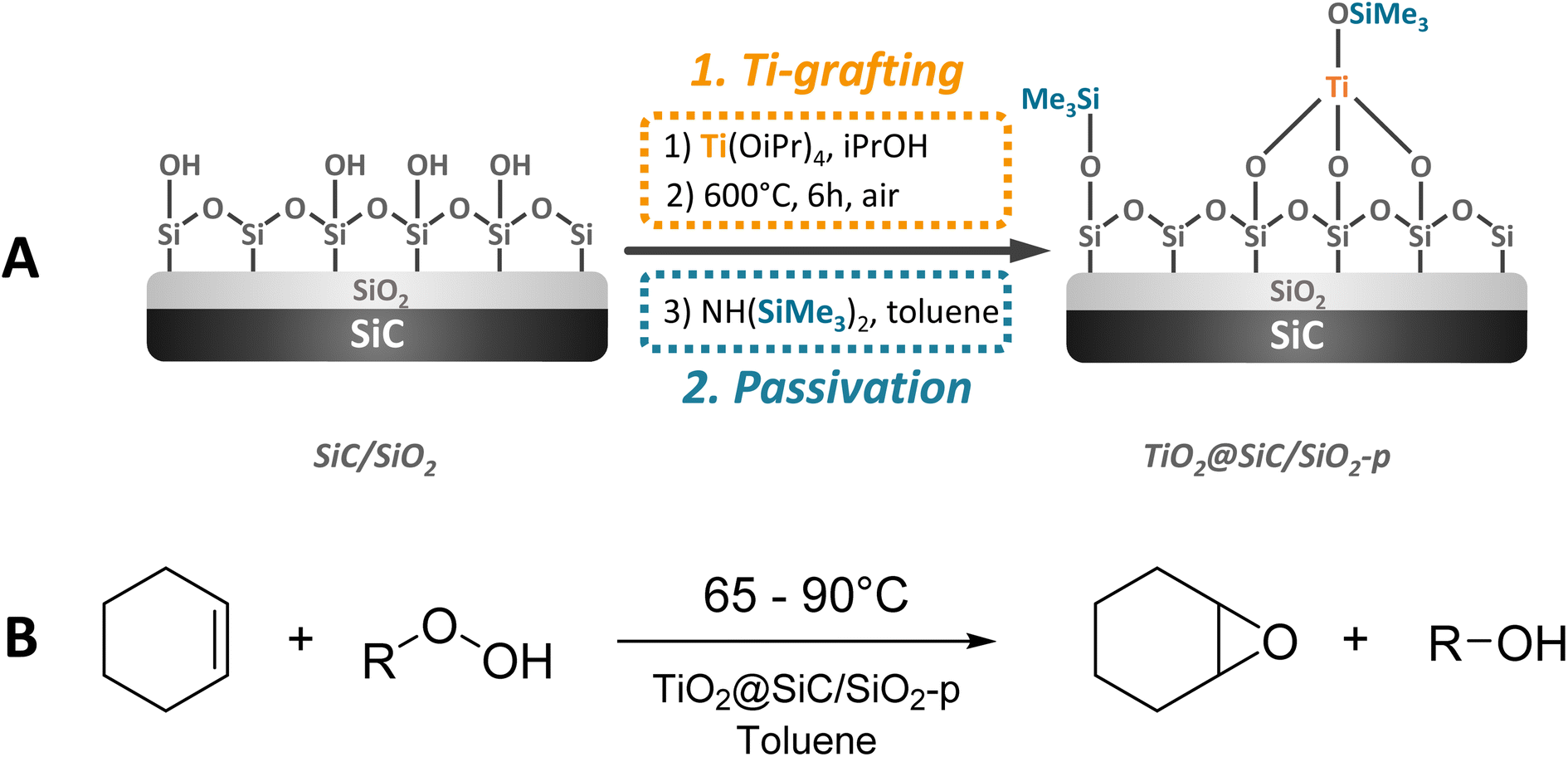 | ||
| Scheme 1 (A) Synthesis protocol for the formation of the SiC-based TiO2 catalyst. (B) Epoxidation reaction of cyclohexene. | ||
The materials were characterized in order to monitor various modifications. We mainly focused on the determination of the porosity in terms of specific surface area SBET by nitrogen sorption. Nitrogen adsorption–desorption isotherms on the silica before and after surface modifications were obtained at 77 K using a Micromeritics Tristar II Plus analyzer. The samples were degassed under reduced pressure at 200 °C and 40 °C, respectively. The specific surface area was calculated using the Brunauer–Emmett–Teller (BET) theory in the 0.05–0.30 P/P0 range. Total pore volume was measured at P/P0 = 0.99. The mesopore volume and average pore diameter were estimated with the Barrett–Joyner–Halenda (BJH) method.
The amount of grafted titanium was determined via energy-dispersive X-ray spectroscopy (EDX). All materials were probed using a field emission gun-scanning electron microscope (JEOL JSM 7100F FEG-SEM) equipped with an energy-dispersive X-ray spectrometer (Oxford ASDD X-Max EDX detector). The amount determined using this method is 1.2 wt%.
The identification of the Ti species was performed via X-ray photoelectron spectroscopy (XPS). X-ray photoelectron emission spectra (XPS) were recorded on a Thermo Scientific K-Alpha spectrometer, using a monochromatic source with an aluminum anode (hυ = 1486.6 eV) and a spot size of 400 μm (CIRIMAT, Toulouse). The calibration energy of the spectrometer was performed using the reference peak C 1s (284.9 ± 0.1 eV). High-resolution scans of C 1s, O 1s, Si 2p, and Ti 2p were acquired with a 30 eV pass energy with an increment value of 0.1 eV.
The environment of Si centers was investigated by solid-state NMR. Solid-state Nuclear Magnetic Resonance (NMR) analyses were carried out at the NMR platform of ICGM. 29Si solid-state NMR spectra were recorded on a 300 MHz Varian VNMRS300 (7.05 Tesla Wide Bore magnet), with a Varian T3 MAS (Magic Angle Spinning) probe and 7.5 mm ZrO2 rotors. Measurements were carried out using the non-quantitative CPMAS technique with 1H decoupling (Cross Polarisation Magic Angle Spinning/magnetization transfer from 1H to 29Si followed by 1H decoupling), with a recycling time of 3 s, a π/2 pulse of 5 μs and a contact time of 5 ms. The chemical shift was calibrated using a secondary reference of Q8M8H (octakis(dimethylsiloxy)octasilsesquioxane), whose left-hand signal was fixed at −2.25 ppm.
Finally, Scanning Electron Microscopy (SEM) was used to study the evolution of the morphology of the materials during the modification steps.
Energy-dispersive X-ray spectroscopy on the titania-grafted SiC materials TiO2@SiC/SiO2-p indicated a Si/Ti atomic ratio of 93/1. Besides titanium, only silicon, carbon and oxygen could be detected in the sample (see the ESI, Fig. S1 and Table S1†).
Nitrogen adsorption–desorption at −196 °C using a Micromeritics TriStar II plus surface analyzer was used to study the porosity of the materials. We investigated the parent SiC and partially oxidized SiC/SiO2 materials as well as the supported and passivated material TiO2@SiC/SiO2-p. All samples were degassed for 15 h under reduced pressure (1.33 × 10−3 mbar) prior to the experiment. The nitrogen sorption isotherms of the three materials are given in the ESI, Fig. S2.† The specific surface areas and total pore volumes, determined at a relative pressure of 0.98 based on the adsorbed nitrogen volume, are reported in Table 1. To determine the specific surface area (SSA) in the range of relative pressure (P/P0) between 10−5 and 0.1, the Brunauer–Emmett–Teller model (BET) was applied. The isotherms of all materials are of type II, indicating non-porous and/or macroporous materials. Both specific surface area and total pore volume of the materials decrease during the oxidation and grafting/passivation steps. More specifically, the specific surface area drops from 35 m2 g−1 for the SiC material to 21 m2 g−1 for the material TiO2@SiC/SiO2-p. Regarding the total pore volume, a decrease can be observed from 0.39 for the SiC material to 0.15 for the material TiO2@SiC/SiO2-p. The oxidation reaction has a stronger impact on the textural properties of the material as it leads to a stronger decrease of both specific surface area and total pore volume compared to the grafting/passivation step. This may be explained by sintering processes, as the oxidation step was performed at high temperatures.
Scanning Electron Microscopy (SEM) images of the parent SiC and TiO2@SiC/SiO2-p are given in Fig. 1. The SiC material consists of agglomerated particles of nanometric size with diameters of around 60 nm. The morphology did not change during the modification steps, as we found identical features in the case of the material.
 | ||
| Fig. 1 SEM images of the parent SiC (left) and Ti-grafted and passivated TiO2@SiC/SiO2-p (right). | ||
29Si solid-state cross-polarization (CP)MAS NMR has been used to study the environment of the silicon centers in the SiC-based materials towards the synthesis steps. This method was particularly used to observe the silicon oxycarbide species which are in the minority compared to silicon carbide species.24Fig. 2 shows the three 29Si CP MAS NMR spectra of the SiC material (commercial silicon carbide carrier in the β-phase purchased from SiCAT, France), the SiC/SiO2 sample, and the TiO2@SiC/SiO2-p material. All spectra were deconvoluted using the Dmfit program25 to identify the different silicon species.
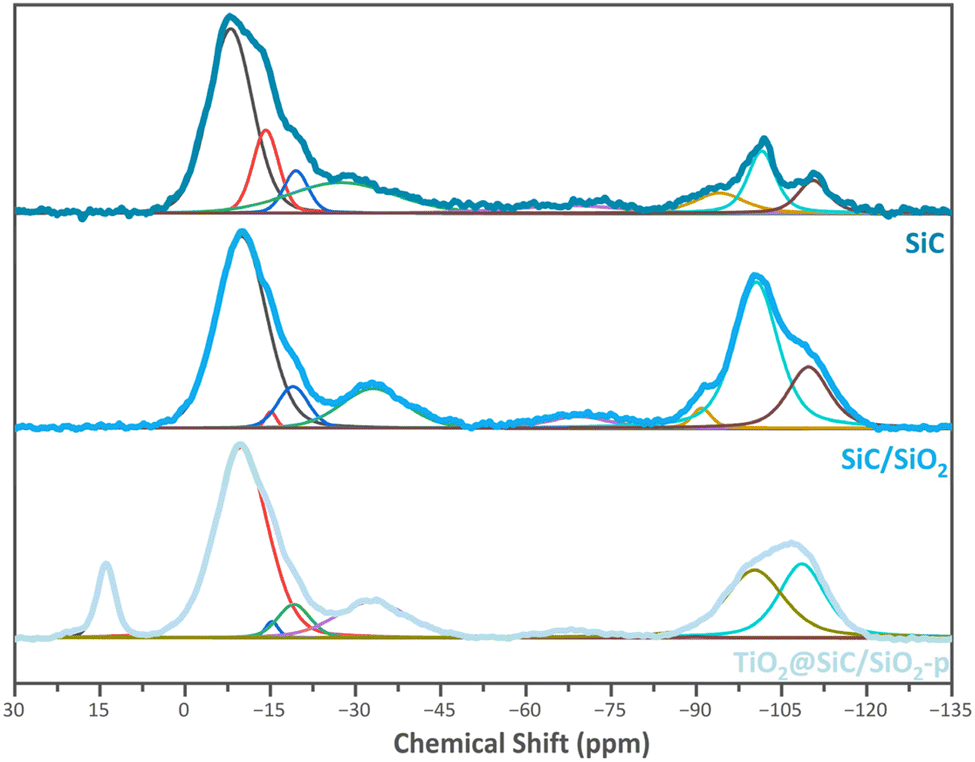 | ||
| Fig. 2 29Si CP-MAS solid-state NMR spectra of the SiC, SiC/SiO2 and TiO2@SiC/SiO2 materials (top to bottom). | ||
In the spectra of the materials SiC and SiC/SiO2, 8 peaks can be found: 2 signals at −29, and −8 ppm corresponding respectively to silicon oxycarbides SiO2C2 and SiOC3; 2 signals around −14 and −19 ppm for Si nucleus coordinated with 4 carbons (SiC4); 1 signal at −74 ppm for SiO3C species; and 3 signals at −91, −100 and −110 ppm corresponding respectively to Q2, Q3 and Q4 species of silicon oxide Si(OSi)n(OH)4−n. The presence of the Qn species on the commercial SiC spectrum demonstrates the existence of a partial oxidation of SiC. It is worth noting that the Qn signals that can be attributed to Si(OSi)n(OH)4−n centers are more intense in the spectrum of the material SiC/SiO2 compared to the parent SiC, indicating successful oxidation of the SiC surface and resulting in the formation of silicon oxide species during the calcination step. The 29Si solid-state CP-NMR spectrum of the material TiO2@SiC/SiO2-p also displays eight signals. However, two main changes can be observed. First, the spectrum of the catalyst shows a new signal at 14 ppm. This signal can be attributed to SiMe3 groups and indicates the capping of the residual hydroxyl groups and therefore a successful passivation of the surface via trimethylsilylation. Second, in the spectrum of the material TiO2@SiC/SiO2-p, we observe a strong decrease in the intensity of the Q3 silicon centers and a complete disappearance of the Q2 centers whereas the intensity of the Q4 centers is nearly unchanged. This trend can be explained by the replacement of most of the hydroxyl groups by –SiMe3 groups and therefore is also indicative of the trimethylsilylation of residual silanol groups. Regarding the set of signals in the range of 5 to −45 ppm and at −68 ppm, characteristic of SiC and SiOC species, no significant changes can be observed.
To investigate the chemical composition and oxidation states of the samples, X-ray photoelectron spectroscopy (XPS) experiments were carried out. XPS is particularly powerful for the determination of the species located at the surface of the samples. The C 1s, O 1s, Si 2p, and Ti 2p XPS spectra of the TiO2@SiC/SiO2-p material are displayed in Fig. 3A–D. XPS spectra of the parent SiC before and after oxidation and titanium-grafting are displayed in Fig. S3 and S4.†
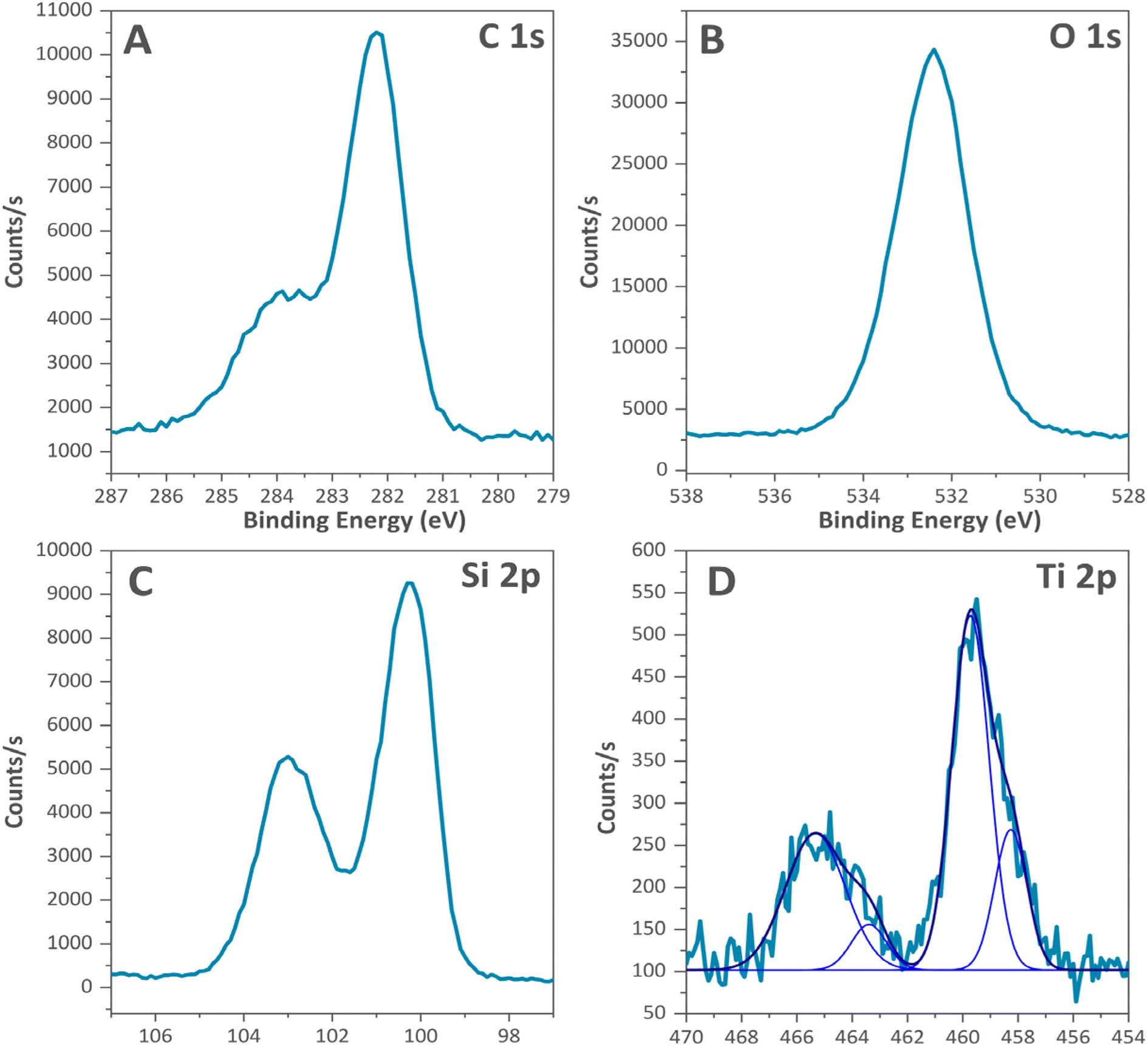 | ||
| Fig. 3 XPS spectra of Ti-grafted SiC supports “TiO2@SiC/SiO2-p”. (A: C1S, B: O1s, C: Si2p, D: Ti 2p). | ||
In Fig. 3A, corresponding to C 1s, two signals can be observed. The most intense signal at 282.4 eV corresponds to the Si–C bond of silicon carbide,26 whereas the shoulder at 284.2 eV can be attributed to Si–C bonds of the trimethylsilyl groups, originating from the passivation reaction.27 The environment of the oxygen centers can be deduced from the XPS spectra given in Fig. 3B. One single signal is observed at 532.3 eV, corresponding to the oxygen of the silica layer (SiO2) at the surface of the material. Even though Netterfield et al. observed a shift towards lower energies of the O 1s photoelectron peak for silica modified with titanium oxide,28 our results do not show this shift. The silicon environment (Fig. 3C) displays two signals centered at 100.1 and 102.9 eV, corresponding to the Si–C bonds in SiC and the Si–O bonds originating from the oxidized silicon carbide layer. The trimethylsilyl groups are not detected, probably due to their low amount or overlapping with other signals. Finally, Fig. 3D displays the XPS spectra of Ti species. The two signals centered at 464.7 and 459.0 eV in Fig. 3D were deconvoluted using OriginPro 2022b software into two peaks each. The peaks at 465.5 and 459.6 eV are attributed to Ti 2p1/2 and Ti 2p3/2 of a tetrahedrally coordinated titanium. The other signals at 463.5 and 458.3 eV correspond to Ti 2p1/2 and Ti 2p3/2 of an octahedrally coordinated titanium.29
After having ascertained the successful grafting of titanium dioxide on the surface of silicon carbide particles, we focused on the evaluation of the catalytic properties of the material. The reaction conditions for the epoxidation reactions are detailed in the ESI.† More specifically, we investigated the epoxidation of cyclohexene (Scheme 1B) using either TBHP or CHP as oxidizing agents. Indeed, our SiC-based catalyst allows a good diffusion of cyclic olefins onto the porosity and provide the accessibility of the reactants on the materials' active sites unlike zeolite-supported catalysts such as TS-1. The reactions were carried out under several conditions: at 90 °C and 65 °C, with and without catalysts. Blank tests first demonstrated that no epoxide formation occurred in the absence of the catalyst. On the other side, the passivated material TiO2@SiC/SiO2-p displayed significant catalytic activity. The conversion of cyclohexene as well as the selectivity of cyclohexene oxide formation as a function of reaction time are displayed in Fig. 4. We observed that the material efficiently promotes the epoxidation of cyclohexene with both oxidizing agents and leads to a cyclohexene conversion of 83% and 72% from TBHP and CHP, respectively, after a reaction time of 20 h. At the end of the reaction time, 2-cyclohexen-1-one and 2-cyclohexen-1-ol were detected as by-products (less than 3% and 1.4% for TBHP and CHP, respectively). These by-products were also detected in the cyclohexene epoxidation reaction with hydrogen peroxide in the vapor and liquid phases at various temperatures.30 They are formed from radical side reactions.31 It may be observed that the selectivity to epoxide increased with longer reaction times (Fig. 4A) and lower temperatures (Fig. 4B), in accordance with the formation of this product being an exothermic reaction. The slightly higher cyclohexene conversion and lower selectivity observed with TBHP may result from the higher reaction temperature.
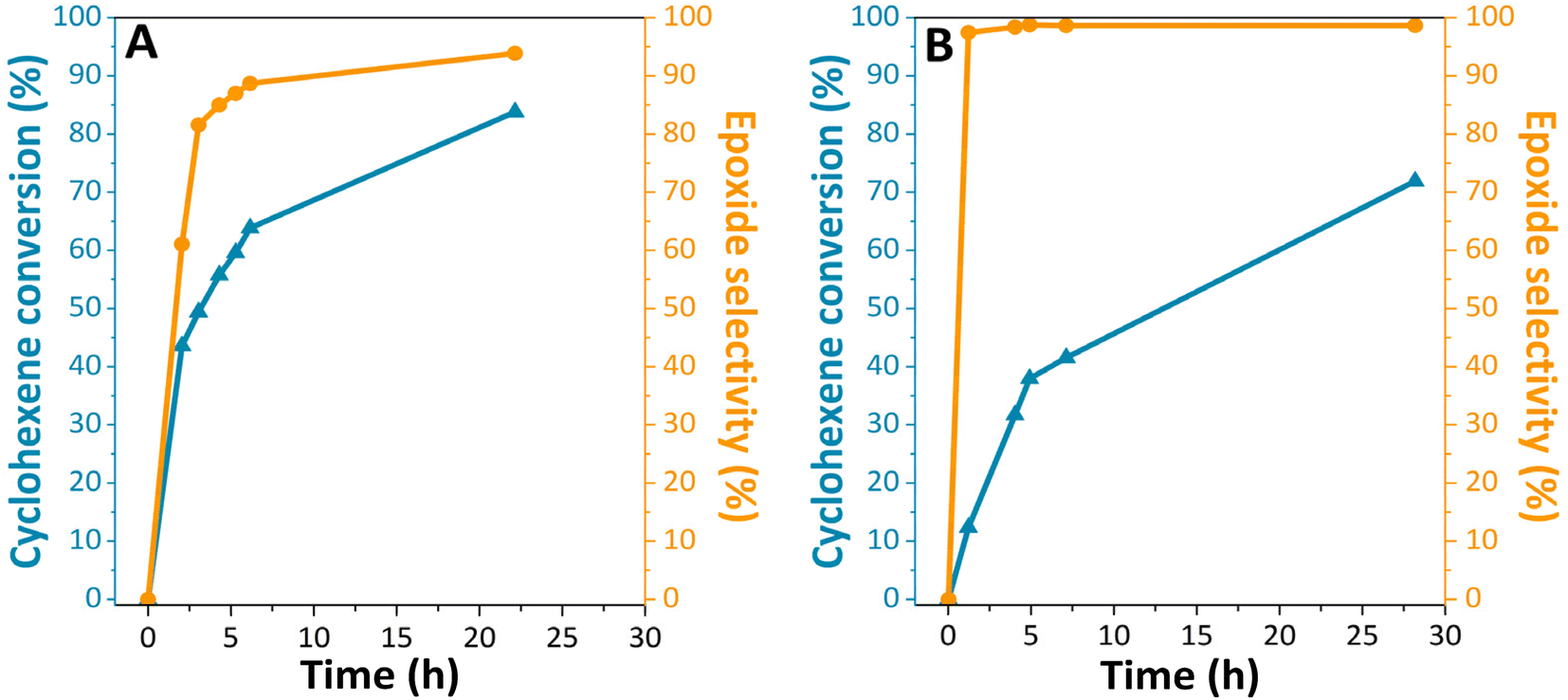 | ||
| Fig. 4 Conversion of cyclohexene (blue) and epoxide selectivity (yellow) results from catalytic tests of the TiO2@SiC/SiO2-p catalyst with (A) tert-butyl hydroperoxide and (B) cumene hydroperoxide as the oxidant. | ||
Concerning the state of art, the literature is rich with different types of heterogeneous epoxidation catalysts.9 The experimental conditions applied for the catalytic tests (type of catalyst, oxidizing agent, starting materials, solvent, temperature, concentrations…) are as diverse as the number of references. For example, a silica-supported titanium catalyst exhibited 34.5% conversion with dilute hydrogen peroxide at 80 °C after 24 h.31 On the other side, almost complete conversion was obtained with a mesoporous hybrid titania–silica xerogel after 30 min at 90 °C for the epoxidation of cyclohexene by CHP.5 Regarding the materials reported here, we observed a surprisingly high catalytic activity considering the low amount of grafted titanium dioxide and the low porosity of the support. We attribute this result to a homogeneous dispersion of Ti species on the external surface of the hydrophobized SiC support as shown by the EDX mapping of TI of the TiO2@SiC/SiO2-p material (see the ESI, Fig. S5†).
4. Conclusions
We report a novel heterogeneous epoxidation catalyst based on silicon carbide support. The material was obtained by a three-step sequence. The successful grafting of TiO2 is triggered by the presence of a silica shell at the surface of the SiC materials which allows anchoring of Ti species via surface sol–gel reactions. The characterization of the materials showed successful immobilization of TiO2 on the surface of the SiC based material. The catalyst displays very promising performances in terms of catalytic activity and selectivity in the epoxidation reaction of cyclohexene. Our work shows that SiC is a versatile support material for the immobilization of epoxidation catalysts. We believe that our approach has high potential for the development of SiC based heat exchange reactors.Data availability
All data are available following this link: https://drive.google.com/drive/folders/1ObbC2uW9mR-_XLLrEgtvWND19C4DoeD_?usp=sharing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was part of the CATASIC project, funded by the French National Research Agency under the grant ANR-20-CE08-0029.References
- G. Sienel, R. Rieth and K. T. Rowbottom, Epoxides, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2000 Search PubMed.
- M. L. Mohammed and B. Saha, Recent Advances in Greener and Energy Efficient Alkene Epoxidation Processes, Energies, 2022, 15(8), 2858, DOI:10.3390/en15082858.
- A. Corma and H. García, Lewis acids as catalysts in oxidation reactions:: From homogeneous to heterogeneous systems, Chem. Rev., 2002, 102(10), 3837–3892, DOI:10.1021/cr010333u.
- I. W. C. E. Arends and R. A. Sheldon, Activities and stabilities of heterogeneous catalysts in selective liquid phase oxidations: recent developments, Appl. Catal., A, 2001, 212(1), 175–187, DOI:10.1016/S0926-860X(00)00855-3.
- O. Lorret, V. Lafond, P. H. Mutin and A. Vioux, One-step synthesis of mesoporous hybrid titania-silica xerogels for the epoxidation of alkenes, Chem. Mater., 2006, 18(20), 4707–4709, DOI:10.1021/cm061478q.
- A. S. Sharma, V. S. Sharma, H. Kaur and R. S. Varma, Supported heterogeneous nanocatalysts in sustainable, selective and eco-friendly epoxidation of olefins, Green Chem., 2020, 22(18), 5902–5936, 10.1039/D0GC01927E.
- M. Taramasso, G. Perego and B. Notari, Preparation of porous crystalline synthetic material comprised of silicon and titanium oxides, US Pat. US4410501, 1983 Search PubMed.
- J. Přech, Catalytic performance of advanced titanosilicate selective oxidation catalysts - a review, Catal. Rev. Sci. Eng., 2018, 60(1), 71–131, DOI:10.1080/01614940.2017.1389111.
- V. Smeets, E. M. Gaigneaux and D. P. Debecker, Titanosilicate Epoxidation Catalysts: A Review of Challenges and Opportunities, ChemCatChem, 2022, 14(1), e202101132, DOI:10.1002/cctc.202101132.
- J. Gao, M. Liu, X. S. Wang and X. W. Guo, Characterization of Ti-ZSM-5 Prepared by Isomorphous Substitution of B-ZSM-5 with TiCl4 and Its Performance in the Hydroxylation of Phenol, Ind. Eng. Chem. Res., 2010, 49(5), 2194–2199, DOI:10.1021/ie901360y.
- J. Přech and J. Cejka, UTL titanosilicate: An extra-large pore epoxidation catalyst with tunable textural properties, Catal. Today, 2016, 277, 2–8, DOI:10.1016/j.cattod.2015.09.036.
- W. Tong, J. P. Yin, L. Y. Ding, H. Xu and P. Wu, Modified Ti-MWW Zeolite as a Highly Efficient Catalyst for the Cyclopentene Epoxidation Reaction, Front. Chem., 2020, 8, 585347, DOI:10.3389/fchem.2020.585347.
- M. Moliner and A. Corma, Advances in the synthesis of titanosilicates: From the medium pore TS-1 zeolite to highly-accessible ordered materials, Microporous Mesoporous Mater., 2014, 189, 31–40, DOI:10.1016/j.micromeso.2013.08.003.
- T. Blasco, A. Corma, M. T. Navarro and J. P. Pariente, Synthesis, characterization, and catalytic activity of Ti-MCM-41 structures, J. Catal., 1995, 156(1), 65–74, DOI:10.1006/jcat.1995.1232.
- F. Bérubé, B. Nohair, F. Kleitz and S. Kaliaguine, Controlled Postgrafting of Titanium Chelates for Improved Synthesis of Ti-SBA-15 Epoxidation Catalysts, Chem. Mater., 2010, 22(6), 1988–2000, DOI:10.1021/cm9030667.
- J. Jarupatrakorn and J. D. Tilley, Silica-supported, single-site titanium catalysts for olefin epoxidation. A molecular precursor strategy for control of catalyst structure, J. Am. Chem. Soc., 2002, 124(28), 8380–8388, DOI:10.1021/ja0202208.
- M. J. Ledoux and C. Pham-Huu, Silicon carbide – a novel catalyst support for heterogeneous catalysis, Cattech, 2001, 5(4), 226–246, DOI:10.1023/a:1014092930183.
- G. Tuci, Y. Liu, A. Rossin, X. Guo, C. Pham, G. Giambastiani and C. Pham-Huu, Porous Silicon Carbide (SiC): A Chance for Improving Catalysts or Just Another Active-Phase Carrier?, Chem. Rev., 2021, 121(17), 10559–10665, DOI:10.1021/acs.chemrev.1c00269.
- C. Duong-Viet, H. Ba, Z. El-Berrichi, J. M. Nhut, M. J. Ledoux, Y. F. Liu and C. Pham-Huu, Silicon carbide foam as a porous support platform for catalytic applications, New J. Chem., 2016, 40(5), 4285–4299, 10.1039/c5nj02847g.
- M. Cozzolino, M. Di Serio, R. Tesser and E. Santacesaria, Grafting of titanium alkoxides on high-surface SiO2 support: An advanced technique for the preparation of nanostructured TiO2/SiO2 catalysts, Appl. Catal., A, 2007, 325(2), 256–262, DOI:10.1016/j.apcata.2007.02.032.
- Y. C. Lin, C. C. Chang, K. H. Sung, J. F. Lee and S. Cheng, Importance of solvents in preparing highly active Ti-SBA-15 catalysts by grafting method, Microporous Mesoporous Mater., 2018, 272, 276–285, DOI:10.1016/j.micromeso.2018.06.046.
- M. P. Coles, C. G. Lugmair, K. W. Terry and T. D. Tilley, Titania-silica materials from the molecular precursor Ti [OSi(OtBu)3]4: Selective epoxidation catalysts, Chem. Mater., 2000, 12(1), 122–131, DOI:10.1021/cm990444y.
- R. Hutter, T. Mallat and A. Baiker, Titania-silica mixed oxides. 2. Catalytic behavior in olefin epoxidation, J. Catal., 1995, 153(1), 177–189, DOI:10.1006/jcat.1995.1119.
- H. Marsmann, 29Si NMR Spectroscopic Results, in NMR Basic Principles and Progress, ed. Kintzinger, J.-P. and Marsmann, H., Springer, 1981, vol. 17, pp. 65–235 Search PubMed.
- D. Massiot, F. Fayon, M. Capron, I. King, S. Le Calvé, B. Alonso, J. O. Durand, B. Bujoli, Z. H. Gan and G. Hoatson, Modelling one- and two-dimensional solid-state NMR spectra, Magn. Reson. Chem., 2002, 40(1), 70–76, DOI:10.1002/mrc.984.
- J. Guo, K. Song, B. Wu, X. Zhu, B. Zhang and Y. Shi, Atomically thin SiC nanoparticles obtained via ultrasonic treatment to realize enhanced catalytic activity for the oxygen reduction reaction in both alkaline and acidic media, RSC Adv., 2017, 7(37), 22875–22881, 10.1039/C7RA01701D.
- V. Jankauskaite, A. Balciunaitiene, R. Alexandrova, N. Buskuviene and K. Zukiene, Effect of Cellulose Microfiber Silylation Procedures on the Properties and Antibacterial Activity of Polydimethylsiloxane, Coatings, 2020, 10(6), 567, DOI:10.3390/coatings10060567.
- R. P. Netterfield, P. J. Martin, C. G. Pacey, W. G. Sainty, D. R. McKenzie and G. Auchterlonie, Ion-assisted deposition of mixed TiO2-SiO2 films, J. Appl. Phys., 1989, 66(4), 1805–1809, DOI:10.1063/1.344352.
- M. C. Capel-Sanchez, G. Blanco-Brieva, J. M. Campos-Martin, M. P. de Frutos, W. Wen, J. A. Rodriguez and J. L. G. Fierro, Grafting Strategy to Develop Single Site Titanium on an Amorphous Silica Surface, Langmuir, 2009, 25(12), 7148–7155, DOI:10.1021/la900578u.
- T. U. Yoon, S. Ahn, A. R. Kim, J. M. Notestein, O. K. Farha and Y. S. Bae, Cyclohexene epoxidation with H2O2in the vapor and liquid phases over a vanadium-based metal-organic framework, Catal. Sci. Technol., 2020, 10(14), 4580–4585, 10.1039/d0cy00833h.
- J. M. Fraile, J. I. García, J. A. Mayoral and E. Vispe, Optimization of cyclohexene epoxidation with dilute hydrogen peroxide and silica-supported titanium catalysts, Appl. Catal., A, 2003, 245(2), 363–376, DOI:10.1016/s0926-860x(02)00643-9.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4na00575a |
| This journal is © The Royal Society of Chemistry 2024 |