
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Albumin nanocapsules and nanocrystals for efficient intracellular drug release†
Sharafudheen Pottanam
Chali‡
a,
Jaana
Westmeier‡
b,
Franziska
Krebs
c,
Shuai
Jiang
a,
Friederike Pauline
Neesen
b,
Doğa
Uncuer
b,
Mario
Schelhaas
d,
Stephan
Grabbe
c,
Christian
Becker
 b,
Katharina
Landfester§
*a and
Kerstin
Steinbrink§
*b
b,
Katharina
Landfester§
*a and
Kerstin
Steinbrink§
*b
aMax Planck Institute for Polymer Research, Mainz, Germany. E-mail: landfest@mpip-mainz.mpg.de
bDepartment of Dermatology, University Hospital Münster, University of Münster, Münster, Germany. E-mail: kerstin.steinbrink@ukmuenster.de
cDepartment of Dermatology, University Medical Center Mainz, Johannes Gutenberg-University, Mainz, Germany
dInstitute of Cellular Virology associated with the Center for Molecular Biology of Inflammation, University of Münster, Münster, Germany
First published on 29th August 2024
Abstract
In order to achieve a therapeutic effect, many drugs have to reach specific cellular compartments. Nanoscale drug delivery systems extend the circulation time, reduce adverse effects and thus improve tolerability compared to systemic administration. We have developed two types of albumin-coated nanocarriers equipped with built-in dyes to track their cellular uptake and intracellular enzymatic opening. Using the approved antiprotozoal drug and STAT3 inhibitor Atovaquone (Ato) as prototype for a hydrophobic small molecule, we show that Ato-loaded ovalbumin-coated nanocapsules (Ato-nCap) preferentially enter human myeloid cells. In contrast, Ato nanocrystals coated with human serum albumin (Ato-nCry) distribute their cargo in all different immune cell types, including T and B cells. By measuring the effect of Ato nanocarriers on induced STAT3 phosphorylation in IL-10-primed human dendritic cells and constitutive STAT3 phosphorylation in human melanoma cells, we demonstrate that the intracellular Ato release is particularly effective from Ato nanocrystals and less toxic than equal doses of free drug. These new nanocarriers thus represent effective systems for intracellular drug delivery.
New conceptsThe intracellular administration of active substances to specific cells or tissues is a central pharmacological problem. Nanodrug systems enable the precise intracellular release of therapeutic drugs, which can enhance the therapeutic effect while reducing the number of side effects. We have developed protein nanocarrier systems for the intracellular release of drugs with limited solubility, which are frequently used in patients with inflammatory and infectious diseases and cancer. Using the approved antiprotozoal small molecule Atovaquone (Ato) we generate two types of nanodrugs: (i) protein nanocapsules with a liquid drug core and (ii) protein-coated drug nanocrystals. Nanocapsules were preferentially taken up by myeloid immune cells, while nanocrystals were taken up by different immune cell populations and non-immune cells. Both systems were intracellularly opened and showed lower toxicity than the free drug. Measuring Ato-mediated STAT3 inhibition demonstrated that nanocrystals are particularly effective both in cells with cytokine-induced and constitutive STAT3 phosphorylation. We have thus developed a new prototype of protein nanocrystals that is superior to conventional particles in terms of intracellular release and toxicity. Our novel nanocarrier engineering strategy can be transferred to other drugs with similar chemical properties and could support the translation from research to clinical application. |
Introduction
Nanocarriers can enable drugs to overcome physiological barriers and reach their target sites safely and sustainably by creating a stable, biocompatible environment, promoting controlled release at the target site, specifically increasing the concentration of a drug at the site of action.1 Therefore, nanocarrier systems can improve the bioavailability and pharmacological activity of drugs combined with reduced off-target toxicity and alleviated systemic effects through spatiotemporal control over drug release.2,3Various parameters such as size, charge, surface, composition and shape of the nanovehicles must be taken into account, as these are related to cell uptake, circulation time, renal clearance and drug stability.4 Nanocarriers can be engineered from different organic and inorganic material resulting in different forms like nanocapsules, -tubes, -rods, -emulsion, or -spheres which specifically interact with cells due to their unique physicochemical characteristics.5 In order to fully exploit this potential, the interaction between nanomaterial and cells must be understood with the main aim of ensuring an effective concentration of a drug at the cellular site of action. However, despite these potential advantages, only a relatively small number of nanoparticle-based medicines have been approved for clinical use, with numerous challenges and hurdles at different stages of development.
In this study, we developed and tested albumin-containing nanocarriers for intracellular release of hydrophobic drugs with limited solubility like small molecules. These compounds are frequently administered in therapeutic approaches for infectious, and inflammatory diseases and cancer but are associated with several side effects due to systemic and untargeted administration. Therefore, nanocarrier-based approaches with high drug loading and profound encapsulation efficiency may overcome the disadvantages of small drug treatments.
Here, we encapsulated the approved antiprotozoal drug and signal transducer and activator of transcription 3 (STAT3) inhibitor Atovaquone (Ato).6 By incorporating two dyes, one to track the nanocarriers and the other to stain the recipient cell after enzymatic intracellular opening of the nanocarriers, we show that Ato-loaded ovalbumin nanocapsules (Ato-nCap) are preferentially taken up and released in human myeloid cells. However, STAT3 phosphorylation in IL-10-primed human dendritic cells (IL-10 DC) remained only partially affected. By encapsulating crystalline Ato with human serum albumin (Ato-nCry), we produced a second generation of nanocarriers that are internalized much more effectively in various human immune cell types, including lymphocytes and efficiently inhibit STAT3 phosphorylation in IL-10 DC and melanoma cells without having a toxic effect.
Our results thus provide an initial basis for the targeted introduction of water-insoluble inhibitors with intracellular activity such as Ato into immune cells.
Material and methods
Materials
Albumin from chicken egg white (lyophilized powder, ≥98%, Sigma Aldrich), human serum albumin (lyophilized powder, ≥97%, Sigma Aldrich), dimethyl sulfoxide (DMSO, <50 ppm water content, Acros Organics), sodium chloride (NaCl, ≥99.0%, Sigma Aldrich), cyanine 5-5′-CCA CTC CTT TCC AGA AAA CT (Cy5-oligo, 0.1 nmol μL−1 in PBS buffer, IBA Lifesciences), cyclohexane (HPLC grade, VWR), chloroform (anhydrous, ≥99%, Sigma Aldrich), 2,4-toluene diisocyanate (TDI, 99%, Sigma Aldrich), sodium dodecyl sulfate (SDS, 99%, Alfa Aesar), and Atovaquone (≥98%, Sigma Aldrich), were used as received. The surfactant poly((ethylene-co-butylene)-block-(ethylene oxide)), P((E/B)-b-EO), consisting of a poly(ethylene-co-butylene) block (NMR: Mn = 3900 g mol−1) and a poly(ethylene oxide) block (NMR: Mn = 2700 g mol−1) was synthesized according to a reported procedure.7 Milli-Q water was used throughout the experiments.Synthesis of STAT3 inhibitor Atovaquone loaded ovalbumin nanocapsules (Ato-nCap)
The nanocapsules (nCap) were synthesized in a water-in-oil miniemulsion. Firstly, the dispersed phase was prepared by dissolving 50 mg ovalbumin and other ingredients (as shown in Table 1) in 500 μL water to form the water phase. For encapsulating Ato in the capsules (Ato-nCap), 200 μL of Ato solution (10 mg mL−1 in DMSO) were used as part of the dispersed phase. The amount of water was reduced accordingly to maintain 500 μL as the total volume for the dispersed phased.| Sample | nCap-Cy5 | nCap-CMFDA | nCap-Cy5-CMFDA | Ato-nCap-Cy5-CMFDA |
|---|---|---|---|---|
| OVA | 50 mg | 50 mg | 50 mg | 50 mg |
| TDI | 3 mg | 3 mg | 3 mg | 3 mg |
| Water | 450 μL | 490 μL | 440 μL | 240 μL |
| NaCl | 7.2 mg | 7.2 mg | 7.2 mg | 7.2 mg |
| Cy5-oligo | 50 μL (136 μM) | 50 μL (136 μM) | 50 μL (136 μM) | |
| CMFDA | 10 μL in DMSO (21.5 mM) | 10 μL in DMSO (21.5 mM) | 10 μL in DMSO (21.5 mM) | |
| Atovaquone | 200 μL (10 mg mL−1 in DMSO) | |||
| KLE | 35.7 mg + 10.7 mg | 35.7 mg + 10.7 mg | 35.7 mg + 10.7 mg | 35.7 mg + 10.7 mg |
| CH | 7.5 g + 5 g | 7.5 g + 5 g | 7.5 g + 5 g | 7.5 g + 5 g |
Next, 35.7 mg surfactant P((E/B)-b-EO) was dissolved in 7.5 g cyclohexane as the oil phase. The oil phase was poured to the aqueous phase under stirring at 500 rpm. The pre-emulsion was homogenized by ultrasonication for 180 s (30 s ultrasonication, 10 s pause) with ice cooling at 70% amplitude using a Branson 450W sonifier and a 1/2′ tip. Separately, 10.7 mg P((E/B)-b-EO) was dissolved in 5 g cyclohexane and 3 mg crosslinker (TDI) was added to the solution. This TDI solution was added dropwise to the obtained miniemulsion for 5 min and the reaction was allowed to proceed for 24 h at 25 °C. Afterwards, excessive surfactant was removed from the dispersion by repetitive centrifugation and replacement of the supernatant with fresh cyclohexane.
Next, the obtained nanocapsules (in cyclohexane) were transferred to an aqueous medium. 600 μL dispersion from cyclohexane was added dropwise to 5 mL 0.1 wt% SDS aqueous solution placed in an ultrasound bath. Subsequently, the sample was stirred in an open vial overnight to evaporate the cyclohexane (Fig. S1, ESI†). Excess SDS was removed via four centrifugation steps by replacing the supernatant with water.
For the preparation of Ato-nCap with dyes Cy5 and/or the cell tracker CMFDA, 50 μL Cy5-oligo (136 μM) and/or 10 μL CMFDA in DMSO (21.5 mM) was added in to the dispersed phase (Table 1).
Preparation of HSA-coated Ato nanocrystals (Ato-nCry)
Human serum albumin (HSA) Ato nanocrystals (Ato-nCry) were synthesized using a miniemulsion solvent evaporation approach as shown in Table 2. Firstly, 50 mg of Ato was dissolved in 1.5 g chloroform to form the oil phase. In the second step, 5 mL of 1 mg mL−1 SDS aqueous solution was poured into the oil solution under stirring at 1000 rpm. After a pre-emulsification step by stirring the oil/water mixture at 1000 rpm for 1 h, the obtained emulsion was sonicated by using a Branson 450 W sonifier with a 1/2′′ tip at 70% amplitude for 180 s (30 s of sonication, 10 s of pause) with ice cooling. The resulting miniemulsion was stirred in an open vial at 1000 rpm for 24 h in order to evaporate the chloroform. The dissolved Ato was crystallized during the solvent evaporation process to form nanocrystals. 27 mg of the protein HSA solution (dissolved in DPBS) was added and the mixture was stirred at 1000 rpm for 1 h and subsequently subjected to a dialysis process (MWCO = 1 kDa) in order to remove the surfactant SDS. Finally, the dialyzed dispersion was centrifuged at 4000 rpm and redispersed in water in order to remove the excess proteins.| Sample | Ato-nCry | Ato-nCry-Cy5 | Ato-nCry-CMFDA | Ato-nCry-Cy5-CMFDA |
|---|---|---|---|---|
| Atovaquone | 50 mg | 50 mg | 50 mg | 50 mg |
| Chloroform | 1.5 g | 1.5 g | 1.5 g | 1.5 g |
| Cy5-NHS | 0.3 mg | 0.3 mg | ||
| CMFDA | 0.1 mg in 10 μL DMSO | 0.1 mg in 10 μL DMSO | ||
| SDS | 5 mL (1 mg mL−1) | 5 mL (1 mg mL−1) | 5 mL (1 mg mL−1) | 5 mL (1 mg mL−1) |
| HSA | 27 mg | 27 mg | 27 mg | 27 mg |
| DPBS | 6 mL | 6 mL | 6 mL | 6 mL |
For the preparation of Ato-nCry with dyes Cy5 and/or the cell tracker CMFDA, 0.3 mg Cy5-NHS was dissolved in chloroform together with Ato, 0.1 mg CMFDA was added in 10 μL DMSO to the Ato in chloroform before adding the SDS solution.
Characterization of nanocapsules and nanocrystals
Dynamic light scattering (DLS) was used to determine the average size and poly dispersity (PDI) of both nanocarriers, nCap and nCry. The nanocarriers were diluted (2 μL sample in 300 μL in cyclohexane or 50 μL nanocarriers in water diluted in 300 μL H2O) and was measured on a Malvern Zetasizer Nano S (Malvern Panalytical) equipped with a detector at 90° scattering mode at 20 °C for cyclohexane and 25 °C for water. The morphology of nanocarriers was examined with a Gemini 1530 (Carl Zeiss AG, Oberkochem, Germany) scanning electron microscope (SEM) operating at 0.35 kV and a JEOL 1400 (Jeol Ltd, Tokyo, Japan) transmission electron microscope (TEM) operating at an accelerating voltage of 120 kV. SEM and TEM samples of nanocarriers were prepared by casting the diluted dispersions on silicon wafers and carbon layer-coated copper grids, respectively. Zeta potential was measured in 1 × 10−3 M potassium chloride solution at 25 °C using a Malvern Zeta sizer (Malvern Instruments, UK). Multiangle DLS measurements were performed using an instrument from ALV (Langen, Germany) with an electronically controlled goniometer and an ALV-5000 multiple τ full-digital correlator with 320 channels for measurements in the range between 10−7 s and 103 s. The source of light was a helium–neon laser of the Type 1145 P from JDS Uniphase (Milpitas, USA) with 632.8 nm wavelength and 25 mV output power. Prior to use, the quartz cuvettes were cleaned with acetone using a Thurmond apparatus. For data analysis, a robust multicomponent fit method reported by ref. 8 was used.Biodegradability and payload release
Biodegradability of nCap was studied by incubating the nanocarriers with serine protease trypsin. 1 mL of dispersions with a solid content of 2 wt% were mixed with 5, 20, or 50 mg trypsin at 37 °C. The corresponding mass ratio between nCap and trypsin was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.25, 1:1, and 1:2.5, respectively. The mixture solutions were placed in dialysis tubes with molecular weight cut-off of 14 kDa. The dialysis tubes were then immersed in 20 mL Milli-Q water and incubated at 37 °C in a shaking culture incubator. During the release experiment, 1 mL of dialysis medium was taken at given interval and equal volume of water was added to keep the volume constant. The nCap were treated in the same way in the absence of trypsin as a control group (OVA:trypsin = 1:0). The release of Cy5 in dialysis medium was quantified by measuring its absorbance at λ = 649 nm by using an Infinite M1000 plate reader (Tecan, Austria).
:0.25, 1:1, and 1:2.5, respectively. The mixture solutions were placed in dialysis tubes with molecular weight cut-off of 14 kDa. The dialysis tubes were then immersed in 20 mL Milli-Q water and incubated at 37 °C in a shaking culture incubator. During the release experiment, 1 mL of dialysis medium was taken at given interval and equal volume of water was added to keep the volume constant. The nCap were treated in the same way in the absence of trypsin as a control group (OVA:trypsin = 1:0). The release of Cy5 in dialysis medium was quantified by measuring its absorbance at λ = 649 nm by using an Infinite M1000 plate reader (Tecan, Austria).
Dendritic cell culture
PBMC were isolated from leukocyte reduction system cones by means of Lymphoprep™ (density 1.077 g mL−1; STEMCELL Technologies, Canada) density gradient centrifugation, and 1.5 × 107 cells per well were incubated for 45 min in a 6-well plate (Corning, USA) in RPMI-1640 medium supplemented with 2% heat-inactivated autologous serum at 37 °C and 5% CO2. After the nonadherent cells were washed off with pre-warmed PBS, the remaining monocytes (purity, >90% CD14+) were incubated in 3 mL per well of IMDM medium (Gibco, USA) supplemented with 2% heat-inactivated autologous plasma. After 24 h, cell culture medium was replaced with IMDM supplemented with 2% autologous plasma, 150 U mL−1 IL-4 (ImmunoTools, Germany), and 400 U mL−1 GM-CSF (LEUKINE®/Sargramostrim; Tanner Pharma, USA). On day 3, 1 mL of medium was exchanged with 1 mL of IMDM containing 2% autologous plasma, 150 U mL−1 IL-4, and 800 U mL−1 GM-CSF. After 7 days of incubation, the resulting immature dendritic cells (DC) were detached with ice-cold PBS and 0.5 × 106 cells were re-seeded into 12-well plates. To induce full DC maturation, cells were stimulated with 6.67 ng mL−1 TNF-α, 6.67 ng mL−1 IL-1β, 66.7 IU mL−1 IL-6 (Strathmann Biotech GmbH, Germany), and 1.34 μg mL−1 PGE2 (Prostaglandin E2; Cayman Chemical, USA). For differentiation of interleukin-10 modulated DC (IL-10 DC), the cell culture medium additionally was supplemented with 20 ng mL−1 of IL-10 (Peprotech, Germany). 48 hours after stimulation, IL-10 DC were washed twice and medium was replaced by IMDM containing 2% autologous serum. For STAT3 activation, IL-10 DC were additionally stimulated with IL-10 (20 ng mL−1) prior to nanocarrier or soluble drug treatment.Melanoma cell culture
G361, A375, and SK-Mel-28 melanoma cell lines were cultured in RPMI-1640 medium supplemented with 10% FCS (fetal calf serum), 1% penicillin/streptomycin, 1% L-glutamine, and 1% sodium pyruvate (Gibco, USA). Cell line MNT-1 was maintained in DMEM supplemented with 10% FCS, 1% penicillin/streptomycin, 1% HEPES, 1% non-essential amino acids, 1% sodium pyruvate, and 1% L-glutamine. All cell lines were split three times weekly depending on confluency.Treatment of IL-10 DC, and melanoma cell lines with Ato nanocarriers
Cells were incubated in IMDM containing 2% autologous plasma (IL-10 DC) or 10% FCS (melanoma cell lines) with 25 μM, 50 μM or 100 μM free Ato, or DMSO alone (as free Ato solvent), or equivalent Ato-nCap or Ato-nCry for 4 hours at 37 °C. DMSO was used as a solvent control. Cells were harvested, transferred into FACS tubes, and analysed.Flow cytometry
Surface phenotyping of PBMC was performed by staining 5 × 104 cells with specific mouse anti-human mAbs for 20 min on ice. Antibodies used were CD11b-APC (ICRF44; Biolegend, USA), CD15-PE-Cy7 (W6D3; Biolegend, USA), TCR αβ-PE (WT31; Biolegend, USA), CD19-BV785 (HIB19; Biolegend, USA). For assessment of intracellular STAT3 expression and phosphorylation, permeabilized and fixed IL-10 DC and melanoma cells were incubated for 30 min at room temperature with specific mouse anti-human STAT3-PE (M59-50; BD Phosflow, USA) or mouse anti-human phosphorylated STAT3-PE (4/P-STAT3; BD Phosflow, USA) antibodies. After being washed, cells were analyzed with a NL-3000 spectral cytometer (Cytek, USA) using unstained cells and single-stained controls.Confocal microscopy
Cells were seeded onto 18-well ibidi glass bottom slides and adhered for at least 2 h at 37 °C and 5% CO2. Cells were washed once with pre-warmed PBS and fixed by adding 50 μL of 0.4% PFA per well for 15 min at RT in the dark. Fixed cells were washed three times and permeabilized with 50 μL of 0.1% Triton X-100® (Merck, Germany). Permeabilized cells were washed 5× with PBS and once with PHEM buffer. For storage, 100 μL of PBS was added to each well to prevent drying out. Images were acquired on a Zeiss Axiovert Z.1 microscope with a Yokogawa CSU22 spinning disk module and a Cool SnapHQ camera (Visitron Systems GmbH, Germany).Statistical analyses
The analyses were performed with GraphPad Prism 9.0. Results are expressed as mean ± SEM (standard error of the mean). Statistical significance was calculated by using an unpaired t-test with a confidence interval of 95%. P-Values lower than 0.05 were considered significant, lower than 0.01 were considered highly significant and lower than 0.001 are of highest significance (* = P ≤ 0.05; ** = P ≤ 0.01; *** = P ≤ 0.001).Results
Nano-encapsulation of dissolved Atovaquone
Ato-loaded ovalbumin (OVA) nanocapsules (Ato-nCap), consisting of an aqueous core and a cross-linked albumin shell, were synthesized in an inverse (water-in-oil) miniemulsion by an interfacial polyaddition reaction (Fig. 1(A)): the STAT3 inhibitor Ato was first dissolved in DMSO and added to the aqueous phase, which was then emulsified in cyclohexane to form a water-in-oil miniemulsion. The use of DMSO allowed us to increase the concentration of Ato inside the droplets from 0.74 μg mL−1 (solubility of Ato in pure water) to 36.7 μg mL−1 in the DMSO/water mixture. The aqueous nanodroplets were stabilized with a block copolymer surfactant P(E/B-b-EO). Upon addition of the crosslinker 2,4-toluenediisocyanate to the emulsion, a polyaddition reaction between the hydroxyl and amino groups of ovalbumin and the isocyanate groups of the crosslinker took place at the nanodroplet–cyclohexane interface, resulting in nanocapsules with a densely crosslinked polypeptide shell. The solubilized STAT3 inhibitor was entrapped in the capsules during their formation. Ato-nCap were then transferred from cyclohexane to an aqueous medium for biological experiments. During this transfer, DMSO diffuses out of the nanocapsules in the continuous aqueous phase leading to a precipitation of the Ato inside the nanocapsules, most probably associated to the shell due to the interaction to albumin.9 Resulting capsules contained 0.63 mg mL−1 Ato. Ato-nCap had a core–shell structure with a hydrodynamic diameter of 300 nm (Fig. 1(B)–(D) and Table 3), were stable in PBS and did not release the drug within 4 weeks of storage.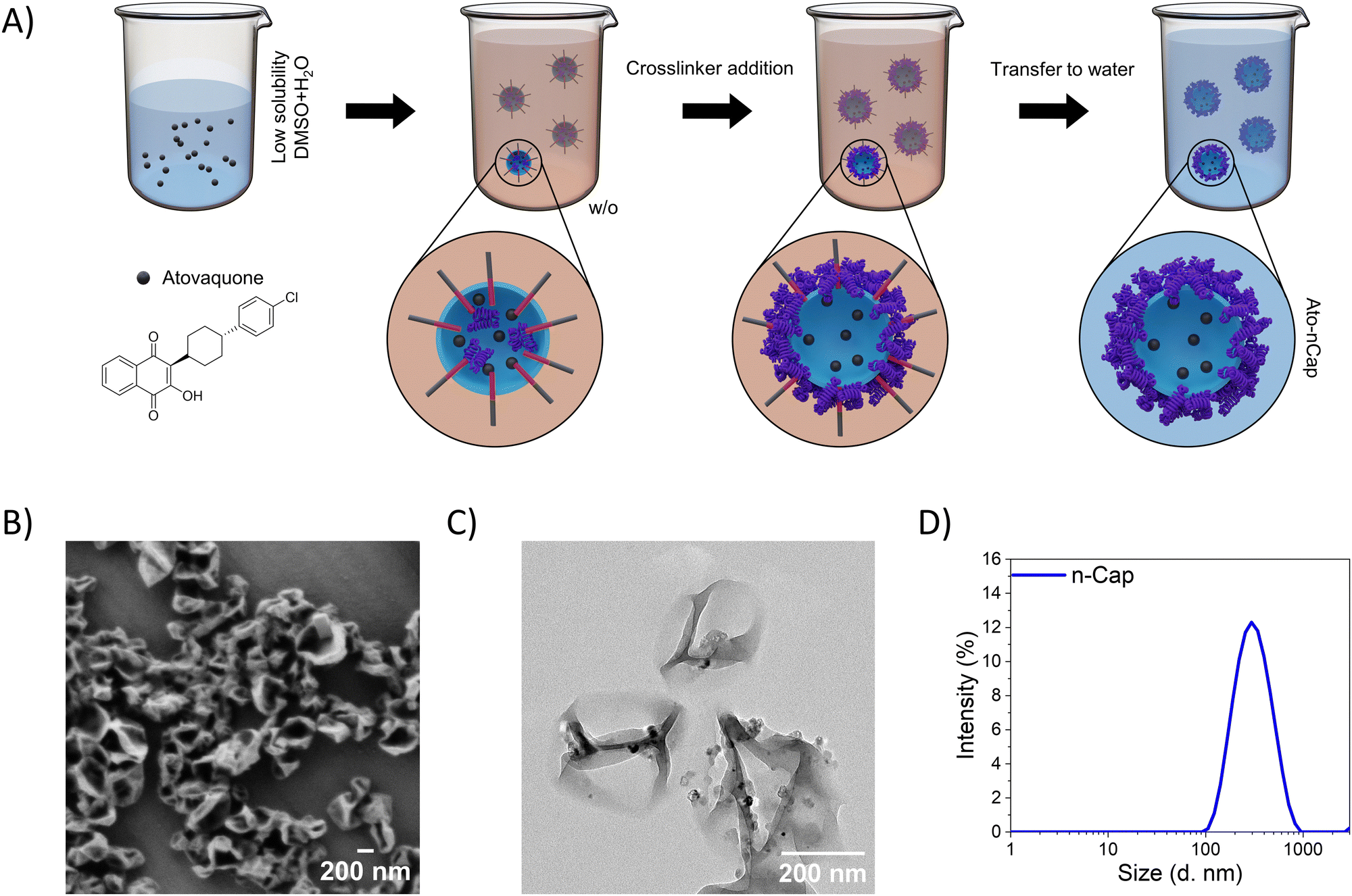 | ||
| Fig. 1 Ato-nCap preparation and characterization. (A) Schematic representation of the generation of Ato-loaded OVA nanocapsules (Ato-nCap) using water-in-oil miniemulsion. (B) SEM (C) TEM micrographs showing the capsules which collapse because of the vacuum in the microscope chamber. This collapse clearly corroborates the hollow structure of the capsules. (D) DLS measurement of nCap. | ||
| Payloads | D h (PDI) (nm) | ζ-Potential (mV) | |
|---|---|---|---|
| OVA-nCap | — | 311 (0.31) | −22.9 |
| OVA-Ato-nCap | Atovaquone | 466 (0.35) | −22.9 |
| HSA-Ato-nCap | Atovaquone | 460 (0.41) | −38.2 |
The core–shell structure and highly cross-linked polypeptide shell (around 12 nm) are advantageous for suppressing non-specific payload release. To investigate enzymatic inhibitor release from the capsules, Ato-nCap were incubated with different concentrations of the serine protease trypsin. Whereas in absence of trypsin Ato-nCap did not release the contained fluorophore Cy5, trypsin digestion resulted in its enzyme concentration and time-dependent release (Fig. S2A, ESI†).
nCap are preferentially taken up by myeloid cells
In order to track the binding or uptake and intracellular opening by cells, nCap contained the dyes Cy5 and CMFDA (5-chloromethylfluorescein diacetate). The latter only fluoresces after hydrolysis by unspecific cellular esterases and therefore represents the intracellular release of the cargo, while Cy5 allows to track the binding of nCap to the cell. Flow cytometric measurement of cellular nCap-mediated Cy5 and CMFDA expression was set up using nCap containing either only Cy5 or CMFDA (Fig. S2B, ESI†). In PBMC, numerous cells bound nCap (Fig. 2(A) and (B)). However, while all myeloid cells and granulocytes interacted with the nCap, only some of the lymphocytes (T cells, B cells) did so. In contrast, CMFDA could only be detected in myeloid cells, which shows that the opening of the nCap and release of encapsulated agent only takes place in these cells (Fig. 2(A) and (C)).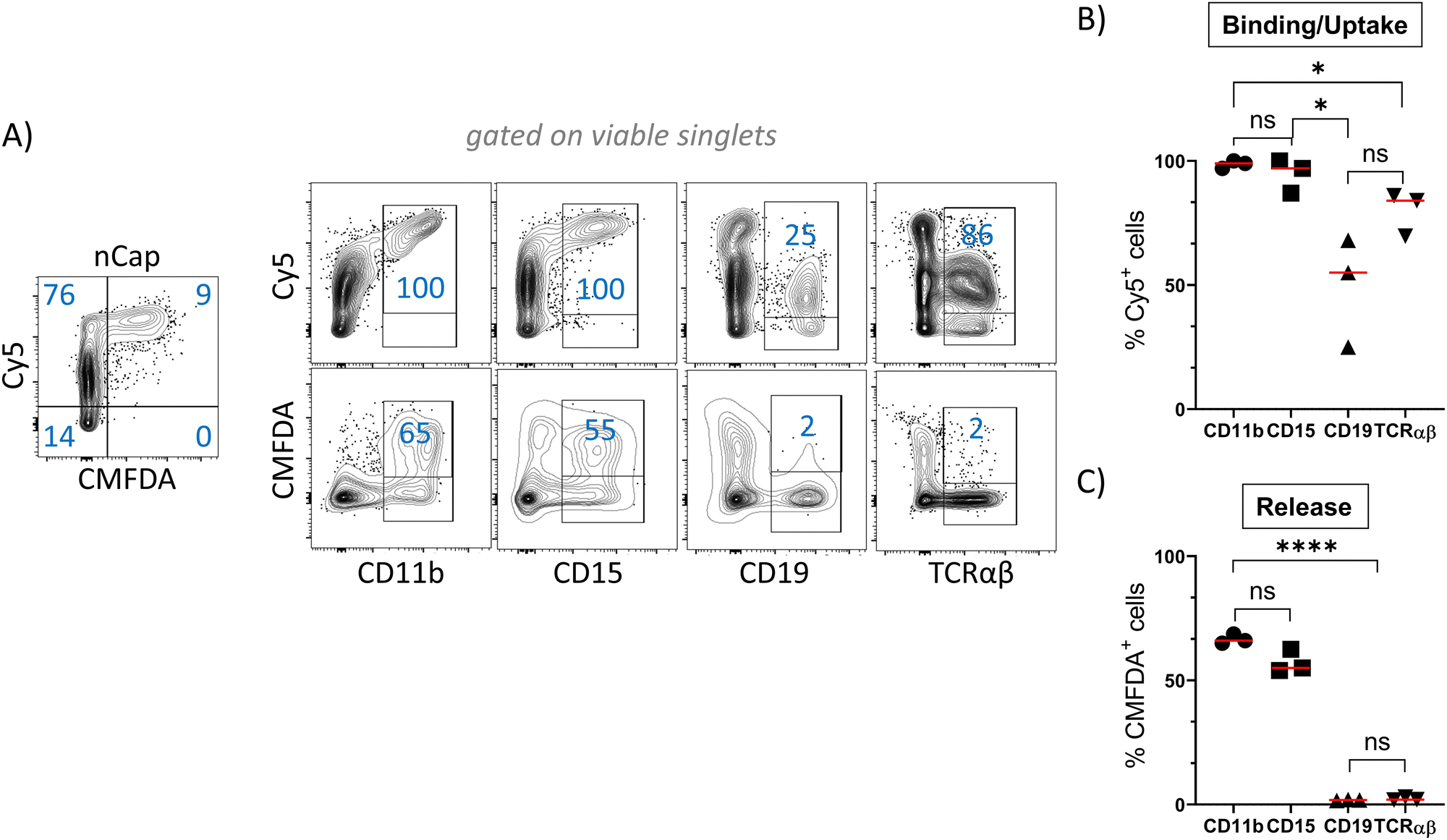 | ||
| Fig. 2 Ato-nCap are preferentially taken up by human myeloid cells. PBMC were incubated for 10 h with Ato-nCap containing Cy5 and CMFDA. The interaction of myeloid cells (CD11b), granulocytes (CD15), B cells (CD19) and T cells (TCRαβ) with the Ato-nCap was then determined by flow cytometry. (A) Representative flow cytometric plots, (B) and (C) statistics. Data shown as mean ± SEM, n = 3 individual donors, numbers represent frequency in gates. Figures and graphs representative of three independent experiments each. | ||
Ato-nCap only partially repress STAT3 phosphorylation in IL-10-primed dendritic cells
To test the efficacy of the incorporated drug, we differentiated human dendritic cells (DC) in the presence of IL-10 to induce activation and phosphorylation of the transcription factor STAT3, which is only lowly expressed in primary cells. After incubation with Ato-nCap or control nCap, DC became Cy5+ and CMFDA+, indicating uptake of nCap and intracellular release of their content (Fig. 3(A) and (B)). However, confocal microscopy images showed that both dyes remained localized in intracellular compartments and that CMFDA in particular did not stain the entire cytoplasm, meaning that the Ato-nCap possibly only opened in endosomes without the dye reaching the entire cell (Fig. 3(A)). At the same time, equal doses of the inhibitor affected cell viability less than the free drug (Fig. 3(B), right panel). Similarly, the STAT3 phosphorylation induced by IL-10 was only slightly affected (Fig. 3(C), right panel). Both observations suggest that Ato-nCap are readily taken up by myeloid cells, but only incompletely opened intracellularly and the stored drug does not reach its full efficacy.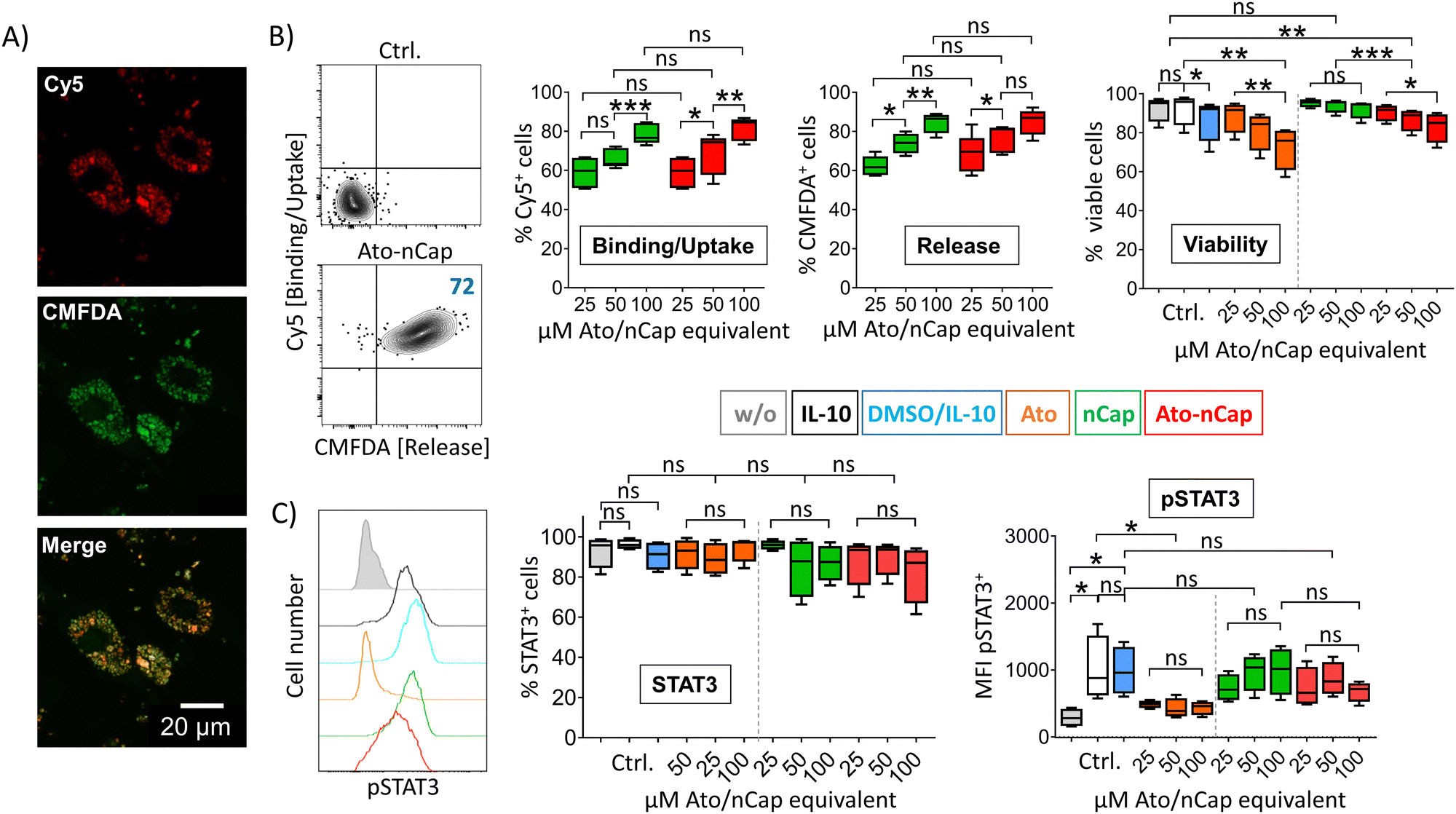 | ||
| Fig. 3 Ato-nCap only partially repress STAT3 phosphorylation in IL-10 primed human dendritic cells. (A) Representative confocal light scanning microscopy images of IL-10 dendritic cells (IL-10 DC) 4 h after incubation with Ato-nCap. Visualization of Cy5 and CMFDA fluorescence introduced by the nanocapsules. (B and C, left panel) Representative flow cytometric plots of IL-10 DC 4h after incubation with Ato-nCap, numbers represent cell frequencies in gates. (B) Frequencies of Cy5+ (binding/uptake), CMFDA+ (intracellular nanocapsule opening) and viable cells. (C) STAT3 expression and STAT3 pY705 phosphorylation 4 h after incubation with indicated amounts of Ato-nCap, non-loaded nCap or Ato. Control cells were left untreated (w/o), treated with IL-10 only (IL-10) or IL-10 and solvent (DMSO). Data shown as mean ± SEM, n = 5 per treatment. All figures and graphs are representative of five independent experiments each. | ||
Nano-encapsulation of crystalline Ato
Due to its low solubility in water, only limited amounts of Ato could be encapsulated in Ato-nCap. To increase drug content, we modified the synthesis to encapsulate crystalline Ato. We also switched to an human serum albumin (HSA) envelope (thickness of around 4 nm) due to its better biocompatibility in humans and large number of cellular receptors compared to OVA.10HSA Ato nanocrystals (Ato-nCry) were synthesized using a solvent evaporation process based on an oil-in-water mini-emulsion (Fig. 4(A)). First, Ato was dissolved in chloroform as a disperse phase, which was emulsified in water to form chloroform nanodroplets. Upon evaporation of the chloroform (confirmed by 1H NMR spectroscopy, Fig. S3A, C and D, ESI†), Ato formed nanocrystals that were stabilized by adding HSA, subsequently, the surfactant SDS was removed by dialysis (Fig. S3B, ESI†). Ato-nCry had the shape of short sticks (Fig. 4(B), (D) and (E)). The hydrodynamic radius (Rh) of Ato-nCry was measured by dynamic light scattering (Fig. 4(F)) and the radius of gyration (Rg) by static light scattering (Fig. 4(G)). The obtained ρ value (ρ = Rg/Rh) of 1.11 reflects the asymmetric rod shape of the nanocrystal. Albumin coating increased the stability of nanocrystals in blood plasma compared to nanocrystals stabilized with SDS and the PEG-based surfactant Lutensol AT50 (Fig. S4, ESI†). Due to the manufacturing method, no active substance-free control particles could be produced. The nanocrystals contained 3.4 mg mL−1 Ato.
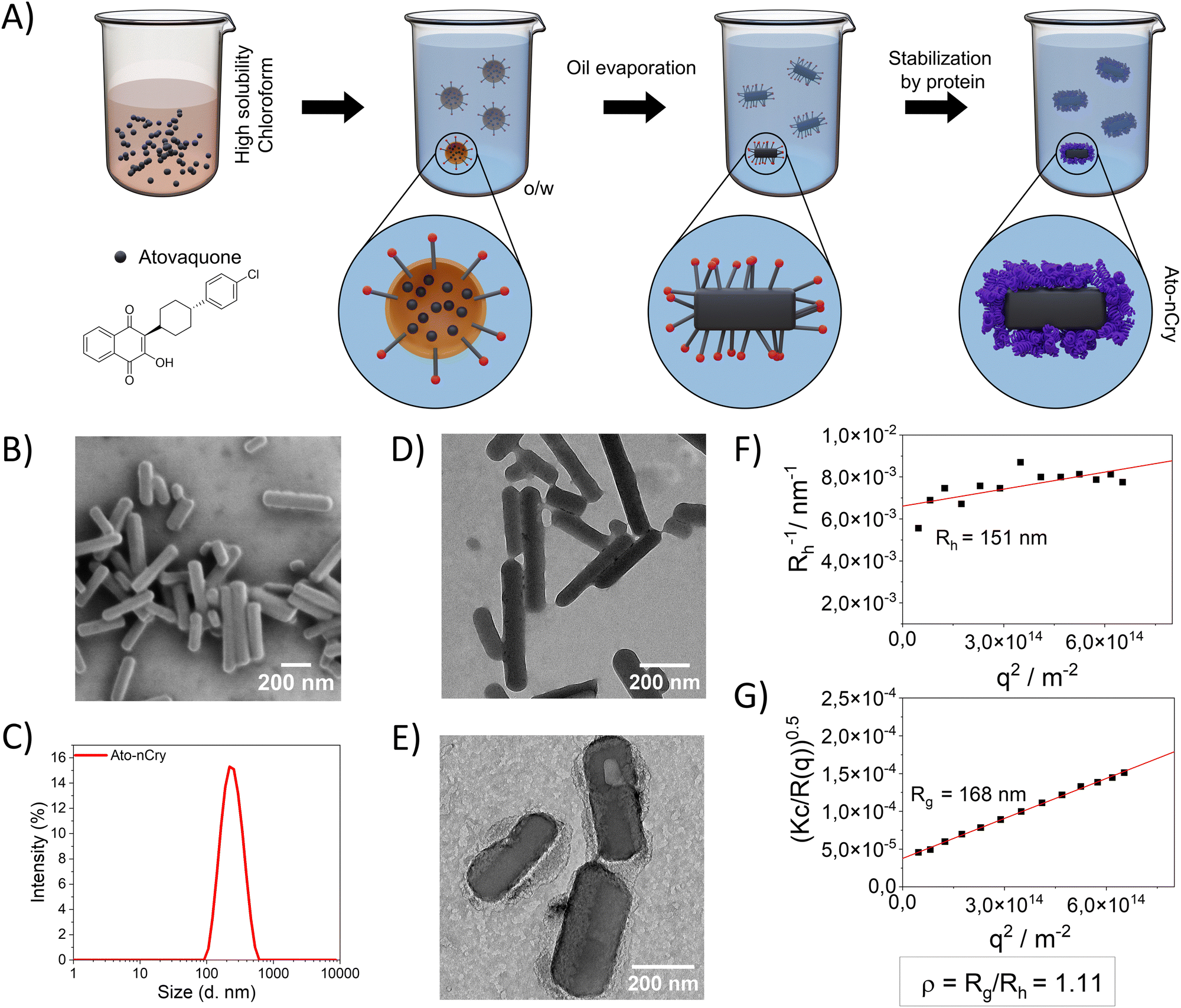 | ||
| Fig. 4 Ato-nCry preparation and properties. (A) Schematic representation of the generation of HSA-coated Ato-nanocystals (Ato-nCry) by oil-in-water mini-emulsion/solvent evaporation method. (B) SEM image, (C) DLS measurement, (D) TEM image of Ato-nCry. (E) High resolution TEM image showing protein adsorbed on the Ato-nCry. (F) Multiangle DLS results showing hydrodynamic radius (Rh) (G) and static light scattering results showing radius of gyration (Rg). The ρ value (ρ = Rg/Rh) reflects the asymmetric degree of nanocrystals. | ||
Ato-nCry are taken up by various immune cells and effectively repress STAT3 phosphorylation in IL-10-primed dendritic cells and melanoma cells
As measured by Cy5 and CMFDA fluorescence, Ato-nCry were readily taken up by all kinds of cells in PBMC including lymphocyte subsets (Fig. 5(A) and (B)). Unlike Ato-nCap, Ato-nCry interacted indiscriminately with all immune cells (Cy5). CMFDA fluorescence showed that the Ato-nCry were opened in all myeloid cells (CD11b+), and surprisingly also in a high proportion of T cells and B cells (CD3+, CD19+). CD15+ cells (granulocytes) appeared to release the dye only partly (Fig. 5(A) and (C)).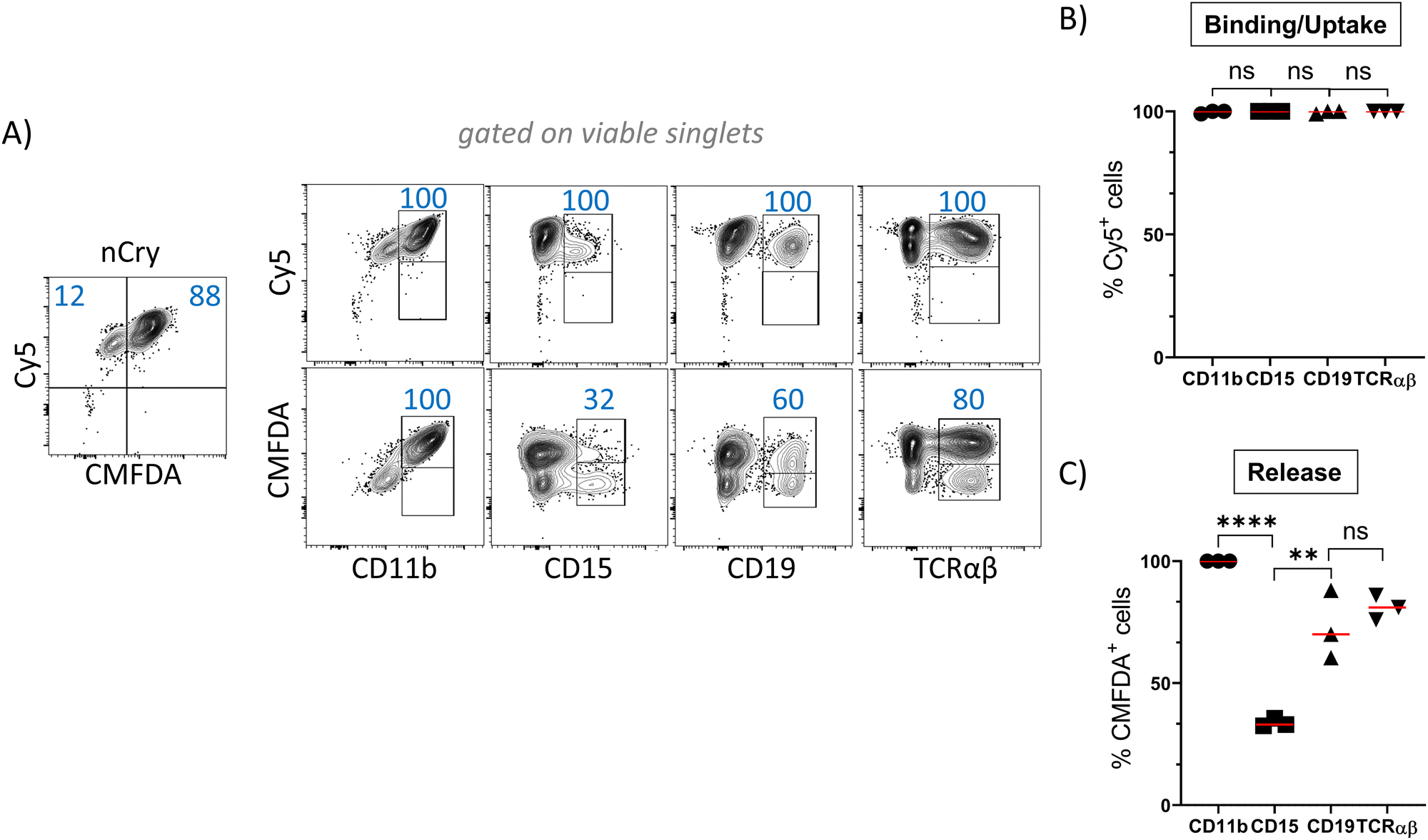 | ||
| Fig. 5 Ato-nCry are taken up by various human immune cells. PBMC were incubated for 10 hours with Ato-nCry containing Cy5 and CMFDA. The interaction of myeloid cells (CD11b), granulocytes (CD15), B cells (CD19) and T cells (TCRαβ) with the Ato-nCry was then determined by flow cytometry. (A) Representative flow cytometric plots, (B) and (C) statistics. Data shown as mean ± SEM, n = 3 individual donors, numbers represent frequencies in gates. Figures and graphs representative of three independent experiments each. | ||
IL-10 primed DC interacted with Ato-nCry like primary myeloid cells: all cells took up the crystals and released the dye CMFDA (Fig. 6(A) and (B)). Here, the confocal images showed that the dye was released into the cytoplasm (Fig. 6(A)). Ato-nCry showed little cellular toxicity that was significantly lower compared to toxic effects induced by the free drug (Fig. 6(B), right panel). At the same time, they suppressed STAT3 phosphorylation as effectively as equal amounts of the free inhibitor, demonstrating its effective intracellular release into the DC (Fig. 6(C), right panel).
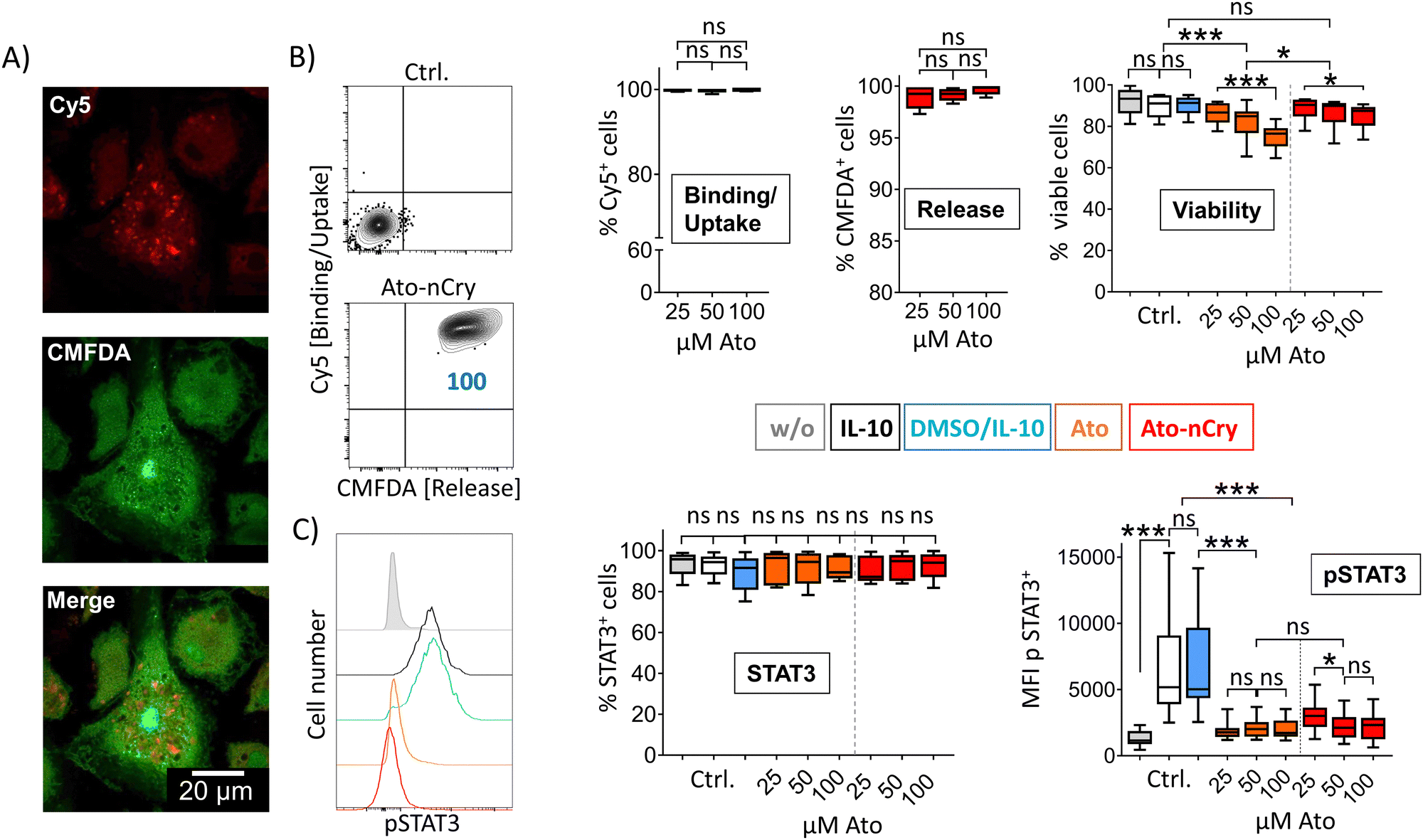 | ||
| Fig. 6 Ato-nCry repress STAT3 phosphorylation in IL-10 primed human dendritic cells. (A) Representative confocal light scanning microscopy images of IL-10 dendritic cells (IL-10 DC) 4 h after incubation with Ato-nCry. Visualization of Cy5 and CMFDA fluorescence introduced by the particles. (B and C, left panel) Representative flow cytometric plots of IL-10 DC 4h after incubation with Ato-nCry, numbers represent cell frequencies in gates. (B) Frequencies of Cy5+ (binding/uptake), CMFDA+ (intracellular particle opening) and viable cells; (C) STAT3 expression and STAT3 pY705 phosphorylation 4 h after incubation with indicated amounts of Ato-nCry or Ato. Control cells were left untreated (w/o), treated with IL-10 only (IL-10) or IL-10 and solvent (DMSO). Data shown as mean ± SEM, n = 5 per treatment. All figures and graphs are representative of five independent experiments each. | ||
While DC express and phosphorylate STAT3 only in response to certain signals such as IL-10, it is constitutively overexpressed and activated in numerous tumor cells.11 To test the effectiveness of particle-mediated STAT3 inhibition on constitutive STAT3+ cells, we incubated four different human melanoma cell lines with Ato-nCry. All lines absorbed the nanocarriers and released CMFDA intracellularly, here shown by representative experiments with the melanoma cell line G361 (Fig. 7(A) and (B)). As before in IL-10 primed DC, Ato-nCry were able to measurably reduce STAT3 phosphorylation (Fig. 7(C)). Fig. 7 shows results with the melanoma cell line G361, representative of the four melanoma cell lines examined.
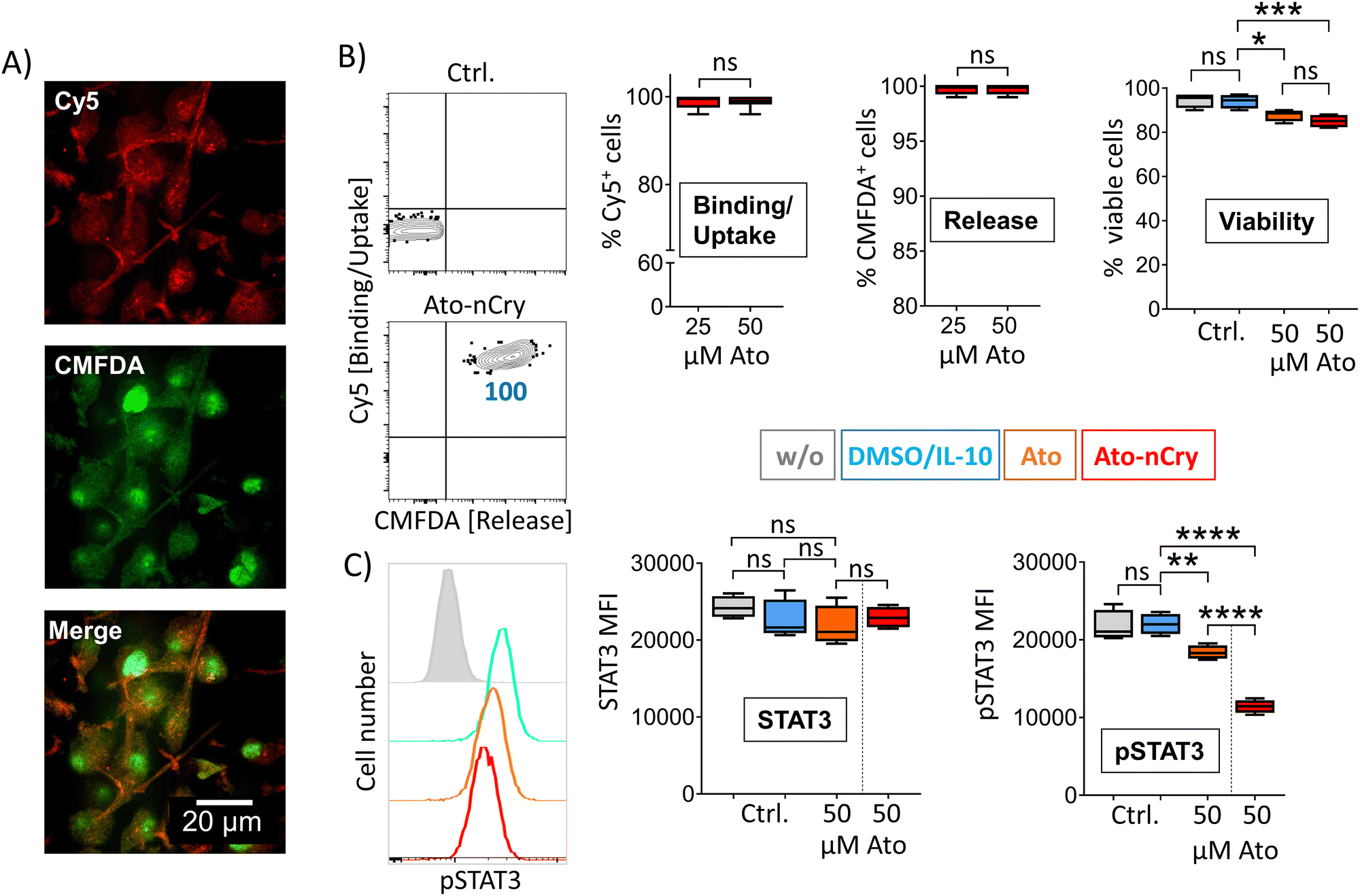 | ||
| Fig. 7 Ato-nCry repress STAT3 phosphorylation in G361 human melanoma cells. (A) Representative confocal light scanning microscopy images of G361 cells, 4 h after incubation with Ato-nCry. Visualization of Cy5 and CMFDA fluorescence introduced by the particles. (B and C, left panel) Representative flow cytometric plots of G361 cells 4h after incubation with Ato-nCry, numbers represent cell frequencies in gates. (B) Frequencies of Cy5+ (binding/uptake), CMFDA+ (intracellular particle opening) and viable cells; (C) STAT3 expression and STAT3 pY705 phosphorylation 4 h after incubation with indicated amounts of Ato-nCry or Ato. Control cells were left untreated (w/o) or treated with solvent (DMSO). Data shown as mean ± SEM. All figures and graphs are representative of three independent experiments each. | ||
Nanocarrier uptake and intracellular opening in immune cells is not determined by the envelope protein
The uptake of ovalbumin coated nCap by myeloid cells and DC presumably occurred via the mannose receptor.12–14 In addition, DC can also take up ovalbumin nanoparticles mannose receptor-independently.12 To test the role of the coat protein in the interaction of the nanocarriers with immune cells, we also prepared HSA-nCap and OVA-coated Ato-nCry (OVA-Ato-nCry) and compared their binding and uptake in PBMC. However, binding and intracellular opening of both nanocarriers were found to be independent of the coat protein (Fig. S5, ESI†), suggesting that other properties such as shape, size and zeta potential determine the type of interaction with immune cells. To simulate different hydrophobic conditions in cells, we investigated the release of Ato from Ato-nCry in the presence of different DMSO concentrations. Ato release increased with DMSO percentage, (Fig. S6, ESI†), suggesting preferential drug release from nCry in hydrophobic cellular micro-compartments.Discussion
Drugs that have shown good therapeutic potential in preclinical trials, often cannot be used clinically due to limited solubility and bioavailability, poor targeting, off-target toxicity or rapid degradation. Therefore, nanocarrier systems are considered a promising technology that enhances the potency of (immuno-) therapy by capitalizing on the superior physicochemical properties of nanovehicles, thereby creating cell delivery systems.15–17 The advantageous features of nanocarriers include encapsulation of drugs for reduction of side effects and toxicity, and a finely modifed chemistry enabling the achievement of controlled and sustained release properties.Numerous review articles unanimously agree that STAT3 is a point of convergence for numerous oncogenic signaling pathways and that its inhibition should have synergistic therapeutic effects in cancer.18–21 A number of studies have examined the effect of STAT3 inhibitor-loaded nanoparticles on tumor immunotherapy.22–29 In one study, Ato-loaded nanocapsules could not affect the growth of colon carcinomas and melanomas in mice, but showed effect in combination with antibodies against the checkpoint molecule PD-1.29 Overall, the studies on STAT3 inhibitor particles underscore the important role of STAT3 in tumor growth, despite the large number of different approaches. However, STAT3 is required for the self-renewal of normal stem cells30 and is significantly involved in embryogenesis and development of the mammary gland,31 so that undesirable effects of STAT3 inhibitors are expected upon systemic interference.
Thus, the clinical usefulness of STAT3 inhibitors is limited by their toxicity32 and unfavorable pharmacological properties such as hydrophobicity33 and no STAT3 inhibitor developed as such has yet been approved for clinical use. In our study using the limited water-soluble drug Atovaquone, we demonstrate that Atovaquone-nanocrystals (Ato-nCry) display a real high drug loading and are superior to regular nanocapsules (nCap) in terms of intracellular release, drug efficacy and toxicity. Due to a lack of traceability of the drug, dyes Cy5 and CMFDA were employed to analyze nanocarrier binding and intracellular cargo release.
We previously have reported the generation of protein nCap with aqueous inner core by inverse miniemulsion process.34 This technique enables the multicomponent encapsulation of cargos with varying size and water solubility in high encapsulation efficiency, making this technique superior to other nanocarrier synthesis approaches. We have already shown the potential of OVA nanocapsules, loaded with two adjuvants Resiquimod (R848) and muramyl dipeptide (MDP), in cancer immunotherapy.35 However, due to the limited water solubility of Ato, the loading in nCap is relatively low. Therefore, nCry with defined size are an alternative and allow a very high loading.
The low water solubility of hydrophobic small molecules like Ato leads to their poor bioavailabilty and as consequence to great limitation of their clinical utility. Design of a reliable and proper delivery system will address the problems of toxicity and restricted bioavailabilty. As demonstrated in our study, generation of nCry can overcome these problems through encapsulation of high concentration of hydrophobic agents in a crystallized core, resulting in opening of the nanocarriers and effective intracellular action of the released compound.
Among various materials used for nanocarrier generation, serum albumin based nanovehicles have been widely developed due to their prominent superiors, including good biocompatibility and biodegradability, non-toxicity, non-immunogenecity and high stability, favoring this protein as carrier for drug delivery.36–39 One example is albumin-bound paclitaxel that has been used for cancer therapy in clinic for years.40 Our study shows that encapsulation with albumins enables an efficient intracellular drug release, combined with low toxicity. In particular, Ato-nCry induce a significantly reduced toxic effect on immune cells compared to the free drug at similar concentrations. Furthermore, our experiments reveal that the nanocrystals are taken up by myeloid cells and DC as well as by T and B cells, followed by high release of the crystallized drug also in lymphocytes. In contrast to phagocytic immune cells, targeting of T cells by nanocarriers for biomedical applications still remains an obstacle as they disclose reduced endocytic activities. For achievement of relevant uptake rates in T cells, nanoparticles often have to be equipped with antibodies (e.g. anti-CD8) or molecules like the cytokine IL-2, thereby enhancing the intracellular incorporation.41,42
Confocal microscopic images of DC after interaction with Ato-nCap show co-localized red and green granules, presumably endosomes, but only slight CMFDA staining of the cytoplasm, suggesting that the nCap are incompletely opened. In contrast to OVA, HSA is non-immunogenic in humans, can be bound by seven membrane-associated receptors43 and has been previously shown to enhance the cellular uptake of anticancer medications.44 In contrast to OVA-coated Ato-nCap, HSA-coated Ato-nCry lead to a significant release of CMFDA into the cytoplasm in all investigated cells, so that the drug release from Ato-nCap and Ato-nCry is expected to differ. In comparison to Ato-nCap, Ato-nCry have a higher drug content. While the rod-shaped nanocrystal itself dissolves only slowly, the thicker crosslinked protein shell of OVA-Ato-nCap slows down the release of the drug from the nanocapsules, leading to the assumption that more drug can be released from Ato-nCry compared to Ato-nCap over time. Moreover, HSA Ato-nCry are less toxic compared to the free drug, possibly because the drug gradually becomes available in cells from the crystal. In addition, we found that the interaction with immune cells of nCap and nCry are independent of the coat protein, suggesting other physicochemical properties as relevant for uptake, opening and toxicity of both nanocarriers.
In our study, we have developed a new prototype of albumin nanocrystals that are superior to conventional nanocapsules in terms of intracellular release and toxicity. Our novel engineering strategy for nanocarriers can be transferred to other drugs with similar chemical properties and may promote the application of nanomedicine in clinical use.
Conclusions
Encapsulation with albumin enables the intracellular release of hydrophobic drugs like small molecules with an intracellular site of action. Drug crystals covered with albumin are superior to conventional nanocapsules in terms of intracellular release, effect and toxicity. While albumin nanocapsules are almost selectively taken up by phagocytes of the immune system, encapsulated drug crystals reach all cell types.Author contributions
Sharafudheen Pottanam Chali: resources, visualization, investigation, methodology, writing – original draft, writing – review & editing. Jaana Westmeier: investigation, writing – review & editing. Franziska Krebs: investigation. Shuai Jiang: investigation. Friederike Pauline Neesen: investigation. Doğa Uncuer: investigation. Mario Schelhaas: resources, supervision. Stephan Grabbe: resources, supervision. Christian Becker: supervision, methodology, visualization, conceptualization, formal analysis, writing – original draft, writing – review & editing. Katharina Landfester: conceptualization, funding acquisition, project administration, methodology, writing – review & editing. Kerstin Steinbrink: conceptualization, funding acquisition, project administration, writing – original draft, writing – review & editing.Data availability
Data for this article, including all measured values and images, are available at figshare at https://figshare.com/projects/Source_data_Nanoscale_Horizon_2024/214063.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the German Research Foundation (DFG): SFB1066/B06-213555243 (K. S., K. L.), SFB1066/B08-213555243 (C. B.), SFB1450/C06-431460824 (K. S., K. L.), TR156/C05-246807620 (K. S.), SFB1009/B11-194468054 (K. S.). Open Access funding provided by the Max Planck Society.Notes and references
- C. Azevedo, M. H. Macedo and B. Sarmento, Drug Discovery Today, 2018, 23, 944–959 CrossRef CAS PubMed.
- B. T. N. Dang, T. K. Kwon, S. Lee, J. H. Jeong and S. Yook, J. Controlled Release, 2024, 365, 773–800 CrossRef CAS PubMed.
- K. Johann, T. Bohn, F. Shahneh, N. Luther, A. Birke, H. Jaurich, M. Helm, M. Klein, V. K. Raker, T. Bopp, M. Barz and C. Becker, Nat. Commun., 2021, 12, 5981 CrossRef CAS.
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941–951 CrossRef CAS PubMed.
- R. Singh, M. Kumawat, H. Gogoi, H. Madhyastha, E. Lichtfouse and H. K. Daima, ACS Appl. Bio Mater., 2024, 7, 727–751 CrossRef CAS PubMed.
- M. Xiang, H. Kim, V. T. Ho, S. R. Walker, M. Bar-Natan, M. Anahtar, S. Liu, P. A. Toniolo, Y. Kroll, N. Jones, Z. T. Giaccone, L. N. Heppler, D. Q. Ye, J. J. Marineau, D. Shaw, J. E. Bradner, T. Blonquist, D. Neuberg, C. Hetz, R. M. Stone, R. J. Soiffer and D. A. Frank, Blood, 2016, 128, 1845–1853 CrossRef CAS.
- H. Schlaad, H. Kukula, J. Rudloff and I. Below, Macromolecules, 2001, 34, 4302–4304 CrossRef CAS.
- K. Rausch, A. Reuter, K. Fischer and M. Schmidt, Biomacromolecules, 2010, 11, 2836–2839 CrossRef CAS.
- H. Kathpalia, S. Juvekar and S. Shidhaye, Colloids Interface Sci., 2019, 29, 26–32 CrossRef CAS.
- Y. Ishima, T. Maruyama, M. Otagiri, V. T. G. Chuang and T. Ishida, Chem. Pharm. Bull., 2022, 70, 330–333 CrossRef CAS PubMed.
- M. Tolomeo and A. Cascio, Int. J. Mol. Sci., 2021, 22, 603 CrossRef CAS PubMed.
- S. Burgdorf, V. Lukacs-Kornek and C. Kurts, J. Immunol., 2006, 176, 6770–6776 CrossRef CAS PubMed.
- L. East and C. M. Isacke, Biochim. Biophys. Acta, Gen. Subj., 2002, 1572, 364–386 CrossRef CAS PubMed.
- A. J. Engering, M. Cella, D. M. Fluitsma, E. C. M. Hoefsmit, A. Lanzavecchia and J. Pieters, Adv. Exp. Med. Biol., 1997, 417, 183–187 CrossRef CAS PubMed.
- B. N. Dang, T. K. Kwon, S. Lee, J. H. Jeong and S. Yook, J. Controlled Release, 2024, 365, 773–800 CrossRef CAS PubMed.
- H. Li, Y. G. Yang and T. Sun, Front. Bioeng. Biotechnol., 2022, 10, 889291 CrossRef PubMed.
- W. Poon, B. R. Kingston, B. Ouyang, W. Ngo and W. C. W. Chan, Nat. Nanotechnol., 2020, 15, 819–829 CrossRef CAS PubMed.
- S. L. Zou, Q. Y. Tong, B. W. Liu, W. Huang, Y. Tian and X. H. Fu, Mol. Cancer, 2020, 19, 145 CrossRef PubMed.
- H. Lee, S. K. Pal, K. Reckamp, R. A. Figlin and H. Yu, Curr. Top. Microbiol., 2011, 344, 41–59 CAS.
- H. Yu, D. Pardoll and R. Jove, Nat. Rev. Cancer, 2009, 9, 798–809 CrossRef CAS PubMed.
- M. Kortylewski, R. Jove and H. Yu, Cancer Metastasis Rev., 2005, 24, 315–327 CrossRef CAS PubMed.
- L. Molavi, A. Mahmud, S. Hamdy, R. W. Hung, R. Lai, J. Samuel and A. Lavasanifar, Mol. Pharmaceutics, 2010, 7, 364–374 CrossRef PubMed.
- Y. H. Ma, X. L. Zhang, X. X. Xu, L. Shen, Y. Yao, Z. Y. Yang and P. S. Liu, PLoS One, 2015, 10, e0124924 CrossRef PubMed.
- S. Labala, A. Jose, S. R. Chawla, M. S. Khan, S. Bhatnagar, O. P. Kulkarni and V. V. K. Venuganti, Int. J. Pharm., 2017, 525, 407–417 CrossRef CAS PubMed.
- S. K. S. S. Pindiprolu, P. T. Krishnamurthy, P. K. Chintamaneni and V. V. S. R. Karri, Artif. Cells, Nanomed., Biotechnol., 2018, 46, 885–898 CrossRef CAS PubMed.
- Y. H. Huang, M. R. Vakili, O. Molavi, Y. Morrissey, C. S. Wu, I. Paiva, A. H. Soleimani, F. Sanaee, A. Lavasanifar and R. Lai, Cancers, 2019, 11, 248 CrossRef CAS PubMed.
- M. Ashrafizadeh, Z. Ahmadi, N. G. Kotla, E. G. Afshar, S. Samarghandian, A. Mandegary, A. Pardakhty, R. Mohammadinejad and G. Sethi, Cells, 2019, 8, 1158 CrossRef CAS PubMed.
- S. H. Bao, H. L. Zheng, J. Y. Ye, H. R. Huang, B. Zhou, Q. Yao, G. Y. Lin, H. L. Zhang, L. F. Kou and R. J. Chen, Front. Pharmacol., 2021, 12, 625084 CrossRef CAS PubMed.
- S. M. Wang, X. R. Zhou, Z. K. Zeng, M. J. Sui, L. H. Chen, C. Feng, C. Huang, Q. Yang, M. J. Ji and P. Hou, J. Nanobiotechnol., 2021, 19, 302 CrossRef CAS PubMed.
- H. Niwa, T. Burdon, I. Chambers and A. Smith, Genes Dev., 1998, 12, 2048–2060 CrossRef CAS PubMed.
- K. Hughes and C. J. Watson, Int. J. Mol. Sci., 2018, 19, 1695 CrossRef PubMed.
- J. D. Beebe, J.-Y. Liu and J.-T. Zhang, Pharmacol. Ther., 2018, 191, 74–91 CrossRef CAS PubMed.
- J. Dong, X.-D. Cheng, W.-D. Zhang and J.-J. Qin, J. Med. Chem., 2021, 64, 8884–8915 CrossRef CAS PubMed.
- S. Ritz, S. Schöttler, N. Kotman, G. Baier, A. Musyanovych, J. Kuharev, K. Landfester, H. Schild, O. Jahn, S. Tenzer and V. Mailänder, Biomacromolecules, 2015, 16, 1311–1321 CrossRef CAS PubMed.
- D. Passlick, K. Piradashvili, D. Bamberger, M. Y. Li, S. Jiang, D. Strand, P. R. Wich, K. Landfester, M. Bros, S. Grabbe and V. Mailänder, J. Controlled Release, 2018, 289, 23–34 CrossRef CAS.
- X. Shen, X. Y. Liu, T. T. Li, Y. Chen, Y. Chen, P. Wang, L. Zheng, H. Yang, C. H. Wu, S. Q. Deng and Y. Y. Liu, Front. Chem., 2021, 9, 746646 CrossRef CAS PubMed.
- M. Hirose, A. Tachibana and T. Tanabe, Mater. Sci. Eng., C, 2010, 30, 664–669 CrossRef CAS.
- S. Gaca, S. Reichert, C. Rödel, F. Rödel and J. Kreuter, J. Microencapsulation, 2012, 29, 685–694 CrossRef CAS PubMed.
- S. Bae, K. Ma, T. H. Kim, E. S. Lee, K. T. Oh, E. S. Park, K. C. Lee and Y. S. Youn, Biomaterials, 2012, 33, 1536–1546 CrossRef CAS PubMed.
- I. Cucinotto, L. Fiorillo, S. Gualtieri, M. Arbitrio, D. Ciliberto, N. Staropoli, A. Grimaldi, A. Luce, P. Tassone, M. Caraglia and P. Tagliaferri, J. Drug Delivery, 2013, 2013, 905091 Search PubMed.
- D. Schmid, C. G. Park, C. A. Hartl, N. Subedi, A. N. Cartwright, R. B. Puerto, Y. Zheng, J. Maiarana, G. J. Freeman, K. W. Wucherpfennig, D. J. Irvine and M. S. Goldberg, Nat. Commun., 2017, 8, 1747 CrossRef PubMed.
- S. U. Frick, M. P. Domogalla, G. Baier, F. R. Wurm, V. Mailänder, K. Landfester and K. Steinbrink, ACS Nano, 2016, 10, 9216–9226 CrossRef CAS PubMed.
- A. M. Merlot, D. S. Kalinowski and D. R. Richardson, Front. Physiol., 2014, 5, 299 Search PubMed.
- C. Bolling, T. Graefe, C. Lubbing, F. Jankevicius, S. Uktveris, A. Cesas, W. H. Meyer-Moldenhauer, H. Starkmann, M. Weigel, K. Burk and A. R. Hanauske, Invest. New Drugs, 2006, 24, 521–527 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nh00161c |
| ‡ Sharafudheen Pottanam Chali and Jaana Westmeier contributed equally as first authors. |
| § Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |