
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTargeting drug resistance in breast cancer: the potential of miRNA and nanotechnology-driven delivery systems
Aditi
Verma
 ,
Krunal
Patel
and
Ashutosh
Kumar
*
,
Krunal
Patel
and
Ashutosh
Kumar
*
Biological and Life Sciences, School of Arts & Sciences, Ahmedabad University, Central Campus, Navrangpura, Ahmedabad 380009, Gujarat, India. E-mail: ashutosh.kumar@ahduni.edu.in
First published on 12th November 2024
Abstract
Breast cancer is the second leading cause of cancer-related deaths in females worldwide. Despite significant advancements in treatment, drug resistance remains a major challenge, limiting the effectiveness of therapies and leading to dismal outcomes. Approximately 50% of HER2+ breast cancer patients develop resistance to trastuzumab, and patients with triple-negative breast cancer often experience resistance to first-line therapies. The drug resistance mechanisms involve altered drug uptake, enhanced DNA repair, and dysregulated apoptosis pathways. MicroRNAs are essential in regulating cellular processes involved in both homeostasis and disease. Recent data suggest that microRNAs can overcome drug resistance by regulating the pathways that confer drug resistance. Combining different conventional anticancer agents with microRNA therapies holds promise for enhancing treatment effectiveness against drug resistant breast cancer. Advancements in nano-drug delivery systems have facilitated the effective delivery of microRNAs by improving their stability, targeting specific cells, and enhancing cellular uptake. This review elucidates the recent advancements in microRNA-based therapies, their effects on gene expression, and their clinical efficacy in overcoming drug resistance in breast cancer.
1. Introduction
Breast cancer remains a significant global health challenge, with over 3.13 million new cases and an estimated 42![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 780 deaths in the U.S. alone in 2024.1 Breast cancer is classified into four molecular subtypes: luminal A, luminal B, HER2-positive, and triple-negative, based on receptor expression. Despite decades of advances, overcoming drug resistance remains the top hurdle in reducing breast cancer's significant mortality rate in women.2 Chemoresistance refers to the phenomenon of drug resistance in cancer cells, which can arise due to mechanisms such as drug efflux, alteration of drug targets, DNA repair, loss of cell death, epithelial to mesenchymal transition (EMT) or epigenetic changes.3 The mechanisms of resistance can be broadly classified into intrinsic and acquired categories. Intrinsic resistance in breast cancer involves various pathways and alterations, contributing to treatment insensitivity, hindrance of antitumor immunity, and promotion of cancer progression.4
780 deaths in the U.S. alone in 2024.1 Breast cancer is classified into four molecular subtypes: luminal A, luminal B, HER2-positive, and triple-negative, based on receptor expression. Despite decades of advances, overcoming drug resistance remains the top hurdle in reducing breast cancer's significant mortality rate in women.2 Chemoresistance refers to the phenomenon of drug resistance in cancer cells, which can arise due to mechanisms such as drug efflux, alteration of drug targets, DNA repair, loss of cell death, epithelial to mesenchymal transition (EMT) or epigenetic changes.3 The mechanisms of resistance can be broadly classified into intrinsic and acquired categories. Intrinsic resistance in breast cancer involves various pathways and alterations, contributing to treatment insensitivity, hindrance of antitumor immunity, and promotion of cancer progression.4
The major factors of intrinsic resistance include (1) inherited genetic alterations in tumor cells leading to unresponsiveness to both traditional chemotherapy and targeted therapies, (2) tumor heterogeneity, which encompasses subsets of cancer stem cells capable of regenerating new tumor cells and causing recurrence, and (3) pharmacokinetic factors such as absorption, distribution, metabolism, and excretion (ADME), as well as pharmacogenetic patterns, which can limit the drug's efficacy in reaching and acting upon tumor cells.5 Acquired resistance in cancer refers to the development of resistance to treatment over time, driven by genetic or environmental factors, leading to reduced drug efficacy, therapy failure, and tumor progression. This resistance can arise from (1) activation of secondary proto-oncogenes, resulting in the emergence of a new driving gene, (2) alterations in the tumor microenvironment following treatment, and (3) modifications in drug targets due to changes in gene expression or mutation. Notably, CDK4/6 inhibition demonstrates early adaptation and acquired resistance in estrogen receptor-positive breast cancer.6,7
Multidrug resistance (MDR) is a phenomenon where cancer cells become less responsive to multiple chemotherapeutic drugs, often leading to treatment failure. This resistance is frequently associated with drug resistance-associated membrane proteins (DRAMPs). DRAMPs operate through two primary mechanisms: they can influence drug accumulation through physicochemical processes or actively expel drug molecules from cells, thereby decreasing intracellular drug concentration. Consequently, addressing drug resistance in breast cancer may involve using microRNAs (miRs), which are 18–22 nucleotide long, single-stranded, non-coding RNA molecules.8
Several factors contribute to the development of resistance to immunotherapy in breast cancer, including genetic modifications and downregulation of cancer antigens. Additionally, changes in the immune cell composition within the tumor microenvironment further impede the effectiveness of immunotherapeutic interventions. These factors collectively hinder the immune system's ability to detect and eliminate cancer cells, reducing treatment efficacy. The paper highlights the use of miR-based combinational therapy to combat drug resistance in breast cancer.
2. Drug resistance in breast cancer
A significant number of cancer patients show resistance to treatment.9 In triple-negative breast cancer, resistance to first-line therapy is particularly high, impeding further treatment efforts. Common breast cancer drugs work through various mechanisms: hormonal treatments like tamoxifen, estradiol, and letrozole modulate hormone activity involved in cell proliferation; monoclonal antibodies such as trastuzumab target HER2-positive cancer; and chemotherapeutic agents like taxanes (e.g., paclitaxel and docetaxel) and anthracyclines (e.g., doxorubicin and daunorubicin) address different aspects of cancer growth.10 Breast cancer cells can become resistant to chemotherapeutic drugs with different mechanisms (Fig. 1), as discussed in the subsequent section.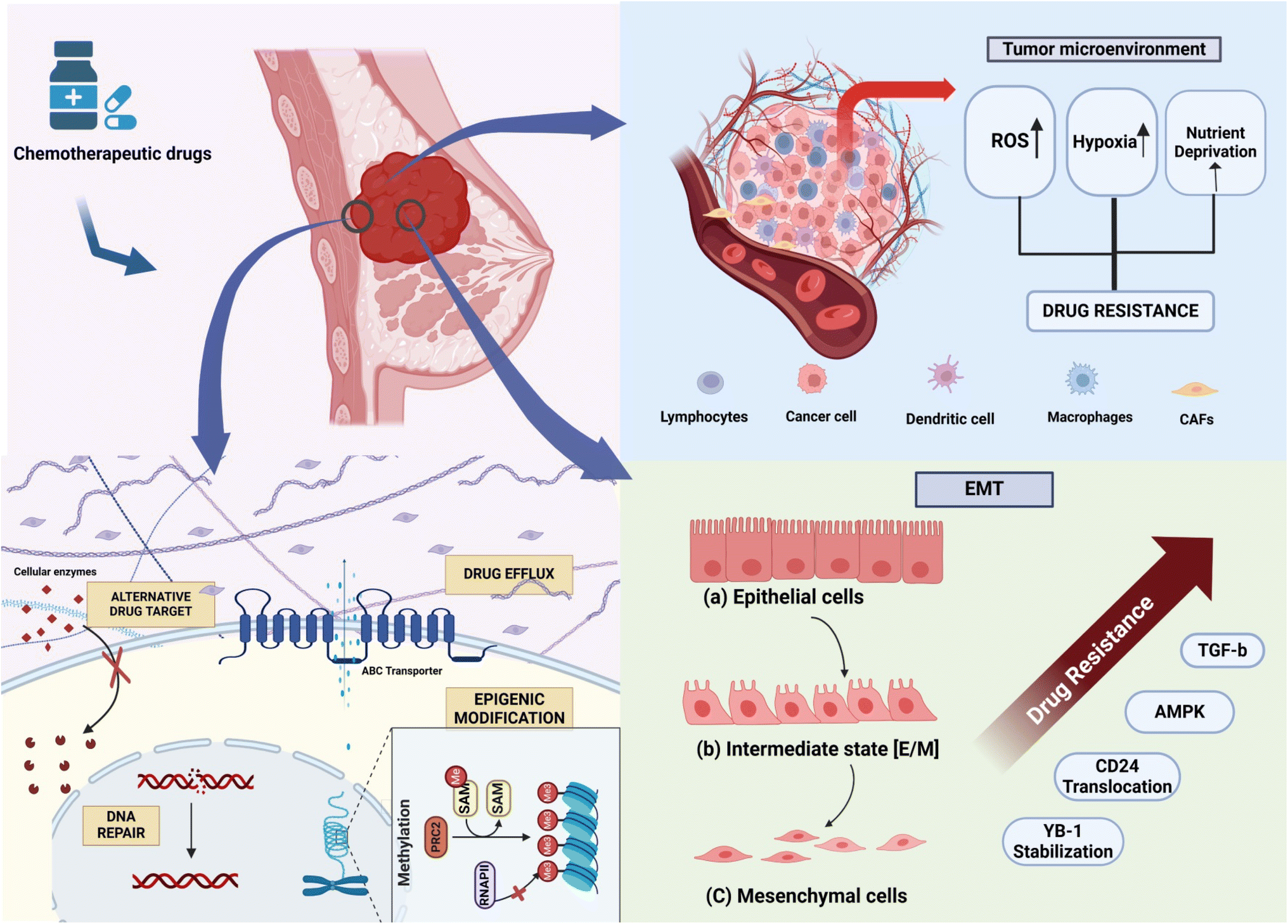 | ||
| Fig. 1 Schematic representation of the mechanism of drug resistance in breast cancer, depicting changes in the cellular matrix, tumor microenvironment, and EMT. | ||
2.1 Increased drug efflux
A primary problem associated with chemotherapy resistance is decreased net drug accumulation within the cells caused by the upregulation of drug efflux pumps.11 The ATP-binding cassette (ABC) transporter superfamily is central to the drug efflux in these cells. There are nine ABC genes in seven subfamilies: ABCA, ABCD, ABCE, ABCB, ABCC, ABCF, and ABCG, and three particular ABC transporters, i.e., ABCB1, ABCC1, and ABCG2, have been implicated in MDR.12 ABCB1, MDR1/P-glycoprotein (P-gp), is expressed in normal tissues but its higher expression in various cancers has been reported. The expression of ABCB1 in breast tumors varies among individuals and has been observed in cancers of the breast, kidneys, colon, adrenals, pancreas, liver, prostate, and ovaries. The overexpression of ABCB1 is associated with chemo-resistance. For instance, when ABCB1 is silenced, its protein expression doubles in drug-resistant SKBR3 and MCF-7 breast cancer cells.13Epigenetic regulation affects both the expression and transport of ABCB1. CpG islands in the proximal promoter region of ABCB1 are often hyper-methylated, which may be linked to decreased transcription of ABCB1 and improved overall survival in individuals with ovarian and breast cancer.12 ABCG2, also known as breast cancer resistance protein (BCRP), contributes to anticancer therapy resistance and is a marker of cancer stem cells (CSCs). ABCG2 is a drug transporter capable of expelling both positively and negatively charged drugs. The overexpression of ABCC1 and ABCG2 is notably documented in triple-negative breast cancer (TNBC) compared to other breast cancer subtypes.14
2.2 Epigenetic modification
Epigenetic mechanisms can contribute to chemotherapy drug resistance, including methylation at cytosine-guanine sites, which enhance gene silencing, hypomethylation that upregulates oncogenes, histone modifications, and non-coding RNA alterations.15 Resistance can also develop against target-directed therapies due to mutations in the causative proteins or epigenetic modifications that may change the proteins' expression levels. The demethylation of an oncogene's promoter region can lead to a pharmacological response by upregulating the gene.16 Conversely, hyper-methylation of genes related to DNA repair can result in cell cycle arrest. Some miRs, such as miR-21, target the PTEN gene, promoting cancer cell proliferation; miR-10 modulates the PTEN/AKT pathway, indicating positive progression of breast cancer; and miR-27b-3p acts as a tumor suppressor targeting the PPARG gene, which is responsible for promoting TNBC metastasis.17–19 The Food and Drug Administration (FDA) has approved epigenetic drugs, such as DNA methylation inhibitors (DNA methyltransferase inhibitors) like AD-738, which is made from azacitidine (decitabine; DAC), and histone deacetylase inhibitors (HDACi), including vorinostat. 5-Azacitidine and entinostat were put together as an epigenetic duo in a phase II clinical test for women with advanced breast cancer.18 On the other hand, doxorubicin administration to breast cancer tumors was shown to possibly be connected to an increase in the risk of metastasis through an increase of SIPA1 expression that can facilitate the epithelial-mesenchymal transition in cancer cells.202.3 DNA repair
DNA damage is the primary mechanism through which cisplatin, 5-fluorouracil, gemcitabine, and methotrexate exert their effects in cancer therapy. The inability to evoke an effective DNA damage response (DDR) contributes to mutagenic events, resulting in the loss of genetic information and leading to cancer and neurological diseases. The DDR activates specific DNA repair processes, potentially restoring the DNA lesions introduced by these drugs.21 Key players in the DDR pathway, especially the major ones like poly(ADP-ribose) polymerase (PARP), are crucial targets for overcoming resistance. PARP inhibitors have been widely used for cancer patients with BRCA1/2 mutations and showed promising clinical activity.22,23 Consequently, cells have developed numerous DDR pathways that cover different lesions' detection, signaling, and repair.24 These pathways include (1) mismatch repair (MMR), (2) translesion synthesis (TLS), (3) nucleotide excision repair (NER), (4) single-strand break (SSB) repair, (5) base excision repair (BER), (6) non-homologous end-joining, and (7) Fanconi Anemia (FA) pathway.25 Understanding these DDR pathways is essential for elucidating the mechanisms behind drug resistance.2.4 Alteration of drug targets
Mutations or alterations in the target are among the most prevalent mechanisms through which drug resistance to a therapeutic molecule can develop. Mutations in the primary drug target or alternations in target proteins can modulate their structure or function, reducing the binding affinity or efficacy of the targeted therapy.26 The detection of specific miRs that can control the expression of these receptors and their associated cofactors has made it possible to understand the mechanisms by which miRs respond to hormonal and targeted cancer therapies. Anticancer drugs were developed to target topoisomerase II, an enzyme that prevents DNA from becoming too coiled or loosened.27 In most cases, the contact between DNA and topoisomerase II is temporary. Consequently, these drugs bind the complex and thwart its function, leading to DNA damage, arrest of DNA synthesis, and disconnect from the mitotic process critical for cell division. Breast cancer treatment with estrogen receptors often leads to adaptations in the drug target and results in drug resistance development. Tamoxifen (TAM) is a drug that is usually given to patients with estrogen receptor-positive (ER-positive) breast cancer to compete with estrogen for the ligand-binding site of the estrogen receptor.28 However, long-term exposure to TAM can lead to drug resistance through mechanisms such as mutations and the reduction of estrogen receptor expression. This issue has prompted the development of new drugs, such as aromatase inhibitors (AIs).292.5 Epithelial-mesenchymal transformation (EMT)
Epithelial-mesenchymal transformation (EMT) is a process in which epithelial cells become depolarised and lose their characteristic adhesive nature, attaining migratory characteristics similar to mesenchymal cells.30 During EMT, epithelial cells undergo changes in protein expression and transcriptional events in response to extracellular stimuli, which can be reversible or irreversible.31 Epithelial cells lose their characteristic cell-to-cell adhesion and acquire similar features to the mesenchymal stem cells (MSCs). Treatment with docetaxel enriched for CD24+ cells in breast cancer cell lines suggests the generation of new cancer stem cells from non-stem cancer cells. This establishes a link between CD24 expression, chemotherapy resistance, and stem cell-like properties in cancer cells.32 The E-cadherin gene (CDH1), highly expressed in epithelial cells but lowly expressed in mesenchymal cells, is a controlling factor in EMT. When the expression level of E-cadherin is reduced, non-invasive epithelial cells gain invasiveness, increasing the metastatic potential of tumor cells.33 Breast cancer cells with a mesenchymal phenotype are more prone to developing resistance to chemotherapy drugs and are associated with cancer stem cells. EMT mediates resistance to apoptosis with cyclophosphamide administration in an EMT-tracing mouse model.34 Recent studies have clarified that there is a resemblance between EMT cells and cancer stem cells (CSCs), and these two cell types contribute to drug resistance in different ways. The possible mechanism is that EMT cells share many signaling molecules with CSCs, such as those from the Wnt, Notch, and Hedgehog pathways.35 Thus, cells acquire resistance against anticancer drugs and circumvent apoptosis induced by drugs. For instance, TGF-β, a central cytokine extensively studied in the context of EMT, has also been explored for its role in the induction of drug resistance. The inhibition of TGF-β can reverse the EMT process and, notably, increase the sensitivity of cancer cells to chemotherapies. This highlights the potential of targeting TGF-β to overcome drug resistance in cancer treatment.362.6 Tumor microenvironment
The tumor microenvironment comprises components that involve stromal cells (fibroblasts, immune system cells, and vascular cells), an extracellular matrix, soluble factors (growth factors, transcription factors, hormones, and cytokines), signaling molecules, hypoxia, and mechanical stress.37 These factors create a supportive environment for metastasis, alter the gene expression of normal cells, facilitate the transfer of miR, cytokines, and P-glycoprotein (P-gp) from resistant cells, and enhance their survival. Recent studies has shown a cross talk between myeloid derived suppressor cells (MDSC) and miRs, suggesting that miRs are able to promote cancer cells and invasion. In some studies, it was reported that MDSCs were able to overcome chemoresistance (doxorubicin resistance) in breast cancer.38 DOX-MDSCs released exosomal miR-126a, which encouraged the development of IL-13+ Th2 T cells. These T cells secreted IL-13, further enhancing the growth of DOX-MDSCs and the production of exosomal miR-126.39 Anticancer drugs disrupt cancer cell functions through various pathways, while also modifying the tumor microenvironment (TME). These treatments stimulate cancer-associated fibroblasts (CAFs) to release a range of cytokines that trigger signaling pathways, helping cancer cells evade destruction and potentially leading to recurrence.40Tumors typically grow under hypoxic conditions and rely on glycolysis for energy.41 As a result, the expression levels of metabolic enzymes increase, making cancer cells more resilient to chemotherapeutic drugs through changes in the cellular environment and metabolic pathways. The tumor microenvironment's pH can affect antitumor drug efficiency, which may help cell growth, metastasis, and drug resistance.42 In an acidic microenvironment, some drugs like doxorubicin, mitoxantrone, and epirubicin lose their ability to penetrate the tissue, reducing their efficacy in binding to target cancer cells.43 Hypoxia contributes to an acidic tumor microenvironment, leading to chemotherapy resistance. Therefore, the tumor microenvironment's pH conditions can influence chemotherapeutics' effectiveness, affecting cell proliferation, metastasis, and drug resistance.44,45
3. miR dysregulation in drug resistance
MicroRNAs (miR) are small, non-coding RNA molecules that can inhibit the translation of specific messenger RNA targets by binding to their 3′ untranslated region (UTR).46 The first miR was discovered in C. elegans in 1993.47 The biogenesis of miR and the mechanism of regulation can be stated as follows: (1) the miR gene is transcribed to Pri-miR by polymerase II in the nucleus, (2) Pri-miR is processed by the Drosha/DGCR8 (DiGeorge Syndrome Critical Region Gene) complex to release the intermediate precursor miR, (3) Pre-miR binds to the Exportin5 complex, (4) Pre-miR is then processed into double-stranded RNA by the Dicer complex in the cytoplasm, (5) the miR duplex is unwound into a single strand by the action of helicase, (6) the RNA strand with lower stability at the 5′ end will bind with the RNA-induced silencing complex (RISC), which forms mature RNA and (7) MiRISC binds to the 3′ untranslated region (UTR) of target miR, thus inhibiting translation.46 During the processing of miR, the expression and activity of the involved proteins are tightly regulated. miRs play a crucial role in regulating the expression of target genes, influencing various physiological and pathological processes. This regulatory mechanism significantly impacts tumor progression and the development of resistance to oncogenic therapies.48miRs are implicated in various human diseases, particularly in the development and progression of cancers, including ovarian, lung, gastric, colon, prostate, colorectal, pancreatic, cervical, and breast.49 Their dysregulation can occur genetically or epigenetically. Studies by Fazi et al. and Saito et al. underscore the significant role of epigenetic modifications, such as DNA methylation and histone acetylation, in regulating miR expression and their potential implications in cancer development and treatment. Fazi et al. discovered that the AML1/ETO fusion protein, commonly associated with acute myeloid leukemia, epigenetically silenced the expression miR-223 through CpG methylation.50 Saito et al. found that treating T24 bladder cancer cells with DNA methylation and histone acetylation inhibitors led to a significant upregulation of 17 out of 313 human miRs, including miR-127, which was accompanied by the downregulation of the proto-oncogene BCL6.51 These findings suggest that the epigenetic silencing of tumor suppressor miRs contributes to cancer development. Reversing these epigenetic modifications through inhibitors could potentially reactivate the expression of these miRs, thereby exerting therapeutic effects (Fig. 2).50
 | ||
| Fig. 2 Comparative diagram of the biogenesis of regulatory miRs: normal regulation (blue) and dysregulated process (red). | ||
RNase III enzymes Drosha and Dicer cleave miRs, crucially regulating miR production. The downregulation of these enzymes is observed in various malignancies, including breast cancer.52 Dicer is particularly significant in breast cancer progression, where lower protein levels serve as an independent predictive factor in both primary and metastatic stages. Studies highlight distinct miR profiles between cancerous and normal cells.53 Dysregulated miRs are implicated in tumor progression and resistance to cancer treatments. High-throughput sequencing and microarrays of miRs in breast cancer cell lines or tissues have identified multiple miRs linked to drug resistance, underscoring their role in cancer onset and progression.54 Approximately 25% of breast cancer cases in humans exhibit amplification or overexpression of the HER2 gene.55 HER2-positive breast cancer cells have a known correlation between the miR-21 expression and HER2/neu overexpression. Elevated miR-21 levels in these cells downregulate the expression of the tumor suppressor gene programmed cell death 4 (PDCD4), potentially through HER2/neu pathways, promoting invasiveness and metastasis. PDCD4, a critical tumor suppressor, is commonly downregulated in cancers and functions to inhibit neoplastic events. Various studies on the regulatory role of miRs in breast cancer drug resistance are shown in Table 1.
| MicroRNA | Expression levels | Resistant drug | Target | Function | Reference |
|---|---|---|---|---|---|
| miR-221 | Upregulated | Tamoxifen | P27 kip1 | Enhances proliferation and invasion | 56 |
| miR-155 | Upregulated | Doxorubicin | FOXO | Promotes proliferation and reduces apoptosis | 57 |
| miR-30b | Upregulated | Trastuzumab | CCNE2 | Induces the apoptosis pathway | 58 |
| miR-26a | Upregulated | Trastuzumab | CCNE2 | Induces the apoptosis pathway | 58 |
| miR-21 | Upregulated | Trastuzumab | PTEN, PDCD4 | Cell survival, apoptosis, resistance to systemic therapy | 59 |
| miR-107 | Downregulated | Paclitaxel | E2F1 | Promotes apoptosis | 60 |
| miR-7 | Downregulate | Trastuzumab | EGFR | Induces cell proliferation and metastasis | 61 |
| miR-128 | Upregulated | Letrozole | TGBR1 | Growth inhibitory effect of TGFβ | 62 |
| miR-214 | Downregulated | Tamoxifen | UCP2 | Inhibition of autophagy | 63 |
| miR-159 | Upregulated | Doxorubicin | Wnt | Promotes apoptosis | 64 |
| miR-16 | Upregulated | Doxorubicin | Bcl-2 | Induces tumor cell apoptosis | 65 |
| miR-298 | Upregulated | Doxorubicin | P-gp | Alters MDR modulation | 66 |
| miR-302b | Downregulated | Cisplatin | E2F1 | Inhibit cell cycle progression | 67 |
| Let-7a | Upregulated | Paclitaxel | ABCB10 | Promotes invasion and autophagy | 68 |
| miR-375 | Downregulated | Trastuzumab | IGF1R | Reverse EMT | 69 |
| miR-125 | Downregulated | Trastuzumab | ERBB2, ERBB3 | Reverse EMT | 70 |
| miR-200c | Downregulated | Trastuzumab | ZNF217, ZEB1 | Inhibit TGF-β signaling | 71 |
| miR-27b | Downregulated | Tamoxifen | HMGB3 | Promotes apoptosis | 72 |
| miR-124 | Upregulated | Doxorubicin | STAT3 | Resists the mechanism of cancer stem cells | 73 |
| miR-199b | Downregulated | Trastuzumab | HER2 | Inhibits ERK1/2 and AKT pathways | 74 |
3.1 Breast cancer subtypes
Breast cancer is categorized into four primary types based on their molecular characteristics, proliferation rate, and prognosis.75 (1) Luminal A: these receptor-positive tumors (with estrogen and progesterone receptors and a low proliferation rate) grow slower and have a better prognosis accordingly. (2) Luminal B: they, too, have a similar expression of receptors but with a higher proliferation rate, which indicates a more aggressive form of the disease and is not always considered curable.76 (3) Triple-negative: these tumors are characterized by the absence of expression of the estrogen receptor, progesterone receptor, and human epidermal growth factor receptor 2 (HER2). The cancer is typically more aggressive, and there is a tendency to have a poor prognosis.77 (4) HER2-overexpressing: in these tumor cells, the HER2 gene is markedly overexpressed, and hence, the HER2 protein accumulates in excess. HER2-positive breast cancers are usually the aggressive ones that have more chance of recurrence.78 Several treatments for breast cancer are currently available, including chemotherapeutic drugs, targeted therapies, and hormonal therapies.793.2 Clinical management of breast cancer
In the clinical management of breast cancer, surgery is typically succeeded by chemotherapy. Patients exhibiting over-expressed or amplified HER2 gain significant benefits from targeted therapeutic approaches. These include monoclonal antibodies against HER2, such as trastuzumab, and small molecule tyrosine kinase inhibitors, which are administered in conjunction with chemotherapeutic agents like doxorubicin, paclitaxel, docetaxel, cyclophosphamide, and carboplatin. Despite substantial progress in delineating the molecular profiles of luminal and HER2-positive breast cancers, the mechanistic foundations of TNBC remain inadequately defined. Consequently, chemotherapy remains the sole effective treatment modality for TNBC.80 TNBC patients are usually given chemotherapy treatments that include drugs like doxorubicin (DOX), cyclophosphamide (CFA), paclitaxel (PTX), or docetaxel (DTX). The common chemotherapy protocols for TNBC are DOX/CFA, DOX/CFA/PTX, and DTX/CFA.81 Despite the promising outcomes of second-generation anti-HER2 therapies in HER2-positive metastatic breast cancer, resistance to these treatments remains a significant challenge for the majority of patients. Advancing our understanding of the underlying mechanisms of drug resistance in HER2-targeted therapies is essential for improving patient outcomes.82The mechanisms of drug resistance are extensively studied, revealing that the activation of various signaling pathways, including Notch-1, RAS-MAPK, PI3K/mTOR, PI3K/AKT, and estrogen receptors (ERs), can induce resistance to a range of therapies.83 For example, Notch-1 activation in BRAF (V600E) melanoma cells and breast cancer cells results in acquired resistance to MAPK inhibitors and tamoxifen, respectively. Similarly, resistance to chemotherapy is associated with the epidermal growth factor receptor (EGFR) pathway, and the inhibition of PI3K/mTOR and EGFR has been shown to enhance drug sensitivity.84 Before the advent of targeted HER2 therapies, patients with HER2-positive breast cancer experienced worse clinical outcomes and prognoses. Resistance to HER2-targeted treatments can arise from poor binding to HER2, insufficient receptor inhibition, increased signaling from alternative receptors, and activation of the PIK3CA pathway. The non-specific nature of traditional chemotherapy and its concomitant toxicity to normal tissues underscore the critical need for alternative approaches. miR-based therapeutics present a compelling option, offering precise gene suppression and the ability to address resistance by concurrently modulating multiple signaling pathways.
4. Advantages of miR-based therapeutics
Traditional chemotherapy inhibits DNA replication in cancer cells but lacks specificity, causing significant harm to normal tissues. Therefore, the issue of low specificity and side effects on healthy tissues is a significant concern.85 In contrast, small noncoding miRs offer a significant advantage by effectively suppressing multiple target genes simultaneously, thereby modulating the expression of several genes of interest.86 Traditionally, most miR-based cancer research has concentrated on restoring or inhibiting the expression of miRs that are dysregulated in cancer cells. Recent advancements, however, have explored using miRs for targeted therapy delivery to specific cells or tissues, a strategy known as miR-based detargeting.87 miR-based therapy has two main approaches: miR replacement and miR inhibition.4.1 miR replacement
The strategy to deal with metastasis in cancer is to regain the levels of miRs that are under-expressed in the metastatic cancer cells. This can be achieved using miR mimics, small, synthetically produced double-stranded RNA molecules. These mimics include an antisense strand that matches the sequence of the downregulated endogenous miR.88 By applying these miR mimics, the expression and function of the targeted miR can be restored in metastatic cells, potentially halting or reversing the metastatic process.miR mimics hold significant therapeutic promise due to several key advantages. Firstly, as smaller molecules, miR mimics can efficiently enter the cytoplasm of target cells through systemic delivery methods. Secondly, numerous miRs are downregulated in cancer, indicating that their tumor-suppressive functions generally outweigh their oncogenic roles. Thirdly, miR mimics can target the same regulatory mRNAs as endogenous miRs due to their sequence similarity. For instance, miR-148b-3p mimics promote the expression of M2 macrophage-related genes (CD163, CD206, IL-10, and arginase-1), while reducing M1 macrophage-related genes (IL-1β and iNOS). Similarly, miR-365 mimics play a critical role in reducing breast cancer growth and enhancing chemo-resistance by targeting GALNT4, which is involved in glycosylation-based post-transcriptional protein modification essential for cell proliferation and tumor progression.74,89,90 Additionally, the tumor suppressor BRCA1 induces the expression of miR-145 and miR-205; thus, miR-145 and miR-205 mimics could be valuable therapeutic strategies for BRCA1-related breast cancer.91
4.2 miR inhibitors
An alternative approach to combating metastasis involves inhibiting the activity of cancer-promoting miRs that are upregulated during the metastatic process. By blocking these oncogenic miRs, the expression of tumor suppressor genes, typically silenced by these miRs, can be restored. miR inhibitors, single-stranded oligonucleotides designed to complement the sequence of the targeted miR, achieve this by binding to and sequestering the oncogenic miRs, preventing their incorporation into the RNA-induced silencing complex (RISC) and thus nullifying their gene-silencing effects.92One method of inhibiting miR progression is using antagomiRs, oligonucleotides 100% complementary to mature miRs. These molecules obstruct the miRISC complex, preventing miR degradation and allowing translation to proceed. Anti-miR oligonucleotides (AMOs), chemically modified, single-stranded DNA-like molecules with 17 to 22 nucleotides, are designed to specifically inhibit selected miRs.93 Current research is focused on enhancing the robustness, delivery, and targeting of AMOs in vivo, aiming to improve their precision and efficacy in clinical applications. AntagomiRs, particularly relevant in breast cancer, inhibit well-established oncogenic miRs (oncomiRs) such as miR-9 and miR-21. miR-21, for example, promotes cancer cell signaling by activating the phosphatidylinositol 3-kinase pathway via PTEN suppression and preventing apoptosis through the anti-apoptotic protein Bcl-2. Studies have shown that antagomiR-21, which inhibits miR-21 expression, increases apoptosis and reduces proliferation rates in breast cancer cells.94
miR sponges, artificially produced RNA molecules with multiple binding sites for specific miRs, offer another means of regulating miR levels.95 These sponges absorb endogenous miRs, reducing their availability and activity within the cell, thereby providing a valuable tool for modulating miR levels and understanding their roles in cellular processes.96
Modulating the expression of miRs represents a potential anti-cancer therapeutic strategy by inhibiting oncogenic miRs (oncomiRs) or restoring tumor suppressor miRs.97 Three prominent modulation approaches are (1) miR sponges, synthetic vectors expressing transcripts with binding sites complementary to the targeted miR and sequestering it from its miR targets. (2) Antisense oligonucleotides like antagomirs and anti-miRs are chemically modified oligonucleotides that can bind and inhibit miRs with high affinity. (3) Genetic knockouts utilizing the CRISPR/Cas9 genome-editing system to delete specific miR sequences.98
4.3 Challenges of miR based therapeutics in cancer
miR-based treatment options are gaining researchers' interest due to their potential in cancer therapy. miRs can target multiple proteins that play critical roles in tumor progression, drug resistance, and metastasis. However, significant challenges remain in translating this potential into effective anti-cancer therapies. The first challenge is related to the stability of miRs, as they are prone to degradation by nucleases present in the blood. This issue can be addressed through chemical modification of anti-miR oligonucleotides (e.g., 2′-O-methylation and phosphorothioate linkages).99 The involvement of a single miRNA in regulating multiple signaling pathways presents both opportunities and challenges. Off-target effects may lead to adverse outcomes.Immunogenicity against synthetic miRs or viral vectors is another concern. MRX34 trials for solid tumors were closed due to the immunological adverse effect in the phase 1 trials.100 Tumors often contain heterogeneous cell populations, and the impact of a single miR may not be sufficient across various tumor subtypes. Effective targeting of cancer cells remains a key challenge in cancer therapeutics, and the precise delivery of miRs to cancer cells is critical. Additionally, miR processing in cancer cells is frequently dysregulated, complicating their therapeutic use. Restoring miR function in such tumors presents further difficulties.99 Clinical data on the use of miRs in breast cancer treatment are limited, and mutations in cancer cells may pose challenges, potentially leading to resistance against miR-based treatments. Addressing these challenges will be essential for unlocking the full potential of miR-based therapies. Various strategies, such as more precise delivery systems, a deeper understanding of miR biology, and combination therapies, are being explored to overcome these obstacles.
5. miR delivery systems
miR based drug delivery systems constitute a key element of treatment approaches for breast cancer in a specific and targeted fashion. Delivery systems can deliver miRs to tumor cells, which allows miRs to modulate gene expression effectively. Enhancing the expression of tumor-suppressive miRs or silencing oncogenic miRs through miR therapy can disrupt pathways critical for tumor proliferation and metastasis. Modified carriers such as nanoparticles, liposomal vectors, and viral vectors are employed to deliver miRs effectively, ensuring better stability, bioavailability, and minimal off-target effects, thereby improving therapeutic outcomes.101 There are two main types of vectors used in miR delivery systems: viral and non-viral vectors, which are depicted in Table 2.| Advantages | Viral and non-viral based vectors | Disadvantages | References |
|---|---|---|---|
| Provides long-term gene expression by integrating into the host genome |

|
Risk of disrupting genes, potentially causing cancer | 102 |
| Can deliver genes into both dividing and non-dividing cells with long-term expression |
|
Risk of insertional mutagenesis and potential immune responses | 103 |
| Low immunogenicity and stable long-term expression |
|
Limited transgene capacity compared to other viral vectors | 104 |
| High specificity for bacterial targets, making them useful for treating bacterial infections |
|
Limited effectiveness in eukaryotic cells, restricting their use in gene therapy for human cells | 105 |
| Can encapsulate both hydrophilic and hydrophobic drugs, improving drug solubility and stability |

|
Prone to rapid clearance by the immune system, limiting circulation time | 106 |
| High surface area allows for versatile drug loading and targeted delivery |

|
Potential toxicity and accumulation in tissues, raising safety concerns | 107 |
| Enhances the solubility of poorly water-soluble drugs, improving bioavailability |

|
Potential instability in the bloodstream, leading to premature drug release | 108 |
| Naturally derived, making them biocompatible and less likely to trigger immune responses |
|
Challenges in large-scale production and standardization for therapeutic use | 109 |
| High drug-loading capacity and ability to penetrate cells efficiently |

|
Potential toxicity and difficulty in biodegradation, raising safety concerns | 110 |
| Highly branched structure allows for precise drug delivery and targeting capabilities |

|
High production costs and potential toxicity due to their size and surface chemistry | 111 |
| Provides a supportive framework for cell growth and tissue regeneration, mimicking natural extracellular matrices |

|
Complex fabrication processes and potential issues with biocompatibility and integration into surrounding tissues | 112 |
| Provides controlled and sustained drug release, enhancing therapeutic efficiency |

|
Potential for limited drug loading capacity and challenges in large-scale production | 113 |
5.1 Viral vector delivery
Viral vector based delivery systems for miR therapeutics commonly employ modified retroviruses, lentiviruses, adenoviruses, or adeno-associated viruses (AAVs) as vectors. These vectors are genetically modified to be replication-deficient, enhancing their safety profiles.114 The key advantage of using viral vectors is their high transfection efficiency, resulting in stable and elevated expression of the delivered miR mimics or antagomirs within the target cells. By leveraging viruses' natural cellular entry mechanisms, these platforms can effectively introduce and express desired miR modulators, enabling sustained gene expression modulation for therapeutic purposes.Retroviral vectors (RVs), derived from RNA viruses like the Moloney murine leukemia virus, integrate therapeutic genes into dividing cells. RVs have shown promise in regenerative medicine, such as enhancing induced pluripotent stem (iPS) cell production through miR-138 overexpression. However, integration can cause insertional inactivation of critical genes, posing a significant risk.115
Lentiviral vectors (LVs), developed from lentiviruses like HIV-2, can actively translocate across intact nuclear membranes, targeting both quiescent and non-quiescent cells.116 LVs have a reduced risk of insertional oncogenesis compared to RVs, as they preferentially integrate within actively transcribing units. They have been used effectively for delivering therapeutic miR mimics or antagonists, such as mitigating chronic metastasis using miR-494 sponges. Lentiviral vectors related to RNA interference can easily silence CLDN1 expression in breast cancer, leading to reduced cell proliferation, migration, invasion, and inhibition of epithelial-mesenchymal transition (EMT).117
Adenoviruses (Ads) and adeno-associated viruses (AAVs), non-enveloped viruses with double-stranded or single-stranded DNA genomes, are also used in miR delivery.118 Unlike other viral vectors, AAVs are non-toxic and can achieve prolonged gene expression. For example, AAV vector-mediated delivery of miR-196a has been shown to attenuate spinal and bulbar muscular atrophy phenotypes by silencing CELF2 and reducing androgen receptor mRNA stability.97
5.2 Non-viral delivery
Non-viral delivery systems aim to facilitate the successful delivery of miR molecules or miR-expressing vectors into target cells while protecting them from nuclease degradation. These approaches include chemical methods such as lipid, polymer, inorganic, and extracellular vesicle-based carrier systems.119 Non-viral systems are less toxic, more biocompatible, and cost-effective compared to viral vector-based delivery systems.120Lipid-based carriers like liposomes and nanoparticles can encapsulate and protect miR molecules, delivering them into cells via membrane fusion or endocytosis.121 Polymer carriers, such as PEI and PLGA, can condense and complex with miRs, prolonging their lifespan, preventing degradation, and supporting cellular uptake. Inorganic nanocarriers, including gold nanoparticles, carbon nanotubes, 3-D scaffolds, and mesoporous silica nanoparticles, are notable for miR transfers. Nanovesicles can be target-specific by labeling them with targeting moieties and loading them with miR molecules using, for instance, surface adsorption and encapsulation inside the porous structure.122
Extracellular vesicles, such as exosomes and microvesicles, have shown great potential as natural carriers for miR delivery.123 These vesicles can incorporate therapeutic miR and efficiently deliver the cargo to target cells with low immunogenicity and toxicity. Researchers focus on improving non-viral delivery systems to enhance safety and biocompatibility for precise gene expression regulation in various disease models, including cancer. Gold nanoparticles conjugated with miR-206 have a potential role in targeted drug delivery in overcoming breast cancer (Fig. 3).107
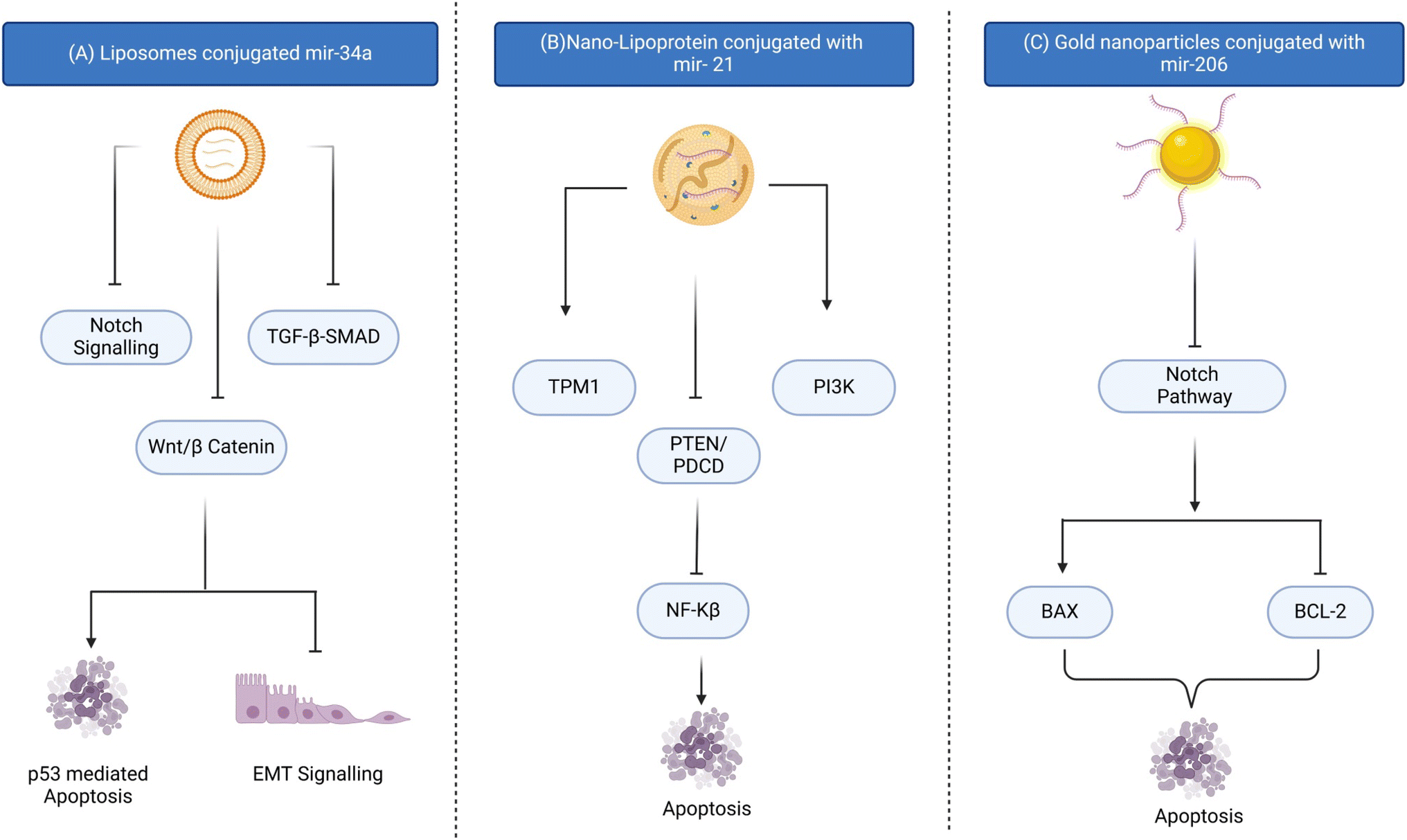 | ||
| Fig. 3 Schematic of recent studies related to nanocarriers conjugated with miRs. (A) Liposomes conjugated with the miR-34a mimic; miR-34a can induce apoptosis and inhibit EMT signalling and enhance sensitivity to chemotherapy by inhibiting the notch signalling, TGF-β-SMAD and Wnt/β catenin pathways, (B) lipoprotein conjugated with the miR-21 inhibitor; it induces TPM1 and PI3K inhibits PTEN/PDCD, inducing apoptosis, and (C) gold nanoparticles conjugated with the mir-206 mimic; miR-206 when given via the gold nano-complex stops cell proliferation, induces G0–G1 cell arrest and changes the mitochondrial membrane potential. | ||
6. Combination treatment strategies
Multi-modal therapy offers the potential to increase treatment effectiveness, reduce side effects, and prevent drug resistance by combining multiple medications.124 miRs regulate approximately 30% of human genes, with about 50% related to tumor development or progression. The contrasting mechanisms of miRs and targeted agents can simultaneously control and modify multiple pathways and targets, making combination therapies more efficacious and beneficial for cancer patients. Multi-arm clinical trials are also ongoing to check the efficacy of combination therapies, including pevonedistat (vorinostat), belinostat, and romidepsin for the treatment of breast cancer, metastatic breast cancer, metastatic castration-resistant prostate cancer, and metastatic ovarian cancer. Romidepsin has also been clinically evaluated as a candidate in combination with cisplatin nivolumab against metastatic TNBC patients.125 In recent years, miR profiling in various cancers has revealed that these molecules can be either overexpressed or have lost their function in tumor tissues compared to normal tissues.126 OncomiRs, overexpressed in tumors, promote abnormal growth by negatively regulating tumor suppressor genes. Conversely, anti-oncomiRs suppress tumor growth and exhibit anti-cancer effects. Multiple studies have demonstrated that miRs are crucial in regulating resistance to chemotherapy drugs by targeting and regulating mRNAs involved in chemo-resistance.127 By altering key cell signaling pathways, miRs can significantly influence a cancer cell's response to chemotherapeutic agents, decreasing resistance or increasing sensitivity.128In the field of cancer treatment, the term “multidrug resistance (MDR)” refers to the capability of cancer cells to survive exposure to a wide range of anticancer drugs.129,130 MDR is a complex phenomenon where cancer cells become nearly invulnerable or insensitive to various drugs, regardless of their molecular targets. This resistance significantly obstructs chemotherapy success and often leads to treatment failure.123 However, the cellular pathways and biomarkers defining MDR and the interactions between these factors remain largely unknown, necessitating further investigation. Research has highlighted the regulatory roles of microRNAs (miRs) in initiating the MDR phenotype by modulating various biological processes. Trastuzumab and lapatinib, for instance, have been found to alter the types and levels of miRNAs in different breast cancer cell lines.131
Using miR mimics (synthetic miRs) and inhibitors alongside specific anticancer drugs is another strategy to overcome drug resistance by targeting multiple possible resistance mechanisms.132 For example, miR-21 is one of the miRs that confer drug resistance by significantly altering the expression of many genes and pathways. Nanoparticle-based drug delivery mechanisms and miR therapy help prolong the treatment duration and combat drug resistance by modifying oncogenes and molecular pathways in tumor development.133 Smart drug delivery systems (SDDSs) can also be co-delivered to cancer cells or cells with an immunosuppressive role to improve their efficacy at the target site by increasing drug localization.134 This method addresses problems like the immunosuppressive microenvironment and resistance to immunomodulatory medicines, thereby enhancing cancer immunotherapy efficiency. The co-delivery of miRs and chemical drugs provides a powerful platform to overcome drug resistance in cancer treatment by simultaneously targeting multiple pathways involved in resistance.135 For instance, the combination of miR-34a and docetaxel with core–shell nanocarriers has shown a synergistic effect on overcoming breast cancer proliferation.136
While monotherapy treatments have success in some cases, patients with metastatic breast cancer often do not respond to single treatments. Developing resistance and ineffectiveness in specific patient populations necessitate more effective therapeutics for better disease outcomes. Researchers have demonstrated clinical benefits associated with combining miR mimics/inhibitors with diverse breast cancer therapies. For instance, miR mimics and chemotherapeutic agents can achieve high treatment efficacy by targeting cancer stem cells and increasing chemotherapy sensitivity.137 For example, miRs targeting HER2 were revealed to make cancer cells sensitive to antibodies against HER2 (trastuzumab) and tyrosine kinase inhibitors (lapatinib), which might improve the treatment outcome.138 Additionally, combining multiple miRs, such as miR-124, miR-128, and miR-137, has shown superior efficacy in disrupting cancer phenotypes and restraining glioma stem cell growth. These results suggest that multi-miR combinations could serve as important biomarkers for early detection of breast cancer and identification of cancer subtypes.19
MicroRNAs (miRs) represent promising candidates for novel therapeutic agent development, considering their multifaceted advantages. Their small size and structural stability, conserved across species, facilitate targeted molecule synthesis, offering significant potential in oncological and molecular biology applications.101 Furthermore, the co-administration of letozole (an aromatase inhibitor) with sorafenib (a multi-kinase inhibitor) has demonstrated synergistic effects by inhibiting cancer cell proliferation in hormone-dependent breast cancer.139 This indicates that combining these targeted drug approaches could be highly effective for treating this type of breast cancer. When integrated with targeted therapies, miR strategies promise to enhance treatment outcomes across all types of cancer, adding significant value to cancer treatment.
7. Preclinical and clinical evidence
Over the past two decades, research has highlighted the crucial role of miRs in development, evolution, and disease. Several preclinical and clinical studies have been carried out to decipher the role of different malignancies.140,141 Joshi et al.142 investigated miRNA-mediated gene regulation in tamoxifen-resistant breast cancer cells (TamR). This analysis found 22 miRs that are typically changed across TamR lines. It highlights miR-regulated genes such as ESR1, PGR1, FOXM1, and 14-3-3, as well as SNAI2 and FYN, which contribute to TamR proliferation. Furthermore, the expression of miRs such as miR-190b and miR-516a-5p may aid in predicting treatment outcomes in ER+ breast cancer patients, providing insights for future therapeutic interventions. miRNAs like miR-128 and miR-223, which regulate metabolism, could be targeted to reduce tamoxifen resistance by depleting excess cholesterol.143Researchers have also explored the synergistic effect of miR mimics with trastuzumab and lapatinib in breast cancer cell lines. Here, they found that higher expression of miR-101-5p was associated with better prognosis in HER2-positive breast cancer patients, underscoring its tumor-suppressing role and potential in overcoming treatment resistance.131 The study by Yixue Hu et al. demonstrated that the combination therapy of triphenylphosphine-tocopherol polyethylene glycol succinate nanoparticles encapsulated with chloride e6 and miR-34a effectively enhanced photodynamic therapy (PDT) in TNBC. This co-delivery system synergistically improved the anti-tumor effects of miRNA and PDT in aggressive tumors, which is evident by enhanced apoptosis of cancer cells and reduced tumor cell migration and invasion.144 This combination had enhanced tumor targeting compared to chloride e6 alone.
The treatment of miR-143 mimics inhibited cell proliferation and migration and increased sensitivity to cisplatin therapy in breast cancer cells. It reduced the levels and phosphorylation of oncoproteins in key pathways, like AKT, WNT/β-catenin, SAPK/JNK, FAK, and JAK/STAT. This suggested that miR-143 may predict response to neoadjuvant therapy and regulate signaling networks to suppress cell proliferation and migration in breast cancer.145 Those results from preclinical research strongly suggest the tremendous efficacy of miR-based combination therapy in circumventing drug resistance. It will thus be revolutionary for breast cancer clinical care, as represented in Fig. 4.
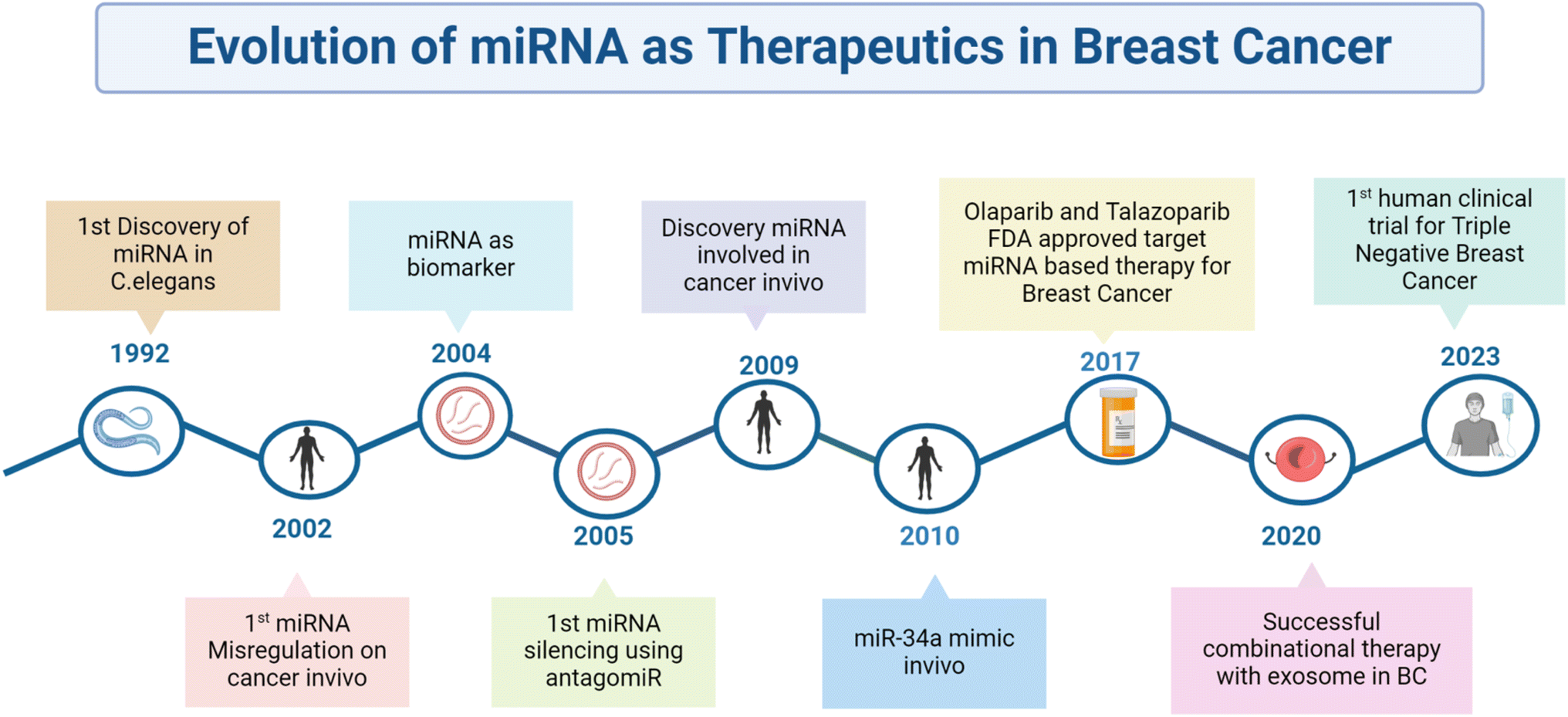 | ||
| Fig. 4 Schematic representation of the timeline in which miRs have evolved to therapeutic use in breast cancer. | ||
The potential of miRs in regulating cell signaling pathways paves the way for an in-depth study of potential miRs in a clinical setup. However, there are limited studies on miRs and breast cancer; here, we have covered some of the promising clinical studies that have been done for various cancers. A randomized clinical trial was carried out to study the differential expression profile of 84 miRs in normal and breast tumor tissues (ClinicalTrials.gov identifier: NCT04516330). Similarly, miR-155 was explored for its diagnostic potential in bladder cancer (ClinicalTrials.gov identifier: NCT03591367). In another study, researchers explored new miR markers for colorectal cancer detection using fecal samples (ClinicalTrials.gov identifier: NCT05346757) (Table 3).151
| MicroRNA name | Targeted miRNA | Mode of action | Background disease | Route of administration | Clinical trial number(s) | References |
|---|---|---|---|---|---|---|
| MRX34 | miR-34a | miRNA mimic | Solid tumors | Intravenously/vehicle transfer (liposomal) | NCT01829971, NCT02862145 | 146 |
| MesomiR 1d | miR-16 | miRNA mimic | Malignant pleural mesothelioma, non-small cell lung cancer | Intravenously/vehicle transfer (non-living minicells) | NCT02369198 | 147 |
| MRG-106 (cobomarsen) | miR-155 | miRNA mimics | Adult T-cell leukaemia/lymphoma (ATLL) | Intravenously/vehicle | NCT03713320 | 148 |
| INT-1B3 | miR-193a-3p | miRNA mimics | Triple-negative breast cancer (TNBC), NSCLC, melanoma, colon cancer, and HCC | Intravenously/vehicle lipid nanoparticles | NCT04675996 | 149 |
| TTX-MC138 | miR-10b | miRNA inhibitor | Brain cancer, breast cancer | Dextran-coated iron oxide nanoparticles | NCT01849952 | 150 |
The potential of miRs as predictors of cancer therapy efficacy has been widely researched. In metastatic castration-resistant prostate cancer, miRs are clinically profiled to serve as predictive markers for chemotherapy effectiveness (ClinicalTrials.gov identifier: NCT04662996), and the correlation between miR-141 and miR-375 with radiation resistance is also being studied in prostate cancer in another study (ClinicalTrials.gov identifier: NCT02391051). Additionally, the capability of miRs to predict the success of targeted immunotherapies has been explored, with exosomal miR profiling in lung cancer patients receiving immunotherapy targeting PD-1 or PD-L1 (ClinicalTrials.gov identifier: NCT04427475). The relationship between blood miR levels and resistance to chemotherapy drugs (epirubicin, cyclophosphamide, paclitaxel, and carboplatin) is under investigation in TNBC patients (ClinicalTrials.gov identifier: NCT04771871). Additionally, the serum levels of microRNA (miR-M371) were assessed as indicators of resistance to chemotherapy and radiotherapy in testicular germ cell tumors (ClinicalTrials.gov identifier: NCT05529251).
miR-34a is crucial in cancer treatment due to its regulation by the tumor suppressor p53, which is often altered in cancers. The MRX34, a miR-34a mimic, showed promise in the first phase of the trial but faced setbacks due to immune-related adverse events in the advanced phase of clinical trials.100 A key observation from the study was that miR-34a effectively reduced oncogene expression, highlighting its potential in overcoming drug resistance in breast cancer.152 Future advancements in synthetic miR mimics and delivery systems could establish miR-34a as a vital component of breast cancer therapy while reducing the target effects of miRNA-based therapeutic agents. Given the extensive evidence linking miRs to resistance against various cancer treatments, there is a compelling need for more clinical trials to validate the utility of miRs as predictive and prognostic markers in cancer therapy.
8. Future perspectives
Recent advancements in gene therapy have revolutionized the field of cancer treatment, offering new hope for patients. They can enhance treatment outcomes by directly targeting and modifying genes playing a role in tumor growth and survival pathways. Innovations such as CRISPR, miR mimics, and viral vector technologies have significantly improved the precision and efficacy of these treatments, paving the way for personalized and more effective cancer therapies. However, conditions like inflammatory diseases and cancer necessitate precise control over effectively delivering the miR to the target site to minimize systemic toxicity.153 miRs have emerged as a crucial tool in predicting and treating drug-resistant cells in breast cancer and downregulating or upregulating gene expression directly affecting the drugs' pharmacokinetics.154In the quest to combat drug resistance in breast cancer, adopting a comprehensive approach that involves repurposing existing drugs, pinpointing new therapeutic targets, and employing advanced treatment techniques is highly warranted. The successful transition of miR-based therapies from laboratory research to clinical application relies heavily on key steps, like enhancing the stability of miR mimics or anti-miR oligonucleotides and developing safe and efficient delivery systems. Nanotechnology emerges as a promising avenue, empowering the creation of nano-carriers capable of reducing adverse effects on normal cells by delivering drugs directly to breast cancer tissues. Nanoparticles, including liposomes and lipid carriers, have successfully targeted drug delivery, particularly when modified with HER-2 antibodies to create immunoliposomes. These nanostructures, loaded with miRs and anticancer drugs like doxorubicin (DOX), demonstrate enhanced specificity for HER-2 positive breast cancer cells, thanks to HER-2-mediated endocytosis. Clinical trials for lipid nanoparticles containing the INT-1B3 drug and mir-193a-3p are in the initial phase of a clinical trial for different cancer types, including TNBC.152
Furthermore, synergistic anticancer effects are observed with nanocarriers such as pegylated liposomes, aptamers, exosomes, and nanoflowers. These carriers efficiently target cancer cells through specific receptors and release their cargo directly into the nucleus of proliferating tumor cells, showing great promise in enhancing breast cancer treatment outcomes.
9. Conclusion
Drug resistance is the major obstacle to successful patient outcomes, necessitating strategies to manage it effectively. The role of miRs is well established in various diseases; as a result, there are preclinical and clinical studies exploring their potential in disease management. miR-based bio-therapeutics hold promise against MDR. The involvement of miRs in breast cancer is well-established and supported by an extensive and continuously growing body of scientific literature. In this review, we examined the role of miRs in drug resistance, emphasizing breast cancer and their possible role in combination therapy. miRs interact with various cellular components, including proteins, messenger RNA, and non-encoding RNA.The use of miRs as biomarkers for disease diagnosis and monitoring treatment outcomes has been extensively investigated. In cancer therapy, miRs hold immense potential, especially in overcoming challenges like chemo resistance, as outlined in the previous sections. Recent advancements in molecular biology and bioengineering are now poised to address the limitations associated with miR-based therapies. One such advancement is the bio-switchable miR mimic delivery system (BiRDS), loaded with miR-27a mimics, which offers advantages, including efficient synthesis, improved storage stability, biocompatibility, and biosafety.155 Integrating miR delivery systems with chemotherapeutic agents could enhance therapeutic outcomes, particularly in breast cancer management. Continued research and development in these areas will be critical to harnessing the full potential of miR-based treatments. Despite the hurdles, with continued innovation and refinement, miR therapies, combined with conventional approaches, could significantly advance cancer treatment, especially in the battle against chemo resistance.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
Aditi Verma: writing and image curation. Krunal Patel: conceptualization and editing. Ashutosh Kumar: conceptualization, supervision, editing and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Indian Council of Medical Research (ICMR), Government of India [grant number: EM/Dev/SG/217/1078/2023]. The figures are created using https://www.biorender.com/.References
- R. L. Siegel, A. N. Giaquinto and A. Jemal, Ca-Cancer J. Clin., 2024, 74(1), 12–49 CrossRef PubMed.
- M. Nikolaou, A. Pavlopoulou, A. G. Georgakilas and E. Kyrodimos, Clin. Exp. Metastasis, 2018, 35, 309–318 CrossRef CAS.
- M. Abolghasemi, S. S. Tehrani, T. Yousefi, A. Karimian, A. Mahmoodpoor, A. Ghamari, F. Jadidi-Niaragh, M. Yousefi, H. S. Kafil and M. Bastami, J. Cell. Physiol., 2020, 235, 5008–5029 CrossRef CAS.
- C. Falato, F. Schettini, T. Pascual, F. Brasó-Maristany and A. Prat, Cancer Treat. Rev., 2023, 112, 102496 CrossRef CAS.
- X. Li, M. T. Lewis, J. Huang, C. Gutierrez, C. K. Osborne, M.-F. Wu, S. G. Hilsenbeck, A. Pavlick, X. Zhang and G. C. Chamness, J. Natl. Cancer Inst., 2008, 100, 672–679 CrossRef CAS.
- P. Fan and V. C. Jordan, Cancer Drug Resist., 2019, 2, 198 Search PubMed.
- M. T. Herrera-Abreu, M. Palafox, U. Asghar, M. A. Rivas, R. J. Cutts, I. Garcia-Murillas, A. Pearson, M. Guzman, O. Rodriguez and J. Grueso, Cancer Res., 2016, 76, 2301–2313 CrossRef CAS PubMed.
- J. Tang, A. Ahmad and F. H. Sarkar, Int. J. Mol. Sci., 2012, 13, 13414–13437 CrossRef CAS.
- B. Kinnel, S. K. Singh, G. Oprea-Ilies and R. Singh, Cancers, 2023, 15, 1320 CrossRef CAS.
- K. Jamialahmadi, F. Zahedipour and G. Karimi, J. Pharm. Pharmacol., 2021, 73, 997–1006 CrossRef PubMed.
- R. Krishna Vadlapatla, A. Dutt Vadlapudi, D. Pal and A. K. Mitra, Curr. Pharm. Des., 2013, 19, 7126–7140 CrossRef.
- K. Natarajan, Y. Xie, M. R. Baer and D. D. Ross, Biochem. Pharmacol., 2012, 83, 1084–1103 CrossRef CAS.
- J. He, E. Fortunati, D.-X. Liu and Y. Li, Int. J. Mol. Sci., 2021, 22, 3199 CrossRef CAS.
- M. Nedeljković, N. Tanić, M. Prvanović, Z. Milovanović and N. Tanić, Breast Cancer, 2021, 28, 727–736 CrossRef PubMed.
- K. Bukowski, M. Kciuk and R. Kontek, Int. J. Mol. Sci., 2020, 21, 3233 CrossRef CAS.
- J. Zhu, Z. Zou, P. Nie, X. Kou, B. Wu, S. Wang, Z. Song and J. He, Cell Death Dis., 2016, 7, e2454 CrossRef.
- H. Pan, H. Peng, Y. Dai, B. Han, G. Yang, N. Jiang and P. Zhou, J. Oncol., 2023, 8, 1244067 Search PubMed.
- R. M. Connolly, H. Li, R. C. Jankowitz, Z. Zhang, M. A. Rudek, S. C. Jeter, S. A. Slater, P. Powers, A. C. Wolff and J. H. Fetting, Clin. Cancer Res., 2017, 23, 2691–2701 CrossRef CAS PubMed.
- S.-J. Shen, Y. Song, X.-Y. Ren, Y.-L. Xu, Y.-D. Zhou, Z.-Y. Liang and Q. Sun, Front. Oncol., 2020, 10, 1371 CrossRef PubMed.
- S. Mohammed, A. A. Shamseddine, B. Newcomb, R. S. Chavez, T. D. Panzner, A. H. Lee, D. Canals, C. M. Okeoma, C. J. Clarke and Y. A. Hannun, Breast Cancer Res., 2021, 23, 76 CrossRef CAS PubMed.
- M. Wang, S. Chen and D. Ao, MedComm, 2021, 2, 654–691 CrossRef CAS.
- D. Jurkovicova, C. M. Neophytou, A. Č. Gašparović and A. C. Gonçalves, Int. J. Mol. Sci., 2022, 23, 14672 CrossRef CAS.
- P. Ter Brugge, P. Kristel, E. Van Der Burg, U. Boon, M. De Maaker, E. Lips, L. Mulder, J. De Ruiter, C. Moutinho and H. Gevensleben, J. Natl. Cancer Inst., 2016, 108, djw148 CrossRef.
- Z. Nikitaki, I. Michalopoulos and A. G. Georgakilas, Future Med. Chem., 2015, 7, 1543–1558 CrossRef CAS.
- F. Dietlein, L. Thelen and H. C. Reinhardt, Trends Genet., 2014, 30, 326–339 CrossRef CAS PubMed.
- R. A. Ward, S. Fawell, N. Floc’h, V. Flemington, D. McKerrecher and P. D. Smith, Chem. Rev., 2020, 121, 3297–3351 CrossRef PubMed.
- J. M. Fortune and N. Osheroff, 2000.
- Q. Feng, Z. Zhang, M. J. Shea, C. J. Creighton, C. Coarfa, S. G. Hilsenbeck and B. W. O’malley, Cell research, 2014, 24(7), 809–819 CrossRef CAS.
- C. J. Fabian, Int. J. Clin. Pract., 2007, 61, 2051–2063 CrossRef CAS PubMed.
- A. Modi, P. Purohit, D. Roy, J. R. Vishnoi, P. Pareek, P. Elhence, P. Singh, S. Sharma, P. Sharma and S. Misra, Mol. Biol. Rep., 2022, 49, 2877–2888 CrossRef CAS.
- J. Felipe Lima, S. Nofech-Mozes, J. Bayani and J. M. Bartlett, J. Clin. Med., 2016, 5, 65 CrossRef.
- H. W. Huth, T. Castro-Gomes, A. M. de Goes and C. Ropert, Sci. Rep., 2021, 11, 17077 CrossRef CAS PubMed.
- J. Yang, P. Antin, G. Berx, C. Blanpain, T. Brabletz, M. Bronner, K. Campbell, A. Cano, J. Casanova and G. Christofori, Nat. Rev. Mol. Cell Biol., 2020, 21, 341–352 CrossRef CAS PubMed.
- F. Lüönd, N. Sugiyama, R. Bill, L. Bornes, C. Hager, F. Tang, N. Santacroce, C. Beisel, R. Ivanek and T. Bürglin, Dev. Cell, 2021, 56, 3203–3221 CrossRef.
- S.-s. Wang, J. Jiang, X.-h. Liang and Y.-l. Tang, OncoTargets Ther., 2015, 2973–2980 CAS.
- M. Zhang, Y. Y. Zhang, Y. Chen, J. Wang, Q. Wang and H. Lu, Front. Cell Dev. Biol., 2021, 9, 786728 CrossRef PubMed.
- S. Taylor, E. P. Spugnini, Y. G. Assaraf, T. Azzarito, C. Rauch and S. Fais, Drug Resist. Updates, 2015, 23, 69–78 CrossRef.
- L. Liang, X. Xu, J. Li and C. Yang, Front. Immunol., 2022, 13, 883683 CrossRef CAS.
- Z. Deng, Y. Rong, Y. Teng, X. Zhuang, A. Samykutty, J. Mu, L. Zhang, P. Cao, J. Yan and D. Miller, Oncogene, 2017, 36, 639–651 CrossRef CAS.
- L. Bu, H. Baba, T. Yasuda, T. Uchihara and T. Ishimoto, Cancer Sci., 2020, 111, 3468–3477 CrossRef CAS.
- R. Wang, A. Hussain, Q. Q. Guo, X. W. Jin and M. M. Wang, World J. Oncol., 2024, 15, 28 CrossRef CAS.
- P. Swietach, R. D. Vaughan-Jones, A. L. Harris and A. Hulikova, Phil. Trans. Roy. Soc. Lond. B, 2014, 369, 20130099 CrossRef PubMed.
- O. Trédan, C. M. Galmarini, K. Patel and I. F. Tannock, J. Natl. Cancer Inst., 2007, 99, 1441–1454 CrossRef.
- Y. Li, L. Zhao and X.-F. Li, Technol. Cancer Res. Treat., 2021, 20, 15330338211036304 CrossRef CAS PubMed.
- N. A. Seebacher, M. Krchniakova, A. E. Stacy, J. Skoda and P. J. Jansson, Antioxidants, 2021, 10, 1801 CrossRef CAS PubMed.
- B. M. Hussen, H. J. Hidayat, A. Salihi, D. K. Sabir, M. Taheri and S. Ghafouri-Fard, Biomed. Pharmacother., 2021, 138, 111528 CrossRef CAS PubMed.
- B. Wightman, I. Ha and G. Ruvkun, Cell, 1993, 75, 855–862 CrossRef CAS.
- W. Hu, C. Tan, Y. He, G. Zhang, Y. Xu and J. Tang, OncoTargets Ther., 2018, 1529–1541 CrossRef.
- E. van Schooneveld, H. Wildiers, I. Vergote, P. B. Vermeulen, L. Y. Dirix and S. J. Van Laere, Breast Cancer Res., 2015, 17, 1–15 CrossRef.
- F. Fazi, S. Racanicchi, G. Zardo, L. M. Starnes, M. Mancini, L. Travaglini and C. Nervi, Cancer cell, 2007, 12(5), 457–466 CrossRef CAS.
- Y. Saito, G. Liang, G. Egger, J. M. Friedman, J. C. Chuang, G. A. Coetzee and P. A. Jones, Cancer Cell, 2006, 9, 435–443 CrossRef CAS PubMed.
- M. Yan, H.-Y. Huang, T. Wang, Y. Wan, S.-D. Cui, Z.-z. Liu and Q.-X. Fan, Pathol. Oncol. Res., 2012, 18, 343–348 CrossRef CAS.
- L. Bhagtaney, A. Dharmarajan and S. Warrier, Cancers, 2024, 16, 957 CrossRef CAS.
- K. R. Kutanzi, O. V. Yurchenko, F. A. Beland, V. F. Checkhun and I. P. Pogribny, Clin. Epigenet., 2011, 2, 171–185 CrossRef CAS PubMed.
- X. Wang, H. Zhang and X. Chen, Cancer Drug Resist., 2019, 2, 141 Search PubMed.
- A. Amiruddin, M. N. Massi, A. A. Islam, I. Patellongi, M. Y. Pratama, N. Sutandyo, R. Natzir, M. Hatta, N. H. M. Latar and S. Wahid, Ann. Med. Surg., 2022, 73, 103092 Search PubMed.
- W. Kong, L. He, M. Coppola, J. Guo, N. N. Esposito, D. Coppola and J. Q. Cheng, J. Biol. Chem., 2010, 285, 17869–17879 CrossRef CAS.
- E. Tormo, A. Adam-Artigues, S. Ballester, B. Pineda, S. Zazo, P. González-Alonso, J. Albanell, A. Rovira, F. Rojo and A. Lluch, Sci. Rep., 2017, 7, 41309 CrossRef CAS.
- D. Bautista-Sánchez, C. Arriaga-Canon, A. Pedroza-Torres, I. A. De La Rosa-Velázquez, R. González-Barrios, L. Contreras-Espinosa, R. Montiel-Manríquez, C. Castro-Hernández, V. Fragoso-Ontiveros and R. M. Álvarez-Gómez, Mol. Ther. Nucleic Acids, 2020, 20, 409–420 CrossRef PubMed.
- C. Ma, X. Shi, W. Guo, J. Niu and G. Wang, Open Med., 2019, 14, 456–466 CrossRef PubMed.
- F. C. Huynh and F. E. Jones, PLoS One, 2014, 9, e114419 CrossRef PubMed.
- S. Masri, Z. Liu, S. Phung, E. Wang, Y.-C. Yuan and S. Chen, Breast Cancer Res. Treat., 2010, 124, 89–99 CrossRef CAS PubMed.
- X. Yu, A. Luo, Y. Liu, S. Wang, Y. Li, W. Shi, Z. Liu and X. Qu, Mol. Cancer, 2015, 14, 1–16 Search PubMed.
- C. Gong, J. Tian, Z. Wang, Y. Gao, X. Wu, X. Ding, L. Qiang, G. Li, Z. Han and Y. Yuan, J. Nanobiotechnol., 2019, 17, 1–18 CrossRef CAS.
- X. Jiang, Y. Liu, G. Zhang, S. Lin, J. Wu, X. Yan, Y. Ma and M. Ma, Evid. base Compl. Alternative Med., 2020, 2020, 5108298 CrossRef.
- L. Bao, S. Hazari, S. Mehra, D. Kaushal, K. Moroz and S. Dash, Am. J. Pathol., 2012, 180, 2490–2503 CrossRef CAS.
- A. Cataldo, D. G. Cheung, A. Balsari, E. Tagliabue, V. Coppola, M. V. Iorio, D. Palmieri and C. M. Croce, Oncotarget, 2016, 7, 786 Search PubMed.
- W. Yang, P. Gong, Y. Yang, C. Yang, B. Yang and L. Ren, Cancer Manag. Res., 2020, 2327–2337 CrossRef CAS PubMed.
- X.-M. Ye, H.-Y. Zhu, W.-D. Bai, T. Wang, L. Wang, Y. Chen, A.-G. Yang and L.-T. Jia, BMC Cancer, 2014, 14, 1–12 Search PubMed.
- G. Bertoli, C. Cava and I. Castiglioni, Theranostics, 2015, 5, 1122 CrossRef CAS PubMed.
- W. D. Bai, X. M. Ye, M. Y. Zhang, H. Y. Zhu, W. J. Xi, X. Huang, J. Zhao, B. Gu, G. X. Zheng and A. G. Yang, Int. J. Cancer, 2014, 135, 1356–1368 CrossRef CAS PubMed.
- X. Li, Y. Wu, A. Liu and X. Tang, Biochem. Biophys. Res. Commun., 2016, 477, 768–773 CrossRef CAS PubMed.
- C. Liu, H. Xing, C. Guo, Z. Yang, Y. Wang and Y. Wang, Cell Cycle, 2019, 18, 2215–2227 CrossRef CAS PubMed.
- L. Yuan, Y. Liu, Y. Qu, L. Liu and H. Li, Front. Oncol., 2019, 9, 1076 CrossRef.
- B. Weigelt, F. C. Geyer and J. S. Reis-Filho, Mol. Oncol., 2010, 4, 192–208 CrossRef CAS PubMed.
- P. Maisonneuve, D. Disalvatore, N. Rotmensz, G. Curigliano, M. Colleoni, S. Dellapasqua, G. Pruneri, M. G. Mastropasqua, A. Luini and F. Bassi, Breast Cancer Res., 2014, 16, 1–9 CrossRef PubMed.
- P. Zagami and L. A. Carey, NPJ Breast Cancer, 2022, 8, 95 CrossRef CAS PubMed.
- E. Hamilton, M. Shastry, S. M. Shiller and R. Ren, Cancer Treat. Rev., 2021, 100, 102286 CrossRef CAS PubMed.
- N. Jayaraj, S. Kar, S. Mohammed, K. Sabarimurugan, G. Priyadharshini, G. Ramesh, S. Baxi and S. Krishnan, Cells, 2019, 8, 1250 CrossRef PubMed.
- H. Muley, R. Fado, R. Rodriguez-Rodriguez and N. Casals, Biochem. Pharmacol., 2020, 177, 113959 CrossRef CAS PubMed.
- H. A. Wahba and H. A. El-Hadaad, Cancer Biol Med., 2015, 12, 106 CAS.
- S. Pernas and S. M. Tolaney, Ther. Adv. Med. Oncol., 2019, 11, 1758835919833519 CrossRef CAS PubMed.
- C. Dong, J. Wu, Y. Chen, J. Nie and C. Chen, Front. Pharmacol., 2021, 12, 628690 CrossRef CAS.
- J. Villanueva, A. Vultur, J. T. Lee, R. Somasundaram, M. Fukunaga-Kalabis, A. K. Cipolla, B. Wubbenhorst, X. Xu, P. A. Gimotty and D. Kee, Cancer Cell, 2010, 18, 683–695 CrossRef CAS.
- S. Nasra, R. Chaudhari and A. Kumar, in Nanomedicine for Cancer Diagnosis and Therapy, Springer, 2021, pp. 115–132 Search PubMed.
- M. Ratti, A. Lampis, M. Ghidini, M. Salati, M. B. Mirchev, N. Valeri and J. C. Hahne, Targeted Oncol., 2020, 15, 261–278 CrossRef.
- B. Dhungel, C. A. Ramlogan-Steel and J. C. Steel, Molecules, 2018, 23, 1500 CrossRef.
- E. Van Rooij and S. Kauppinen, EMBO Mol. Med., 2014, 6, 851–864 CrossRef CAS.
- W. Dai, J. He, L. Zheng, M. Bi, F. Hu, M. Chen, H. Niu, J. Yang, Y. Luo, W. Tang and M. Sheng, J. Breast Cancer, 2019, 22, 219 CrossRef PubMed.
- J. Zhang, Z. Zhang, Q. Wang, X.-J. Xing and Y. Zhao, Eur. Rev. Med. Pharmacol. Sci., 2016, 20, 4710–4718 CAS.
- M. Ishida and F. M. Selaru, Curr. Pathobiol. Rep., 2013, 1, 63–70 CrossRef PubMed.
- D.-D. Nguyen and S. Chang, Int. J. Mol. Sci., 2017, 19, 65 CrossRef PubMed.
- D.-K. Chae, E. Ban, Y. S. Yoo, J. H. Baik and E. J. Song, Anal. Bioanal. Chem., 2016, 408, 4829–4833 CrossRef CAS.
- M. Han, M. Liu, Y. Wang, X. Chen, J. Xu, Y. Sun, L. Zhao, H. Qu, Y. Fan and C. Wu, PLoS One, 2012, 7, e39520 CrossRef CAS PubMed.
- M. S. Ebert and P. A. Sharp, RNA, 2010, 16, 2043–2050 CrossRef CAS PubMed.
- A. H. Alkan and B. Akgül, miRNomics: MicroRNA Biology and Computational Analysis, 2022, pp. 91–104 Search PubMed.
- N. Arghiani and K. Shah, Cancer Biol. Med., 2022, 19, 289 CAS.
- G. Aquino-Jarquin, Cancer Res., 2017, 77, 6812–6817 CrossRef CAS PubMed.
- M. Pagoni, C. Cava, D. C. Sideris, M. Avgeris, V. Zoumpourlis, I. Michalopoulos and N. Drakoulis, J. Personalized Med., 2023, 13, 1586 CrossRef PubMed.
- D. S. Hong, Y.-K. Kang, M. Borad, J. Sachdev, S. Ejadi, H. Y. Lim, A. J. Brenner, K. Park, J.-L. Lee and T.-Y. Kim, Br. J. Cancer, 2020, 122, 1630–1637 CrossRef CAS.
- I. Dasgupta and A. Chatterjee, Methods Protoc., 2021, 4, 10 CrossRef CAS.
- H. Yin, R. L. Kanasty, A. A. Eltoukhy, A. J. Vegas, J. R. Dorkin and D. G. Anderson, Nat. Rev. Genet., 2014, 15, 541–555 CrossRef CAS PubMed.
- B. Gentner, G. Schira, A. Giustacchini, M. Amendola, B. D. Brown, M. Ponzoni and L. Naldini, Nat. Methods, 2009, 6, 63–66 CrossRef CAS PubMed.
- M. Shayestehpour, S. Moghim, V. Salimi, S. Jalilvand, J. Yavarian, B. Romani and T. Mokhtari-Azad, Virus Res., 2017, 240, 207–214 CrossRef CAS PubMed.
- F. Haque, H. Hu and P. Guo, in RNA Nanotechnology and Therapeutics, CRC Press, 2022, pp. 473–483 Search PubMed.
- M. T. Luiz, J. A. P. Dutra, L. B. Tofani, J. T. C. de Araújo, L. D. Di Filippo, J. M. Marchetti and M. Chorilli, Pharmaceutics, 2022, 14, 821 CrossRef CAS PubMed.
- R. Chaudhari, S. Nasra, N. Meghani and A. Kumar, Sci. Rep., 2022, 12, 4713 CrossRef CAS PubMed.
- A. Chaudhuri, K. Ramesh, D. N. Kumar, D. Dehari, S. Singh, D. Kumar and A. K. Agrawal, J. Drug Delivery Sci. Technol., 2022, 77, 103886 CrossRef CAS.
- D. N. Kumar, A. Chaudhuri, F. Aqil, D. Dehari, R. Munagala, S. Singh, R. C. Gupta and A. K. Agrawal, Cancers, 2022, 14, 1435 CrossRef CAS.
- B. F. Dizaji, A. Farboudi, A. Rahbar, M. H. Azarbaijan, and M. R. Asgary, Ther. Delivery, 2020, 11(10), 653–672 CrossRef CAS.
- S. K. Dubey, M. Kali, S. Hejmady, R. N. Saha, A. Alexander and P. Kesharwani, Eur. J. Pharm. Sci., 2021, 164, 105890 CrossRef CAS PubMed.
- H. P. Dang, A. Shafiee, C. A. Lahr, T. R. Dargaville and P. A. Tran, Adv. Therap., 2020, 3, 2000056 CrossRef CAS.
- V. Sriram and J.-Y. Lee, Colloids Surf., B, 2021, 208, 112061 CrossRef CAS.
- N. Yang, Int. J. Pharm. Investig., 2015, 5, 179 CrossRef CAS PubMed.
- N. Nayerossadat, T. Maedeh and P. A. Ali, Adv. Biomed. Res., 2012, 1, 27 CrossRef.
- D. Escors and K. Breckpot, Arch. Immunol. Ther. Exp., 2010, 58, 107–119 CrossRef CAS.
- X. Zhao, Y. Zou, Q. Gu, G. Zhao, H. Gray, L. M. Pfeffer and J. Yue, Viruses, 2015, 7, 2965–2979 CrossRef CAS PubMed.
- S. Daya and K. I. Berns, Clin. Microbiol. Rev., 2008, 21, 583–593 CrossRef CAS.
- M. Scheideler, I. Vidakovic and R. Prassl, Chem. Phys. Lipids, 2020, 226, 104837 CrossRef CAS.
- A. E. Labatut and G. Mattheolabakis, Eur. J. Pharm. Biopharm., 2018, 128, 82–90 CrossRef CAS PubMed.
- N. Naimi, H. Seyedmirzaei, Z. Hassannejad and A. S. Khaboushan, Biomed. Pharmacother., 2024, 175, 116691 CrossRef CAS.
- Q. Liu, D. Li, X. Pan and Y. Liang, J. Nanobiotechnol., 2023, 21, 334 CrossRef.
- R. Rahbarghazi, N. Jabbari, N. A. Sani, R. Asghari, L. Salimi, S. A. Kalashani, M. Feghhi, T. Etemadi, E. Akbariazar and M. Mahmoudi, Cell Commun. Signal., 2019, 17, 1–17 Search PubMed.
- J. Foucquier and M. Guedj, Pharmacol. Res. Perspect., 2015, 3, e00149 CrossRef.
- R. Rupaimoole and F. J. Slack, Nat. Rev. Drug Discov., 2017, 16, 203–222 CrossRef CAS.
- S. Deng, G. A. Calin, C. M. Croce, G. Coukos and L. Zhang, Cell Cycle, 2008, 7, 2643–2646 CrossRef CAS PubMed.
- E. Giovannetti, A. Erozenci, J. Smit, R. Danesi and G. J. Peters, Crit. Rev. Oncol. Hematol., 2012, 81, 103–122 CrossRef.
- X.-J. Liang, C. Chen, Y. Zhao and P. C. Wang, Multi-drug Resistance in Cancer, 2010, pp. 467–488 Search PubMed.
- S. Dallavalle, V. Dobričić, L. Lazzarato, E. Gazzano, M. Machuqueiro, I. Pajeva, I. Tsakovska, N. Zidar and R. Fruttero, Drug Resist. Updates, 2020, 50, 100682 CrossRef PubMed.
- T. B. Emran, A. Shahriar, A. R. Mahmud, T. Rahman, M. H. Abir, M. F. R. Siddiquee, H. Ahmed, N. Rahman, F. Nainu, E. Wahyudin, S. Mitra, K. Dhama, M. M. Habiballah, S. Haque, A. Islam and M. M. Hassan, Front. Oncol., 2022, 12, 891652 CrossRef PubMed.
- L. S. Normann, M. R. Aure, S.-K. Leivonen, M. H. Haugen, V. Hongisto, V. N. Kristensen, G. M. Mælandsmo and K. K. Sahlberg, Sci. Rep., 2021, 11, 10893 CrossRef CAS.
- X. Dai and C. Tan, Adv. Drug Deliv. Rev., 2015, 81, 184–197 CrossRef CAS PubMed.
- K. S. Smalley, N. K. Haass, P. A. Brafford, M. Lioni, K. T. Flaherty and M. Herlyn, Mol. Cancer Therapeut., 2006, 5, 1136–1144 CrossRef CAS.
- R. Chaudhari, P. Patel, N. Meghani, S. Nasra and A. Kumar, 3 Biotech, 2021, 11, 175 CrossRef.
- X. Deng, M. Cao, J. Zhang, K. Hu, Z. Yin, Z. Zhou, X. Xiao, Y. Yang, W. Sheng and Y. Wu, Biomaterials, 2014, 35, 4333–4344 CrossRef CAS PubMed.
- L. Zhang, X. Yang, Y. Lv, X. Xin, C. Qin, X. Han, L. Yang, W. He and L. Yin, Sci. Rep., 2017, 7, 46186 CrossRef CAS.
- S. Dwivedi, P. Pareek, J. R. Vishnoi, P. Sharma and S. Misra, World J. Stem Cell., 2022, 14, 310–313 CrossRef.
- S. Miroshnichenko and O. Patutina, Front. Pharmacol., 2019, 10, 488 CrossRef CAS.
- S. Daldorff, R. M. R. Mathiesen, O. E. Yri, H. P. Ødegård and J. Geisler, Br. J. Cancer, 2017, 116, 10–20 CrossRef CAS PubMed.
- T. Xie, M. Huang, Y. Wang, L. Wang, C. Chen and X. Chu, Cell. Physiol. Biochem., 2016, 40, 62–76 CrossRef CAS.
- V. Angerilli, F. Galuppini, G. Businello, L. Dal Santo, E. Savarino, S. Realdon, V. Guzzardo, L. Nicolè, V. Lazzarin and S. Lonardi, Biomedicines, 2021, 9, 318 CrossRef CAS PubMed.
- T. Joshi, D. Elias, J. Stenvang, C. L. Alves, F. Teng, M. B. Lyng, A. E. Lykkesfeldt, N. Brünner, J. Wang and R. Gupta, Oncotarget, 2016, 7, 57239 CrossRef PubMed.
- G. B. H. Palma and M. Kaur, IUBMB Life, 2023, 75, 743–764 CrossRef CAS PubMed.
- Y. Hu, D. Wang, T. Zhang, M. Lei, Y. Luo, Z. Chen, Y. Li, D. Duan, L. Zhang and Y. Zhu, Int. J. Nanomed., 2024, 1809–1825 CrossRef CAS.
- R. García-Vázquez, L. A. Marchat, E. Ruíz-García, H. Astudillo-De la Vega, A. Meneses-García, C. Arce-Salinas, E. Bargallo-Rocha, Á. Carlos-Reyes, J. S. López-González and C. Pérez-Plasencia, Technol. Cancer Res. Treat., 2019, 18, 1533033819827309 CrossRef PubMed.
- M. S. Beg, D. S. Hong, J. C. Sachdev, A. J. Brenner, M. J. Borad, H. Y. Lim, T.-Y. Kim, C. Becerra, K. Park and A. G. Bader, J. Clin. Oncol., 2016, 34, 15 Search PubMed.
- N. Van Zandwijk, N. Pavlakis, S. C. Kao, A. Linton, M. J. Boyer, S. Clarke, Y. Huynh, A. Chrzanowska, M. J. Fulham and D. L. Bailey, Lancet Oncol., 2017, 18, 1386–1396 CrossRef CAS.
- E. Anastasiadou, A. G. Seto, X. Beatty, M. Hermreck, M.-E. Gilles, D. Stroopinsky, L. C. Pinter-Brown, L. Pestano, C. Marchese and D. Avigan, Clin. Cancer Res., 2021, 27, 1139–1149 CrossRef CAS.
- M. T. van den Bosch, S. Yahyanejad, M. F. Alemdehy, B. J. Telford, T. de Gunst, H. C. den Boer, R. M. Vos, M. Stegink, L. A. van Pinxteren and R. Q. Schaapveld, Mol. Ther. Nucleic Acids, 2021, 23, 1161–1171 CrossRef CAS.
- A. M. Krichevsky and E. J. Uhlmann, Neurotherapeutics, 2019, 16, 319–347 CrossRef CAS.
- S. Duran-Sanchon, L. Moreno, J. M. Augé, M. Serra-Burriel, M. Cuatrecasas, L. Moreira, A. Martín, A. Serradesanferm, À. Pozo and R. Costa, Gastroenterology, 2020, 158, 947–957 CrossRef CAS.
- T. Kim and C. M. Croce, Exp. Mol. Med., 2023, 55, 1314–1321 CrossRef CAS PubMed.
- Q. Cheng, L. Farbiak, A. Vaidya, E. Guerrero, E. E. Lee, E. K. Rose, X. Wang, J. Robinson, S. M. Lee and T. Wei, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2313009120 CrossRef CAS.
- S. Majumder and S. T. Jacob, Gene Expr., 2011, 15, 141 CrossRef.
- Y. Jiang, S. Li, R. Shi, W. Yin, W. Lv, T. Tian and Y. Lin, Adv. Sci., 2024, 11, 2305622 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |