
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRuC nanosheet as a promising biosensing material for detecting the aromatic amino acids: a DFT study†
Mahmoud A. A.
Ibrahim
 *ab,
Nada K. M.
Ahmed
a,
Amna H. M.
Mahmoud
a,
Mohamed A.
El-Tayeb
c,
Ashraf M. M.
Abdelbacki
d,
Shahzeb
Khan
e,
Mahmoud E. S.
Soliman
f and
Tamer
Shoeib
*g
*ab,
Nada K. M.
Ahmed
a,
Amna H. M.
Mahmoud
a,
Mohamed A.
El-Tayeb
c,
Ashraf M. M.
Abdelbacki
d,
Shahzeb
Khan
e,
Mahmoud E. S.
Soliman
f and
Tamer
Shoeib
*g
aComputational Chemistry Laboratory, Chemistry Department, Faculty of Science, Minia University, Minia 61519, Egypt. E-mail: m.ibrahim@compchem.net
bSchool of Health Sciences, University of KwaZulu-Natal, Westville Campus, Durban 4000, South Africa
cDepartment of Botany and Microbiology, College of Science, King Saud University, P.O. Box 2455, Riyadh 11451, Saudi Arabia
dDeanship of Skills Development, King Saud University, P.O. Box 2455, Riyadh 11451, Saudi Arabia
eCentre for Pharmaceutical Engineering Science, Faculty of Life Science, School of Pharmacy and Medical Sciences, University of Bradford, BD7 1DP, UK
fMolecular Bio-computation and Drug Design Laboratory, School of Health Sciences, University of KwaZulu-Natal, Westville Campus, Durban 4000, South Africa
gDepartment of Chemistry, The American University in Cairo, New Cairo 11835, Egypt. E-mail: t.shoeib@aucegypt.edu
First published on 25th October 2024
Abstract
Density functional theory (DFT) calculations were performed to examine the potential of the RuC nanosheet as a biosensor towards the aromatic amino acids (AAA; tryptophan (TRP), histidine (HIS), tyrosine (TYR), and phenylalanine (PHE)). The AAA molecules were placed vertically and horizontally with respect to the RuC surface and then subjected to geometrical relaxation. According to the geometry relaxation results, it was found that all AAA molecules preferred to be adsorbed on the RuC surface in a horizontal configuration rather than a vertical one, except the HIS molecule, which desired to be vertically adsorbed on the RuC nanosheet. From the energy manifestations, the adsorption process within the TRP⋯RuC complexes had the greatest desired negative adsorption energy (Eads), followed by HIS⋯, TYR⋯, and then PHE⋯RuC complexes (Eads = −40.22, −36.54, −23.95, and −16.62 kcal mol−1, respectively). As indicated by the FMO data, changes in the EHOMO, ELUMO, and Egap values of the RuC nanosheet following the adsorption process demonstrated the capacity of the RuC nanosheet to adsorb the AAA molecules. The outcomes of Bader charge transfer revealed that the RuC nanosheet had the ability to donate electrons to the AAA molecules during the adsorption process, supported by the positive Qt values. Consistent with the Eads conclusions, the TRP⋯RuC complexes had the largest Qt values, indicating the potential affinity of the RuC nanosheet to adsorb the TRP molecule. Following the adsorption of AAA molecules on the RuC nanosheet, new peaks and bands were discovered based on the DOS and the band structure plots, respectively, revealing the validity of the adsorption process. Additionally, the current adsorption findings on the RuC nanosheet were compared to those on the graphene (GN) nanosheet. The outcomes of the comparison demonstrated the outperformance of the RuC nanosheet over the GN nanosheet in adsorbing the AAA molecules. These outcomes provide a solid foundation for further research on the RuC nanosheets to detect small biomolecules.
1. Introduction
The efficient exfoliation of the graphene (GN) nanosheet has paved an essential path toward the creation of low-dimensional nanomaterials.1–3 Motivated by the potential of GN in a wide range of practical applications, several free-standing 2D structures, planar and non-planar, have been constructed from their bulk equivalents.4,5 Eventually, this resulted in the formation of layered double hydroxides,6,7 MXenes,8,9 vertical and lateral van der Waals heterostructures,10 transition metal dichalcogenides (TMDs),11 borophene,12–15 and several families of 2D materials.3,16 The distinct characteristics of the 2D materials have prompted additional research into the possibility of producing novel 2D materials, including transition metals and groups III and IV elements.9,17 Among these, carbides based on transition metals have been attracting much interest due to their promising characteristics, including energy storage and superconductivity.18 The ruthenium carbide (RuC), a novel structure of 2D transition metal carbide formed by combining carbon and ruthenium atoms in a planar surface, was computationally hypothesized by Gorkan et al. in 2020.19 The RuC nanosheet has outstanding structural, mechanical, and electrical stabilities and is a very stable type of 2D transition metal carbide.19 The history of RuC goes back to the 1960s when the bulk ruthenium carbides were reported as superhard materials that could be synthesized at high temperatures and ambient pressure.20 When using a laser-heated diamond anvil process at high temperature and pressure, Kumar et al. announced later in 2012 that they had synthesized Ru2C stoichiometry in Fe2N type hexagonal structure with the P3m1 space group.21 Using a simple hydrothermal synthesis approach, the most recent study by Cored et al. examined the processes for fabricating and stabilizing surface ruthenium carbide species on a metallic Ru core.22 Moreover, no additional experimental research on ruthenium carbides has been completed. Computational analysis of bulk ruthenium carbides has been defined, involving studies of different stoichiometries such as rhombohedral RuC23–25 and hexagonal RuC, Ru2C, and Ru3C.23,26Utilization of the RuC nanosheet as a detector towards small molecules, such as NH3, NO, and NO2, was examined using density functional theory (DFT) computations.27 The adsorption energy (Eads) outcomes found that the NO adsorption over the RuC nanosheet had the most favorable negative Eads of −1.718 eV.27 According to our knowledge, no further studies have been carried out to investigate the potential of RuC nanosheets to adsorb biomolecules.
The way that biological molecules interact with nanomaterials is of great interest to researchers studying biomedical and biosensing applications.28–30 Among the biological molecules, the adsorption of nucleic acid molecules and proteins has garnered significant interest.31,32 Amino acids (AAs) are essential for all living things due to their important function as the building blocks of enzymes and proteins.31–33 These AAs also serve as important metabolic intermediates, such as in the synthesis of vitamins, hormones, and neurotransmitters.34,35 Among the AAs, the biogenic aromatic α-amino acids (AAA), including histidine (HIS), tyrosine (TYR), phenylalanine (PHE), and tryptophan (TRP), are crucial residues containing aromatic systems that determine how protein residues interact with the environment.36–38
The application of 2D nanosheets as a biosensor for AAA molecules has garnered significant attention. Therefore, the current work was dedicated to evaluating the adsorption features of the AAA molecules (AAA; tryptophan (TRP), histidine (HIS), tyrosine (TYR), and phenylalanine (PHE)) on the RuC nanosheet by means of DFT calculations (Fig. 1). To determine the minimal structures, relax computations were first applied to the geometric structures of AAA⋯RuC complexes. Subsequently, the adsorption energies were determined for each complex under investigation. To clarify how the adsorption process affects the properties of the studied RuC nanosheet, computations of Bader charge transfer, density of state (DOS), frontier molecular orbital (FMO), and band structures were carried out. In addition, a comparison with the GN nanosheet provided more insight into the efficiency of the RuC nanosheet in adsorbing the AAA molecules. Thus, the AAA⋯GN complexes were relaxed, and their corresponding Eads were evaluated. The outcomes of this investigation will provide insightful information on the potential applications of the RuC nanosheet in sensing biological molecules in the future.
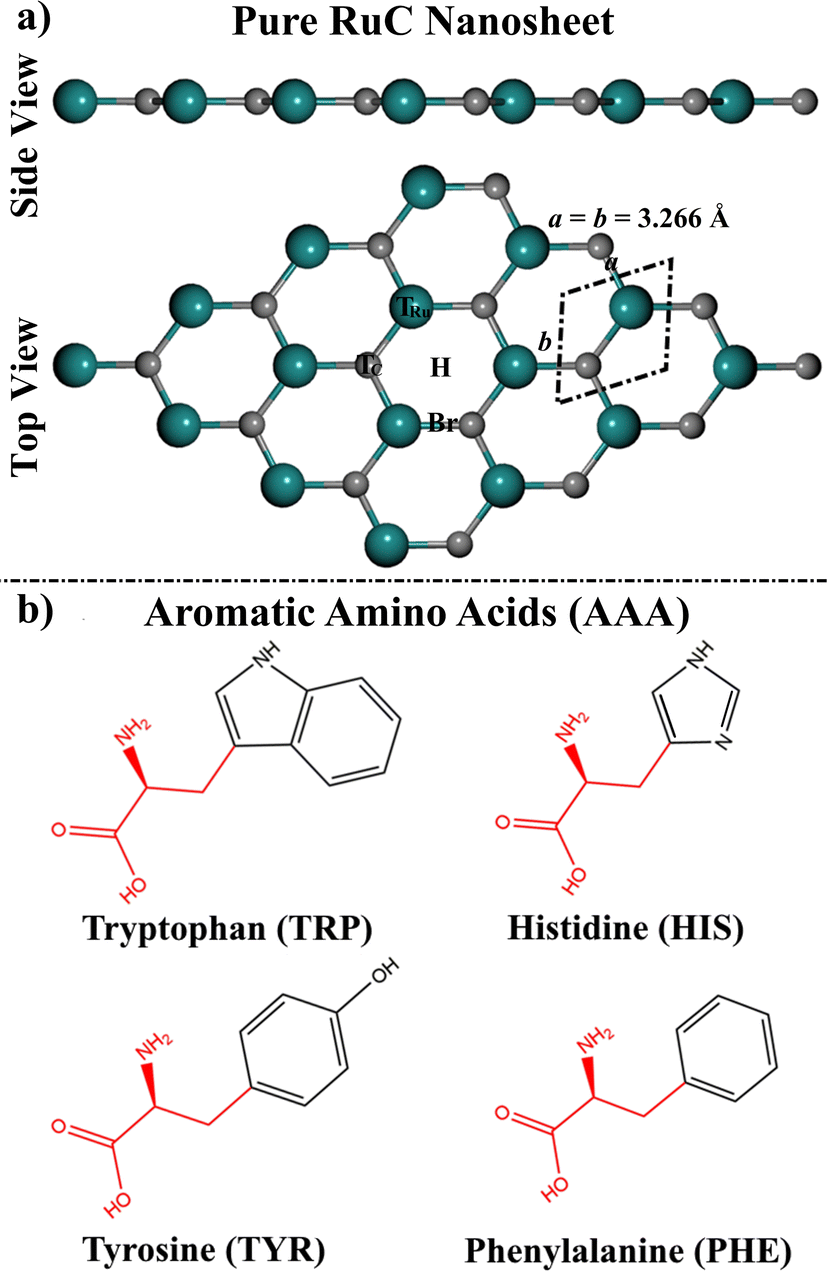 | ||
| Fig. 1 (a) Side and top perspectives of a 4 × 4 × 1 supercell of the RuC nanosheet. The dashed rhombus represents the unit cell of the RuC. The adsorption sites of the top, bridge, and hollow are represented as T, H, and Br, respectively. (b) 2D chemical structures of the studied aromatic amino acids (AAA; tryptophan (TRP), histidine (HIS), tyrosine (TYR), and phenylalanine (PHE)). In our models, the red fragments were replaced by a hydrogen atom. | ||
2. Computational methodology
All computations were carried out using Quantum ESPRESSO 7.1 package39,40 utilizing Density Functional Theory (DFT) methods.41,42 DFT calculations were employed by using the projector augmented wave (PAW).43 Implementing the Perdew–Burke–Ernzerhof (PBE) exchange–correlation function, Generalized Gradient Approximation (GGA) was utilized.44 All calculations were supported with the Grimme-D3 correction to adjust the van der Waals interactions.45,46 The charge density and energy cutoffs were determined at 400 Ry and 50 Ry, respectively. The force and energy convergence criteria for all computations were 10−4 eV Å−1 and 10−5 eV, respectively. For structure relaxation and density of state computation, the Brillouin zone was sampled using Monkhorst–Pack grids of 5 × 5 × 1 and 15 × 15 × 1 k-points, respectively. The Marzari–Vanderbilt smearing technique was implemented in relaxation computations.47 For all calculations, a 20 Å vacuum was placed along the vertical direction of the RuC nanosheet to prevent interactions between the periodic images.For the purpose of investigating the adsorption of AAA molecules over the RuC nanosheet, the optimized RuC unit cell was widened to a 4 × 4 × 1 supercell with 16 Ru and 16 C atoms (Fig. 1). The AAA molecules were then placed vertically and horizontally with respect to the RuC surface to explore all possible adsorption configurations. Adsorption energies (Eads) were then assessed as follows:
| Eads =EAAA⋯RuC − (EAAA + ERuC) | (1) |
| Qt = Q(combined RuC) − Q(isolated RuC) | (2) |
| Δρ = ρAAA⋯RuC − ρAAA − ρRuC | (3) |
| Egap = ELUMO − EHOMO | (4) |
Band structure and the density of states (DOS) calculations were also investigated to comprehend the electronic properties of the RuC nanosheet. The desorption of the AAA molecules from the RuC nanosheet was assessed by computing the recovery time (τ) for the AAA⋯RuC complexes as follows:
τ = v−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−Eads/KT) exp(−Eads/KT) | (5) |
Further comparative analysis of the adsorption efficiency of the RuC and GN nanosheets toward AAA molecules was carried out. Therefore, geometrical relaxation, Eads, and Bader charge calculations were conducted for the AAA⋯GN complexes.
3. Results and discussion
3.1. Geometric structures
Prior to the adsorption process of AAA molecules, the 4 × 4 × 1 supercell of the RuC nanosheet was modeled and relaxed. The optimal structure of the RuC nanosheet is presented in Fig. 1a.From the optimized RuC unit cell, it was found that the Ru–C bond length and lattice constant were 1.906 Å and a = b = 3.266 Å, respectively. The aforementioned findings were in line with previous investigations, in which the lattice parameter was a = 3.25 and 3.24 Å.19,27 According to the relaxed RuC nanosheet, four adsorption sites can be observed: two top (TRu and TC), bridge (Br), and hollow (H) sites (Fig. 1).
3.2. Adsorption energy
The adsorption of AAA molecules over the RuC nanosheet was investigated at all possible adsorption sites (Fig. S1†). The adsorption energies (Eads) and associated equilibrium adsorption distances for the relaxed AAA⋯RuC complexes were computed (Fig. S1†). The structures and the atomic coordinates of the most stable relaxed AAA⋯RuC complexes are given in Fig. 2 and Table S1,† respectively. All AAA molecules preferred to be adsorbed on the RuC nanosheet through their aromatic rings except the HIS molecule, which showed more desirability to be adsorbed through the lone pair electrons on its nitrogen atom.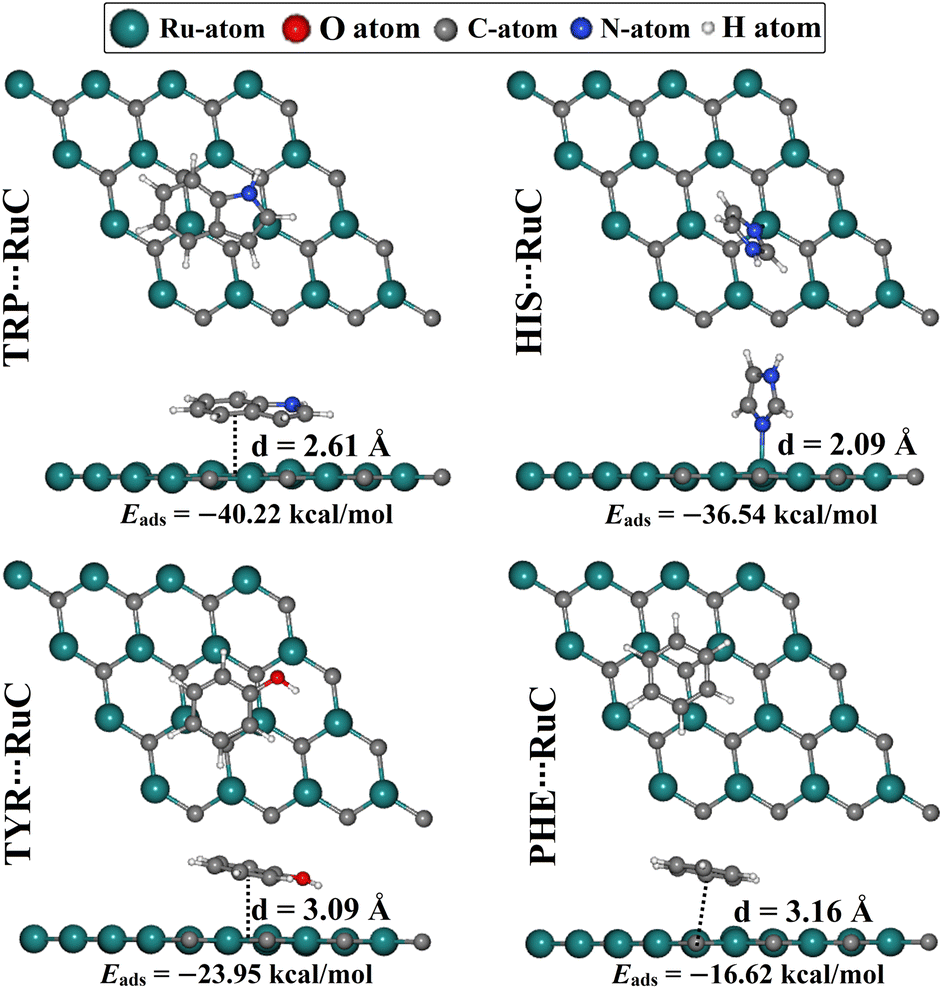 | ||
| Fig. 2 Side and top representations of the relaxed AAA⋯RuC complexes (AAA; tryptophan (TRP), histidine (HIS), tyrosine (TYR), and phenylalanine (PHE)). The adsorption energies (Eads) and the equilibrium adsorption distances (d) between the AAA molecules and the RuC nanosheet are in kcal mol−1 and Å, respectively. The d values were measured between the centroid of the AAA rings and the RuC surface, except for the HIS⋯RuC, d was measured between the N atom of the HIS molecule and the RuC surface. | ||
The considerable capability of the RuC nanosheet to adsorb AAA molecules was confirmed by the negative values of Eads for all AAA⋯RuC complexes under investigation. Among the adsorption processes of the AAA molecules over the RuC nanosheet, the most significant negative Eads value of −40.22 kcal mol−1 was denoted for the TRP⋯RuC complex. The superior adsorption behavior of TRP, compared to the other AAA molecules, aligns with the results of an earlier study on the adsorption of AAA molecules on a graphene nanosheet.52 The size of the π-system of the studied AAA molecules might be the reason for their favorability of being adsorbed on the RuC nanosheet.53 In this regard, the RuC nanosheet was more suitable for adsorbing molecules containing two aromatic rings (i.e., TRP) than those containing a single aromatic ring (i.e., HIS, TYR, and PHE). Turning to the second potent AAA molecule, the HIS adsorption over the RuC nanosheet showed a negative Eads value of −36.54 kcal mol−1 and an equilibrium adsorption distance of 2.09 Å. For TYR, its oxygen atom enhanced the adsorption process on the RuC nanosheet, demonstrating the third promising negative Eads value of −23.95 kcal mol−1. Finally, the PHE⋯RuC complex had the smallest negative Eads value of −16.62 kcal mol−1 since the PHE molecule contains only carbon and hydrogen as heteroatoms. Remarkably, it was observed that the adsorption distance within the AAA⋯RuC complexes decreased as the negative Eads increased. Numerically, the equilibrium adsorption distances were 3.16, 3.09, and 2.61 Å within PHE⋯, TYR⋯, and TRP⋯RuC complexes, with Eads values of −16.62, −23.95, and −40.22 kcal mol−1, respectively (Fig. 2). For the latter complexes, the correlation coefficient (R2) value between the Eads values and the adsorption distances was 0.93. However, the HIS⋯RuC complex showed the smallest adsorption distance of 2.09 Å, due to the adsorption of HIS via the lone pair of electrons on its nitrogen atom over the RuC nanosheet (Fig. 2).
3.3. Frontier molecular orbital (FMO) calculations
Frontier molecular orbitals (FMO) theory was utilized to better understand how the adsorption process of the AAA molecules impacted the electronic features of the RuC nanosheet. The energies of the highest occupied molecular orbitals (EHOMO), the lowest unoccupied molecular orbitals (ELUMO), and energy gap (Egap) values were determined (Table 1). Furthermore, the HOMO and LUMO distributions prior to and following the adsorption process were produced to comprehend the electron transfer regioselectivity for the systems under investigation (Fig. 3).| System | E HOMO (eV) | E LUMO (eV) | E gap (eV) |
|---|---|---|---|
| a The relaxed AAA⋯RuC complexes are shown in Fig. 2. | |||
| Isolated systems | |||
| Pure RuC | −3.412 | −3.341 | 0.071 |
| Tryptophan (TRP) | −4.794 | −1.032 | 3.762 |
| Histidine (HIS) | −5.582 | −0.705 | 4.876 |
| Tyrosine (TYR) | −5.342 | −1.071 | 4.271 |
| Phenylalanine (PHE) | −6.139 | −1.039 | 5.100 |
|
|||
| Combined systemsa | |||
| TRP⋯RuC | −2.534 | −2.479 | 0.056 |
| HIS⋯RuC | −2.488 | −2.470 | 0.016 |
| TYR⋯RuC | −2.873 | −2.866 | 0.007 |
| PHE⋯RuC | −2.899 | −2.790 | 0.109 |
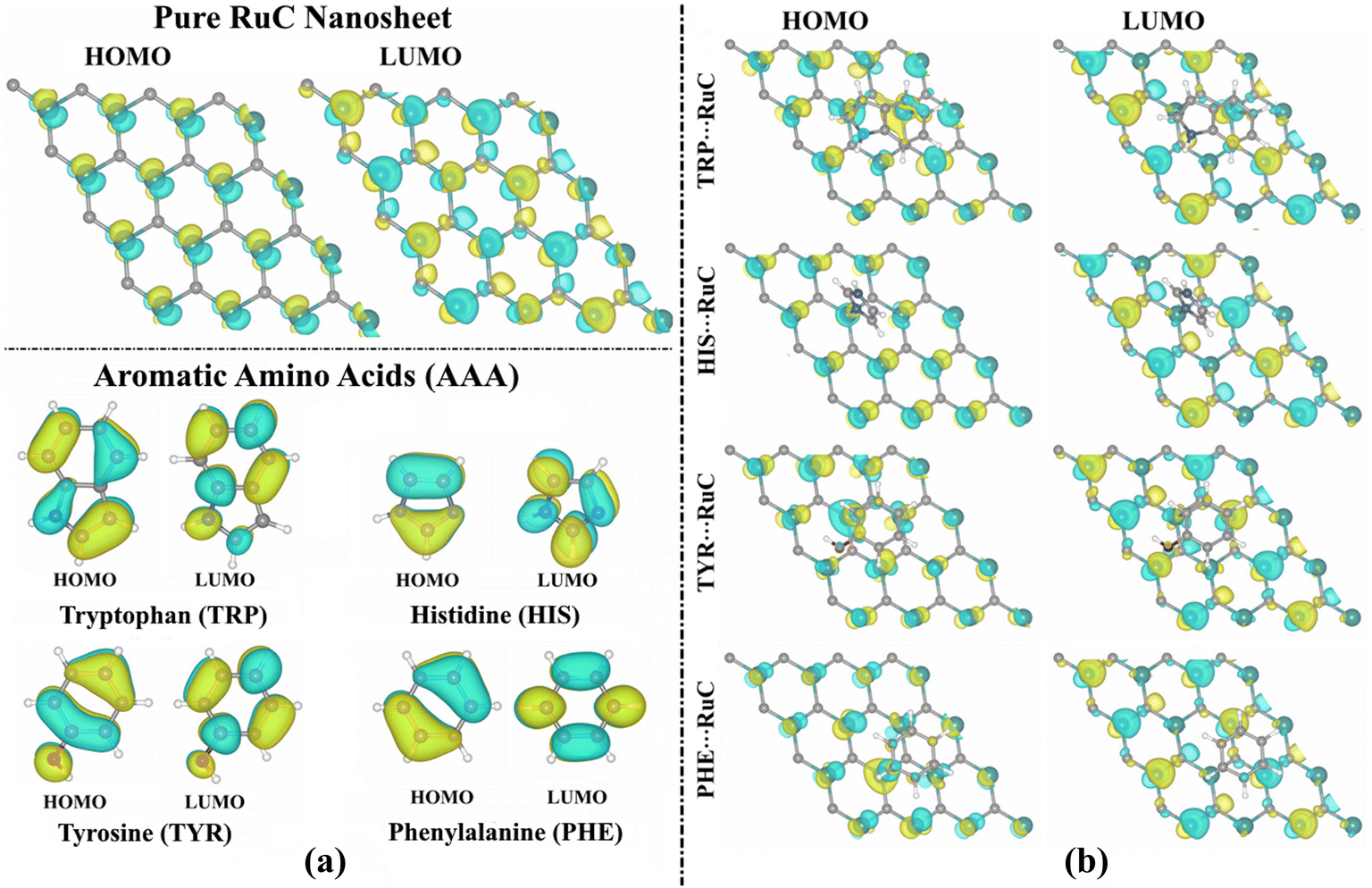 | ||
| Fig. 3 HOMO and LUMO distributions of (a) pure RuC nanosheet and isolated AAA molecules, and (b) the relaxed AAA⋯RuC complexes (AAA; tryptophan (TRP), histidine (HIS), tyrosine (TYR), and phenylalanine (PHE)). | ||
According to the findings in Table 1, the EHOMO, ELUMO, and Egap values of the studied systems showed significant changes before and following the adsorption process. As an example, the EHOMO value of the pure RuC nanosheet was −3.412 eV, while the TRP⋯, HIS⋯, TYR⋯, and PHE⋯RuC complexes had EHOMO values of −2.534, −2.488, −2.873, and −2.899 eV, respectively (Table 1). Further, the Egap values of the AAA molecules and RuC nanosheet altered following the adsorption process, revealing the validity of the adsorption process. In this regard, the Egap of the pure RuC nanosheet had a value of 0.071 eV that decreased following the adsorption of TRP to 0.056 eV (Table 1). Interestingly, the Egap was indicated with modest values, implying that the charge might be transferred within the complex.
The propensity of the molecule to donate electrons is shown by a high EHOMO value, whereas a low ELUMO value reveals the tendency of the molecule to accept electrons. Besides, a small Egap value demonstrates a considerable chemical reactivity of the molecule. According to data listed in Table 1, TRP showed the highest chemical reactivity, followed by TYR, HIS, and then PHE, with Egap values of 3.762, 4.271, 4.876, and 5.100 eV, respectively.
From Fig. 3a, the LUMO distributions were located on the C–N, N–H, C–H, and C–O bonds of the AAA molecules, suggesting that these atoms served as locations for accepting electrons during the adsorption process over the RuC nanosheet. The amount of the LUMO distribution on the TRP molecule was more than that of the other AAA molecules, indicating its significant electron-accepting property. For the relaxed AAA⋯RuC complexes, the LUMO orbitals distribution can be observed on the Ru atoms of the nanosheet, demonstrating their significant role in the adsorption process (Fig. 3b).
3.4. Charge transfer calculation
Bader charge analysis is regarded as a useful technique for figuring out how charges are transferred during the adsorption process.48,49 Consequently, the charge transfer (Qt) was determined for all relaxed AAA⋯RuC complexes (Fig. 4).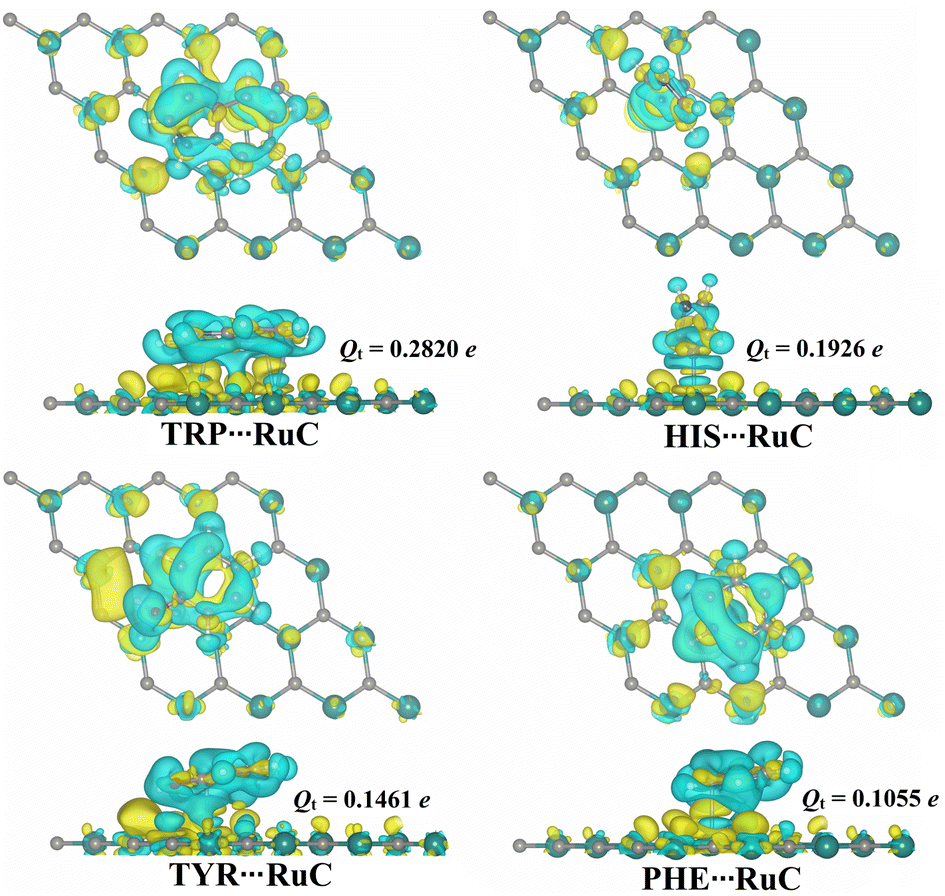 | ||
| Fig. 4 Top and side perspectives of the charge density difference (Δρ) maps for the relaxed AAA⋯RuC complexes (AAA; tryptophan (TRP), histidine (HIS), tyrosine (TYR), and phenylalanine (PHE)). Sites of electron depletion and accumulation are shown by cyan and yellow-colored regions, respectively. Charge transfer difference (Qt) values for the RuC nanosheet before and after the adsorption of AAA molecules are in e. | ||
According to the charge transfer calculations, all estimated Qt values were positive, indicating that the charge was moved from the RuC nanosheet to the adsorbed AAA molecules. In line with the Eads results, the Qt values increased in order: PHE⋯ < TYR⋯ < HIS⋯ < TRP⋯RuC complexes. In terms of values, the Qt values of the PHE⋯, TYR⋯, HIS⋯, and TRP⋯RuC complexes were 0.1055, 0.1461, 0.1926, and 0.2820e, respectively (Fig. 4). Overall, the RuC nanosheet within the AAA⋯RuC complexes showed to have an electron-donating nature by the positive Qt values. To examine the distribution of charge for the relaxed AAA⋯RuC complexes, maps of the charge density difference (Δρ) were plotted and shown in Fig. 4.
In Fig. 4, positive (yellow) and negative (cyan) regions represent the regions of electron accumulation and depletion, respectively. The most remarkable electron accumulation region was observed in the TRP⋯RuC complex, demonstrating the superior ability of the TRP molecule to be adsorbed on the studied RuC nanosheet compared to the other AAA molecules. In all investigated AAA⋯RuC complexes, the electron depletion region was observed above the surface of the RuC nanosheet, demonstrating its electron-donating property (Fig. 4).
3.5. Recovery time
Recovery time (τ) computations can also be carried out to assess the desorption of AAA molecules from the RuC nanosheet. As a result, the τ was estimated for the relaxed AAA⋯RuC complexes at two different temperatures, and the findings are listed in Table 2.Based on eqn (5), the τ results in Table 2 demonstrated the direct correlation between the τ and the Eads outcomes; τ increased as negative Eads values increased. For instance, the TRP⋯RuC complex, which had the most significant negative Eads with a value of −40.22 kcal mol−1, exhibited the longest τ with a value of 2.75 × 1011 and 2.00 × 1010 s at the room and human body temperatures, respectively. As observed in Table 2, the τ values decreased as the temperature increased. For example, the τ of the PHE⋯RuC complex was 1.46 × 10−6 s at room temperature (i.e., 298.15 K) and became 4.95 × 10−7 s at human body temperature (i.e., 310.15 K). The τ results validated the affinity of the RuC nanosheet to adsorb AAA molecules effectively.
3.6. Band structure
Band structures were carried out before and after the adsorption of the AAA molecules over the RuC nanosheet to find out how the adsorbates changed the electronic characteristics of the adsorbent (Fig. 5).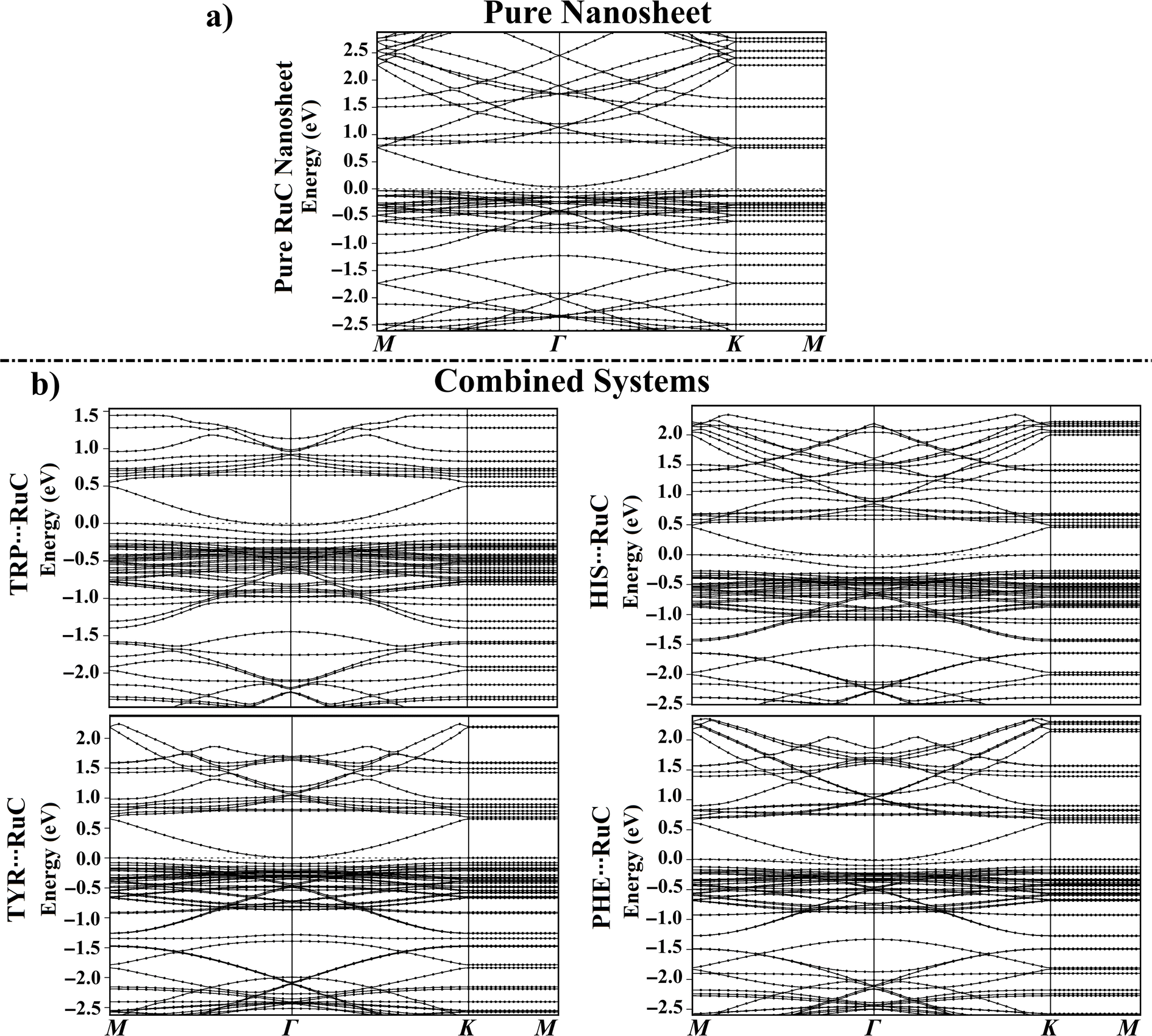 | ||
| Fig. 5 Electronic band structure along the high symmetry path of the Brillouin zone (M–Γ–K–M) for (a) the pure RuC nanosheet and (b) the relaxed AAA⋯RuC complexes (AAA; tryptophan (TRP), histidine (HIS), tyrosine (TYR), and phenylalanine (PHE)). The Fermi level is shown at zero. | ||
For all AAA⋯RuC complexes, the density of valence bands near the Fermi level increased as the adsorption energy increased following the order PHE⋯ < TYR⋯ < HIS⋯ < TRP⋯RuC complexes (Fig. 5b). Upon the adsorption of the TRP molecule over the RuC surface, additional valence and conduction bands were observed in its band structure. For instance, additional bands were noted in the valence region at around −1.60 and −1.80 eV (Fig. 5b). A significant shift in valence bands (from −0.03 to 0.00 eV) can also be observed for the TRP⋯RuC complex (Fig. 5b).
New valence bands appeared in the band structure of the HIS⋯RuC complex, reflecting the interaction of the HIS molecule with the pure RuC nanosheet. Additional valence bands were observed at around −1.65 eV and between −0.25 to −0.80 eV, whereas new conduction bands were identified at approximately 1.75 and 1.07 eV (Fig. 5b). The valence and conduction bands of the HIS⋯RuC complex were moved nearer the Fermi level, indicating the considerable of the HIS adsorption over the RuC nanosheet. As noted in the band structures depicted in Fig. 5, the electronic characteristics of the RuC nanosheet seemed to be impacted by the adsorption of TYR and PHE molecules, exhibiting an increase in valence and conduction bands. The Egap findings supported the observed band shifts; for example, the Egap of the pure RuC nanosheet was 0.071 eV, which shifted to 0.056, 0.016, 0.007, and 0.109 eV within the TRP⋯, HIS⋯, TYR⋯, and PHE⋯RuC complexes (Table 1).
3.7. Density of state (DOS) calculations
The total density of states (TDOS) and projected density of states (PDOS) plots for the RuC nanosheet prior to and following the adsorption were generated to determine how the electrical characteristics of the RuC nanosheet were affected during the adsorption of the AAA molecules (Fig. 6).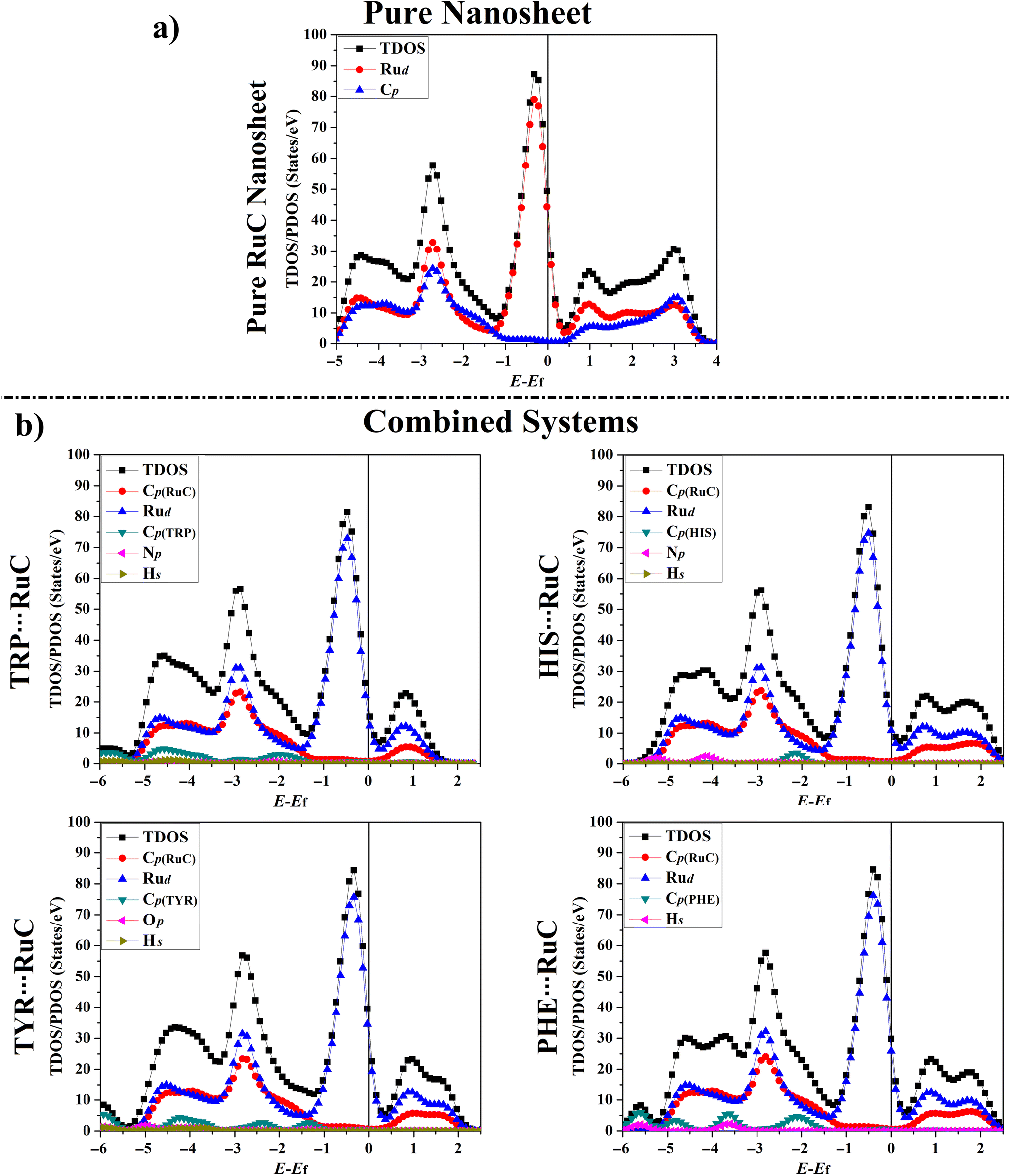 | ||
| Fig. 6 TDOS and PDOS plots for (a) the pure RuC nanosheet and (b) the relaxed AAA⋯RuC complexes (AAA; tryptophan (TRP), histidine (HIS), tyrosine (TYR), and phenylalanine (PHE)). The contributions of the d-orbital of ruthenium (Rud), p-orbital of carbon (Cp), nitrogen (Np), oxygen (Op), and the s-orbital for hydrogen (Hs) in the adsorption process. The Fermi level is located at zero. | ||
As seen in Fig. 6a, the PDOS plot of the pure RuC nanosheet revealed that the d-orbitals of the Ru atoms are primarily responsible for the TDOS near the Fermi level. According to Fig. 6b, it can be seen that the Rud, Np, Op, and Cp had significant contributions to the adsorption process within the relaxed AAA⋯RuC complexes, and their PDOS peaks appeared between −6.00 and 2.50 eV. As an example, the PDOS plot of the TRP⋯RuC complex showed that the Rud and Cp of the RuC nanosheet were the primary origins of the adsorption process. The Cp of the TRP molecule had the largest contribution to the adsorption process on the RuC nanosheet, appearing in the valence region at energy ranges of −1.50 to −2.50, −3.50 to −5.00, and −5.50 to −7.00 eV (Fig. 6b). The Hs and Np of the TRP molecule within the TRP⋯RuC complex showed minor contributions that can be observed in the valence region from −1.50 to −7.00 eV. Obviously, an overlap between the Cp of the RuC nanosheet and the Cp, Np, and Hs of the TRP molecule was noted from −0.50 to −1.40 eV in the valence region. The latter observation confirmed the strong interaction between the TRP molecule and the RuC nanosheet.
3.8. Adsorption process on graphene (GN) nanosheet
As a point of comparison, the AAA⋯GN complexes were relaxed at the same configuration as the AAA⋯RuC complexes, and then their corresponding Eads values were evaluated (Fig. 7). The atomic coordinates of the relaxed AAA⋯GN complexes are given in Table S1.†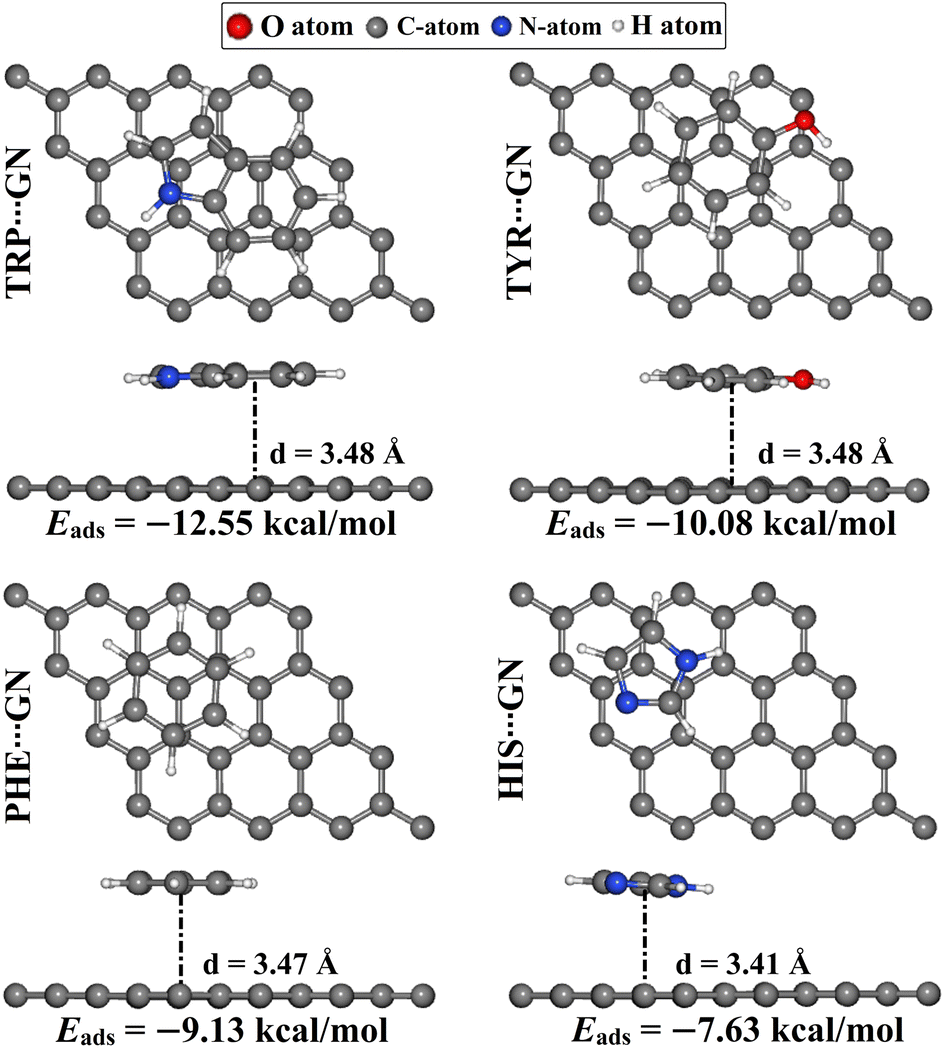 | ||
| Fig. 7 Top and side representations of the relaxed AAA⋯GN complexes (AAA; tryptophan (TRP), histidine (HIS), tyrosine (TYR), and phenylalanine (PHE)). The adsorption energies (Eads) and the equilibrium adsorption distances (d) between the AAA molecules and the GN nanosheet are in kcal mol−1 and Å, respectively. The d values are measured between the centroid of the AAA rings and the GN surface. | ||
According to the results, the negative Eads of the AAA⋯GN complexes increased in the order of HIS⋯ < PHE⋯ < TYR⋯ < TRP⋯GN complexes with Eads values of −7.63, −9.13, −10.08, and −12.55 kcal mol−1, respectively. The latter results were consistent with the prior study.52 In terms of comparison, the Eads values of the AAA⋯GN complexes in the modeled configurations were less negative than those of the AAA⋯RuC complexes, demonstrating the superior adsorption efficacy of the RuC nanosheet toward the AAA molecules. For instance, the Eads values of the TRP⋯RuC and TRP⋯GN complexes were −40.22 and −12.55 kcal mol−1, respectively.
Upon the relaxed AAA⋯GN complexes, the Qt values were computed within the Bader charge method (Fig. 8). Based on the Bader charge results, all AAA⋯GN complexes showed positive Qt values, demonstrating that GN nanosheet acted, similarly to RuC nanosheet, as an electron donor during the adsorption process. In this spirit, the Qt values of the TRP⋯, TYR⋯, PHE⋯, and HIS⋯GN complexes were 0.0156, 0.0053, 0.0072, and 0.0065e, respectively (Fig. 8). Additionally, the Δρ maps were produced for the relaxed AAA⋯GN complexes and are illustrated in Fig. 8. As shown in Fig. 8, cyan regions were observed above the GN nanosheet within all relaxed AAA⋯GN complexes, demonstrating the electron-donating nature of the GN nanosheet during the adsorption process. Similar to the AAA⋯RuC complexes, the TRP⋯GN complex also exhibited the largest amount of the transferred charge supported by the highest Qt value of 0.0156e (Fig. 8).
 | ||
| Fig. 8 Top and side perspectives of the charge density difference (Δρ) maps for the relaxed AAA⋯GN complexes (AAA; tryptophan (TRP), histidine (HIS), tyrosine (TYR), and phenylalanine (PHE)). Sites of electron depletion and accumulation are shown by cyan and yellow-colored regions, respectively. Charge transfer difference (Qt) values for the GN nanosheet prior to and following the adsorption of AAA molecules are in e. | ||
4. Conclusion
AAA molecules are building blocks for synthesizing numerous biologically active compounds necessary to preserve regular biological processes. Based on their significance, the propensity of the RuC nanosheet as a biosensor for different AAA molecules was assessed within both horizontal and vertical configurations employing the density functional theory (DFT). Based on the relaxed structures, it was found that the AAA molecules favored their horizontal configuration within the adsorption process on the RuC nanosheet, while the HIS molecule desired to be adsorbed vertically. As per Eads outcomes, the TRP⋯RuC complexes showed the greatest negative Eads value of −40.22 kcal mol−1. From the FMO data, notable changes in the EHOMO, ELUMO, and Egap values of the RuC nanosheet following the adsorption process demonstrated the capability of the RuC nanosheet to adsorb the AAA molecules. According to Bader charge transfer analysis, the RuC nanosheet functioned as an electron donor during the adsorption process, supported by positive Qt values. In line with the Eads values, the TRP⋯RuC complex had the largest Qt value of 0.2820e. The adsorption of AAA molecules on the RuC nanosheet was also confirmed by the band structure and DOS analysis, supported by the appearance of additional bands and peaks in their plots, respectively. The outcomes of a comparative investigation revealed that the RuC nanosheet outperformed the GN nanosheet in adsorbing the AAA molecules. These findings lay a solid basis for further investigations into the potential of the RuC nanosheet as an effective biosensor for small biomolecules.Data availability
The data supporting this article have been included as ESI.†Author contributions
Conceptualization, M. A. A. I. and T. S.; methodology, M. A. A. I., A. H. M. M., and M. E. S. S.; software, M. A. A. I.; formal analysis, N. K. M. A. and A. H. M. M.; investigation, N. K. M. A., A. H. M. M., and S. K.; resources, M. A. A. I., M. A. E.-T., A. M. M. A. and T. S.; data curation, N. K. M. A. and A. H. M. M.; visualization, N. K. M. A. and A. H. M. M., and M. E. S. S.; supervision, M. A. A. I.; project administration, M. A. A. I. and A. H. M. M.; writing—original draft, N. K. M. A. and A. H. M. M.; writing—review and editing, M. A. A. I., M. A. E.-T., A. M. M. A., S. K., M. E. S. S. and T. S.; all authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare no competing interests.Acknowledgements
The authors extend their appreciation to the Researchers Supporting Project number (RSPD2024R978), King Saud University, Riyadh, Saudi Arabia, for funding this work. The computational work was completed with resources provided by the Center for High-Performance Computing (Cape Town, South Africa, https://www.chpc.ac.za), and Bibliotheca Alexandrina (https://hpc.bibalex.org).References
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 Search PubMed.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 Search PubMed.
- K. S. Novoselov, D. Jiang, F. Schedin, T. J. Booth, V. V. Khotkevich, S. V. Morozov and A. K. Geim, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10451–10453 Search PubMed.
- M. A. A. Ibrahim, M. H. A. Hamad, A. H. M. Mahmoud, G. A. H. Mekhemer, S. R. M. Sayed, M. K. A. El-Rahman, P. A. Sidhom, E. Dabbish and T. Shoeib, Pharmaceutics, 2023, 15, 1640 Search PubMed.
- M. A. A. Ibrahim, A. H. M. Mahmoud, K. A. Soliman, G. A. H. Mekhemer, M. N. Ahmed, A. M. Shawky, M. A. S. Abourehab, E. B. Elkaeed, M. E. S. Soliman and N. A. M. Moussa, Nanomaterials, 2022, 12, 1028 Search PubMed.
- D. G. Evans and R. C. T. Slade, in Layered Double Hydroxides, ed. X. Duan and D. G. Evans, Springer Berlin Heidelberg, Berlin, Heidelberg, 2005, ch. 5, pp. 1–87, DOI:10.1007/430_005.
- A. I. Khan and D. O'Hare, J. Mater. Chem., 2002, 12, 3191–3198 Search PubMed.
- M. Liao, Z. Zheng, H. Jiang, M. Ma, L. Wang, Y. Wang and S. Zhuang, Sci. Total Environ., 2024, 912, 169014 Search PubMed.
- Y. Gogotsi and B. Anasori, ACS Nano, 2019, 13, 8491–8494 Search PubMed.
- A. K. Geim and I. V. Grigorieva, Nature, 2013, 499, 419–425 Search PubMed.
- S. Manzeli, D. Ovchinnikov, D. Pasquier, O. V. Yazyev and A. Kis, Nat. Rev. Mater., 2017, 2, 17033 Search PubMed.
- A. J. Mannix, X. F. Zhou, B. Kiraly, J. D. Wood, D. Alducin, B. D. Myers, X. Liu, B. L. Fisher, U. Santiago, J. R. Guest, M. J. Yacaman, A. Ponce, A. R. Oganov, M. C. Hersam and N. P. Guisinger, Science, 2015, 350, 1513–1516 Search PubMed.
- M. A. A. Ibrahim, A. H. M. Mahmoud, G. A. H. Mekhemer, A. M. Shawky, M. E. S. Soliman and N. A. M. Moussa, Nanomaterials, 2022, 12, 3411 Search PubMed.
- M. A. A. Ibrahim, A. H. M. Mahmoud, N. A. M. Moussa, G. A. H. Mekhemer, S. R. M. Sayed, M. N. Ahmed, M. K. Abd El-Rahman, E. Dabbish and T. Shoeib, Molecules, 2023, 28, 5476 Search PubMed.
- B. Feng, J. Zhang, Q. Zhong, W. Li, S. Li, H. Li, P. Cheng, S. Meng, L. Chen and K. Wu, Nat. Chem., 2016, 8, 563–568 Search PubMed.
- N. R. Glavin, R. Rao, V. Varshney, E. Bianco, A. Apte, A. Roy, E. Ringe and P. M. Ajayan, Adv. Mater., 2020, 32, e1904302 Search PubMed.
- C. Zhang, Y. L. Ma, X. T. Zhang, S. Abdolhosseinzadeh, H. W. Sheng, W. Lan, A. Pakdel, J. Heier and F. Nüesch, Energy Environ. Mater., 2020, 3, 29–55 Search PubMed.
- Y. Gogotsi, Nat. Mater., 2015, 14, 1079–1080 Search PubMed.
- T. Gorkan, S. Demirci, S. Jahangirov, G. Gokoglu and E. Akturk, Phys. Chem. Chem. Phys., 2020, 22, 15488–15495 Search PubMed.
- C. P. Kempter and M. R. Nadler, J. Chem. Phys., 1960, 33, 1580–1581 Search PubMed.
- N. R. Sanjay Kumar, N. V. Chandra Shekar, S. Chandra, J. Basu, R. Divakar and P. Sahu, J. Phys.: Condens. Matter, 2012, 24, 362202 Search PubMed.
- J. Cored, A. Garcia-Ortiz, S. Iborra, M. J. Climent, L. Liu, C. H. Chuang, T. S. Chan, C. Escudero, P. Concepcion and A. Corma, J. Am. Chem. Soc., 2019, 141, 19304–19311 Search PubMed.
- H. Gopalakrishna Pillai, A. Kulangara Madam, S. Natarajan, S. Chandra and V. Mundachali Cheruvalath, J. Phys. Chem. Solids, 2016, 94, 47–58 Search PubMed.
- G. Harikrishnan, K. M. Ajith, S. Chandra and M. C. Valsakumar, Modell. Simul. Mater. Sci. Eng., 2015, 23, 085006 Search PubMed.
- G. Harikrishnan, K. M. Ajith, S. Chandra and M. C. Valsakumar, Mater. Sci. Semicond. Process., 2015, 40, 484–490 Search PubMed.
- C. Z. Fan, S. Y. Zeng, Z. J. Zhan, R. P. Liu, W. K. Wang, P. Zhang and Y. G. Yao, Appl. Phys. Lett., 2006, 89, 071913 Search PubMed.
- S. Thomas and M. A. Zaeem, Appl. Surf. Sci., 2021, 563, 150232 Search PubMed.
- E. Gonzalez Solveyra, D. H. Thompson and I. Szleifer, Polymers, 2022, 14, 739 Search PubMed.
- P. Singla, M. Riyaz, S. Singhal and N. Goel, Phys. Chem. Chem. Phys., 2016, 18, 5597–5604 Search PubMed.
- D. Thiruppathi, P. Karuppasamy, M. Ganesan, V. K. Sivasubramanian, T. Rajendran and S. Rajagopal, Int. J. Chem. Kinet., 2014, 46, 606–618 Search PubMed.
- C. K. Yang, Comput. Phys. Commun., 2011, 182, 39–42 Search PubMed.
- D. Farmanzadeh and S. Ghazanfary, Struct. Chem., 2013, 25, 293–300 Search PubMed.
- S. Mukhopadhyay, S. Gowtham, R. H. Scheicher, R. Pandey and S. P. Karna, Nanotechnology, 2010, 21, 165703 Search PubMed.
- S. C. Daubner, T. Le and S. Wang, Arch. Biochem. Biophys., 2011, 508, 1–12 Search PubMed.
- T. Fukuwatari and K. Shibata, Int. J. Tryptophan Res., 2013, 6, 3–8 Search PubMed.
- M. El Refaey, C. P. Watkins, E. J. Kennedy, A. Chang, Q. Zhong, K. H. Ding, X. M. Shi, J. Xu, W. B. Bollag, W. D. Hill, M. Johnson, M. Hunter, M. W. Hamrick and C. M. Isales, Mol. Cell. Endocrinol., 2015, 410, 87–96 Search PubMed.
- M. Cao, M. Gao, M. Suastegui, Y. Mei and Z. Shao, Metab. Eng., 2020, 58, 94–132 Search PubMed.
- W. M. Dudek, S. Ostrowski and J. C. Dobrowolski, J. Phys. Chem. A, 2022, 126, 3433–3444 Search PubMed.
- P. Giannozzi, O. Andreussi, T. Brumme, O. Bunau, M. Buongiorno Nardelli, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, M. Cococcioni, N. Colonna, I. Carnimeo, A. Dal Corso, S. de Gironcoli, P. Delugas, R. A. DiStasio Jr, A. Ferretti, A. Floris, G. Fratesi, G. Fugallo, R. Gebauer, U. Gerstmann, F. Giustino, T. Gorni, J. Jia, M. Kawamura, H. Y. Ko, A. Kokalj, E. Kucukbenli, M. Lazzeri, M. Marsili, N. Marzari, F. Mauri, N. L. Nguyen, H. V. Nguyen, A. Otero-de-la-Roza, L. Paulatto, S. Ponce, D. Rocca, R. Sabatini, B. Santra, M. Schlipf, A. P. Seitsonen, A. Smogunov, I. Timrov, T. Thonhauser, P. Umari, N. Vast, X. Wu and S. Baroni, J. Phys.: Condens. Matter, 2017, 29, 465901 Search PubMed.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. Dal Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, J. Phys.: Condens. Matter, 2009, 21, 395502 Search PubMed.
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 Search PubMed.
- G. Kresse and J. Furthmuller, Comput. Mater. Sci., 1996, 6, 15–50 Search PubMed.
- P. E. Blochl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 Search PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 Search PubMed.
- N. Marzari, D. Vanderbilt, A. De Vita and M. C. Payne, Phys. Rev. Lett., 1999, 82, 3296–3299 Search PubMed.
- G. Henkelman, A. Arnaldsson and H. Jonsson, Comput. Mater. Sci., 2006, 36, 354–360 Search PubMed.
- W. Kutzelnigg, Angew. Chem., Int. Ed. Engl., 1993, 32, 128–129 Search PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 Search PubMed.
- B. Tamilselvi, D. S. Bhuvaneshwari, P. Karuppasamy, S. Padmavathy, S. Nikhil, S. B. Siddegowda and H. C. Ananda Murthy, ACS Phys. Chem. Au, 2024, 4, 67–84 Search PubMed.
- H. Vovusha, S. Sanyal and B. Sanyal, J. Phys. Chem. Lett., 2013, 4, 3710–3718 Search PubMed.
- L. R. Rutledge and S. D. Wetmore, J. Chem. Theory Comput., 2008, 4, 1768–1780 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4na00670d |
| This journal is © The Royal Society of Chemistry 2024 |