
Biodegradable persistent ROS-generating nanosonosensitizers for enhanced synergistic cancer therapy by inducing cascaded oxidative stress†
Yue
Chen‡
a,
Tong
Ding‡
b,
Zhengzheng
Qian‡
a,
Zerui
Ma
a,
Liming
Zhou
a,
Zhiling
Li
b,
Runkai
Lv
c,
Yinghui
Xu
a,
Yingjie
Xu
a,
Linhui
Hao
c,
Chen
Zhu
 *d,
Xikuang
Yao
*c,
Wenying
Yu
*b and
Wenpei
Fan
*a
*d,
Xikuang
Yao
*c,
Wenying
Yu
*b and
Wenpei
Fan
*a
aState Key Laboratory of Natural Medicines and Jiangsu Key Laboratory of Drug Discovery for Metabolic Diseases, Center of Advanced Pharmaceuticals and Biomaterials, China Pharmaceutical University, Nanjing 211198, China. E-mail: wenpei.fan@cpu.edu.cn
bJiangsu Key Laboratory of Bioactive Natural Product Research and State Key Laboratory of Natural Medicines, China Pharmaceutical University, Nanjing 211100, China. E-mail: ywy@cpu.edu.cn
cSchool of Flexible Electronics (Future Technologies) and Institute of Advanced Materials (IAM), Nanjing Tech University (Nanjing Tech), Nanjing 211816, China. E-mail: iamxyao@njtech.edu.cn
dDepartment of Orthopedics, The First Affiliated Hospital of USTC, Division of Life Sciences and Medicine, University of Science and Technology of China, Hefei 230001, China. E-mail: zhuchena@ustc.edu.cn
First published on 11th September 2024
Abstract
Sonodynamic therapy (SDT) is gaining popularity in cancer treatment due to its superior controllability and high tissue permeability. Nonetheless, the efficacy of SDT is severely diminished by the transient generation of limited reactive oxygen species (ROS). Herein, we introduce an acid-activated nanosonosensitizer, CaO2@PCN, by the controllable coating of porphyrinic metal–organic frameworks (PCN-224) on CaO2 to induce cascaded oxidative stress in tumors. The PCN-224 doping can generate ROS during SDT to induce intracellular oxidative stress and abnormal calcium channels. Meanwhile, the ultrasound also promotes extracellular calcium influx. In addition, CaO2@PCN sequentially degrades in the tumor cell lysosomes, releasing Ca2+ and H2O2 to induce further abnormal calcium channels and elevate the levels of Ca2+. Insufficient catalase (CAT) in tumor cells promotes intracellular calcium overload, which can induce persistent ROS generation and mitochondrial dysfunction through ion interference therapy (IIT). More importantly, PCN-224 also protects CaO2 against significant degradation under neutral conditions. Hence, the well-designed CaO2@PCN produces synergistic SDT/IIT effects and persistent ROS against cancer. More notably, the acidity-responsive biodegradability endows CaO2@PCN with excellent biosafety and promising clinical potential.
New conceptsOur study introduces a unique approach to enhance the efficacy of sonodynamic therapy (SDT) toward tumors by developing an acidity-responsive degradable CaO2@PCN, through in situ epitaxial growth of porphyrinic metal–organic frameworks (MOFs) onto calcium peroxide (CaO2). Different from the traditional sonodynamic therapy with transient reactive oxygen species (ROS) generation, the CaO2@PCN nanoplatform incorporates CaO2 as a persistent ROS-generating material. CaO2 can be used to elicit tumor cell death by inducing calcium overload through ion interference therapy (IIT). More importantly, the PCN shell on CaO2 prevents it from hydrolysis under physical conditions. Besides, the transient ROS generation during SDT can damage calcium channels, further assisting CaO2 in inducing intracellular calcium overload. Crucially, the calcium overload in cancer cell death leads to mitochondrial dysfunction and persistent ROS generation. This mechanism enables the activation of tumor microenvironment-responsive sonodynamic therapy (SDT) and ion interference therapy (IIT) effects. This innovative strategy capitalizes on the large tissue permeability of ultrasound (US) and the acidity-responsive biodegradability of CaO2@PCN, offering a promising solution for treating tumors while ensuring high biosafety and in vivo elimination. By combining the strengths of sonosensitizers and calcium peroxide, our approach represents a significant advancement in cancer therapy, with potential implications for improving treatment outcomes and expanding the clinical utility of SDT/IIT protocols. |
Introduction
Cancer remains a serious threat to human life and public health.1,2 Conventional clinical cancer treatments, including chemotherapy and radiotherapy, tend to produce considerable systemic toxicity and radiation damage.3,4 Therefore, it is imperative to develop safer therapeutic modalities. Featuring the collective merits of non-invasiveness, ease of manipulation, and negligible physical damage, low-intensity ultrasound (US) has been widely used as a diagnostic imaging tool in clinics.5–7 Additionally, US activates sonosensitizers to generate cytotoxic reactive oxygen species (ROS) to eliminate cancer cells, which brings forth a promising protocol for tumor treatment called sonodynamic therapy (SDT).8,9 Distinct from light-activated photodynamic therapy (PDT), which suffers from shallow tissue penetration depth, US can penetrate deep soft tissues, making it ideal for the precision treatment of deep-seated tumors.As a critical element of SDT, sonosensitizers have a profound effect on therapeutic efficacy, so carefully selecting the appropriate sonosensitizer is essential to achieve an optimal SDT effect. Compared to inorganic sonosensitizers (e.g., MnWOx, TiO2, etc.) which are challenging to degrade and exhibit low sonocatalytic activity,10,11 organic sonosensitizers, particularly porphyrin and its derivatives, are the most frequently employed for SDT due to their definite molecular structure and high sonodynamic efficiency.10 However, porphyrin's poor water solubility, low bioavailability, and susceptibility to self-quenching under physiological conditions usually restrict its clinical application, which emphasizes the essentiality of protective delivery strategies, such as nanovesicle assistance.7,12,13 Functioning as an organic linker within the crystal lattice, the porphyrinic metal–organic frameworks (MOFs) are effective in preventing porphyrin leakage compared to most nanovesicles.14–17 In particular, the porphyrinic MOFs substantially promote both the stability and fluorescence signal intensity of porphyrins by averting self-aggregation and self-quenching.18,19
The reactive oxygen species (ROS) generated by sonosensitizers during SDT can induce oxidative stress within tumors, leading to tumor cell death. However, the transient and restricted production of ROS decreases the therapeutic effect of SDT.20 To induce robust tumor cell death, various protocols have been proposed to elevate the level of ROS by increasing the US intensity or oxygen supply.21–23 Even so, the therapeutic outcome is still far from satisfactory. Consequently, there is a need to develop more effective methods to generate sustaining and sufficient ROS for improving anti-tumor effects. As a persistent ROS-generating material, calcium peroxide (CaO2) has received considerable attention in cancer treatment.14 CaO2 can release Ca2+ and hydrogen peroxide (H2O2) to induce elevation of intracellular Ca2+ levels and dysfunction of calcium channels in tumor cells, ultimately resulting in calcium overload. Conversely, sufficient intracellular CAT protects normal cells by preventing calcium channel dysfunction through the degradation of H2O2. Consequently, calcium overload can trigger cancer cell death by inducing mitochondrial membrane potential disruptions and promoting ROS generation, which represents a safe yet effective treatment paradigm of ion interference therapy (IIT).24–27 Nonetheless, the instability of CaO2 and its tendency to undergo spontaneous hydrolysis are critical drawbacks, necessitating surface modification for stabilization.
In this study, an in situ epitaxial growth approach is proposed for the precise encapsulation of porphyrinic MOFs (PCN-224) onto CaO2 to form a biodegradable CaO2@PCN nanoplatform with acidity-responsive characteristic (Scheme 1). The outer PCN-224 shell effectively prevents CaO2 from spontaneous hydrolysis under physiologically neutral conditions. Upon reaching the tumor cell, the transient ROS generated by porphyrin on PCN-224 during SDT can not only instigate oxidative stress but also exacerbate the disruption of calcium channels. Meanwhile, the PCN-224 shell of CaO2@PCN gradually degrades to permit the hydrolysis of inner CaO2 into Ca2+ and H2O2 to elevate intracellular Ca2+ levels. Consequently, the SDT strengthens the abnormal retention of Ca2+ in the cytoplasm to induce calcium overload. The calcium overload results in mitochondrial dysfunction and persistent ROS generation. As a result, CaO2@PCN is expected to induce acid-activated SDT/IIT effects against deep-seated malignancies. Furthermore, the acidity-responsive biodegradability of CaO2@PCN guarantees in vivo elimination and high biosafety, exhibiting wide-ranging clinical potential.
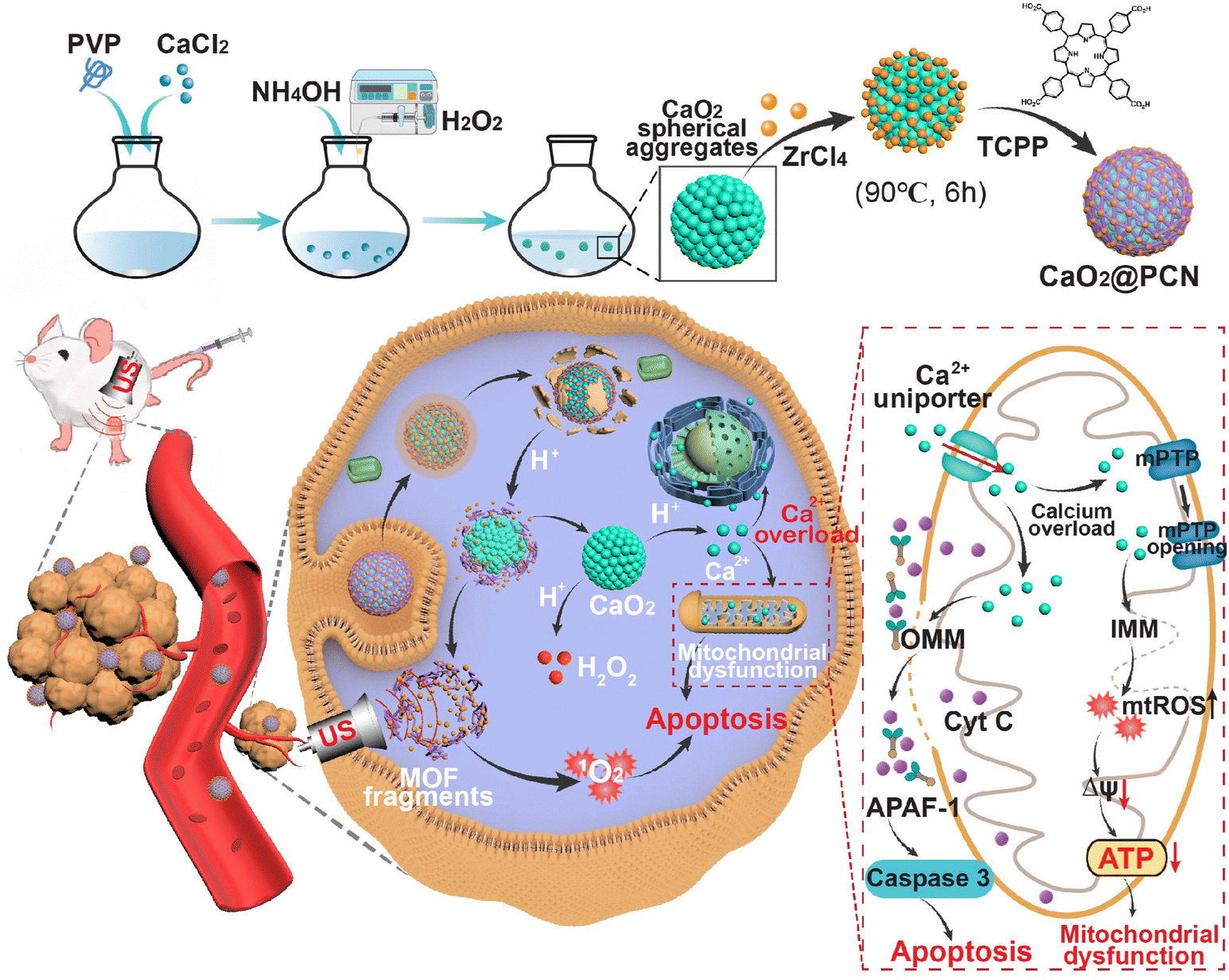 | ||
| Scheme 1 Schematic mechanism of CaO2@PCN for acidity-responsive sequential degradation using SDT/IIT. | ||
Results and discussion
Synthesis and characterization of CaO2
CaO2 particles were synthesized using a hydrolyzation–precipitation method (Fig. 1a) through the reaction of CaCl2 with H2O2 in an ethanol solution aided by polyvinylpyrrolidone (PVP).28–30 Interestingly, there was a significant correlation between the morphology of the as-prepared CaO2 and feeding CaCl2 concentration. As demonstrated in Fig. 1b–e, a lower concentration of CaCl2 gave smaller and more uniform CaO2 nanoparticles. When the feeding CaCl2 concentration was 48 mM, the CaO2 products were shaped irregularly with an average diameter of 99.33 nm (Fig. 1b and f). With a gradual reduction of the CaCl2 concentration from 24 mM to 12 mM and then to 6 mM, the CaO2 products exhibited smoother surfaces and higher dispersities (Fig. 1c–e). Accordingly, the average particle sizes of CaO2 products gradually decreased from 84.40 nm to 74.52 nm to 72.53 nm (based on the measurements of 150 particles in the TEM images) (Fig. 1g–i). However, a further decrease in CaCl2 concentration to 6 mM resulted in a colorless and transparent solution of CaO2 products due to the decreased yield. These findings demonstrated that the lower feeding CaCl2 concentration within a certain range led to fewer CaO2 nanoparticles with a more uniformly smaller size. Therefore, considering the morphology, particle size, dispersity, and yield, the CaO2 nanoparticles prepared with concentrations of 12 mM and 24 mM of CaCl2 were chosen for subsequent studies.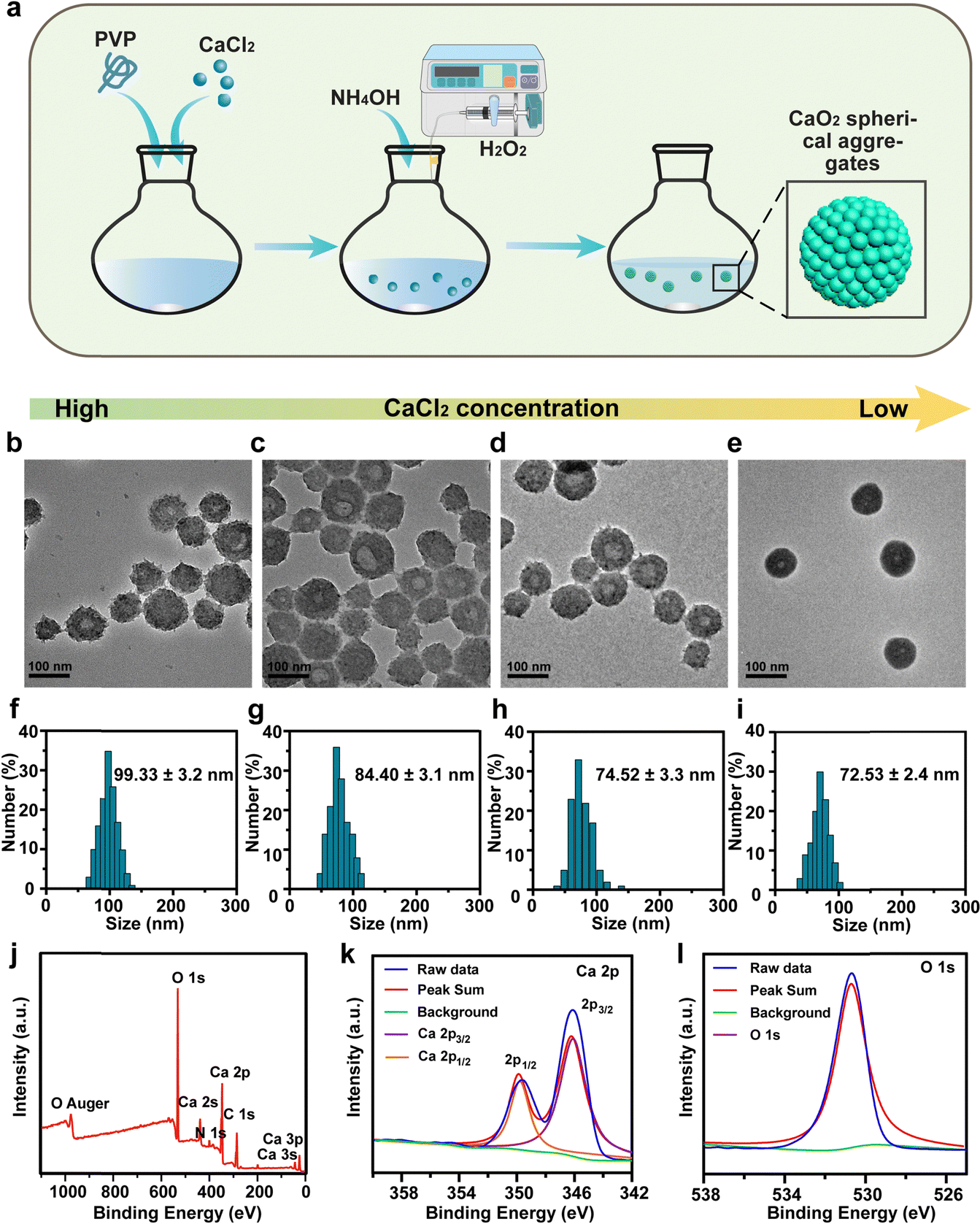 | ||
| Fig. 1 Synthesis and Characterization of CaO2. (a) Synthesis process of CaO2. (b)–(e) TEM images and (f)–(i) particle size distributions of CaO2 prepared in the presence of CaCl2 at different feeding concentrations. Scale bar, 100 nm. (b) and (f) 48 mM, (c) and (g) 24 mM, (d) and (h) 12 mM, and (e) and (i) 6 mM. (j)–(l) XPS spectra of (j) CaO2, (k) Ca element, and (l) O element. | ||
Further research was conducted to determine the influence of PVP on the formation of CaO2. CaO2 nanoparticles produced with PVP displayed uniform and spherical morphologies (Fig. S1a, ESI†). In contrast, those produced without PVP displayed irregular shapes and broad particle sizes, and their solution appeared as a white suspension (Fig. S1b–d, ESI†). This was attributed to PVP acting as a colloidal stabilizer, which could directly assemble small nanocrystals into uniform spherical nanoparticles with controllable sizes.31 Then, the X-ray photoelectron spectroscopy (XPS) spectrum of CaO2 demonstrated the presence and the valence state of Ca, O, N, and C elements (Fig. 1j). The presence of Ca2+ was denoted by a pair of Ca 2p peaks at 346.5 and 350.0 eV, linked to the spin–orbit splitting components of 2p3/2 and 2p1/2 (Fig. 1k). The O 1s peak at 531.1 eV was attributed to the O–O bond, signifying a valence state of −1 for O in CaO2 (Fig. 1l).32 Furthermore, CaO2 was examined for its stability. Obvious solution delamination and particle agglomeration in the CaO2 solution provided evidence for its instability at pH 7.4 (Fig. S2, ESI†).
Synthesis and characterization of CaO2@PCN
The CaO2@PCN nanoplatform with a core/shell structure was obtained by the uniform decoration of a PCN-224 shell onto the surface of CaO2 particles. In brief, a PCN-based MOF shell was deposited onto CaO2, involving the coordination interaction between the zirconium ion (Zr4+) and meso-tetra-(4-carboxyphenyl) porphine (TCPP). During the procedure, Zr4+ from ZrCl4·8H2O was first anchored onto the surface of CaO2 and then coordinated with TCPP to form the PCN-224 shell. Notably, the morphology and size of CaO2@PCN were adjustable by modulating the feeding concentration ratio of CaO2 to TCPP. As illustrated in Fig. 2 and 3a, the resultant CaO2@PCN was obtained with smaller particle sizes and higher dispersity when applying CaO2 prepared at a lower concentration of CaCl2. It was also found that a lower feeding concentration of CaO2 led to a thicker MOF layer formation on the CaO2 surface. In addition, a gradual decrease in the feeding TCPP concentration resulted in CaO2@PCN that exhibited less agglomeration, better uniformity, and a smoother surface (Fig. 3b–e). In the presence of 5 mg mL−1 CaO2 (prepared using 12 mM CaCl2) and 0.0625 mg mL−1 TCPP, CaO2@PCN was obtained with a composition ratio of Ca2+/Zr4+ of 6.27/1. The resulting product displayed a uniform morphology with high dispersity, high stability, and an average particle size of 79.63 nm (from measurements of 150 particles in the TEM images) (Fig. S3 and S4, ESI†), preferentially selected for the following experiments. Due to the hydrated state, the hydrodynamic size of CaO2@PCN was 112.8 nm, which was larger than the particle size in TEM. After covering CaO2 with PCN, the zeta potential of CaO2 changed from 11.17 mV to −11.90 mV (Fig. S5, ESI†). Additionally, the stability of CaO2@PCN was also tested by incubating it with PBS buffer for 60 min. Results indicated that there was no apparent solution delamination and particle agglomeration in the solution, providing evidence of stability at pH 7.4 (Fig. S6, ESI†). To confirm the successful coating of PCN on CaO2, elemental mapping was conducted, which revealed the presence of C, N, O, Ca, and Zr elements in the structure of CaO2@PCN along with the uniform distribution of Zr elements on the surface of CaO2 (Fig. 3f). Additionally, the Fourier transform infrared (FTIR) spectrum analysis exhibited comparable absorption peaks (around 1646 cm−1 and 870 cm−1) in both CaO2@PCN and CaO2 (Fig. 3g). Moreover, Ca, O, and Zr elements were also detected in the XPS spectrum of CaO2@PCN (Fig. 3h). As depicted in Fig. 3i and j, the binding energies of O 1s and Zr 3d were 531.4 eV and 181.9/184.2 eV, indicating that the valence states of O and Zr in CaO2@PCN were −1 and +4, respectively. The results above confirmed that the structure of CaO2 remained intact during the PCN coating process. Besides, the presence of TCPP in CaO2@PCN was determined through the identification of its characteristic absorption peak at 420 nm using an ultraviolet-visible (UV-vis) spectrometer,33 which determined the loading capacity of TCPP in CaO2@PCN to be 13.92% (Fig. S7–S9, ESI†). Moreover, the X-ray diffraction (XRD) pattern of CaO2@PCN exhibited the characteristic peaks (2θ) of both PCN-224 and CaO2 (Fig. S10, ESI†),34 suggesting the successful coating of PCN onto CaO2 and the preparation of CaO2@PCN.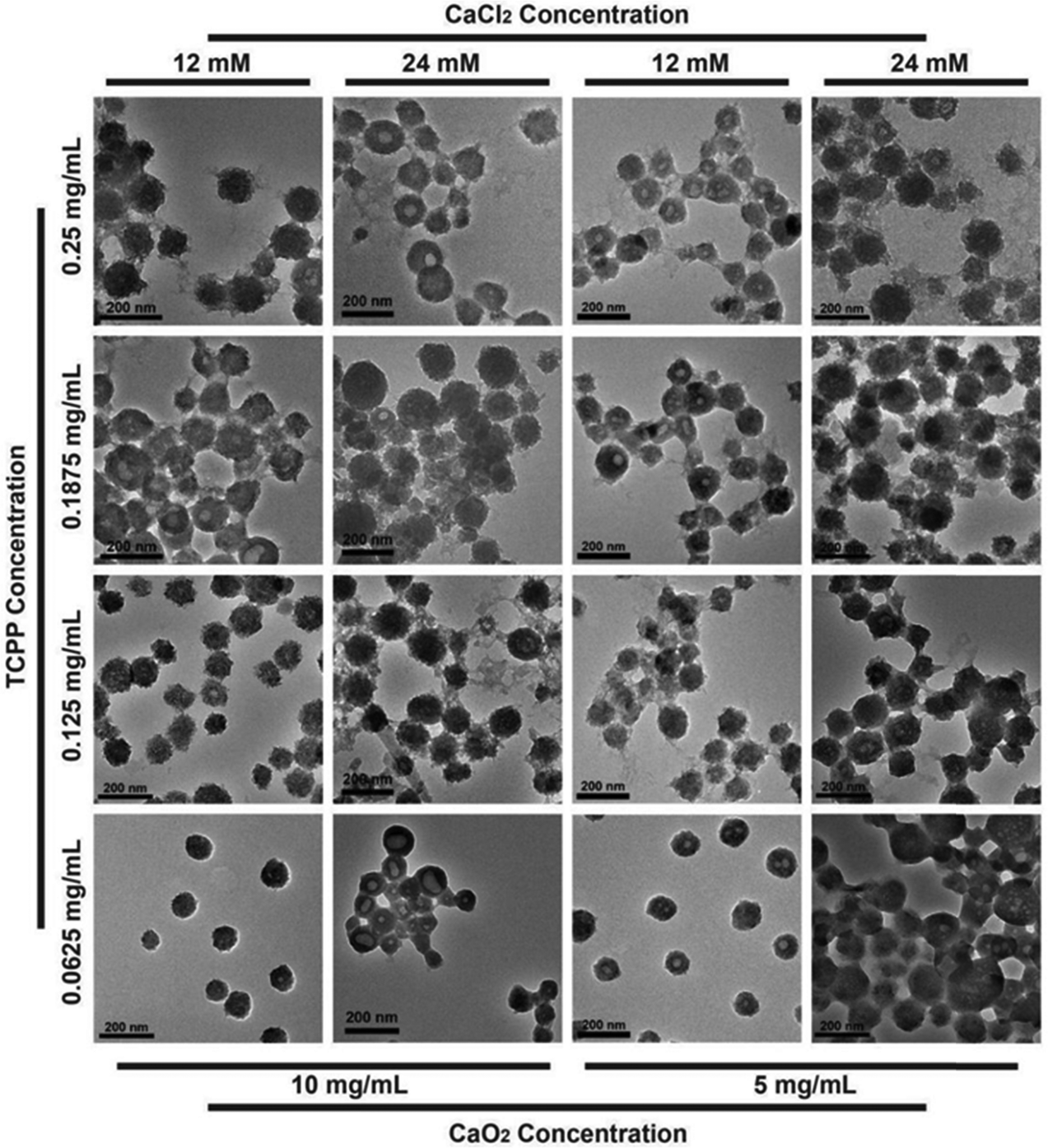 | ||
| Fig. 2 TEM images of CaO2@PCN obtained by modulating the feeding concentration ratio of CaO2 to TCPP. Scale bar, 200 nm. | ||
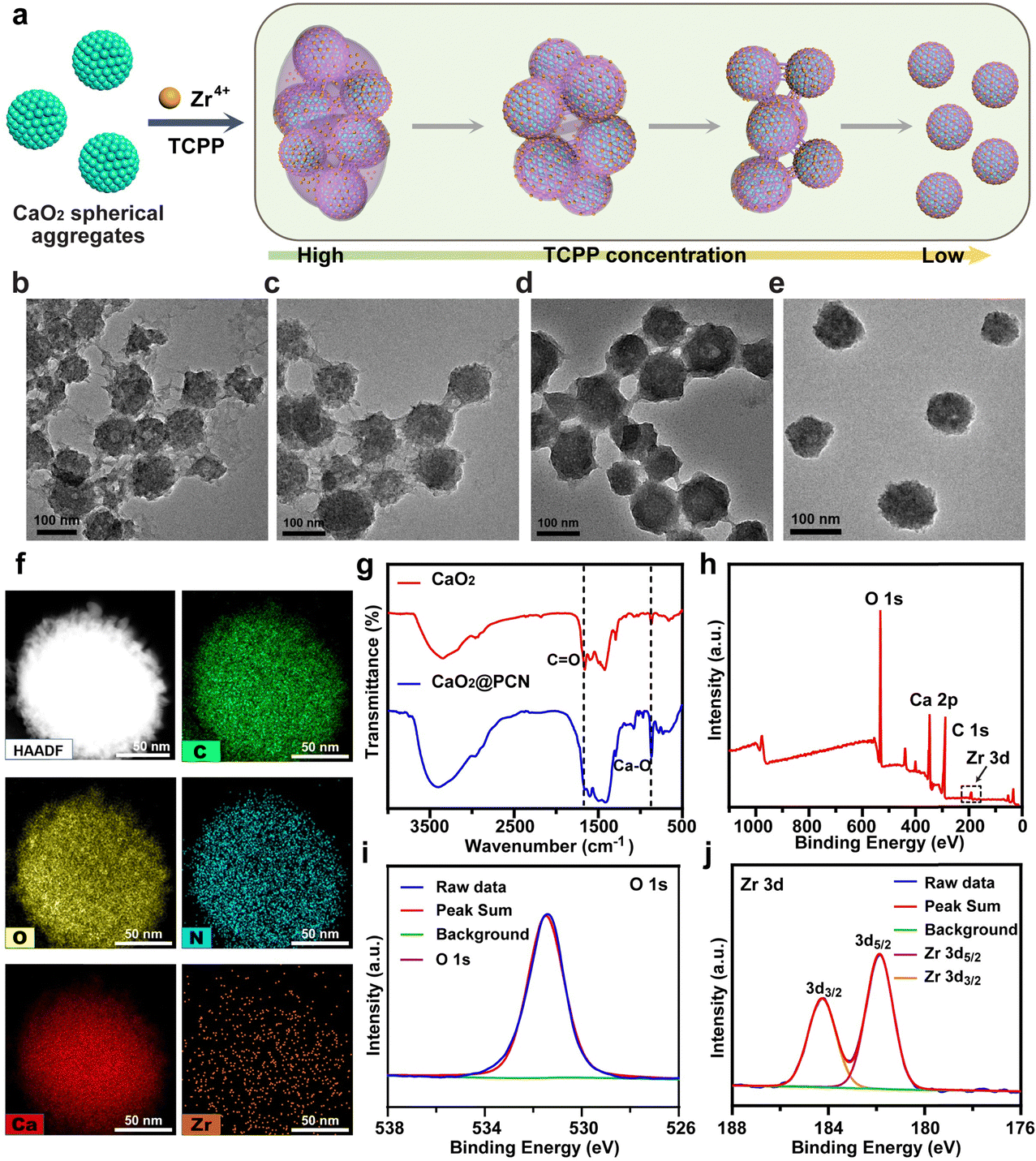 | ||
| Fig. 3 Synthesis and characterization of CaO2@PCN. (a) Scheme of the preparation of CaO2@PCN. (b)–(e) TEM images of CaO2@PCN prepared in the presence of 5 mg mL−1 CaO2 (prepared using 12 mM CaCl2) and different feeding concentrations of TCPP (b. 0.25 mg mL−1; c. 0.1875 mg mL−1; d. 0.125 mg mL−1; e. 0.0625 mg mL−1). Scale bar, 100 nm. (f) Elemental mapping images of CaO2@PCN. Scale bar, 50 nm. (g) FTIR spectra of CaO2 and CaO2@PCN. (h)–(j) XPS spectra of (h) CaO2@PCN, (i) O element, and (j) Zr element. | ||
Due to the charge reversal induced by protonation, the coordination bond between Zr4+ and TCPP was susceptible to acidity,35 which could result in the collapse of the outer PCN-224 shell, leading to the complete degradation of CaO2@PCN (Fig. 4a). To confirm the acidity-triggered biodegradation of CaO2@PCN, TEM was used to reveal the morphological changes of CaO2@PCN in phosphate buffer solution (PBS) with pH 5.5. The outer PCN shell began to collapse and partly fragmented after 1 h of incubation, leaving CaO2@PCN with rough edges. After 12 h of incubation, the original structure of CaO2@PCN disintegrated into pieces, which correlated well with the Ca2+/Zr4+ release curves (Fig. 4b). The time-dependent Ca2+/Zr4+ release from CaO2@PCN in PBS with various pH values was measured through inductively coupled plasma optical emission spectrometry (ICP-OES). As shown in Fig. 4c and d, CaO2@PCN exhibited pH-responsive release of both Zr4+ and Ca2+. After 48 h of incubation, only 26.15% of Zr4+ and 16.25% of Ca2+ were discharged in a neutral solution (pH 7.4), whereas 51.12% of Zr4+ and over 80% of Ca2+ were released in an acidic solution (pH 5.5). All these results corroborated the acidity-triggered biodegradation of CaO2@PCN for controllable release of Ca2+ and Zr4+. Furthermore, the effect of US on calcium ion release was also investigated. As shown in Fig. S11 (ESI†), there was no significant difference in Ca2+ release between the US-treated and non-US-treated groups, indicating that the Ca2+ release was independent of the US treatment.
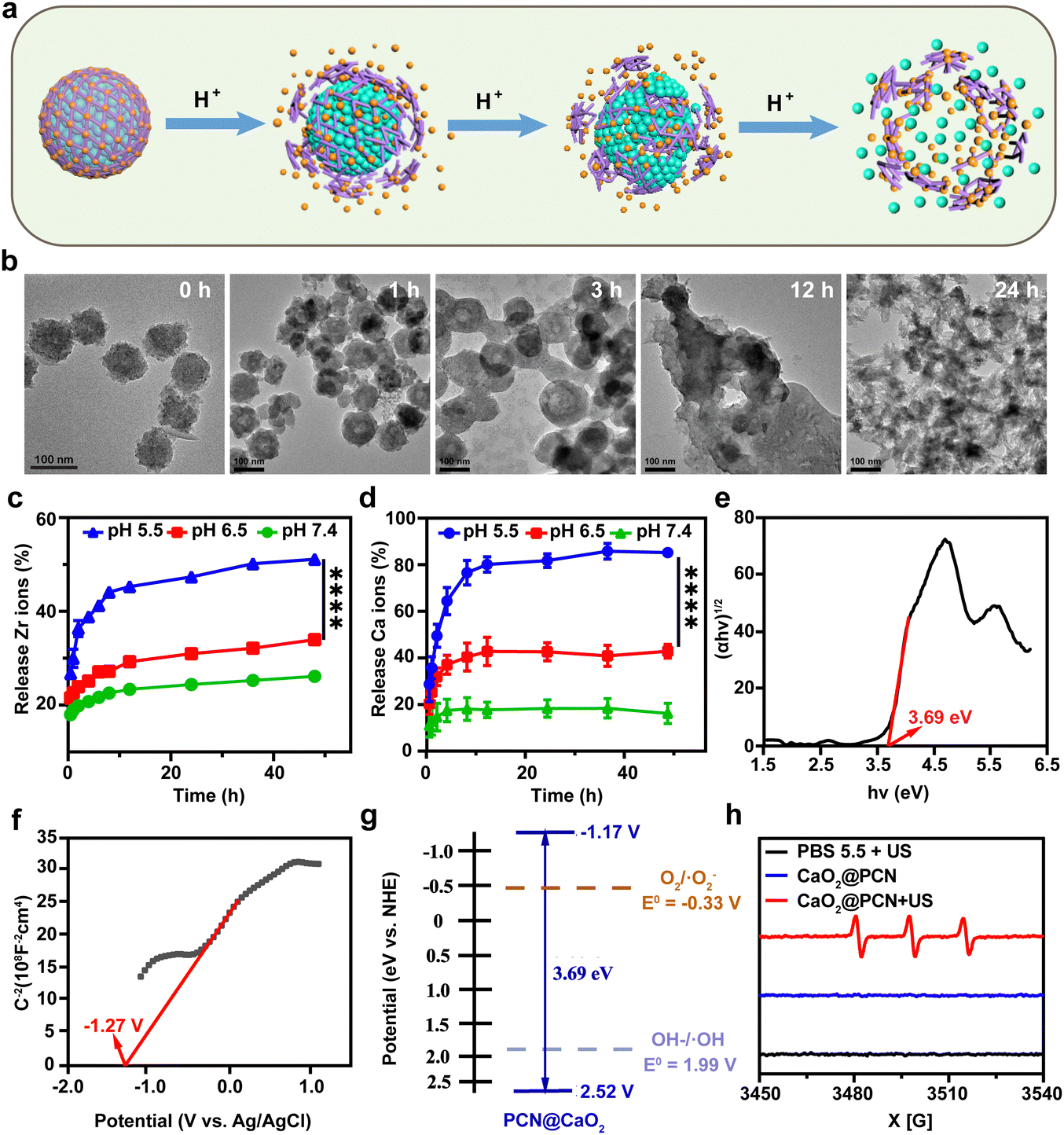 | ||
| Fig. 4 (a) Scheme of the acidity-triggered full degradation process of CaO2@PCN. (b) TEM images of CaO2@PCN after incubation in an acidic solution (pH = 5.5) for 0, 1, 3, 12, and 24 h. Scale bar, 100 nm. (c) and (d) Time-dependent release profiles of (c) Zr4+ and (d) Ca2+ from CaO2@PCN under different pH values. n = 3, mean ± s.d., ****P < 0.0001. (e)–(f) Tauc plots (e), Mott–Schottky plots (f), and band structure (g) of CaO2@PCN. (h) ESR spectra of CaO2@PCN using Temp as a spin trap. | ||
As a typical porphyrin sonosensitizer,36 TCPP in CaO2@PCN could be activated by low-intensity US to generate cytotoxic ROS. To confirm the sonodynamic performance of CaO2@PCN, the conduction band (CB) and valence band (VB) of CaO2@PCN were measured. First, the band gap of CaO2@PCN was calculated using the Kubelka–Munk (KM) function using ultraviolet-visible (UV-vis) diffuse reflectance spectroscopy (Fig. S12, ESI†). According to a Tauc plot, the band gap of CaO2@PCN was calculated to be 3.69 eV (Fig. 4e). Then the Mott–Schottky method was used to determine the CB of CaO2@PCN. As shown in Fig. 4f, the flat band potential vs. Ag/AgCl electrode of CaO2@PCN was determined to be −1.27 V, which demonstrated that the value of flat band potential vs. normal hydrogen electrode (NHE) was −1.07 V. In addition, the positive slope of the Mott–Schottky plot indicated that CaO2@PCN was an n-type semiconductor, and therefore the CB of CaO2@PCN was determined to be −1.17 V. Based on the band gap, the VB of CaO2@PCN could be calculated to be 2.52 V. These results showed that CaO2@PCN could generate ROS during SDT, since its CB edge (around −1.17 V) was more negative than the O2/·O2− redox couple potential (−0.16 V), and produce ·OH since its VB edge (around 2.52 V) was more positive than the OH–/·OH redox couple potential (1.99 V) (Fig. 4g).
To further confirm the sonodynamic performance of CaO2@PCN, the ROS generation after US irradiation (3 MHz, 2 W cm−2, 3 min) was detected by both electron spin resonance (ESR) spectroscopy and 1,3-diphenylisobenzofuran (DPBF). Briefly, 2,2,6,6-tetramethylpiperidine (Temp) was used as a spin trapper to identify singlet oxygen (1O2), and effective generation of 1O2 was observed in CaO2@PCN upon US irradiation (Fig. 4h). As shown in Fig. S13 (ESI†), the characteristic peak of DPBF at 420 nm gradually decreased in the presence of CaO2@PCN with the increase of US irradiation time, also indicating that CaO2@PCN could generate 1O2 upon US stimulation.
In vitro cytotoxicity and anticancer mechanism of CaO2@PCN
Before assessing the in vitro anticancer effect of CaO2@PCN, a study was conducted to evaluate the intracellular uptake efficiency of CaO2@PCN by the mouse breast cancer cells (4T1 cells) via confocal laser scanning microscopy (CLSM) in combination with flow cytometry (FCM). During 6 h of incubation, a significant amount of CaO2@PCN nanoparticles were gradually endocytosed by 4T1 cells with a strengthened TCPP fluorescence signal (Fig. 5a and Fig. S14, ESI†). This was consistent with the FCM analysis, which showed the elevated cellular internalization of CaO2@PCN with the extended incubation time (Fig. S15, ESI†), unambiguously indicating the effective internalization of CaO2@PCN by 4T1 cells. Subsequently, the intracellular degradation of CaO2@PCN was evaluated by investigating its localization inside lysosomes at various time intervals. Fig. 5b and Fig. S16 (ESI†) showed a gradual increase in TCPP fluorescence from 3 to 12 h and a gradual decrease in colocalization between TCPP and lysosomes. These results indicated that the nanoparticles were taken up by cells and subsequently enter the lysosome, which provided a favorable acidic environment for the degradation of CaO2@PCN. Then, the acidity-responsive biodegradability of CaO2@PCN generated free TCPP and Ca2+, which diffuse into the cytoplasm subsequently. Then, the intracellular Ca2+ level was quantified with a commercial fluorescence Ca2+ indicator, Fluo-4 AM. Both CLSM images and FCM analysis showed that the cells treated with CaO2@PCN exhibited time-dependent intensification of Ca2+ fluorescence (Fig. 5c and Fig. S17, S18, ESI†), verifying the efficient uptake of CaO2@PCN by 4T1 cells leading to an increase in intracellular Ca2+ concentration. Cancer cells undergo apoptosis due to intracellular Ca2+ overload, where excessive free Ca2+ enters mitochondria to disrupt mitochondrial functions.37–40 This alteration was visualized with Fluo-4 AM and Mito-Tracker Red using CLSM, and Image J was used to analyze the colocation rate. As shown in Fig. S19 and S20 (ESI†), the colocalization rate of Ca2+ and mitochondria in 4T1 cells was only about 22% after 1 h of co-incubation with CaO2@PCN. However, the colocation rate rose to 84.33% after 12 h of co-incubation, suggesting a significant accumulation of free Ca2+ in mitochondria over time. Based on the above results, massive Ca2+ would flow into the mitochondria during the decomposition of CaO2@PCN, which might affect the mitochondrial membrane potential (MMP). Herein, a commercial JC-1 probe was employed to measure the MMP levels in 4T1 cells. At high MMP levels, the probe formed J-aggregates and exhibited red fluorescence, while at low MMP levels, it existed as monomers and exhibited green fluorescence.41,42 The corresponding fluorescence images showed that the JC-1 probe emitted notable green fluorescence in 4T1 cells treated with CaO2 and CaO2@PCN but predominantly red fluorescence after incubation with PBS (control) and PCN (Fig. 5d and e). These contrasting images indicated that CaO2@PCN caused the most significant damage to mitochondrial function in 4T1 cells, contributing to the most cell apoptosis. In addition, the longer incubation time of CaO2@PCN with 4T1 cells resulted in stronger green fluorescence (Fig. S21 and S22, ESI†), indicating that the prolonged incubation time led to more significant mitochondrial damage.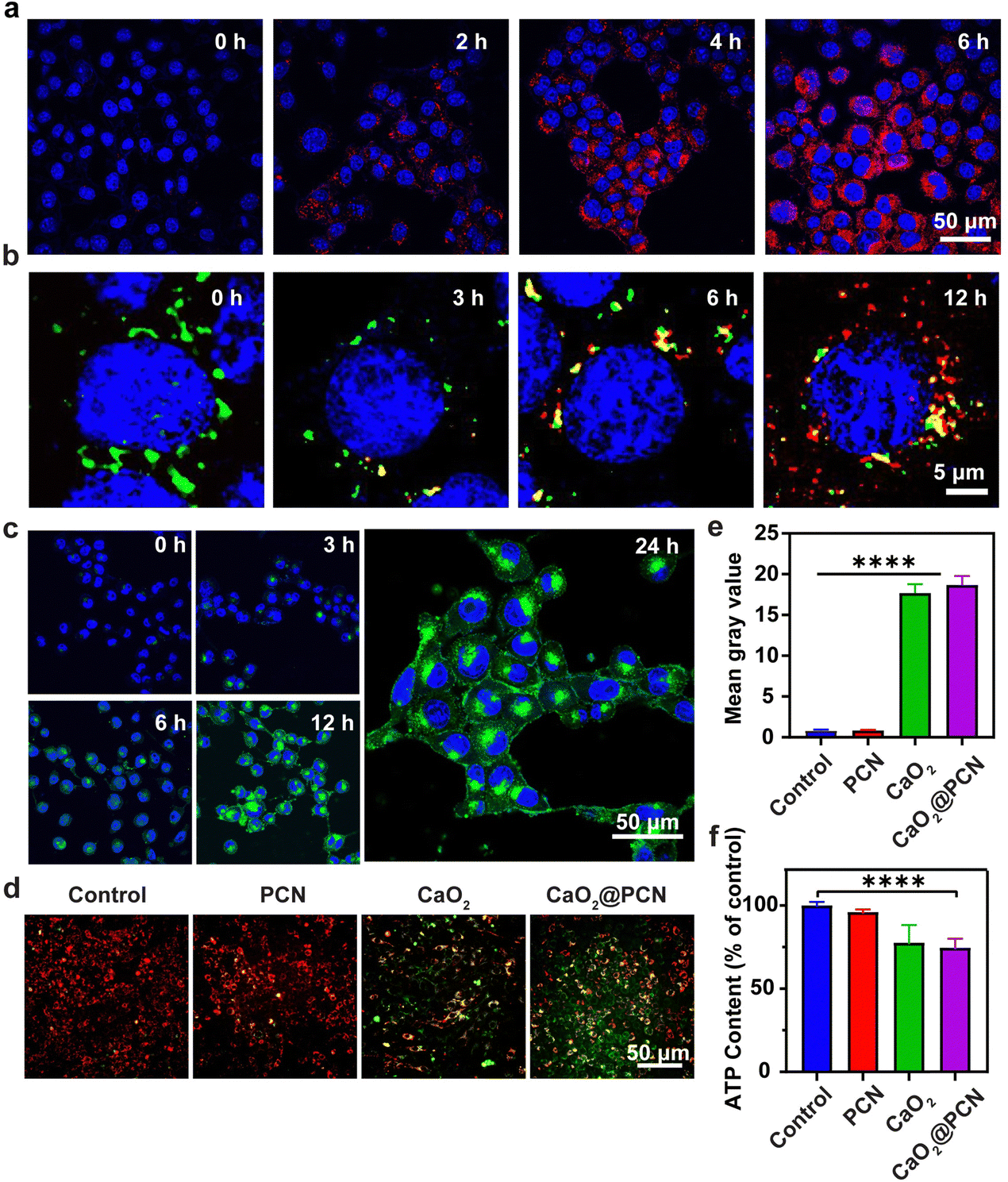 | ||
| Fig. 5 (a) CLSM images (red, TCPP; blue, DAPI) of cellular uptake of 4T1 cells incubated with CaO2@PCN for 0, 2, 4, and 6 h. Scale bar, 50 μm. (b) CLSM images (red, TCPP; green, Lyso-Tracker) of distribution of TCPP in the lysosome of 4T1 cells incubated with CaO2@PCN for 0, 3, 6, and 12 h. Scale bar, 5 μm. (c) CLSM images (green, Fluo-4; blue, DAPI) of intracellular Ca2+ level in 4T1 cells after incubation with CaO2@PCN for 0, 3, 6, 12, and 24 h. Scale bar, 50 μm. (d) Fluorescence images (red, JC-1 aggregates, high membrane potential; green, JC-1 monomer, low membrane potential) and (e) quantitative analysis of the mitochondrial membrane potential of 4T1 cells after different treatments in (d). Scale bar, 50 μm. (f) Intracellular ATP content in 4T1 cells after different treatments. 68.5 μg mL−1 CaO2, 87.2 μg mL−1 PCN, and 75 μg mL−1 CaO2@PCN ([TCPP] = 10 μg mL−1, [Ca2+] = 23 μg mL−1) were used. n = 3, mean ± s.d., ****P < 0.0001. | ||
Furthermore, Ca2+ overload can increase ROS generation, amplifying intracellular oxidative stress. This was detectable with a commercial probe, 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA). Notably, the green fluorescence of 4T1 cells receiving CaO2 and CaO2@PCN was significantly brighter than 4T1 cells in the PCN-treated group. This was attributed to the elevated intracellular oxidative stress, demonstrating that the Ca2+ generated from CaO2 and CaO2@PCN could accumulate in the mitochondria to elevate the ROS yield (Fig. S23 and S24, ESI†). Besides, tumor cells could be starved to death due to the reduction of ATP caused by the intracellular Ca2+-overload-induced mitochondrial dysfunction. Compared to the Control group, ATP contents were markedly decreased in 4T1 cells treated with CaO2 and CaO2@PCN, intuitively suggesting the potential of Ca2+ overload to suppress energy supply in cancer cells (Fig. 5f).
The in vitro cytotoxicities of CaO2, PCN, and CaO2@PCN were evaluated by a typical methyl thiazolyl tetrazolium (MTT) assay. The cytotoxicity of blank PCN was negligible in 4T1 cells even at a large concentration ([TCPP] = 600 μg mL−1) (Fig. S25, ESI†). Comparatively, the viabilities of 4T1 cells were prominently declined after incubation with CaO2 and CaO2@PCN. The survival rates of 4T1 cells decreased to 54.96% and 46.38% after treatment with 100 μg mL−1 CaO2 and CaO2@PCN, respectively (Fig. S26 and S27, ESI†), which might be attributed to the intracellular Ca2+ overload. Fortunately, the results presented in Fig. S28 (ESI†) demonstrated the favorable biocompatibility of CaO2@PCN in vitro, as L929 mouse fibroblast cells displayed a high survival rate of over 90% after incubation with 200 μg mL−1 CaO2@PCN.
To further explore the mechanism of CaO2@PCN-induced cell apoptosis, the expression levels of caspase-3, cytochrome C (Cyt-C), cleaved-caspase-3, and apoptotic protease-activating factor 1 (APAF-1) in 4T1 cells were measured by the western blot assay. Fig. 6a and Fig. S29, S30 (ESI†) display appreciable downregulation of caspase-3 and Cyt-C expression levels, coupled with efficient up-regulation of cleaved-caspase-3 and APAF-1 expression levels in 4T1 cells treated with CaO2 and CaO2@PCN, indicating that Ca2+ overload triggered apoptosis in a mitochondrial dysfunctional manner.
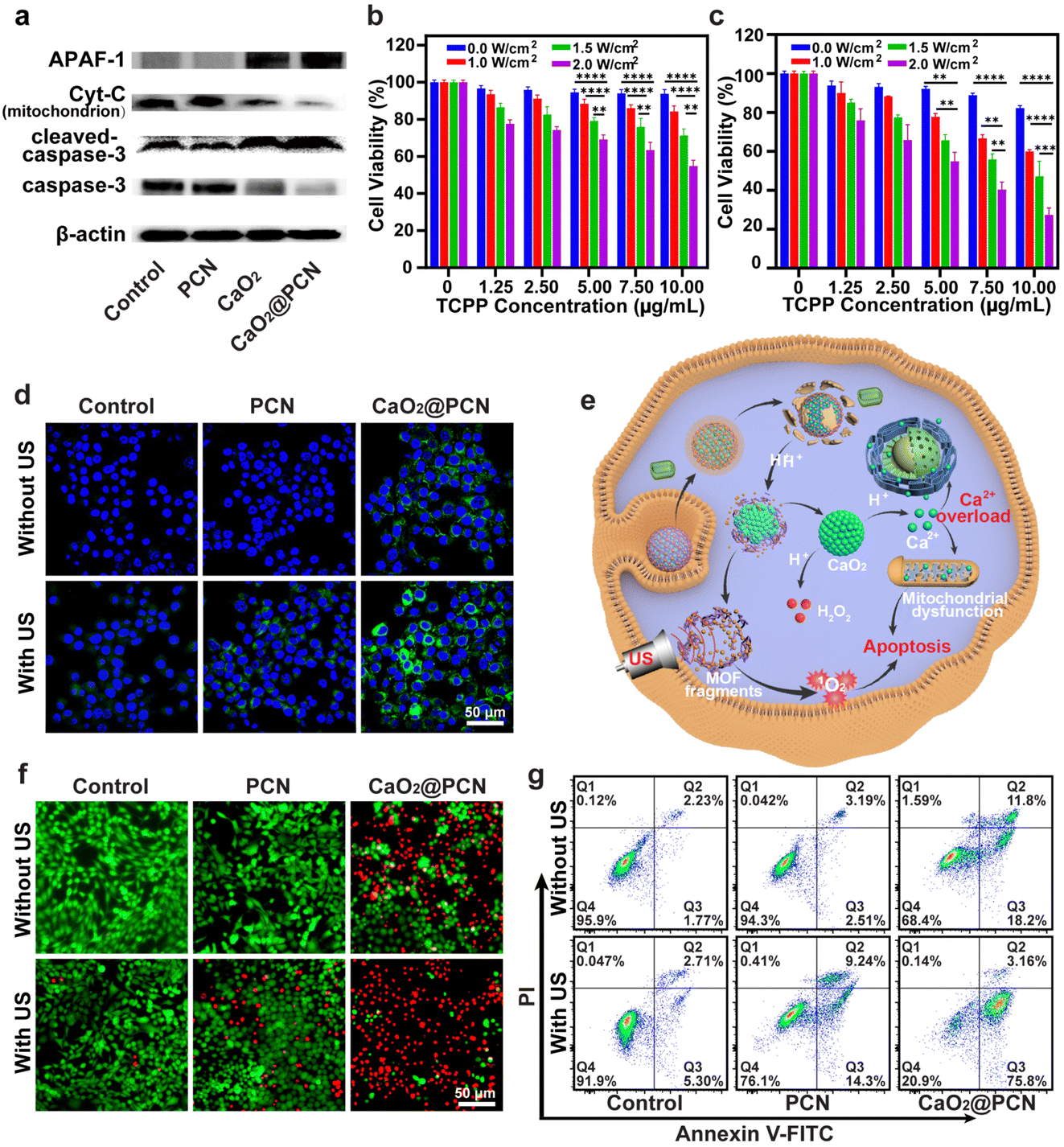 | ||
| Fig. 6 (a) Western blot assay of caspase-3, cleaved-caspase-3, Cyt-C, and APAF-1 expression in 4T1 cells after various treatments. (b) and (c) Relative viabilities of 4T1 cells after treatment with various concentrations of (b) PCN and (c) CaO2@PCN upon varied power densities of US irradiation. n = 6, mean ± s.d., **P < 0.01, ***P < 0.001, ****P < 0.0001. (d) CLSM images of intracellular ROS generation in 4T1 cells after different treatments. DCFH-DA (green, DCF; blue, DAPI). Scale bar, 50 μm. (e) Scheme of the antitumor mechanism of CaO2@PCN via Ca2+-overload-induced IIT and SDT. (f) Fluorescence images of calcein-AM (green, live cells) and PI (red, dead cell) co-stained 4T1 cells after various treatments. Scale bar, 50 μm. (g) Flow cytometry analysis of apoptosis in Annexin V-FITC/PI dual-stained 4T1 cells after different treatments. 68.5 μg mL−1 CaO2, 87.2 μg mL−1 PCN, and 75 μg mL−1 CaO2@PCN ([TCPP] = 10 μg mL−1, [Ca2+] = 23 μg mL−1) were used. | ||
In vitro synergistic treatment of CaO2@PCN
Based on the previously explored in vitro cytotoxicity and anticancer mechanism of CaO2@PCN investigated above, we next studied the synergistic SDT/IIT effect of CaO2@PCN upon US irradiation in vitro by an MTT assay. As shown in Fig. 6b and c, the viabilities of 4T1 cells prominently decreased with the increased TCPP concentration or US power density, demonstrating a controllable SDT effect. Notably, 4T1 cells treated with CaO2@PCN under US irradiation exhibited an elevated mortality rate than those treated with PBS or PCN under the same TCPP concentration and US power density (Fig. S31, ESI†). Moreover, the excess Ca2+ released from CaO2@PCN elevated the ROS level, while the TCPP of CaO2@PCN concurrently produced 1O2 upon US irradiation, both of which cooperated to break the intracellular redox/oxidation balance to kill cancer cells. Then, DCFH-DA was utilized to track intracellular ROS fluctuation induced by various treatments. The highest level of ROS was detected in 4T1 cells treated with CaO2@PCN after US irradiation, confirming that the SDT in this combined treatment strategy induced overwhelming intracellular oxidative stress (Fig. 6d and e and Fig. S32, ESI†).To further verify the therapeutic effect of CaO2@PCN upon US irradiation, cell apoptosis following different treatments was measured using calcein acetoxymethyl ester (calcein-AM, green fluorescence) and propidium iodide (PI, red fluorescence) co-staining assay. Compared with other groups (control, control + US, PCN, PCN + US, and CaO2@PCN), cells treated with CaO2@PCN + US displayed obvious red fluorescence (dead) with negligible green fluorescence (live) (Fig. 6f), suggesting the largest cytotoxicity of synergistic SDT/IIT. Additionally, a similar trend was observed in the flow cytometry analysis of the Annexin V-FITC/PI co-staining assay (Fig. 6g). The CaO2@PCN + US group showed the highest apoptosis rate, at 78.96%, higher than 8.01% of the Control + US group, 23.54% of the PCN + US group, and 30% of the CaO2@PCN group (Fig. S33, ESI†). According to these results, CaO2@PCN upon US irradiation boosted intracellular oxidative stress and yielded a stronger synergistic antitumor effect of SDT/IIT combination than SDT or IIT alone.
In vivo synergistic treatment of CaO2@PCN
The in vivo biodistribution of CaO2@PCN in mice was evaluated by tracking the characteristic TCPP fluorescence signal. Fluorescence images revealed the gradually strengthened TCPP fluorescence in the tumor after intravenous injections of CaO2@PCN (Fig. 7a), which was visible at 2 h post-injection and peaked at 12 h post-injection (Fig. S34a, ESI†). Notably, the remaining fluorescence of TCPP in the tumor site at 48 h post-injection (Fig. 7b and Fig. S34b, ESI†) implied relatively long tumor retention of CaO2@PCN. In addition, the pharmacokinetics of CaO2@PCN demonstrated relatively fast clearance in vivo due to its small particle size (Fig. S35, ESI†).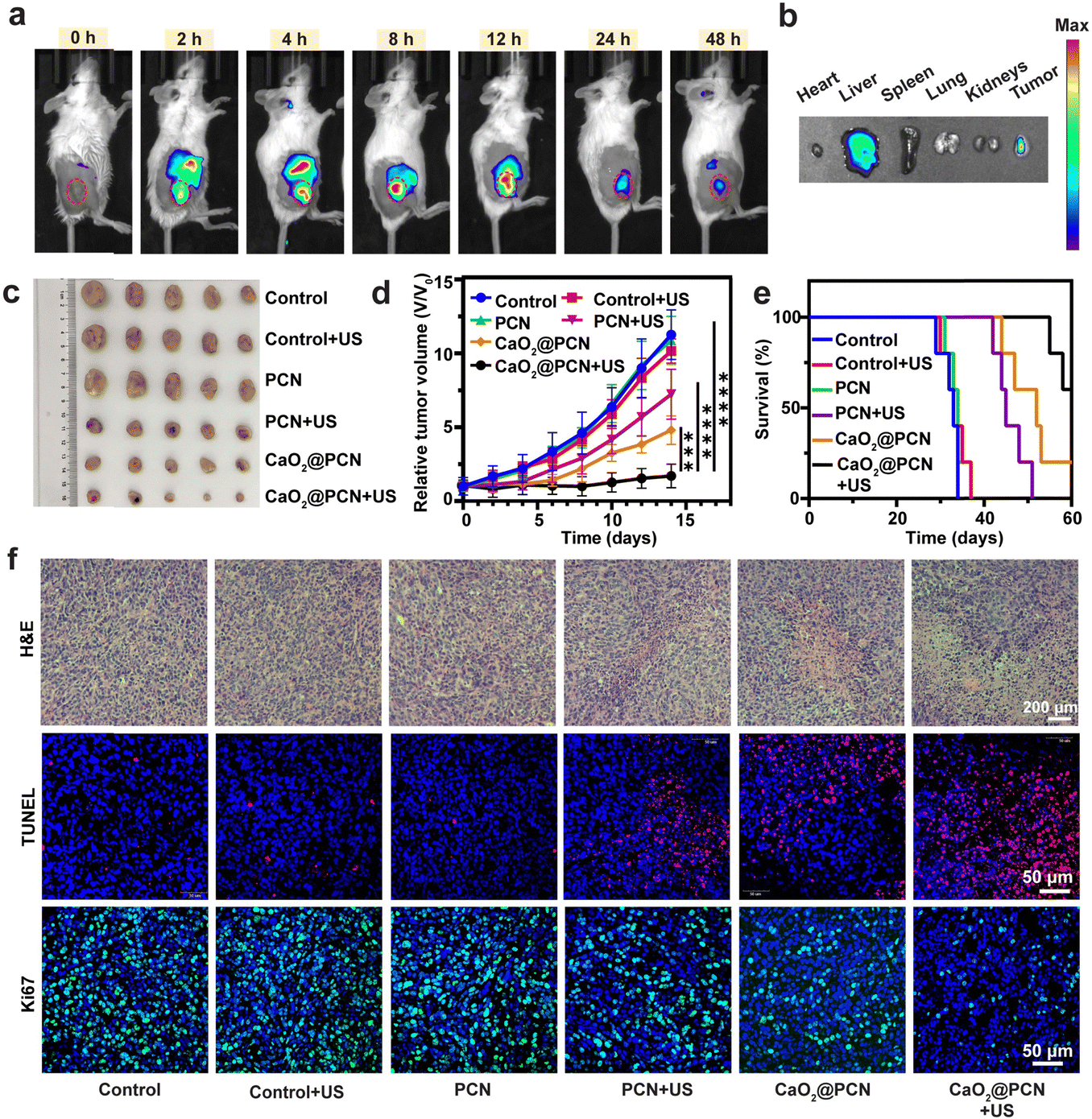 | ||
| Fig. 7 (a) In vivo fluorescence imaging of 4T1 tumor-bearing mice at given time points after i.v. injection of CaO2@PCN. (b) Ex vivo fluorescence imaging of major organs and tumor at 48 h i.v. injection of CaO2@PCN. (c) Tumor images were obtained from the 4T1 tumor-bearing BALB/c mice on the 14th day after treatment. (d) Relative tumor volume of 4T1 tumor-bearing mice after different treatments. n = 5, mean ± s.d., ***P < 0.001, ****P < 0.0001. (e) Survival percentages of the 4T1 tumor-bearing BALB/c mice after different treatments (n = 5 biologically independent samples). (f) Histological analyses with H&E (scale bar, 200 μm), TUNEL (red, TUNEL; blue, DAPI; scale bar, 50 μm), and Ki67 (green, Ki67; blue, DAPI; scale bar, 50 μm) staining of 4T1 tumors after different treatments. 8.72 mg mL−1 PCN and 7.50 mg mL−1 CaO2@PCN ([TCPP] = 1.0 mg mL−1, [Ca2+] = 2.3 mg mL−1) were used. | ||
Subsequently, the in vivo anticancer efficacy of CaO2@PCN upon US irradiation was further evaluated on the same 4T1 tumor-bearing mouse model. Both the US group and the PCN group had a similar negligible effect on tumor growth inhibition as the control group, suggesting that neither was ineffective as a single treatment (Fig. 7c, d and Fig. S36, ESI†). The PCN + US group displayed an inhibition rate of about 47%, which was ascribed to the SDT effect. As a result of Ca2+-induced IIT, the mice treated with CaO2@PCN demonstrated a visible tumor inhibition rate of 61%. More significantly, Ca2+-induced IIT combined with SDT was found to yield the strongest synergistic antitumor effect and the longest survival time, with a large tumor inhibition rate of 85% (Fig. 7e and Fig. S37, ESI†). To further verify the antitumor effect of CaO2@PCN upon US irradiation, the ROS generation in tumor tissues was assessed by dihydroethidium (DHE) immunofluorescence staining. Fig. S38 and S39 (ESI†) showed that the mice treated with CaO2@PCN upon US irradiation exhibited the strongest red fluorescence signal of ROS, indicating the largest intracellular oxidative stress caused by Ca2+-induced IIT and SDT. Finally, all the mice subjected to different treatments showed no apparent body weight fluctuation (Fig. S40, ESI†), as well as no visible pathological changes in the hematoxylin–eosin (H&E)-stained major organs, both of which verified the excellent biosafety of these treatments (Fig. S41, ESI†). Meanwhile, there were no significant changes in markers of liver or kidney damage (Fig. S42, ESI†). Additionally, in hematological blood cell tests, the hemolysis rates of CaO2, PCN, and CaO2@PCN were less than 5% (Fig. S43, ESI†), demonstrating their relatively high biocompatibility.
Furthermore, histopathological analysis of tumor slices, including hematoxylin and eosin (H&E), terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL), and Ki-67 staining, was performed to validate the highest antitumor efficacy of CaO2@PCN upon US irradiation. 4T1 tumors after different treatments were harvested for toxicity assessment using H&E staining. It could be seen that a majority of tumor tissues underwent cell apoptosis and necrosis, while only a small portion of tumor cells survived (Fig. 7f), indicating the superior synergistic tumoricidal effect of CaO2@PCN upon US irradiation. Similarly, the strongest TUNEL fluorescence (red) was observed in the CaO2@PCN + US group (Fig. 7f). Afterward, Ki-67 with specific green fluorescence was utilized to evaluate the proliferation ability of tumor cells. As shown in Fig. 7f, the CaO2@PCN + US group showed the weakest green fluorescence, suggesting the strongest suppression effect on cancer cell proliferation. Overall, all the observations demonstrated that CaO2@PCN upon US irradiation resulted in the most significant tumor suppression through the synergy of Ca2+-induced IIT and SDT.
Conclusion
In summary, we reported a simple and feasible strategy to suppress tumor growth by combining Ca2+-induced IIT and SDT. A pH-responsive degradable CaO2@PCN nanosonosensitizer was successfully prepared by an in situ epitaxial growth method. The further surface modification of the porphyrinic MOF (PCN-224) prevented not only the premature leakage of TCPP but also the spontaneous hydrolysis of CaO2. Upon arriving at the acidic TME, the PCN-224 shell of CaO2@PCN would gradually degrade to allow for the hydrolysis of inner CaO2 into Ca2+ and H2O2. Meanwhile, the concurrent generation of exogenous Ca2+ would efficiently cause intracellular Ca2+ accumulation, ultimately resulting in cell apoptosis via Ca2+-overload-induced mitochondrial dysfunction. Based on the in vitro and in vivo observations, CaO2@PCN upon US irradiation could elicit a remarkable synergistic SDT/IIT effect, evidenced by a large tumor growth inhibition rate of 85% after a 14-day treatment. To summarize, the well-designed CaO2@PCN nanoparticle is expected to guide the future design of clinically promising degradable nanomedicines for precision synergistic cancer therapy.Materials and methods
Materials
Zirconium chloride (ZrCl4, 98%), meso-tetra-(4-carboxyphenyl) porphyrin (TCPP), zirconium oxychloride octahydrate (ZrOCl2·8H2O), benzoic acid (BA) and polyvinyl pyrrolidone (PVP, K-58) were purchased from Aladdin Reagents Company (Shanghai, China). Calcium chloride anhydrous (CaCl2, AR ≥ 96%), hydrogen peroxide (H2O2, 30%), ammonia solution (NH3·H2O, 25–28%), N,N-dimethylformamide (DMF), and ethanol (AR ≥ 99%) were obtained from Nanjing Chemical Reagents Company (Nanjing, China). Methyl thiazolyl tetrazolium (MTT), 2′-7′-dichlorofluoresceindiacetate (DCFH-DA), JC-1 probe, Fluo-4 AM, 2-(4-amidinophenyl)-6-indolecarbamidine dihydrochloride (DAPI), calcein acetoxymethylester (calcein AM), propidiumiodide (PI) and Annexin V-FITC/PI kit were purchased from Solarbio (Beijing, China). Caspase-3, Cyt-C, and APAF-1 antibodies were purchased from Beyotime Biotechnology (Shanghai, China). Cleaved-caspase-3 polyclonal antibody was purchased from Elabscience® Biotechnology Co., Ltd. Singlet oxygen sensor green (SOSG) was obtained from Thermo Fisher Scientific (Shanghai, China). Rapid transfer buffer, universal antibody diluent, and serum-free cell freeze were purchased from New Cell & Molecular Biotech Co., Ltd.Synthesis of CaO2
CaCl2 (40 mg) and PVP (350 mg) were dissolved and mixed in ethanol (15 mL) by sonication. Then 1 mL of NH3·H2O (0.8 M) was added to the above-mixed solution and stirred for 5 min, followed by dropwise addition of 0.2 mL of H2O2 (1 M) solution in 4 min with the help of a syringe pump at a rate of 3 mL h−1 to activate the reaction. The final product was collected by centrifugation and washed with ethanol three times to remove excess free PVP.Synthesis of CaO2@PCN
10 mg of the as-prepared CaO2 nanoparticles were dissolved in 1 mL of ethanol and mixed with 0.4 mg of ZrCl4 dissolved in a mixed solvent of DMF (3 mL) and ethanol (3 mL) for 10 min of sonication at 4 °C, followed by addition of 0.6 mg of TCPP in a mixed solvent of DMF (2.5 mL) and ethanol (2.5 mL). After another 10 min of sonication at 4 °C, the above system was transferred to an oil bath and stirred for 6 h at 90 °C. Finally, the product was harvested after centrifugation and washed with DMF and ethanol several times.Characterization
The morphologies of nanoparticles were observed with a transmission electron microscope (TEM, JEM-2011, Japan). High-angle annular dark field-scanning transmission electron microscopy (HAADF-STEM) images and elemental mapping images of CaO2@PCN were obtained on an FEI Talos F200X microscope. Zeta potential and dynamic light scattering (DLS) were measured using a Malvern instrument Zetasizer Nano system. XRD patterns were collected on an X’Pert MPD X-ray diffractometer. The infrared spectra of nanoparticles were probed using a Nicolet 8700 FTIR spectrometer. UV-vis absorption spectra were recorded on a HITACHI UH 5300 UV-vis spectrophotometer. X-ray photoelectron spectroscopy (XPS) spectra were recorded on a Thermo ESCALAB 250Xi X-ray photoelectron spectrometer. Element concentrations were quantified using inductively coupled plasma optical emission spectra (ICP-OES, ICPE-9000, Japan). UV-vis diffuse reflectance spectroscopy spectra were obtained using a UV-vis-NIR spectrophotometer (Lambda 950, USA). Electrochemical measurements were recorded using an electrochemical workstation (Model CHI-600A, China). The confocal laser scanning microscopy (CLSM) images were obtained on an FV 3000 Olympus confocal laser scanning microscope. Flow cytometry analyses were conducted on an Agilent NovoCyte flow cytometer.The TCPP content of CaO2@PCN
50 μL of the as-prepared CaO2@PCN nanoparticles was dissolved in 4 mL of DMF. Next, the TCPP content of the above solution was quantified by recording the TCPP absorbance at 420 nm with a HITACHI UH 5300 UV-vis spectrophotometer. TCPP content (weight %) = (weight of TCPP in CaO2@PCN)/(weight of CaO2@PCN) × 100%.Biodegradation of CaO2@PCN
1 mg of CaO2@PCN was dispersed in 1 mL of PBS solution (pH 5.5) and incubated at 37 °C in a shaking culture incubator. At given time points (0, 1, 3, 12, and 24 h), 10 μL of the solution was taken out for TEM observation to observe the degradation of CaO2@PCN.Ca2+ and Zr4+ release performance
To study the effect of pH values on Ca2+ and Zr4+ release performance, a dialysis bag (3500 Da) containing 2 mg of CaO2@PCN was immersed in 30 mL of PBS with different pH values (7.4, 6.5, and 5.5) and shaken evenly in a shaking culture incubator at 37 °C. To study the effect of US on Ca2+ release from CaO2@PCN, 2 mg mL−1 of CaO2@PCN was treated with or without US irradiation for 5 min. The solution was placed in a dialysis bag (3500 Da) and then immersed in 30 mL of PBS with pH 5.5 and shaken evenly in a shaking culture incubator at 37 °C. At given time points (0.5, 1, 2, 4, 6, 8, 12, 24, 36, and 48 h), 1 mL of the solution was taken out to measure the released concentration of Ca2+ or Zr4+, and the original mixed solution was replenished with 1 mL of fresh PBS. The released Ca2+ or Zr4+ concentrations were quantified by ICP-OES and the release curve was made on the percentage of cumulative Ca2+ or Zr4+ outside the dialysis bag to the total Ca2+ or Zr4+ within CaO2@PCN.Stability of CaO2@PCN
100 μg of CaO2@PCN was dispersed in 1 mL of PBS (pH 7.4), RMPI 1640 (Shanghai BasalMedia Technologies Co., Ltd, China), and RMPI 1640 with 10% fetal bovine serum (FBS) (Gibco, USA) for incubation at 37 °C. Then, the hydrodynamic size change of CaO2@PCN was measured using a Malvern instrument, the Zetasizer Nano system.In vitro cellular uptake
4T1 cells were seeded at a density of 2.5 × 105 per well in a 6-well plate and incubated for 12 hours to achieve adherence. 75 μg mL−1 CaO2@PCN ([TCPP] = 10 μg mL−1, [Ca2+] = 23 μg mL−1) in RPMI 1640 media was added and incubated with the cells for different periods (2, 4, and 6 h). Then the cells were washed with PBS three times and stained with 4% polyoxymethylene for 30 min. After washing with PBS three times, the cells were stained with DAPI (1 μg mL−1) for 15 min. Finally, the cellular uptake was visualized under a confocal laser scanning microscope (CLSM) and quantitatively analyzed with flow cytometry (FCM).Intracellular Ca2+ level test
4T1 cells were seeded in a 6-well plate with a density of 2.5 × 105 per well and incubated for 12 h. The cells were treated with 75 μg mL−1 CaO2@PCN ([TCPP] = 10 μg mL−1, [Ca2+] = 23 μg mL−1) for different periods (0, 3, 6, 12, and 24 h). Then, the cells were washed with PBS several times and stained with Fluo-4 AM (2 μM) for 30 min. After washing with PBS several times, the cells were stained with DAPI (1 μg mL−1) for 15 min. Finally, the intracellular Ca2+ level was observed by CLSM and detected using flow cytometry.Detection of mitochondrial membrane potential and mitochondrial damage
4T1 cells were seeded in a 6-well plate at a density of 2.5 × 105 per well and cultured overnight. Afterward, the cells were sequentially treated with varying nanoformulations for 12 h in the groups of control, 68.5 μg mL−1 CaO2, 87.2 μg mL−1 PCN, or 75 μg mL−1 CaO2@PCN ([TCPP] = 10 μg mL−1, [Ca2+] = 23 μg mL−1). Next, the cells were stained with JC-1 and incubated for 30 min, and then the changes of mitochondrial membrane potential in cells were visualized using an inverted fluorescence microscope. To further explore the influence of CaO2@PCN on mitochondrial damage, the distribution of mitochondria in 4T1 cells after treatment with 75 μg mL−1 CaO2@PCN ([TCPP] = 10 μg mL−1, [Ca2+] = 23 μg mL−1) for different periods (0, 3, 6, 9, 12, and 24 h) was measured. JC-1 was used as a fluorescent probe and incubated with 4T1 cells for another 30 min. Subsequently, the distribution of mitochondria was visualized using an inverted fluorescence microscope and analyzed by flow cytometry.Measurement of the intracellular ATP content
4T1 cells were seeded in 6-well plates at a density of 1.5 × 105 per well and cultured for 12 h at 37 °C. Then the cells were treated with pure RPMI 1640 media or media containing 68.5 μg mL−1 CaO2, 87.2 μg mL−1 PCN, and 75 μg mL−1 CaO2@PCN ([TCPP] = 10 μg mL−1, [Ca2+] = 23 μg mL−1) for 12 h. Next, the ATP content was estimated with an ATP testing kit for 30 min of incubation and measured using a microplate reader.Western blotting analysis
4T1 cells were seeded in 6-well plates with a density of 1.5 × 105 per well and cultured overnight. Then the cells were treated with different nanoformulations (68.5 μg mL−1 CaO2, 87.2 μg mL−1 PCN, or 75 μg mL−1 CaO2@PCN ([TCPP] = 10 μg mL−1, [Ca2+] = 23 μg mL−1)). To replace nanoformulation suspensions, an equal volume of RPMI 1640 media was set as the control group. The expressions of proteins were evaluated by western blot analysis, while the protein concentration of each group was measured using the BCA Protein Assay Kit (Solarbio, Beijing). The expressions of caspase-3, Cyt-C, APAF-1 and cleaved-caspase-3 proteins were evaluated by western blot analysis.Apoptosis assay
4T1 cells were seeded in a 6-well plate at a density of 2.5 × 105 per well and cultured at 37 °C for 12 h. Then the cells were incubated with 2 mL of a fresh medium supplemented with 87.2 μg mL−1 PCN or 75 μg mL−1 CaO2@PCN ([TCPP] = 10 μg mL−1, [Ca2+] = 23 μg mL−1) for 8 h. Cells without any treatment were set as a control. Then the cells were subjected to US irradiation (2.0 W cm−2, 3 MHz, and 50% duty cycle) and cultured for another 24 h. Finally, the cells in each well were harvested and simultaneously stained with Annexin V-FITC and propidium iodide (PI), and the number of dead cells was further analyzed by flow cytometry.Animals model
BALB/c female mice (∼20 g) were purchased from Yangzhou University (Yangzhou, China). The mice were kept on a 12-hour light–dark cycle and adapted to the experimental conditions for one week before the experiment. All animal experimental protocols were approved by the Animal Experimentation Ethics Committee of China Pharmaceutical University (Nanjing, China). In this study, the animals’ tumor model was established by subcutaneously injecting 5 × 106 4T1 cells into female BALB/c mice at 6–7 weeks of age (∼20 g). When the tumor volumes reached 100 mm3, the mice were randomly divided into several groups for the following experiments.In vivo fluorescence imaging
When the tumor volumes reached 100 mm3, the tumor-bearing mice were used for in vivo fluorescence imaging. A 7.5 mg mL−1 solution of CaO2@PCN ([TCPP] = 1.0 mg mL−1, [Ca2+] = 2.3 mg mL−1) was injected into the mice via the tail vein at a volume of 100 μL. Then the mice were imaged at given time points (0, 1, 2, 4, 8, 12, 24, and 48 h) to evaluate the biodistribution of CaO2@PCN.In vivo tumor therapy
The 4T1 tumor-bearing mice were randomly divided into six groups (n = 5 per group) and subjected to different treatments: control, control + US, 8.72 mg mL−1 PCN, 8.72 mg mL−1 PCN + US, 7.50 mg mL−1 CaO2@PCN, and 7.50 mg mL−1 CaO2@PCN + US ([TCPP] = 1.0 mg mL−1, [Ca2+] = 2.3 mg mL−1). On days 1, 3, and 5, mice were intravenously injected with 100 μL of a solution consisting of PBS, PCN, and CaO2@PCN. Then, the mice in the PBS + US, PCN + US, and CaO2@PCN + US groups were exposed to US irradiation (3 MHz, 2 W cm−2, 10 min, and 50% duty cycle) at 8 h post-injection. After treatment, the tumor volume (V) was monitored by recording the length (L) and width (W) of each tumor every two days and calculated using the formula: V = L × W2/2 (W and L are the shortest and longest diameters of tumors). At the end of the evaluation (day 14), five other mice were killed to take photos. Besides, the mice from different groups were executed and the major organs of mice in each group were collected for H&E staining and TUNEL assays. Finally, the survival duration of the remaining mice in each group was observed until day 60 after receiving the first dose and the survival curves were plotted. Meanwhile, blood was collected from the mice for hematology and AST (alanine aminotransferase), ALT (alanine aminotransferase), BUN (urea nitrogen), and CREA (creatinine) assays.Author contributions
Conceptualization, Y. C., Z. Q., C. Z., X. Y., W. Y., and W. F.; methodology, Y. C., Z. Q., Z. M., and L. Z.; investigation, Y. C., Z. Q., Z. M., Z. L., R. L., Y. X., Y. X., and L. H.; writing – original draft, Y. C., Z. Q., and W. F.; writing – review & editing, Y. C., Z. M., T. D., and W. F.; supervision, Y. C., Z. M., X. Y., and W. F.; and funding acquisition, C. Z., X. Y., W. Y., and W. F.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was financially supported by the National Recruitment Program for Youth Talents, the Jiangsu Specially-Appointed Professors Program, the Key Research and Development Plan of Jiangsu Province (BE2023844), the Funding of Double First-Rate Discipline Innovation Team (CPUQNJC22_04), the Key Research and Development Plan of Jiangsu Province (BE2023844), the National Natural Science Foundation of China (No. 81973180, 82472463), the Fundamental Research Funds for the Central Universities of China (2632023TD01), the Anhui Provincial Key Research and Development Program-Clinical Medical Research Translation Specialization (202304295107020020), and the Natural Science Foundation of Jiangsu Province (BK20200709).Notes and references
- J. Jiang, J. Mei, Y. Ma, S. Jiang, J. Zhang, S. Yi, C. Feng, Y. Liu and Y. Liu, Explor, 2022, 2, 20210144 Search PubMed
.
- Q. Chen, C. Li and Q. Wang, Small Methods, 2023, 7, 2201457 CrossRef CAS PubMed
- D. Luo, K. A. Carter, D. Miranda and J. F. Lovell, Adv. Sci., 2017, 4, 1600106 CrossRef PubMed
- J. Shi, P. W. Kantoff, R. Wooster and O. C. Farokhzad, Nat. Rev. Cancer, 2017, 17, 20–37 CrossRef CAS PubMed
- S. Liang, X. Deng, P. Ma, Z. Cheng and J. Lin, Adv. Mater., 2020, 32, e2003214 CrossRef PubMed
- G. Canavese, A. Ancona, L. Racca, M. Canta, B. Dumontel, F. Barbaresco, T. Limongi and V. Cauda, Chem. Eng. J., 2018, 340, 155–172 CrossRef CAS PubMed
- Z. Gong and Z. Dai, Adv. Sci., 2021, 8, 2002178 CrossRef CAS PubMed
- X. Pan, W. Wang, Z. Huang, S. Liu, J. Guo, F. Zhang, H. Yuan, X. Li, F. Liu and H. Liu, Angew. Chem., Int. Ed., 2020, 59, 13557–13561 CrossRef CAS PubMed
- L. Rengeng, Z. Qianyu, L. Yuehong, P. Zhongzhong and L. Libo, Photodiagn. Photodyn. Ther., 2017, 19(2017), 159–166 CrossRef PubMed
- Y. Zeng, Q. Ouyang, Y. Yu, L. Tan, X. Liu, Y. Zheng and S. Wu, Small Methods, 2023, 7, 2201248 CrossRef CAS PubMed
- S. Bai, N. Yang, X. Wang, F. Gong, Z. Dong, Y. Gong, Z. Liu and L. Cheng, ACS Nano, 2020, 14, 15119–15130 CrossRef CAS PubMed
- M. Xu, L. Zhou, L. Zheng, Q. Zhou, K. Liu, Y. Mao and S. Song, Cancer Lett., 2021, 497, 229–242 CrossRef CAS PubMed
- S. Liang, X. Deng, Y. Chang, C. Sun, S. Shao, Z. Xie, X. Xiao, P. Ma, H. Zhang, Z. Cheng and J. Lin, Nano Lett., 2019, 19, 4134–4145 CrossRef CAS PubMed
- J. Chen, Y. Zhu and S. Kaskel, Angew. Chem., Int. Ed., 2021, 60, 5010–5035 CrossRef CAS PubMed
- P. Gao, Y. Chen, W. Pan, N. Li, Z. Liu and B. Tang, Angew. Chem., Int. Ed., 2021, 60, 16763–16776 CrossRef CAS PubMed
- S. Su, Y. Ding, Y. Li, Y. Wu and G. Nie, Biomaterials, 2016, 80, 169–178 CrossRef CAS PubMed
- X. Wang, F. Yan, X. Liu, P. Wang, S. Shao, Y. Sun, Z. Sheng, Q. Liu, J. F. Lovell and H. Zheng, J. Controlled Release, 2018, 286, 358–368 CrossRef CAS PubMed
- J. Chakraborty, I. Nath and F. Verpoort, Coord. Chem. Rev., 2021, 326, 135–163 CrossRef
- J. M. Park, K. I. Hong, H. Lee and W. D. Jang, Acc. Chem. Res., 2021, 54, 2249–2260 CrossRef CAS PubMed
- J. F. Chen, L. T. Feng, P. L. Jin, J. X. Shen, J. Y. Lu, Y. Song, G. W. Wang, Q. Chen, D. Y. Huang, Y. Zhang, C. Zhang, Y. F. Xu and P. T. Huang, J. Nanobiotechnol., 2022, 20, 283 CrossRef CAS PubMed
- T. T. Wu, Y. Liu, Y. Cao and Z. H. Liu, Adv. Mater., 2022, 34, e2110364 CrossRef PubMed
- M. Pan, D. R. Hu, L. P. Yuan, Y. Yu, Y. C. Li and Z. Y. Qian, Acta Pharm. Sin. B, 2023, 13, 2926–2954 CrossRef CAS PubMed
- W. Jiang, Z. Zhang, Q. Wang, J. Dou, Y. Zhao, Y. Ma, H. Liu, H. Xu and Y. Wang, Nano Lett., 2019, 19, 4060–4067 CrossRef CAS PubMed
- Y. Liu, M. Zhang and W. Bu, View, 2020, 1, e18 CrossRef
- M. Zhang, R. Song, Y. Liu, Z. Yi, X. Meng, J. Zhang, Z. Tang, Z. Yao, Y. Liu, X. Liu and W. Bu, Chem, 2019, 5, 2171–2182 CAS
- C. Dong, X. Dai, X. Wang, Q. Lu, L. Chen, X. Song, L. Ding, H. Huang, W. Feng, Y. Chen and M. Chang, Adv. Mater., 2022, 34, 2205680 CrossRef CAS PubMed
- Y. Li, S. Zhou, H. Song, T. Yu, X. Zheng and Q. Chu, Biomaterials, 2021, 277, 121080 CrossRef CAS
- J. He, L. H. Fu, C. Qi, J. Lin and P. Huang, Bioact. Mater., 2021, 6, 2698–2710 CAS
- S. Shen, M. Mamat, S. Zhang, J. Cao, Z. D. Hood, L. Figueroa-Cosme and Y. Xia, Small, 2019, 15, 1902118 CrossRef PubMed
- Y. Sheng, H. Nesbitt, B. Callan, M. A. Taylor, M. Love, A. P. McHale and J. F. Callan, J. Controlled Release, 2017, 264, 333–340 CrossRef CAS PubMed
- D. Van Haute, J. M. Longmate and J. M. Berlin, Adv. Mater., 2015, 27(35), 5158–5164 CrossRef CAS PubMed
- N. N. Amerhaider Nuar, S. N. A. Md Jamil, F. Li, I. D. Mat Azmi, P. C. Chiang and T. S. Y. Choong, Polymers, 2022, 14, 3866 CrossRef CAS PubMed
- Z. Cai, M. Huang, J. Dai, G. Zhan, F.-l Sun, G.-L. Zhuang, Y. Wang, P. Tian, B. Chen, S. Ullah, J. Huang and Q. Li, ACS Catal., 2022, 12, 709–723 CrossRef CAS
- S. S. Madan, K. L. Wasewar and C. Ravi Kumar, Adv. Powder Technol., 2016, 27, 2112–2120 CrossRef CAS
- Y. Wang, J. Yan, N. Wen, H. Xiong, S. Cai, Q. He, Y. Hu, D. Peng, Z. Liu and Y. Liu, Biomaterials, 2020, 230, 119619 CrossRef CAS PubMed
- I. Rosenthal, J. Z. Sostaric and P. Riesz, Ultrason. Sonochem., 2004, 11, 349–363 CrossRef CAS PubMed
- B. Liu, Y. Bian, S. Liang, M. Yuan, S. Dong, F. He, S. Gai, P. Yang, Z. Cheng and J. Lin, ACS Nano, 2022, 16, 617–630 CrossRef CAS PubMed
- P. Zheng, B. Ding, R. Shi, Z. Jiang, W. Xu, G. Li, J. Ding and X. Chen, Adv. Mater., 2021, 33, 2007426 CrossRef CAS PubMed
- C. Giorgi, F. Baldassari, A. Bononi, M. Bonora, E. De Marchi, S. Marchi, S. Missiroli, S. Patergnani, A. Rimessi, J. M. Suski, M. R. Wieckowski and P. Pinton, Cell Calcium, 2012, 52, 36–43 CrossRef CAS PubMed
- S. Marchi, S. Patergnani, S. Missiroli, G. Morciano, A. Rimessi, M. R. Wieckowski, C. Giorgi and P. Pinton, Cell Calcium, 2018, 69, 62–72 CrossRef CAS
- J. Liu, H. Wang, X. Yi, Y. Chao, Y. Geng, L. Xu, K. Yang and Z. Liu, Adv. Funct. Mater., 2017, 27, 1703832 CrossRef
- P. Zheng, B. Ding, Z. Jiang, W. Xu, G. Li, J. Ding and X. Chen, Nano Lett., 2021, 21, 2088–2093 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nh00189c |
| ‡ Yue Chen, Tong Ding and Zhengzheng Qian contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |