
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChiroptically active quantum nanonails†
Finn
Purcell-Milton
 *ab,
Vera A.
Kuznetsova
*a,
Xue
Bai
a,
Áine
Coogan
a,
Marina
Martínez-Carmona
c,
Jorge A.
Garcia
d,
A. Louise
Bradley
d and
Yurii K.
Gun’ko
*a
*ab,
Vera A.
Kuznetsova
*a,
Xue
Bai
a,
Áine
Coogan
a,
Marina
Martínez-Carmona
c,
Jorge A.
Garcia
d,
A. Louise
Bradley
d and
Yurii K.
Gun’ko
*a
aSchool of Chemistry, CRANN and AMBER Research Centres, Trinity College Dublin, Dublin, Ireland. E-mail: finn.purcellmilton@tudublin.ie; kuznetsv@tcd.ie; igounko@tcd.ie
bSchool of Chemical & BioPharmaceutical Sciences, Technological University Dublin, Grangegorman, Dublin, Ireland
cDepartamento de Didáctica de las Ciencias Experimentales, Universidad de Murcia, 30100 Murcia, Spain
dSchool of Physics, Trinity College Dublin, Dublin 2, Ireland
First published on 2nd April 2024
Abstract
In recent years, extensive research efforts have been dedicated to the investigation of CdSe/CdS-based quantum-confined nanostructures, driven by their distinctive properties. The morphologies of these nanostructures have been shown to directly affect their properties, an area which has proven to be an important field of study. Herein, we report a new morphology of CdSe/CdS core–shell heterostructures in the form of a ‘nanonail’ – a modified nanorod-like morphology, in which a distinctive triangular head can be observed at one end of the structure. In-depth studies of this morphology reveal a material with tuneable rod length and width, as well as exceptional photoluminescent properties. Following this, we have demonstrated the ability to induce chiroptical activity via ligand exchange, revealing the important role of the specific morphology, shell thickness and chiral ligand concentration in the effect of ligand induced chirality. In addition, the cellular uptake and cytotoxicity of obtained chiral nanostructures were evaluated on human lung-derived A549 cancer cells, revealing a significant enantioselectivity in biological activity. Finally, analysis on monolayers of the material demonstrate the complete absence of FRET processes. Overall, this CdSe/CdS heterostructure is another tuneable morphology of a very important nanomaterial, one which shows great advantages and a range of potential applications.
New conceptsWe report a new, anisotropic distinct CdSe/CdS quantum nanostructure morphology – dubbed the ‘nanonail’ (NN) – showcasing a modified dot-in-rod-like form with a discernible triangular head at one end. The position of the CdSe seed is highly controlled due to the growth mechanism, consistently placing it at the ‘head’ of the nanonail. Excellent precision over the resulting morphology has also been demonstrated via seed selection and shell thickness, enabling the length and width of the rod to be finely controlled. In addition, we have studied the optical properties and carried out ligand exchange with chiral ligands, namely L- and D-cysteine, to enable the induction of chirality. The influence of chiral ligand concentration on the optical activity was analysed and the biological activity of L-Cys-NN and D-Cys-NN enantiomers were compared. Furthermore, we also demonstrate enantioselectivity in the biological activity of these chiroptically active NNs, highlighting their versatile potential across various applications. |
Introduction
CdSe/CdS core–shell colloidal quantum dots (QDs) were among some of the first highly photoluminescent (PL) quantum dots synthesised.1 They continue to be some of the most extensively studied core–shell QDs, serving as a model system for understanding a range of photophysical and electronic properties on the nanoscale.2–5 Furthermore, they have been utilised as crucial components in a range of devices including LEDs,6 photovoltaic cells,7 and luminescent solar concentrators.8 CdSe/CdS QDs exhibit outstanding optical properties and can effectively absorb and emit light over the majority of the visible spectrum, via tuning the size of the initial CdSe QD core, as well as the thickness of the CdS shell. In addition, they show large Stokes shifts, and have been reported to achieve near unity fluorescent quantum yields.9 In addition to the simple spherical core–shell structure, CdSe/CdS systems can exist in a range of other morphologies, including dot-in-rod (DiR),10,11 rod-in-rod,12 dot-in-tetrapod (DiT),11,13 dot-in-platelet,14 dot-in-octopod,15 core-in-crown,16 platelet-in-platelet17 and other branched structures.18,19The DiR structures have been particularly interesting, finding diverse applications due to their high quantum yields, large Stokes shifts and versatile morphologies, in addition to the unique shape-dependent photophysics that have been the subject of numerous in-depth studies.20–24 These CdSe/CdS nanostructures have demonstrated morphology-dependent luminescence excitation, with the CdS peak showing a defined peak in certain cases, while in others there was no clear exciton peak.25 Furthermore, the band alignment of this structure has been of particular interest in a number of publications showing transition from type-I to quasi-type-II regimes and type-II behaviour.23,26,27 Optical anisotropy of the electronic states has been investigated relative to the morphology of the DiR structures, revealing an anisotropic dependency on shell thickness on the short side of the nanostructure.28 In another study, it was demonstrated that the exciton localization efficiency in this structure was dependent upon rod length, decreasing as the rod length increased, but interestingly was independent of the band alignment.29 Moreover, another significant study demonstrated that the optical anisotropy of CdSe/CdS DiR does not arise from anisotropic location of the QD core, but rather the local anisotropy of the CdS shell, emphasising the importance of considering both core and shell properties in understanding optical behaviour.30
The DiR morphology is typically produced via the synthesis of wurtzite CdSe QD cores. These cores are used as seeds to grow an anisotropic shell, which occurs at an accelerated rate along the [0001] and [000![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ] directions of the wurtzite seed, resulting in a wurtzite-phase CdS shell. It has also been found that the relative growth in each direction can be distinctly controlled via the use of organic ligands with electron-donating groups. The anion-rich (000) planes exhibit higher growth rates than cation-rich (0001) surfaces, as a result of the difference in binding energy of the ligands to those crystal planes. Interestingly, this growth rate has also been shown to be controllable via the use of precise ratios of the ligand hexylphosphonic acid (HPA) and octadecylphosphonic acid (ODPA) and, in some cases, has even facilitated the production of novel dual diameter DiR structures.31
] directions of the wurtzite seed, resulting in a wurtzite-phase CdS shell. It has also been found that the relative growth in each direction can be distinctly controlled via the use of organic ligands with electron-donating groups. The anion-rich (000) planes exhibit higher growth rates than cation-rich (0001) surfaces, as a result of the difference in binding energy of the ligands to those crystal planes. Interestingly, this growth rate has also been shown to be controllable via the use of precise ratios of the ligand hexylphosphonic acid (HPA) and octadecylphosphonic acid (ODPA) and, in some cases, has even facilitated the production of novel dual diameter DiR structures.31
In addition, these structures have shown significant potential for various applications via orientational arrangement, achieved via a range of approaches such as evaporation on capillary tubes,32 electric field,33 drop casting, spin coating,34 mechanical stretching35 and self-assembly on a water surface.36 This enables the anisotropic properties such as polarized emission and absorption to be attainable on the macroscale in the design of devices such as liquid crystal displays,35 lasers32 and LEDs.36
Therefore, it is clear that with additional advances in synthetic control, new parameters become controllable and unique properties become available. With this in mind, we report an interesting new CdSe/CdS dot in rod morphology, termed the ‘nanonail’ (NN), produced via a seeded hot injection approach. This structure manifests as a modified nanorod-like morphology, in which a distinctive triangular head can be seen in the structure at one end. In this structure, the position of the CdSe seed is highly controlled due to the growth mechanism, consistently placing it at the ‘head’ of the nanonail. Excellent precision over the resulting morphology has also been demonstrated via seed selection and shell thickness, enabling the length and width of the rod to be finely controlled. Following this, we have studied the optical properties and carried out ligand exchange with chiral ligands, namely L- and D-cysteine, to enable the induction of chirality. The influence of chiral ligand concentration on the optical activity was analysed and the biological activity of L-Cys-NN and D-Cys-NN enantiomers were compared. In addition, monolayers of the CdSe/CdS NN based structures have been studied in the solid state, paying specific attention to the Förster resonance energy transfer (FRET).
Preparation of CdSe/CdS NN hetero-nanostructures
Initially, zinc blende (zb)-CdSe QD seeds were synthesised, producing a total of four samples of varying diameters, using a modified synthetic approach reported in literature.5,37 Details of the synthetic procedure, as well as variation of the reaction conditions, are given in ESI† (see Materials and methods, and Table S1). Briefly, the synthesis involved formation of the Cd precursor from CdO in degassed oleic acid at 240 °C. Following this, the selenium precursor was formed from elemental selenium and TOP, to which oleylamine, TDPA and ODE were added, degassed, and heated to 165 °C under an Ar atmosphere. Following this, in a separate vessel, a volume of the cadmium oleate formed previously, was added to ODE, degassed and heated to 240 °C under an Ar atmosphere, after which a volume of the Se-TOP, oleylamine, TDPE solution was quickly introduced using the well-known hot-injection approach. The growth times were varied from 0–5 minutes, after which the reaction vessel was removed from the heating mantle and cooled to RT, giving a number of CdSe QDs of increasing size, dependent upon the growth time.Following this, samples were characterised using TEM and STEM, XRD, UV-Vis and PL. UV-Vis absorption spectra (Fig. 1(A)) show the characteristic first exciton peaks for each CdSe seed, the position of which enables the size of the resulting nanocrystal to be calculated,38 giving a diameter ranging from 2.1–3.65 nm. In addition, the first exciton half-width half-maximum (HWHM) was determined (14.5–18.5 nm) indicating a narrow size distribution in each sample. The resulting PL exciton emission of these samples is given in Fig. 1(B) and show a shifting peak position in-line with the first exciton position, giving a Stokes shift of 4.5–8 nm and a full-width half-maximum (FWHM) of 31.5–42 nm for the samples. Full details of these parameters are provided in the ESI† (Table S2). In addition to the PL exciton emission, results for the two smaller samples, zb-CdSe-1 and -2 showed a broad PL peak stretching to the NIR associated with defect surface states, with the resulting PL full spectra for these samples shown in Fig. S1 of the ESI.† TEM and STEM results confirm the formation of spherical CdSe cores, with diameters in line with that calculated via first exciton positions from UV-Vis absorption spectra, with a representative TEM image shown in Fig. 1(C). Additional TEM and STEM images, as well as size distributions, are provided in the ESI† (Fig. S2–S5). XRD was carried out on samples to confirm the formation of the zinc blende structure, as shown in Fig. 1(D). The characteristic zinc blende associated-reflections are represented by blue lines, corresponding to 2θ values reported in the literature for zinc blende CdSe QDs.39
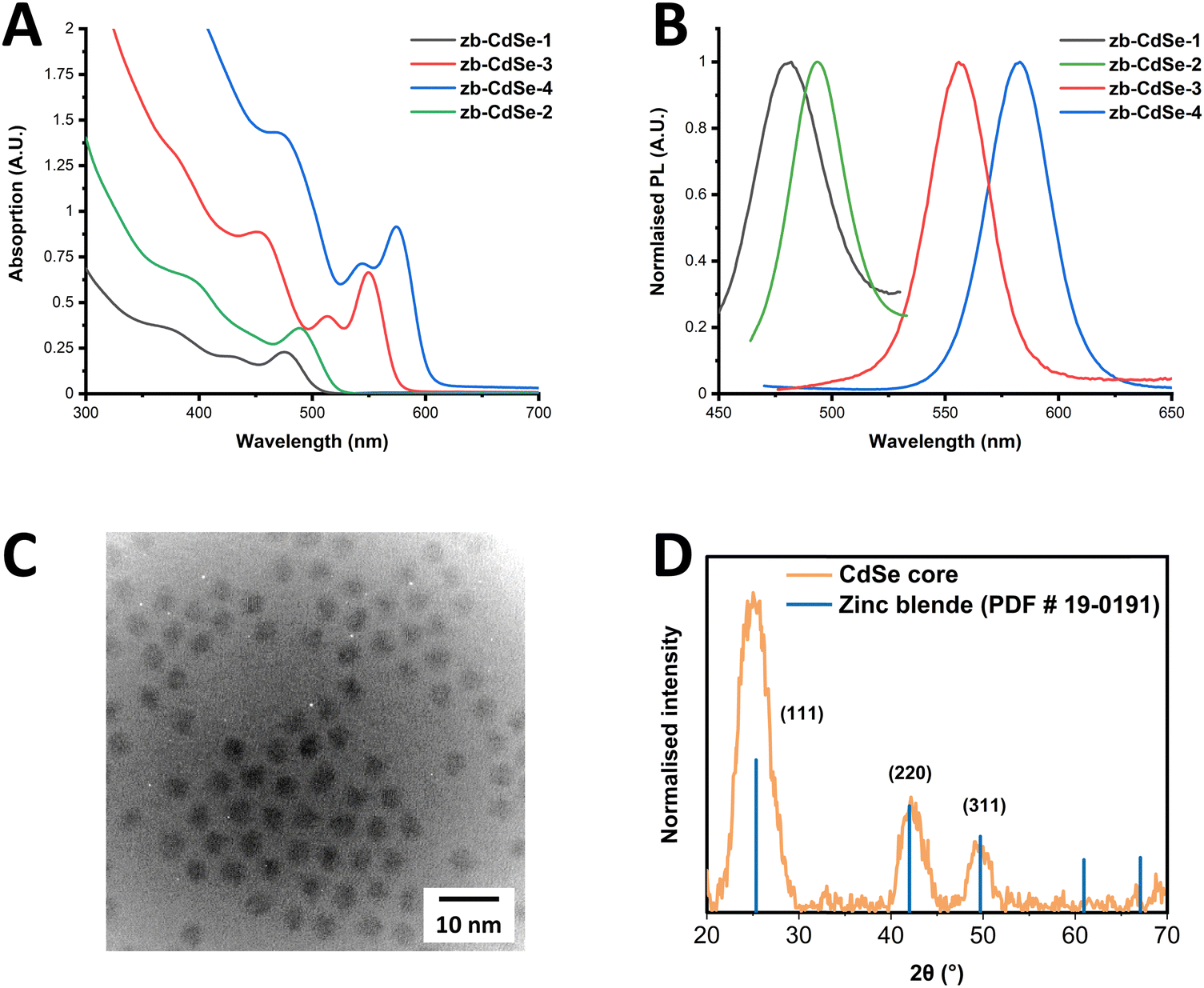 | ||
| Fig. 1 (A) UV-Vis absorption and (B) PL spectra of zb-CdSe core QDs. (C) TEM image, and (D) XRD pattern of zb-CdSe-4. Zinc-blende associated reference reflections are represented by blue lines. | ||
The as-synthesised zb-CdSe core QDs were then used as seeds to grow a CdS shell, resulting in a novel nanonail (NN) CdSe/CdS structure. This involved the use of a modified synthetic method, which was previously reported to be an effective approach in coating CdSe seeds.13 Briefly, the synthesis involved the heating of CdO in OA, ODE and ODPA, under an inert atmosphere to form the cadmium precursor at 350 °C. After this, a volume of TOP was added, followed by a hot injection containing zb-CdSe seeds mixed with the S source (S-TOP). This was swiftly injected, after which the CdS shell was allowed to grow for 10 minutes on the CdSe seeds, cooled to room temperature, and subsequently cleaned. Full synthetic details can be found in the ESI† (Section I – Materials and methods).
A range of samples were produced, using two different concentrations of seeds (7.5–15 nmol of QDs) and four zb-CdSe seeds with differing diameters (2.1–3.65 nm), with the full index of reaction conditions used given in ESI† (Table S3). Following the synthesis, the resultant materials were characterised using TEM, STEM, EDX, UV-Vis, PL emission, PL lifetime decay, and XRD.
TEM was carried out on all CdSe/CdS NN samples, with a select sample of images provided in Fig. 2. TEM images of two samples, CdSe/CdS NN-3 (Fig. 2(A)), and CdSe/CdS NN-6 (Fig. 2(B)), both of which used a CdSe seed of 2.9 nm (sample zb-CdSe-3), clearly depict a novel morphology, consisting of a wider, triangular head structure accompanied by an elongated tail (Fig. 2(C)). Further information including dimension measurements (Table S4), diagram (Fig. S6), additional TEM images (Fig. S7–S13), and size distributions (Fig. S14–S20) are provided in the ESI.† The variable between these two samples is the shell thickness, with half the concentration of seeds used for the synthesis of NN-6 relative to NN-3 (7.5 nmol vs. 15 nmol), resulting in deposition of a larger CdS shell in the case of NN-6. Across all synthesised samples, the resulting structures measured from 23.9 nm to 65.9 nm in length. The top width varied from 10 to 13.3 nm, while the tail width ranged from 5.4 nm to 7.2 nm. The aspect ratio of length to width has been found to vary from 2.4 to 5.5 when measuring the end width, and 3.5 to 12.2 when using the tail width. A tapering ratio was determined from the width at the top of the structure to the end of the tail of the structure and was found to range from 1.5 to 2.2 across the samples.
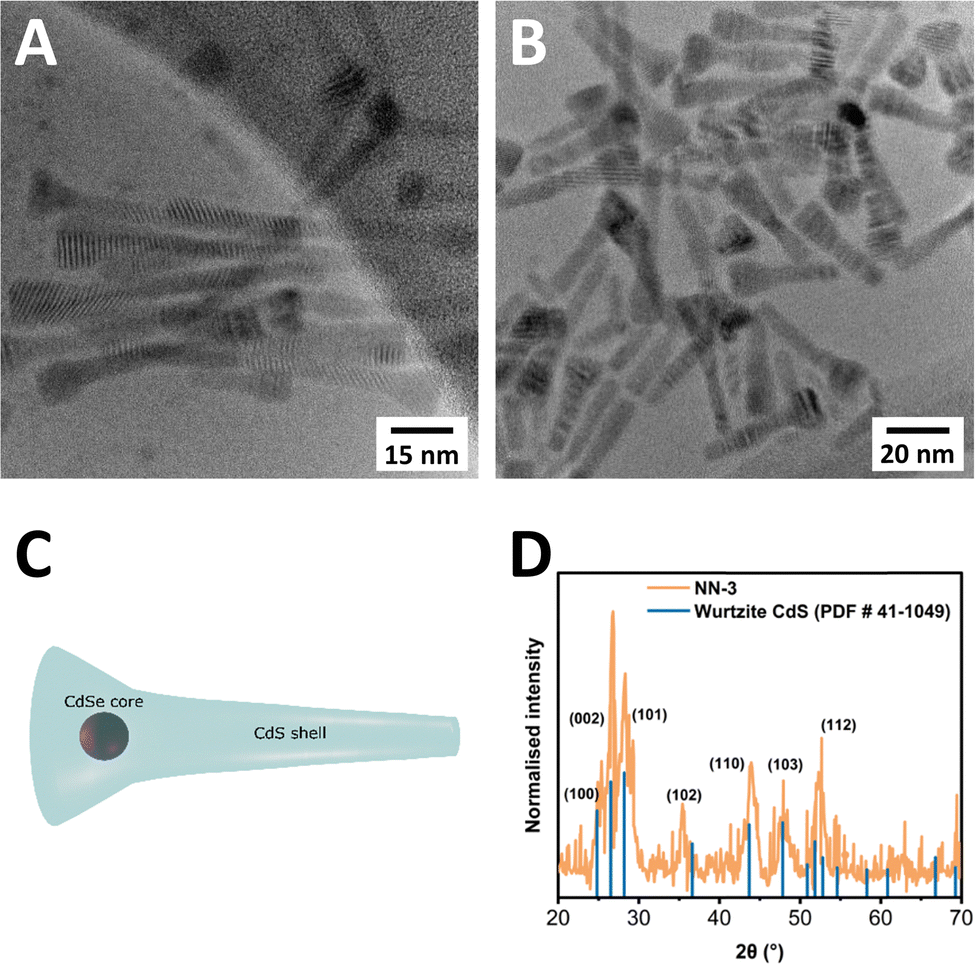 | ||
| Fig. 2 (A) and (B) TEM images of two selected CdSe/CdS NN samples – CdSe/CdS NN-3 (A), and NN-6 (B). (C) Schematic of proposed NN structure. (D) XRD pattern of NN-3. Characteristic wurtzite-associated reference reflections are represented by blue lines. | ||
Furthermore, XRD was carried out to determine the crystal structure of the resultant NNs, as shown in Fig. 2(D), confirming that the NNs adopt a wurtzite crystal structure, with characteristic wurtzite-associated reflections labelled in blue, matching reported values.39
UV-Vis absorption spectra of the NNs are presented in Fig. 3(A), with the samples showing the sharp onset of CdS shelling below 500 nm. Additionally, the position of the CdSe first exciton is still visible in some samples, while in other cases the finer detail has completely disappeared, with the first exciton peaks listed in Table S5 of ESI.† Furthermore, the difference in CdS shelling in samples can be more clearly seen in Fig. S21 (ESI†), in which the comparison of the CdS absorption is clearly shown, with the CdS shell being far more significant in the sample CdSe/CdS NN-6 than NN-3.
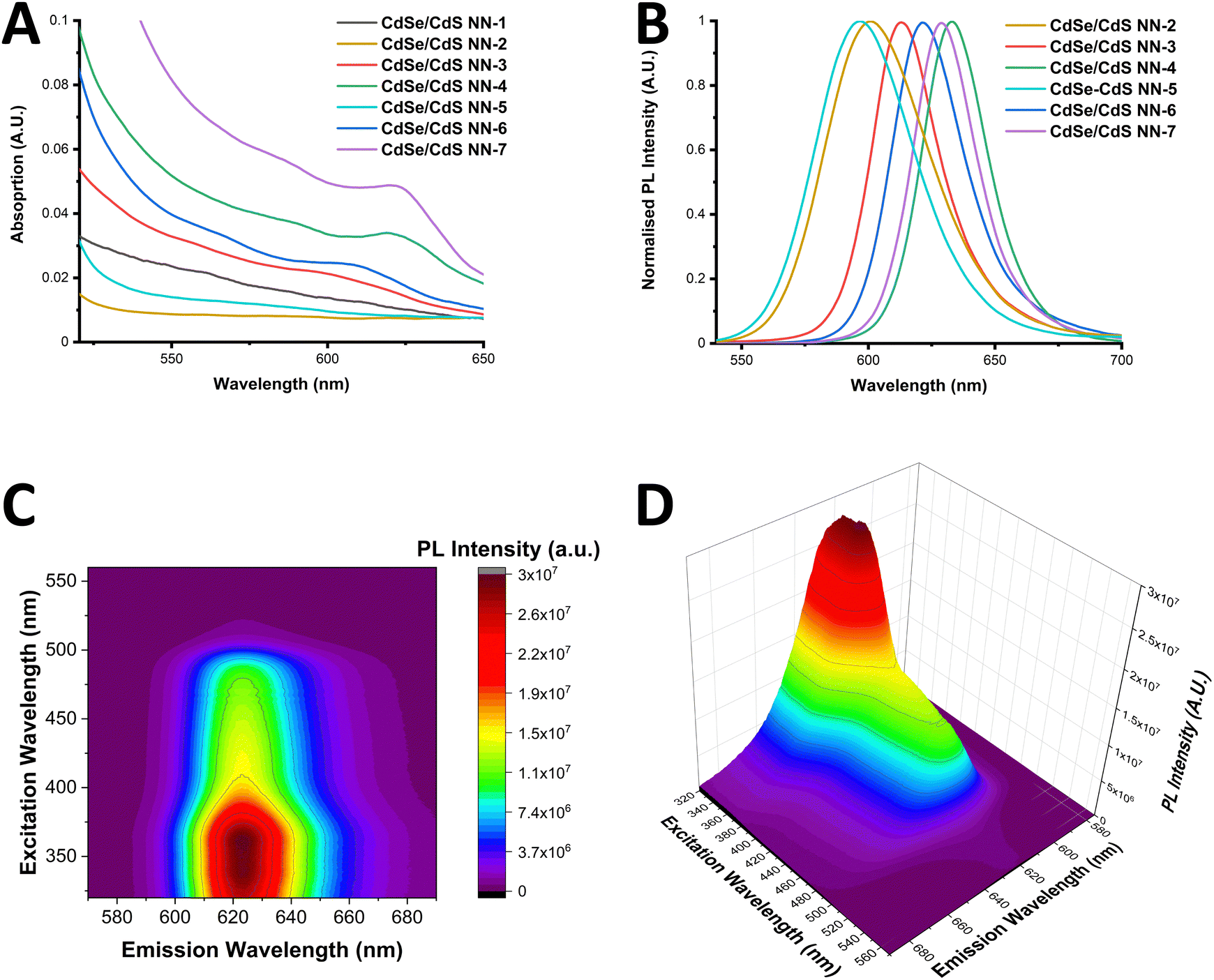 | ||
| Fig. 3 UV-Vis absorption (A) and PL spectra (B) of the range of NN synthesised. (C) and (D): EEM of CdSe/CdS NN-6 shown in a contour map (C) and 3-dimensional plot (D). | ||
PL spectra of the NNs are presented in Fig. 3(B), with samples showing sharp PL emission, ranging from 597–633 nm. Red-shifting of the PL emission is observed to be dependent upon the length of the CdS shell, as well as the initial size of the core. It is worth noting that sample CdSe/CdS NN-1 displayed a distinct spectrum with two emission peaks, most likely due to CdS emission. Results of photoluminescence quantum yield (PLQY) studies of the samples are provided in the ESI† (Table S5), with a maximum PLQY of 19% reported in the case of the CdSe/CdS NN-4 sample. It was noted that a decrease in PLQY was found was observed for samples with longer CdS shells, and that a decrease in PLQY was also found when using smaller CdSe QDs cores, which can be due to a more likely type-II band alignment as the CdSe QD becomes smaller.
Th excitation emission matrix (EEM) of CdSe/CdS NN-6 is shown in a contour map and as a 3-dimensional plot in Fig. 3(C) and (D) respectively. These spectra were adjusted with a correction factor to account for the inner filter effect (calculated using eqn (S1) and (S2) in the ESI†) with the uncorrected spectra provided in Fig. S23 (ESI†). The resulting corrected spectra in Fig. 3(C) and (D) show a distinctive behaviour of the material, exhibiting a strong maximum emission intensity in the range of 620–625 nm upon excitation of approximately 330–370 nm, accompanied by a general decrease in emission intensity with lower energy excitation. In addition, no peak maximum red shifting is found to occur upon excitation variation, which again supports the lack of a broad size distribution of the NN sample. Furthermore, these graphs clearly demonstrate that the shell plays a significant role in photon emission, with PL intensity strongly increasing when excited below 500 nm, the onset of CdS absorption.
PL lifetime decay analysis was carried out on the samples which demonstrated the highest PLQYs, as shown in Fig. S22 of the ESI.† The PL lifetime decay curves were fit with a triexponential decay function, giving an average time of between 27.8–69.6 ns, with the results obtained from fitting of the triexponential decay provided in Table S6 of the ESI.† It is also worth noting that upon comparing the lifetime of NN-3 and NN-6, which differ in the thickness of the CdS shell deposited, we find an increased PL lifetime in NN-6 relative to NN-3, as found in other core/shell CdSe/CdS structures. In general, the lifetimes presented here are typical of CdSe/CdS nanostructures, and indicate the normal quasi-type II band alignment present in these structures.27,40
Discussion of CdSe/CdS NN growth mechanism
Clearly the origins of this novel CdSe/CdS morphology must be strongly connected to previously reported structures, particularly the similar DiT and DiR CdSe/CdS heterostructures, as supported by the examples provided above. The key factor in synthesising these morphologies is based upon the use of either wurtzite or zinc blende seeds of CdSe QDs. These seeds serve as the foundation for the formation of either DiR structures or DiT structures via the growth of an anisotropic CdS shell around these cores, either in one direction in the case of DiR, or in four directions in the case of DiT. This synthesis follows a seeded growth methodology, in which the CdSe core crystal structure plays a crucial role in determining the shape and growth pattern of the CdS shell produced. In this study, zb-CdSe QD seeds have been used, therefore the anticipated outcome would have been the formation of a DiT. However, the result was instead a dot-in-tapered rod structure, or a NN. Therefore, the key differences that led to this result should be investigated.The first unusual element to this synthesis lies in the utilisation of an alternative approach to zb-CdSe QD seed synthesis. This method involves the less widely used approach of simultaneous injection of TDPA, the Se-TOP precursor and oleylamine to a solution of Cd(oleate). Se-TOP is a common precursor for CdSe synthesis, and the addition of oleylamine to it has been shown to decrease the overall reactivity of this precursor via the formation of hydroselenite.41 In this synthesis, it has been demonstrated that the addition of TDPA to the injection solution is the key factor that enables the consistent and reproducible production of zb-CdSe QDs. It is worth noting these zb-CdSe QD seeds have been used to produce CdSe TPs previously.41,42 In contrast, the most common approach to producing zb-CdSe QDs uses an alternative Se source, Se-ODE (selenium powder dispersed in ODE) and usually exclusively fatty acid ligands (e.g. myristic acid, oleic acid).43–46 Therefore, as a means to confirm that the type of zb-CdSe synthesis used is playing a key role here, zb-CdSe QDs were also synthesised using the more common approach, with the method and results provided in the ESI† (see Section VI. CdSe/CdS Dot in Tetrapods), confirming the XRD and optical properties. Two sizes of CdSe seeds, 4.0 nm and 5.6 nm, were then used in the 2nd step of the NN synthesis instead, resulting exclusively in the formation of the CdSe/CdS DiT morphology, as has been reported previously,13 with the resulting morphology and optical characterisation given in the ESI† (see Section VI. CdSe/CdS Dot in Tetrapods). This result therefore confirms that the type of zb-CdSe seeds play an important role in producing the unique NN morphology.
Additionally, in the seeded growth synthetic approach, other factors are key in deciding the exact morphology of the product, particularly the choice of ligands, since the crystal structure and shape of the resulting shell can be strongly influenced by the type and ratio of ligands present. The effects of the choice of ligand on the resulting morphology is three-fold. Firstly, ligands can selectively bind to certain facets of a nanocrystal, and therefore play a role in determining the crystal structure formed. Furthermore, the morphology of the resulting structure is also influenced by certain facets exhibiting higher degrees of surface passivation, resulting in anisotropic growth. Finally, the most recently reported effect is that ligand concentration has a notable impact on the exact concentration of monomer species in solution, which can play a key role in determining 1D or 3D growth of the surface of NCs. This insight has been shown to enable the tuning the dimensions of nanomaterials, such as the width or length of a given nanorod, for example.47,48
When considering the synthesis of CdSe/CdS NN, the use of a lower concentration of ODPA and/or addition of OA to the synthesis, which are used in the highly reported standard CdSe/CdS DiT synthesis, are therefore important parameters. In particular, the CdS shell growth is carried out in the presence of oleic acid and ODPA, which are both partially used to form a mixture of Cd(OA)2 and Cd(ODPA)2 as cadmium precursors. It has been reported that the use of cadmium carboxylate ligands favour the growth of a zinc blende shell, while cadmium phosphonate ligands instead favour the growth of the wurtzite phase.49 In addition, the approach utilizes a volume of TOP which has also been shown to stabilize the zinc blende phase of CdS, while OA has been shown to preference the wurtzite phase of CdS.50 It is also worth noting that zb-CdSe seeds used are synthesised in the presence of TDPA. Consequently, the resulting seed solution contains a small of volume of TDPA, with the seeds being at least partially capped by this ligand. Interestingly, studies have demonstrated that the addition of a small proportion of short chain phosphonic acid to this type of reaction solution, in addition to the ODPA present in the reaction mixture, helps to preserve the zb structure of the CdSe seed in a CdSe/CdS DiT synthesis. On the other hand, in the case of the TDPA being absent, a much higher proportion of DiR is produced even with the use of zb-CdSe seeds, due to the conversion of these seeds from zinc blende to wurtzite phase in the reaction mixture.39
Thus, while CdS has been determined to be in the wurtzite phase at the end of the NN synthesis, it is possible that the CdS shell underwent several steps in alternative intermediate phases before being produced in the final wurtzite form. Unfortunately, due to the rapid growth and high temperature of the synthesis, it was highly challenging to study the intermediate stages of the synthesis, hindering us from fully elucidating the formation mechanism of this unique morphology.19 In summary, this distinctive morphology arises from the specific conditions of zb-CdSe QD produced in the first step, coupled with the specific ligand content and concentration of the CdS shell growth of the 2nd step. Most importantly, these conditions result in the NN morphology being the vastly dominant species present, with only minimal amounts of side products produced.
Ligand-induced chirality in CdSe/CdS NNs
Following this work, the effect of chiral ligand exchange upon the structure was studied, since morphology and shell thickness have been found to play an important role in ligand-induced chirality in previous work.2,19,51 Optical activity was induced by exchanging the original achiral organic ligands with D- and L-cysteine (Cys), an amino acid which has been extensively studied for the induction of chirality in nanomaterials.52–54 The samples were then named relative to the capping ligand as either L-Cys-NNs or D-Cys-NNs. The methodology for this ligand exchange is based on our previous publications.2,53,55,56 UV-Vis, PL and circular dichroism (CD) spectra of the NNs before and after the ligand exchange are presented in Fig. 4. Following ligand exchange, the position and shape of NN absorption and PL emission remain unchanged compared to the original NN sample, as seen Fig. 4(A) and (B). In contrast, the PL intensity decreased after the ligand exchange, with the effect being identical for both L- and D-Cys-NNs, which is in line with previous reports of cysteine capped CdSe/CdS structures.2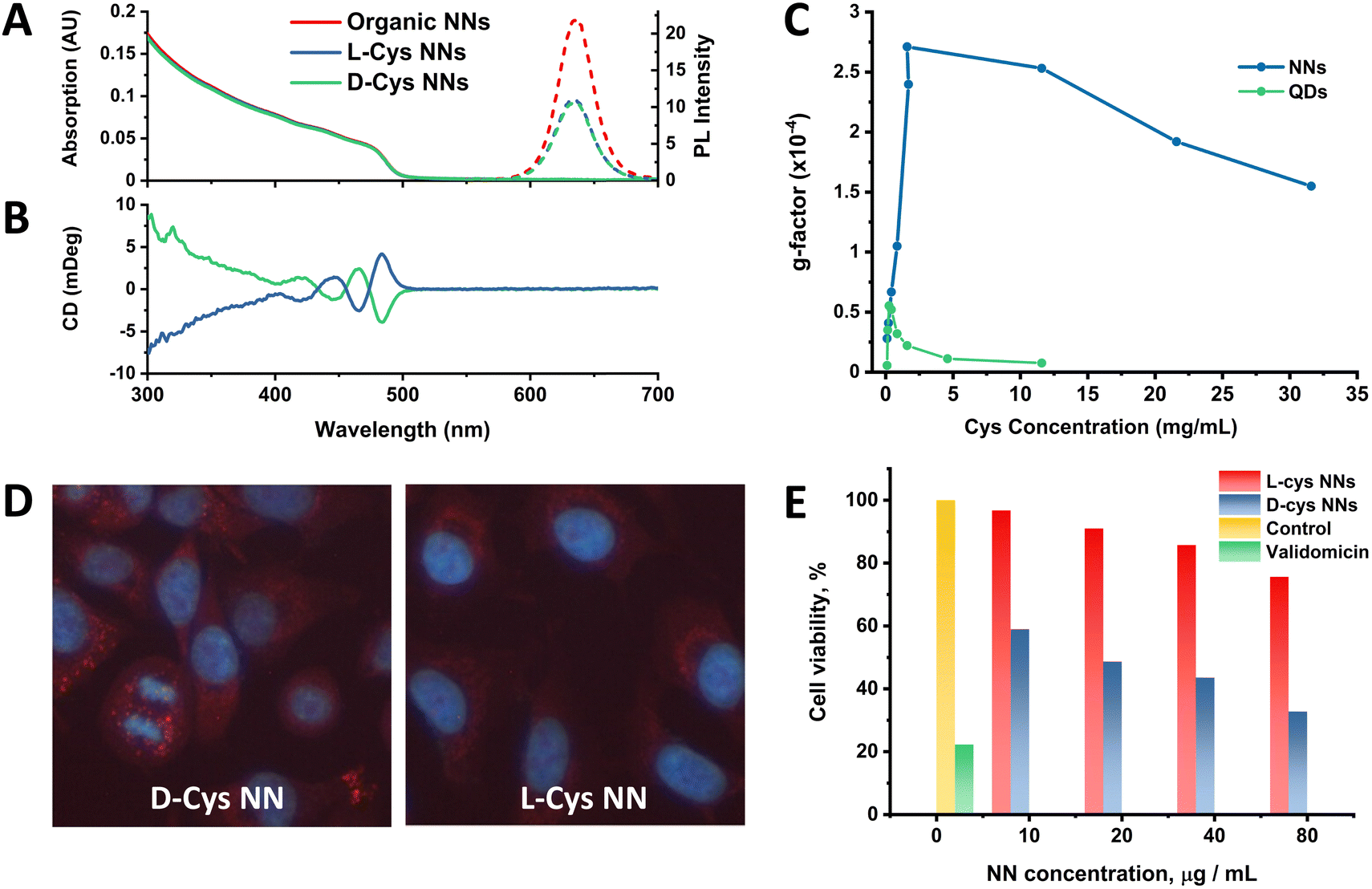 | ||
| Fig. 4 (A) UV-Vis and PL spectra of NN samples before and after ligand exchange with L- and D-cysteine. (B) CD spectra of L- and D-cysteine-capped NNs. (C) The dependence of CdSe/CdS NN and spherical QD g-factor on Cys concentration. (D) PL images of A549 cells incubated with L-Cys and D-Cys NNs for 24 h. (E) Cell viability of A549 Cells after incubation with chiral NNs for 24 h versus NN concentration, represented as the percentage of viable cells. | ||
Following ligand exchange, Fig. 4(B) confirms that L- and D-Cys-capped NNs display mirror image CD signals in the region of the NN CdS shell absorption. Induction of optical activity in the spectral region of the NN CdS shell absorption onset (below 500 nm) after the ligand exchange can be attributed to hybridization of the CdS shell valence band states with the HOMO/LUMO orbitals of the chiral D- and L-Cys ligands.57 Unfortunately, due to instrumentation limitation, it was very difficult to detect the CD signal from the CdSe core absorption onset region (550–650 mm) as has been previously carried out in the study of other CdSe/CdS structures2 due to the very low content of CdSe compared to CdS in the NNs. The concentration range at which NNs are colloidally stable resulted in a UV-Vis absorption contribution from CdSe which was much lower than optimal for CD measurements. Despite this, a CD signal from the CdSe region was registered, as shown in Fig. S27 of the ESI.† However, due to the low absorption in this region, the spectra exhibited poor signal-to-noise ratios, making the data in this region less reliable. It should be noted that the g-factor value obtained from the CdSe core was approximately an order of magnitude higher than the signal from the CdS shell, with the obtained values being on the order of 1 × 10−3 and 1 × 10−4 for core and shell, respectively. g-Factor spectra of the NNs in the CdS absorption region are shown in the ESI† (Fig. S29–S32). A probable explanation for this observation is due to preferable localization of the exciton in the CdSe core, which as a result of electronic interactions with cysteine ligands, leads to the induction of chiroptical activity to a greater extent in the core than in the shell, as has been reported previously.2,58
Previously published work from our group studied the effect of cysteine concentration on the intensity of the CD signal of spherical CdSe/CdS QDs. This work revealed a complex concentration dependence of the chiroptical activity, showing an initial growth in the CD signal value, followed by a subsequent decrease after reaching a certain critical concentration, which was associated with a change in the cysteine binding mode.53 It is of great interest to study the same effect in anisotropic CdSe/CdS nanoparticles, such as the NN structures reported here. For this, ligand exchange was performed with different amounts of cysteine, varying from 0.1 to 31.6 mg ml−1, and the resulting optical activity of the NNs was compared with the previously reported spherical QD data, as shown in Fig. 4(C). For NNs, the same trend was observed as for spherical QDs, where initially a positive correlation was observed with increasing cysteine concentration, until a limiting concentration was reached, and the g-factor intensity subsequently began to decrease. However, the g-factor values of the NNs were in general much higher than those of spherical QDs, with the maximum values differing almost five-fold. This observed enhancement of the g-factor for anisotropic nanostructures compared with their isotropic counterparts is a phenomenon that has been reported previously in great detail for CdSe nanorods,59 as well as for CdSe/CdS tadpoles.19 Gao et al. describe that the origin of enhanced chiroptical activity in anisotropic CdSe nanostructures is a result of the two distinct dipole orientations along the long and short axis of the nanostructure, giving rise to linear polarisation in the excitonic transitions. Thus, polarised excitonic transitions along the long axis of the nanostructure can give rise to enhanced CD signals,59 as has also been observed for other anisotropic CdSe/CdS nanostructures.19 Additionally, the decrease in the g-factor began at much higher Cys concentration compared to the spherical QDs, likely due to the fact there is simply more available area for Cys ligands to bind to the anisotropic NN surface when compared to the spherical QDs. It is worth noting that, unlike with spherical QDs, we were limited to studying the CD signal intensity in the CdS shell absorption region only. As was mentioned previously, due to the low absorption contribution from the CdSe core, it was not possible to compare the effect of the cysteine concentration on its intensity in this region.
Chiral ligand exchange was tested across the range of NN samples produced, and it was found that CD signals varied significantly across samples, as shown in Fig. S28 of the ESI.† In general, a trend can be observed in which the smaller CdSe core (2.25 nm, zb-CdSe-2) seed based NNs display stronger CD signals upon ligand exchange (CdSe/CdS NN-2 and 5), though further studies would be required to fully elucidate the overall pattern. In addition, as has been studied previously,53 a relationship between ligand concentration and CD signal strength was demonstrated (see ESI,† Fig. S29–S32).
Cellular uptake and cytotoxicity studies of chiral CdSe/CdS NNs
The biological activity of the NNs were tested on the human lung-derived A549 cancer cell line, so as to investigate the potential for enantioselective cellular uptake and cytotoxicity as demonstrated in previous studies.52,55,60,61 For the cell accumulation assay, L- and D-Cys-NNs were incubated with cells for 24 hours, and the PL intensity of the chiral NNs inside the cells was estimated by analysis of the PL images (Fig. 4(D)). The images clearly confirm the intracellular uptake and accumulation of the chiral NNs into the cells, showing their characteristic fluorescent labelling. The bright fluorescence spots observed within the cells reflect the intracellular distribution of NNs due to, most likely, their endosomal localization. It can be visually observed that the intensity of the fluorescence of D-Cys NNs is much higher than that of L-Cys NNs, which is evidence of more efficient cellular uptake of D-Cys NNs comparing to L-Cys NNs. For comparison, the cellular uptake of cysteine-capped spherical CdSe/CdS QDs was also explored. Unlike the NNs, spherical QDs have less enantioselectivity in cell internalization and are less localized to endosomes (Fig. S33, ESI†). While further studies are required to fully elucidate the difference in the mechanisms of biological activity between NNs and spherical QDs, this may be a result of the enhanced induction of chirality in cysteine-capped anisotropic NNs compared to spherical QDs, as shown in Fig. 4(C), and as highlighted in recent work from Hao et al.62 This morphology-dependent cellular uptake has also been observed for other anisotropic nanostructures, including chiral gold nanooctopods,63 and asymmetric gold NPs.64 For the cell viability assay, cells were incubated with NNs at concentrations ranging from 10 to 80 μg ml−1 for 24 hours (Fig. 4(E)). The assay revealed a significant difference in cytotoxicity between the two enantiomers, with D-Cys NNs exhibiting 2.3 times higher cytotoxicity levels than L-Cys-NNs at maximum concentration. This is in line with previous works demonstrating enantioselective toxicity in other QDs.52,55,60,61CdSe/CdS NN monolayers and intra-energy transfer
The effects of possible FRET processes in monolayers of CdSe/CdS NN-3 were studied. This sample in solution displays a large Stokes shift of 146 nm (Fig. S34, ESI†), with the Stokes shift being over three times larger than the FWHM, resulting in no significant overlap between the absorption and emission, causing the overlap integral J to vanish. Consequently, no FRET is expected within the ensemble of these NNs (see eqn (S4)–(S7), in ESI†). This suggests therefore that the CdSe/CdS NNs should not suffer from the intra-energy transfer seen in other closely packed QD systems. An additional study was undertaken to verify the absence of intra-energy transfer in monolayers of NNs of varying concentrations. The PL spectra of monolayers can be seen in Fig. 5(A) for three different sample concentrations, termed as concentration 1, 2, and 3, respectively. More information on the concentrations used can be found in the ESI† (see Section I – Materials and methods). The PL peak of the monolayers is centred around 620 nm and is found not to vary between monolayers of different concentrations with samples compared to a reference sample (PE bilayers). The measurements were performed at a number of positions across the sample, with a variation of approximately 10%, represented as the error in the data points in Fig. 5(B).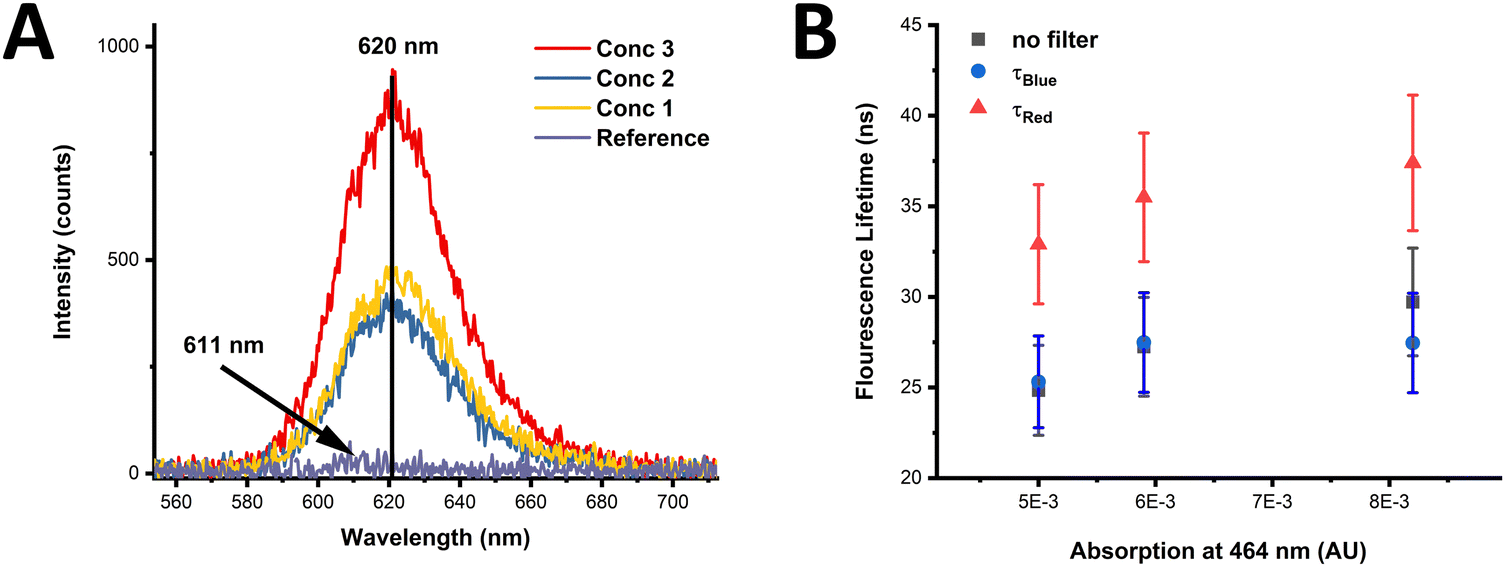 | ||
| Fig. 5 PL spectra of CdSe/CdS NN-3 in monolayers of varying concentration (A) and graph of average fluorescence decay lifetimes of NN monolayers (B) measured at different sides of the spectra: τBlue (measured with 600 nm filter), τRed (measured with 650 nm filter) and the overall emission (no filter) at three difference concentrations. | ||
The normalised spectra for the CdSe/CdS NN-3 monolayers and CdSe/CdS NN-3 in solution confirms that there is no shift of the PL peak or change in the spectral shape with increasing NN concentration, as shown in Fig. S35 of the ESI.† In contrast, previous reports of intra-layer FRET for an inhomogeneous broadened QD distribution show a red shift in the emission peak with increasing concentration.65,66
In addition, the average fluorescence lifetimes of the different concentration monolayers were measured, as shown in Fig. 5(B). In this, the average lifetimes were calculated from fitted data (see ESI,† Fig. S36) of τBlue (measured with 600 nm filter), τRed (measured with 50 nm filter) and a measurement without a filter (termed no filter). From this data, it is clear that no decrease in the average fluorescence lifetime of the smaller NNs, given by τBlue, occurs with increasing concentration. Furthermore, if intra-energy transfer were present, the difference in τRed and τBlue, presented as 1 − τBlue/τRed, would be expected to increase with increasing concentration, however, in this case the lifetime difference is constant over the measured range (see ESI,† Fig. S37). Therefore, the invariance of τBlue and the lifetime difference between the red-side and blue-side of the PL spectrum, as well as constant PL emission peak position and shape, confirms that there is no observable intra-layer FRET in the NN monolayers investigated in this study. Given that suppression of FRET in QD thin films is crucial in improving the efficiency of QD-LEDs, the lack of FRET observed in monolayers of these NNs suggests they may be promising candidates for use in QD-LEDs.67,68 While numerous other factors such as charge transport characteristics, quantum yield, colour purity, and long-term photostability are vital considerations for high-performance QD-LEDs, the absence of FRET in monolayers of these NNs is noteworthy, and thus deserving of further exploration.69,70
Conclusions
Thus, we have demonstrated the highly controllable synthesis of a new nanonail morphology within the extensively studied CdSe/CdS family of nanomaterials. The synthesised NNs exhibit distinct morphology-dependent optical properties, setting them apart from other previously reported CdSe/CdS nanostructures. Through ligand exchange with cysteine, chirality has been successfully induced in these NNs, as demonstrated by the distinctive CD signals observed, as well as the enantioselective cellular uptake and cytotoxicity. Finally, given the absence of FRET in monolayers of these NNs, we believe their potential suitability for QD-LEDs warrants further attention and study. This work demonstrates yet another promising CdSe/CdS nano-heterostructure morphology, one that is readily controllable and tuneable, with a plethora of potential applications, from biological to optical.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to acknowledge Science Foundation Ireland (grant numbers: SFI 20/FFP-A/8904, 12/IA/1300 and 16/TIDA/4122) and the Irish Research Council (grant number: GOIPG/2019/2788) for financial support.References
- X. Peng, M. C. Schlamp, A. V. Kadavanich and A. P. Alivisatos, J. Am. Chem. Soc., 1997, 119, 7019–7029 CrossRef CAS.
- F. Purcell-Milton, A. K. Visheratina, V. A. Kuznetsova, A. Ryan, A. O. Orlova and Y. K. Gun’Ko, ACS Nano, 2017, 11, 9207–9214 CrossRef CAS PubMed.
- A. K. Visheratina, F. Purcell-Milton, R. Serrano-García, V. A. Kuznetsova, A. O. Orlova, A. V. Fedorov, A. V. Baranov and Y. K. Gun’ko, J. Mater. Chem. C, 2017, 5, 1692–1698 RSC.
- I. V. Martynenko, A. S. Baimuratov, V. A. Osipova, V. A. Kuznetsova, F. Purcell-Milton, I. D. Rukhlenko, A. V. Fedorov, Y. K. Gun’Ko, U. Resch-Genger and A. V. Baranov, Chem. Mater., 2018, 30, 465–471 CrossRef CAS.
- B. Mahler, P. Spinicelli, S. Buil, X. Quelin, J. P. Hermier and B. Dubertret, Nat. Mater., 2008, 7, 659–664 CrossRef CAS PubMed.
- B. N. Pal, Y. Ghosh, S. Brovelli, R. Laocharoensuk, V. I. Klimov, J. A. Hollingsworth and H. Htoon, Nano Lett., 2012, 12, 331–336 CrossRef CAS PubMed.
- X. Y. Yu, J. Y. Liao, K. Q. Qiu, D. Bin Kuang and C. Y. Su, ACS Nano, 2011, 5, 9494–9500 CrossRef CAS.
- F. Meinardi, A. Colombo, K. A. Velizhanin, R. Simonutti, M. Lorenzon, L. Beverina, R. Viswanatha, V. I. Klimov and S. Brovelli, Nat. Photonics, 2014, 8, 392–399 CrossRef CAS.
- I. Coropceanu, A. Rossinelli, J. R. Caram, F. S. Freyria and M. G. Bawendi, ACS Nano, 2016, 10, 3295–3301 CrossRef CAS PubMed.
- L. Carbone, C. Nobile, M. De Giorgi, F. Della Sala, G. Morello, P. Pompa, M. Hytch, E. Snoeck, A. Fiore, I. R. Franchini, M. Nadasan, A. F. Silvestre, L. Chiodo, S. Kudera, R. Cingolani, R. Krahne and L. Manna, Nano Lett., 2007, 7, 2942–2950 CrossRef CAS.
- D. V. Talapin, J. H. Nelson, E. V. Shevchenko, S. Aloni, B. Sadtler and A. P. Alivisatos, Nano Lett., 2007, 7, 2951–2959 CrossRef CAS.
- A. Sitt, A. Salant, G. Menagen and U. Banin, Nano Lett., 2011, 11, 2054–2060 CrossRef CAS.
- J. I. Wong, N. Mishra, G. Xing, M. Li, S. Chakrabortty, T. C. Sum, Y. Shi, Y. Chan and H. Y. Yang, ACS Nano, 2014, 8, 2873–2879 CrossRef CAS PubMed.
- E. Cassette, B. Mahler, J. M. Guigner, G. Patriarche, B. Dubertret and T. Pons, ACS Nano, 2012, 6, 6741–6750 CrossRef CAS PubMed.
- A. Corrias, E. Conca, G. Cibin, G. Mountjoy, D. Gianolio, F. De Donato, L. Manna and M. F. Casula, J. Phys. Chem. C, 2015, 119, 16338–16348 CrossRef CAS.
- M. D. Tessier, P. Spinicelli, D. Dupont, G. Patriarche, S. Ithurria and B. Dubertret, Nano Lett., 2014, 14, 207–213 CrossRef CAS PubMed.
- B. Mahler, B. Nadal, C. Bouet, G. Patriarche and B. Dubertret, J. Am. Chem. Soc., 2012, 134, 18591–18598 CrossRef CAS PubMed.
- A. Antanovich, A. Prudnikau, K. Grzhegorzhevskii, P. Zelenovskiy, A. Ostroushko, M. V. Kuznetsov, A. Chuvilin and M. V. Artemyev, Nanotechnology, 2018, 29, 395604 CrossRef PubMed.
- J. Hao, Y. Li, J. Miao, R. Liu, J. Li, H. Liu, Q. Wang, H. Liu, M. H. Delville, T. He, K. Wang, X. Zhu and J. Cheng, ACS Nano, 2020, 14, 10346–10358 CrossRef CAS PubMed.
- Y. Luo and L. W. Wang, ACS Nano, 2010, 4, 91–98 CrossRef CAS PubMed.
- G. Bertoni, V. Grillo, R. Brescia, X. Ke, S. Bals, A. Catellani, H. Li and L. Manna, ACS Nano, 2012, 6, 6453–6461 CrossRef CAS PubMed.
- A. Sitt, I. Hadar and U. Banin, Nano Today, 2013, 8, 494–513 CrossRef CAS.
- C. Segarra, J. I. Climente, A. Polovitsyn, F. Rajadell, I. Moreels and J. Planelles, J. Phys. Chem. Lett., 2016, 7, 2182–2188 CrossRef CAS PubMed.
- A. N. Grennell, J. K. Utterback, O. M. Pearce, M. B. Wilker and G. Dukovic, Nano Lett., 2017, 17, 3764–3774 CrossRef CAS PubMed.
- N. J. Borys, M. J. Walter, J. Huang, D. V. Talapin and J. M. Lupton, Science, 2010, 330, 1371–1374 CrossRef CAS PubMed.
- A. Sitt, F. Della Sala, G. Menagen and U. Banin, Nano Lett., 2009, 9, 3470–3476 CrossRef CAS PubMed.
- H. Eshet, M. Grünwald and E. Rabani, Nano Lett., 2013, 13, 5880–5885 CrossRef CAS PubMed.
- B. T. Diroll, A. Koschitzky and C. B. Murray, J. Phys. Chem. Lett., 2014, 5, 85–91 CrossRef CAS PubMed.
- K. Wu, L. J. Hill, J. Chen, J. R. McBride, N. G. Pavlopolous, N. E. Richey, J. Pyun and T. Lian, ACS Nano, 2015, 9, 4591–4599 CrossRef CAS PubMed.
- B. T. Diroll, N. Gogotsi and C. B. Murray, Chem. Mater., 2016, 28, 3345–3351 CrossRef CAS.
- D. Kim, Y. K. Lee, D. Lee, W. D. Kim, W. K. Bae and D. C. Lee, ACS Nano, 2017, 11, 12461–12472 CrossRef CAS PubMed.
- Y. Gao, V. D. Ta, X. Zhao, Y. Wang, R. Chen, E. Mutlugun, K. E. Fong, S. T. Tan, C. Dang, X. W. Sun, H. Sun and H. V. Demir, Nanoscale, 2015, 7, 6481–6486 RSC.
- M. V. Mukhina, A. S. Baimuratov, I. D. Rukhlenko, V. G. Maslov, F. Purcell Milton, Y. K. Gun’Ko, A. V. Baranov and A. V. Fedorov, ACS Nano, 2016, 10, 8904–8909 CrossRef CAS.
- P. Rastogi, F. Palazon, M. Prato, F. Di Stasio and R. Krahne, ACS Appl. Mater. Interfaces, 2018, 10, 5665–5672 CrossRef CAS PubMed.
- P. D. Cunningham, J. B. Souza, I. Fedin, C. She, B. Lee and D. V. Talapin, ACS Nano, 2016, 10, 5769–5781 CrossRef CAS.
- A. Rizzo, C. Nobile, M. Mazzeo, M. De Giorgi, A. Fiore, L. Carbone, R. Cingolani, L. Manna and G. Gigli, ACS Nano, 2009, 3, 1506–1512 CrossRef CAS.
- M. B. Mohamed, D. Tonti, A. Al-Salman, A. Chemseddine and M. Chergui, J. Phys. Chem. B, 2005, 109, 10533–10537 CrossRef CAS.
- W. W. Yu, L. Qu, W. Guo and X. Peng, Chem. Mater., 2003, 15, 2854–2860 CrossRef CAS.
- J. Huang, M. V. Kovalenko and D. V. Talapin, J. Am. Chem. Soc., 2010, 132, 15866–15868 CrossRef CAS PubMed.
- K. Gong, J. E. Martin, L. E. Shea-Rohwer, P. Lu and D. F. Kelley, J. Phys. Chem. C, 2015, 119, 2231–2238 CrossRef CAS.
- M. B. Mohamed, D. Tonti, A. Al Salman and M. Chergui, ChemPhysChem, 2005, 6, 2505–2507 CrossRef CAS PubMed.
- E. Park, J. Ryu, Y. Choi, K. J. Hwang and R. Song, Nanotechnology, 2013, 24, 145601 CrossRef PubMed.
- J. Lim, W. K. Bae, K. U. Park, L. Zur Borg, R. Zentel, S. Lee and K. Char, Chem. Mater., 2013, 25, 1443–1449 CrossRef CAS.
- L. Liu, Z. Zhuang, T. Xie, Y. G. Wang, J. Li, Q. Peng and Y. Li, J. Am. Chem. Soc., 2009, 131, 16423–16429 CrossRef CAS PubMed.
- J. Jasieniak, C. Bullen, J. Van Embden and P. Mulvaney, J. Phys. Chem. B, 2005, 109, 20665–20668 CrossRef CAS PubMed.
- Y. A. Yang, H. Wu, K. R. Williams and Y. C. Cao, Angew. Chem., Int. Ed., 2005, 44, 6712–6715 CrossRef CAS.
- M. J. Enright and B. M. Cossairt, Chem. Commun., 2018, 54, 7109–7122 RSC.
- M. J. Enright, H. Sarsito and B. M. Cossairt, Mater. Chem. Front., 2018, 2, 1296–1305 RSC.
- Y. Gao and X. Peng, J. Am. Chem. Soc., 2014, 136, 6724–6732 CrossRef CAS PubMed.
- A. Nag, A. Hazarika, K. V. Shanavas, S. M. Sharma, I. Dasgupta and D. D. Sarma, J. Phys. Chem. Lett., 2011, 2, 706–712 CrossRef CAS.
- F. P. Milton, J. Govan, M. V. Mukhina and Y. K. Gun’Ko, Nanoscale Horiz., 2015, 1, 14–26 RSC.
- I. V. Martynenko, V. A. Kuznetsova, I. K. Litvinov, A. O. Orlova, V. G. Maslov, A. V. Fedorov, A. Dubavik, F. Purcell-Milton, Y. K. Gun’ko and A. V. Baranov, Nanotechnology, 2016, 27, 075102 CrossRef CAS PubMed.
- V. A. Kuznetsova, E. Mates-Torres, N. Prochukhan, M. Marcastel, F. Purcell-Milton, J. O’Brien, A. K. Visheratina, M. Martinez-Carmona, Y. Gromova, M. Garcia-Melchor and Y. K. Gun’Ko, ACS Nano, 2019, 13, 13560–13572 CrossRef CAS PubMed.
- M. V. Mukhina, V. G. Maslov, I. V. Korsakov, F. P. Milton, A. Loudon, A. V. Baranov, A. V. Fedorov and Y. K. Gunko, Mater. Res. Soc. Symp. Proc., 2015, 1793, 27–33 CrossRef.
- V. A. Kuznetsova, A. K. Visheratina, A. Ryan, I. V. Martynenko, A. Loudon, C. M. Maguire, F. Purcell-Milton, A. O. Orlova, A. V. Baranov, A. V. Fedorov, A. Prina-Mello, Y. Volkov and Y. K. Gun’Ko, Chirality, 2017, 29, 403–408 CrossRef CAS PubMed.
- D. D. Barnes, V. Kuznetsova, A. Visheratina, F. Purcell-Milton, M. A. Baranov, D. M. Lynch, H. Martin, Y. K. Gun’ko and E. M. Scanlan, Org. Biomol. Chem., 2023, 21, 2905–2909 RSC.
- U. Tohgha, K. K. Deol, A. G. Porter, S. G. Bartko, J. K. Choi, B. M. Leonard, K. Varga, J. Kubelka, G. Muller and M. Balaz, ACS Nano, 2013, 7, 11094–11102 CrossRef CAS PubMed.
- A. Ben-Moshe, A. Teitelboim, D. Oron and G. Markovich, Nano Lett., 2016, 16, 7467–7473 CrossRef CAS PubMed.
- X. Gao, X. Zhang, K. Deng, B. Han, L. Zhao, M. Wu, L. Shi, J. Lv and Z. Tang, J. Am. Chem. Soc., 2017, 139, 8734–8739 CrossRef CAS PubMed.
- Y. Li, Y. Zhou, H. Y. Wang, S. Perrett, Y. Zhao, Z. Tang and G. Nie, Angew. Chem., Int. Ed., 2011, 50, 5860–5864 CrossRef CAS PubMed.
- M. Martínez-Carmona, C. Cela, V. A. Kuznetsova, J. A. Geoghegan and Y. K. Gun’ko, J. Mater. Chem. B, 2021, 9, 3544–3553 RSC.
- J. Hao, D. Hu, P. Chen, L. Ma, M. Xia, M. Chen, H. Xu, H. Liu, Y. Li, J. Cheng, X. Jin, P. Duan and X. Xu, Mater. Des., 2023, 226, 111653 CrossRef CAS.
- N. N. Zhang, H. R. Sun, S. Liu, Y. C. Xing, J. Lu, F. Peng, C. L. Han, Z. Wei, T. Sun, B. Yang and K. Liu, CCS Chem., 2022, 4, 660–670 CrossRef CAS.
- L. Xu, X. Wang, W. Wang, M. Sun, W. J. Choi, J. Y. Kim, C. Hao, S. Li, A. Qu, M. Lu, X. Wu, F. M. Colombari, W. R. Gomes, A. L. Blanco, A. F. de Moura, X. Guo, H. Kuang, N. A. Kotov and C. Xu, Nature, 2022, 601, 366–373 CrossRef CAS PubMed.
- M. Lunz, A. L. Bradley, W. Y. Chen, V. A. Gerard, S. J. Byrne, Y. K. Gun’Ko, V. Lesnyak and N. Gaponik, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 205316 CrossRef.
- A. P. Litvin, E. V. Ushakova, P. S. Parfenov, A. V. Fedorov and A. V. Baranov, J. Phys. Chem. C, 2014, 118, 6531–6535 CrossRef CAS.
- H. Zhang, Q. Su, S. Chen, H. Zhang, Q. Su and S. Chen, Adv. Opt. Mater., 2020, 8, 1902092 CrossRef CAS.
- Z. Qin, Q. Su, S. Chen, Z. Qin, Q. Su and S. Chen, Adv. Opt. Mater., 2023, 11, 2202451 CrossRef CAS.
- S. M. Jung, T. H. Lee, S. Y. Bang, S. D. Han, D. W. Shin, S. Lee, H. W. Choi, Y. H. Suh, X. B. Fan, J. W. Jo, S. Zhan, J. Yang, C. Samarakoon, Y. Kim, L. G. Occhipinti, G. Amaratunga and J. M. Kim, npj Comput. Mater., 2021, 7, 1–11 CrossRef.
- W. Su and F. Yuan, Matter, 2022, 5, 2464–2466 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nh00015c |
| This journal is © The Royal Society of Chemistry 2024 |