
Thermodynamic phase control of Cu–Sn alloy electrocatalysts for selective CO2 reduction†
Soohyun
Go‡
a,
Woosuck
Kwon‡
a,
Deokgi
Hong‡
b,
Taemin
Lee
 a,
Sang-Ho
Oh
b,
Daewon
Bae
a,
Jeong-Heon
Kim
a,
Seolha
Lim
a,
Young-Chang
Joo
b and
Dae-Hyun
Nam
*ac
a,
Sang-Ho
Oh
b,
Daewon
Bae
a,
Jeong-Heon
Kim
a,
Seolha
Lim
a,
Young-Chang
Joo
b and
Dae-Hyun
Nam
*ac
aDepartment of Energy Science and Engineering, Daegu Gyeongbuk Institute of Science and Technology (DGIST), Daegu 42988, Republic of Korea
bDepartment of Materials Science and Engineering, Seoul National University, Seoul 08826, Republic of Korea
cDepartment of Materials Science and Engineering, Korea University, Seoul 02841, Republic of Korea. E-mail: dnam@korea.ac.kr
First published on 10th September 2024
Abstract
In the electrochemical CO2 reduction reaction (CO2RR), Cu alloy electrocatalysts can control the CO2RR selectivity by modulating the intermediate binding energy. Here, we report the thermodynamic-based Cu–Sn bimetallic phase control in heterogeneous catalysts for selective CO2 conversion. Starting from the thermodynamic understanding about Cu–Sn bimetallic compounds, we established the specific processing window for Cu–Sn bimetallic phase control. To modulate the Cu–Sn bimetallic phases, we controlled the oxygen partial pressure (pO2) during the calcination of electrospun Cu and Sn ions-incorporated nanofibers (NFs). This resulted in the formation of CuO–SnO2 NFs (full oxidation), Cu–SnO2 NFs (selective reduction), Cu3Sn/CNFs, Cu41Sn11/CNFs, and Cu6Sn5/CNFs (full reduction). In the CO2RR, CuO–SnO2 NFs exhibited formate (HCOO−) production and Cu–SnO2 NFs showed carbon monoxide (CO) production with the faradaic efficiency (FE) of 65.3% at −0.99 V (vs. RHE) and 59.1% at −0.89 V (vs. RHE) respectively. Cu-rich Cu41Sn11/CNFs and Cu3Sn/CNFs enhanced the methane (CH4) production with the FE of 39.1% at −1.36 V (vs. RHE) and 34.7% at −1.50 V (vs. RHE). However, Sn-rich Cu6Sn5/CNFs produced HCOO− with the FE of 58.6% at −2.31 V (vs. RHE). This study suggests the methodology for bimetallic catalyst design and steering the CO2RR pathway by controlling the active sites of Cu–Sn alloys.
New conceptsIn general, the phase control of Cu–Sn bimetallic catalysts was focused on controlling the composition ratio of Cu and Sn during the fabrication process. This resulted in the limited phase range of the Cu–Sn bimetallic catalyst and CO2 reduction reaction (CO2RR) products. In this work, we developed a methodology for phase control of Cu–Sn bimetallic catalysts based on thermodynamic principles. We applied thermodynamic principles for the fabrication of a nanoscale Cu–Sn alloy with controlled bimetallic phases. We made a thermodynamic prediction of the bimetallic catalyst by modulating the oxygen partial pressure (pO2) during calcination; full oxidation, selective reduction, and full reduction of the Cu–Sn–C-based ternary elemental system. Starting from the thermodynamic understanding about the redox of Cu–Sn–C and the formation conditions of Cu–Sn compounds, we discovered the specific processing window for Cu–Sn bimetallic phase control. This enabled the predictive synthesis of phase-controlled nanomaterials for selective CO2 conversion. |
Introduction
Renewable electricity-powered carbon dioxide (CO2) reduction reaction (CO2RR) is receiving great attention as a promising avenue of carbon capture and utilization (CCU) for carbon neutrality and net zero emission.1 CO2RR enables addressing the fossil fuel-derived energy and environmental issues by converting atmospheric CO2 into value-added chemicals such as carbon monoxide (CO), formate (HCOO−), methane (CH4), ethylene (C2H4), and ethanol (C2H5OH). For the selective conversion of CO2 into specific chemical and to suppress the hydrogen evolution reaction (HER), which produces hydrogen (H2), heterogeneous CO2RR electrocatalysts are essential to steer the reaction pathway by controlling the binding of intermediates over the scaling relationship.2,3CO2RR product selectivities of heterogeneous catalysts can be controlled by introduction of secondary metals.4,5 Compared to bare Cu catalysts, heterogeneous Cu alloy catalysts enable more efficient binding energy control of CO2RR intermediates by tuning the d-band center.6 Among them, bimetallic Cu–Sn alloy catalysts are effective to control the CO2RR pathway because of miscibility between Cu and Sn (activity coefficient (γ) < 1) and Sn has an effect of preventing HER.7,8 Therefore, bimetallic Cu–Sn catalysts can regulate the binding energy of CO2RR intermediates and change the electronic structure of the catalysts for superior activity and selectivity for specific products.9 The primary studies which mainly control the Cu–Sn composition ratio resulted in the limited phase control.6,10–12 These phenomena have been observed for various fabrication methods such as electrochemical deposition13 and thermal annealing (Cu3Sn, Cu6Sn5).14 Their CO2RR products were limited to CO and HCOO−.6,15,16
To intensify the synergistic effect of Cu–Sn-based CO2RR catalysts, it is important to extend the range of controllable Cu–Sn bimetallic phases.17 We applied electrospun nanofibers (NFs), which show one-dimensional (1D) nanostructures with high aspect ratio,6,18–21 as electrocatalysts with active materials and supports.22 After electrospinning to form metal ions-incorporated polymer NFs, Cu–Sn bimetallic catalysts can be produced by calcination. CNFs can act as supports to control the phase and structure of the Cu–Sn alloy active materials during calcination.23
Here, we developed a thermodynamic-based catalysts fabrication methodology to control the phase and structures of Cu–Sn bimetallic catalysts toward selective CO2RR. We categorized the redox of Cu–Sn ions during calcination in terms of full oxidation, selective reduction, and full reduction in an Ellingham diagram, which presents the information about oxidation and reduction of elements according to oxygen partial pressure (pO2) and temperature. The binary phase diagram provides guidelines for the formation of Cu–Sn intermetallic compounds in full reduction. From these two diagrams, we designed various phases of bimetallic catalysts such as metal oxide–metal oxide, metal oxide–metal, metal–metal, and alloys.
Electrospun Cu, Sn ions-incorporated polymer NFs were calcinated in three different pO2 regions to control the oxidation state of Cu–Sn bimetallic catalysts. This resulted in the formation of CuO–SnO2, Cu–SnO2 phases in full oxidation and selective reduction regions. Especially, in the full reduction, we modulated the molar ratio of Cu and Sn to control the phase of Cu–Sn alloys. This resulted in the formation of stoichiometry-controlled Cu–Sn alloys (intermetallic compounds) such as Cu3Sn, Cu41Sn11, and Cu6Sn5 in CNFs (carbon nanofibers). We investigated how the phase control of active sites can steer the CO2RR pathways, related by the material–property relationship. We showcased that CO2RR products can be modulated as CO, HCOO−, and CH4 according to the phases of Cu–Sn bimetallic catalysts.
Results and discussion
Thermodynamic strategies for Cu–Sn bimetallic catalysts formation
For the fabrication of Cu–Sn bimetallic catalysts, we utilized CNFs as a matrix to control the kinetics of Cu–Sn alloy formation during nucleation and growth in the calcination process. We fabricated Cu–Sn bimetallic catalysts by the stepwise electrospinning of Cu, Sn precursors + polymers and the calcination of electrospun NFs. We designed the processing window for Cu–Sn bimetallic catalysts fabrication by the thermochemical computation with FactsageTM program (Fig. 1). | ||
| Fig. 1 Thermodynamic based Cu–Sn bimetallic catalyst design principles. (a) Ellingham diagram to design the Cu–Sn bimetallic catalyst according to the pO2 at 700 °C. The Ellingham diagram shows the Gibbs free energies of oxides for Cu, Sn, and C. (b) Cu–Sn binary phase diagram to predict the Cu–Sn intermetallic compounds. Schematics of (c) Our thermodynamic strategy to fabricate the Cu–Sn bimetallic catalysts by using electrospinning and calcination process in a controlled environment. | ||
Ellingham diagram displays the standard Gibbs free energy of formation (ΔG°) for the metal and carbon (C) oxides according to the calcination temperature (Fig. 1a). Although Sn exhibits higher oxidation tendency than that of Cu, there exists a crossing point of ΔG° between Sn and C oxidation at the temperature of 625 °C. When the calcination temperature exceeds the of 625 °C, Sn shows lower oxidation tendency than that of C. Based on this different oxidation tendencies of Cu, Sn, and C, we set the calcination temperature of Cu, Sn precursors + polymer NFs as 700 °C for the relatively higher oxidation tendency of C than other two metals (2Cu + O2 → CuO: −137.3 kJ mol−1, 4Cu + O2 → 2Cu2O: −194.8 kJ mol−1, Sn + O2 → SnO2: −379.2 kJ mol−1, 2C + O2 → 2CO: −395.7 kJ mol−1).24
The ΔG° in the Ellingham diagram is described as a function of ΔG° = RT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lnpO2, where the R, T, and pO2 stand for gas constant, temperature, and oxygen partial pressure. In the Cu–Sn bimetallic catalyst design system, we can control the Cu–Sn bimetallic phases in terms of full oxidation, selective reduction, and full reduction by controlling the pO2. In the condition of ΔG° > −137.3 kJ mol−1, Cu, Sn, C are oxidized, and Cu and Sn form metal oxides (full oxidation, FO). In the condition of −379.2 kJ mol−1 < ΔG° <−194.8 kJ mol−1 (selective reduction, SR), Sn and C are oxidized, and Cu is reduced to metallic state. When the ΔG° is lower than −395.7 kJ mol−1 (full reduction, FR), both Cu, Sn, and C are reduced, forming intermetallic compounds (Cu, Sn alloys) within CNFs.
lnpO2, where the R, T, and pO2 stand for gas constant, temperature, and oxygen partial pressure. In the Cu–Sn bimetallic catalyst design system, we can control the Cu–Sn bimetallic phases in terms of full oxidation, selective reduction, and full reduction by controlling the pO2. In the condition of ΔG° > −137.3 kJ mol−1, Cu, Sn, C are oxidized, and Cu and Sn form metal oxides (full oxidation, FO). In the condition of −379.2 kJ mol−1 < ΔG° <−194.8 kJ mol−1 (selective reduction, SR), Sn and C are oxidized, and Cu is reduced to metallic state. When the ΔG° is lower than −395.7 kJ mol−1 (full reduction, FR), both Cu, Sn, and C are reduced, forming intermetallic compounds (Cu, Sn alloys) within CNFs.
To investigate the formation trends of Cu–Sn intermetallic compounds according to Cu–Sn ratio and temperature, we investigated Cu–Sn binary phase diagram (Fig. 1b). In the phase diagram, we found that the intermetallic state of Cu6Sn5, Cu3Sn, and Cu41Sn11 can be induced due to the miscibility between Cu and Sn (activity coefficient (γ) < 1).8 Furthermore, we calculated the ΔG° of Cu–Sn alloys (Cu41Sn11, Cu3Sn, Cu10Sn3, Cu6Sn, Cu12Sn10) according to temperature. (Fig. S1, ESI†). At the temperature of 700 °C, we found that the stoichiometry of Cu–Sn alloys can be diversified according to their ΔG°.
Fig. 1c displays our strategy to fabricate the Cu–Sn bimetallic catalysts based on the Ellingham diagram and binary phase diagram. The fabrication of NFs and calcination process in controlled environment were performed to synthesize the Cu–Sn bimetallic catalysts. We utilized uniaxial electrospinning and coaxial electrospinning to fabricate the Cu, Sn ions-incorporated polymer NFs, used as a matrix to control the structures of the Cu–Sn bimetallic catalysts. We fabricated three types of electrospun NFs by utilizing (1) polyacrylonitrile (PAN) as polymer for uniaxial electrospinning (PAN + Cu precursor + Sn precursor, CuSn uniaxial NFs), (2) PAN and poly(styrene-co-acrylonitrile) (SAN) with coaxial electrospinning (core: SAN + Sn precursor, shell: PAN + Cu precursor, Cu shell Sn core NFs), and (3) PAN and SAN with coaxial electrospinning (core: SAN + Cu precursor, shell: PAN + Sn precursor, Sn shell Cu core NFs). The molar ratio of Cu precursor and Sn precursor (1:1) are same in three types of NFs solution. In the coaxial electrospinning, the press rate of shell and core in syringe pumps were 0.5 mL h−1 and 0.25 mL h−1 respectively. Therefore, as-spun NFs with coaxial electrospinning have 2:1 (shell:core) ratio of metal precursor due to the different press speed of core and shell solution, the ratio of Cu precursor:Sn precursor is 1:1, 2:1, and 1:2 in CuSn uniaxial NFs, Cu shell Sn core NFs, and Sn shell Cu core NFs respectively.
The X-ray photoelectron spectroscopy (XPS) analysis was conducted to identify the chemical state of as-spun NFs (Fig. S2, ESI†). The three types of as-spun NFs were composed of Cu2+, Sn2+, and Sn4+ without metallic Cu and Sn. Electrospun CuSn uniaxial NFs were calcined with pO2 controlled environment to modulate the oxidation states of CuSn bimetallic catalysts. Furthermore, we investigated the calcined Cu shell Sn core NFs and Sn shell Cu core NFs, fabricated by the calcination under full reduction to control the Cu–Sn alloy phases.
The CuSn uniaxial NFs were calcined in full oxidation, selective reduction, and full reduction to modulate the Cu–Sn phase. Cu shell Sn core NFs and Sn shell Cu core NFs were calcined in full reduction region to modulate the intermetallic compounds formation in Cu–Sn alloy. In the calcination under full oxidation, Cu oxide–Sn oxide NFs can be formed by the full oxidation of Cu precursor, Sn precursor, and polymer NFs. In the calcination under selective reduction, reduced Cu–Sn oxide NFs can be formed by the oxidation of Sn precursors and polymer NFs and the reduction of Cu precursors. In the calcination under full reduction, Cu–Sn alloys/CNFs can be formed.
Phase evolution and chemical states of Cu–Sn bimetallic catalysts
The phases of Cu–Sn bimetallic catalysts can be affected by the thermochemical environment during calcination.25 The phase and crystallinity of the Cu–Sn bimetallic catalysts were analyzed by X-ray diffraction (XRD) (Fig. 2a). They were determined by comparing with the XRD references of Cu, Sn oxides, metallic Cu, Sn, and Cu–Sn alloys (Fig. S3, ESI†). When we calcined the CuSn uniaxial NFs under full oxidation, CuO–SnO2 NFs were formed by the oxidation of Cu and Sn ions. When we calcined the CuSn uniaxial NFs under selective reduction, Cu–SnO2 NFs were formed by the oxidation of Sn and reduction of Cu. When we calcined electrospun NFs under full reduction, Cu and Sn were not oxidized and three types of Cu–Sn alloys/CNFs were formed according to the Cu:Sn ratio controlled in uniaxial and coaxial electrospinning. Cu3Sn/CNFs were evolved from CuSn uniaxial NFs (Cu:Sn = 1:1), Cu41Sn11/CNFs were evolved from Cu shell Sn core CNFs (Cu:Sn = 2:1), and Cu6Sn5/CNFs were evolved from Sn shell Cu core NFs (Cu:Sn = 1:2).
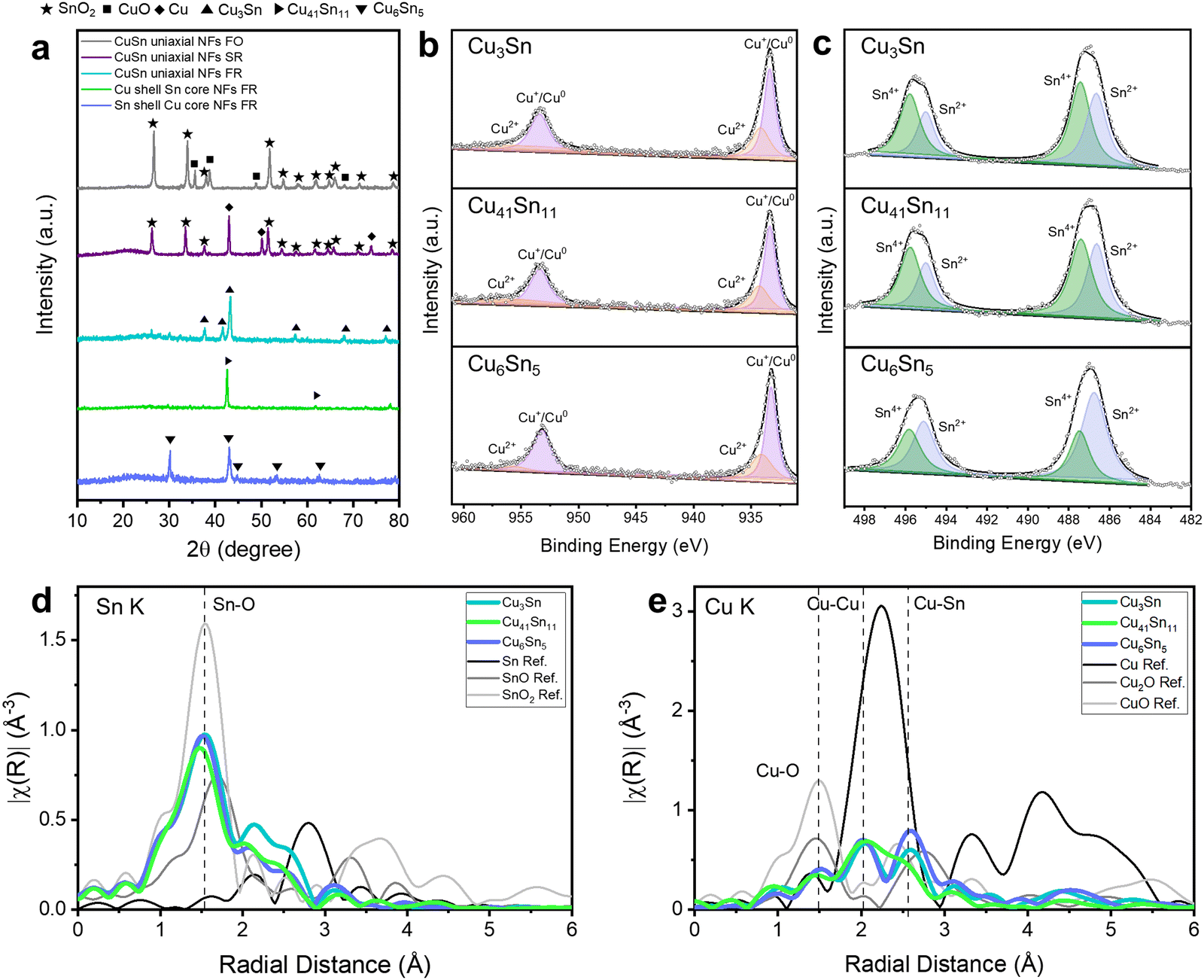 | ||
| Fig. 2 (a) X-ray diffraction (XRD) patterns of synthesized bimetallic Cu–Sn catalysts. XPS spectra for (b) Cu 2p, and (c) Sn 3d of bimetallic Cu–Sn alloys/CNFs to investigate the oxidation states of Cu and Sn in the nanofibers after calcination. The Ar etching was conducted before analysis. EXAFS of bimetallic Cu–Sn alloys/CNFs in terms of (d) Sn K edge and (e) Cu K edge to investigate the atomic coordinative structures according to the types of Cu–Sn alloys. | ||
To investigate the chemical states of Cu–Sn bimetallic catalysts, we analyzed the XPS of Cu 2p and Sn 3d after Ar etching to eliminate the native oxide generated from the air (Fig. 2b, c and Fig. S4, ESI†). The calcination of uniaxial NFs in full oxidation region exhibited the oxidation states of Cu and Sn as Cu2+, Sn2+, and Sn4+, Cu ions were converted to CuO and Sn ions were converted to SnO and SnO2. However, in the XRD pattern, only SnO2 crystalline structure was detected, implying that SnO2 was formed with a high crystallinity while SnO has low crystallinity. The calcination of CuSn uniaxial NFs in the selective reduction exhibited the oxidation states of Cu and Sn as Cu+/Cu0, Sn2+, and Sn4+. This reveals that Cu ions were converted to Cu or Cu2O without CuO and Sn ions were converted to SnO and SnO2. The calcination of uniaxial and coaxial NFs in full reduction exhibited the oxidation states of Cu and Sn as Cu+/Cu0, Cu2+, Sn2+, and Sn4+ due to the Cu–Sn alloy formation.26
To verify more accurate stoichiometry of Cu–Sn alloy/CNFs, inductively coupled plasma optical emission spectroscopy (ICP-OES) analysis was performed (Table S1, ESI†). The Cu–Sn ratios of Cu3Sn/CNFs, Cu41Sn11/CNFs, and Cu6Sn5/CNFs were Cu1.999Sn (Cu2Sn), Cu4.262 Sn (Cu4.3Sn), and Cu1.408Sn (Cu7Sn5) respectively. These similar stoichiometries of XRD pattern and ICP-OES data imply that the phase of Cu–Sn alloys consists of single alloy phase.
Additionally, we conducted XRD and XPS analysis after CO2RR to investigate the phase transformation during CO2RR process (Fig. S5–S8, ESI†). In the CuO–SnO2 NFs and Cu–SnO2 NFs, XRD shows that CuO–SnO2 and Cu–SnO2 were reduced to metallic Cu and Sn after CO2RR (Fig. S7, ESI†). The XPS data of Cu 2p represents that the CuO–SnO2 NFs and Cu–SnO2 NFs were reduced to Cu+/Cu0 after CO2RR. At the Sn 3d, Sn0 state evolved by reduction potential both in CuO–SnO2 NFs and Cu–SnO2 NFs after CO2RR (Fig. S5, ESI†). Combined with these XRD and XPS investigations, we expect that Cu–Sn oxides NFs were reduced to metallic states due to the reduction potential during CO2RR.
In Cu–Sn alloys/CNFs, XRD shows that crystalline structures of Cu–Sn alloys/CNFs were not changed after CO2RR (Fig. S8, ESI†). XPS analysis of Cu 2p and Sn 3d represents that the chemical states of Cu–Sn alloys/CNFs were maintained despite the condition for applying reduction potential during CO2RR. Therefore, the intermetallic structure of Cu–Sn alloys/CNFs was not transformed to another alloy phase or divided metallic phase during the CO2RR process, because of the miscibility between Cu and Sn, Cu–Sn alloys continuously maintain the thermodynamically stable intermetallic phase despite the reduction potential.
We further investigated the Sn K edge and Cu K edge X-ray absorption spectroscopy (XAS) of Cu–Sn alloy/CNFs to investigate their atomic structures and oxidation states. In the X-ray absorption near-edge structure (XANES) spectrum, we confirmed the oxidation states of Sn as Sn4+ and Cu as Cu/Cu+ by comparing the Sn K edge XANES of SnO2 and Cu K edge of Cu2O and Cu (Fig. S9, ESI†). Cu3Sn, Cu41Sn11, and Cu6Sn5 showed the identical oxidation states of Sn and Cu, similar with the results of XPS.
Extended X-ray absorption fine structure spectroscopy (EXAFS) provides the information about atomic coordinates such as coordination number and atomic distance. In the Sn K edge EXAFS, we found that major atomic coordinates are Sn–O bonding for all Cu3Sn, Cu41Sn11, and Cu6Sn5 (Fig. 2d). This might be related with the existence of Sn4+ oxidation states. Interestingly, Cu K edge EXAFS of Cu–Sn alloys/CNFs revealed the atomic bonding of Cu–O, Cu–Cu, and Cu–Sn (Fig. 2e). The highest Cu–Sn coordination number in Cu K edge EXAFS was shown in Cu6Sn5 and it became lower in Cu-rich Cu–Sn alloys such as Cu3Sn and Cu41Sn11. Furthermore, the absence of Sn–Sn peaks after the first shell Sn–O bond peak indicates the atomic distribution of Sn atoms in Cu. The Cu K-edge data analysis included two paths, Cu–Cu and Cu–Sn. (Fig. 2e) These results demonstrate that Cu–Sn alloys are in the states of Cu–Sn bonding.27
We also investigated the effect of forming three different Cu–Sn alloy phases on the CNFs using Raman spectroscopy, which provides the information about the C crystallinity of CNFs (Fig. S10, ESI†).28 We investigated the peak intensity ratio (ID/IG) of C in Cu–Sn alloys/CNFs.24 They all showed ID/IG values of 2.71 for Cu3Sn/CNFs, 2.79 for Cu41Sn11/CNFs, and 2.67 for Cu6Sn5/CNFs. This result indicates similar crystallinity of C in Cu–Sn/CNFs.
Structure investigation of Cu–Sn bimetallic catalysts
The morphologies of the calcined Cu, Sn precursor + polymer NFs were analyzed by scanning electron microscope (SEM) and transmission electron microscope (TEM) (Fig. 3 and Fig. S11, ESI†). When the electrospun NFs calcined under full oxidation and selective reduction region, XRD reveals the formation of CuO + SnO2 and Cu + SnO2 phases (Fig. 2a). SEM images of CuO−SnO2 NFs and Cu–SnO2 NFs exhibited that the NFs are composed of metal oxide or metal grains with hollow structures (Fig. S11, ESI†). There was no existence of C in NFs because of the combustion during the calcination of electrospun NFs by full oxidation and selective reduction. | ||
| Fig. 3 Structure investigation of bimetallic Cu–Sn alloys/CNFs using SEM, TEM, STEM-EDS, and HRTEM images for (a)–(d) Cu3Sn/CNFs, (e)–(h) Cu-rich Cu41Sn11/CNFs, and (i)–(l) Sn-rich Cu6Sn5/CNFs. In the STEM-EDS, red, green, and blue color mappings indicate the distribution of Cu, Sn, and C, respectively. | ||
Fig. 3a–d show the structures of Cu3Sn/CNFs fabricated by the high vacuum calcination of electrospun Cu, Sn uniaxial NFs. SEM and TEM images show that Cu–Sn alloy nanoparticles are formed outside of the CNFs. TEM energy dispersive spectroscopy (TEM EDS) confirms that the precipitated nanoparticles were composed of a distribution of Cu and Sn atoms, forming an alloy (Fig. 3c). High-resolution TEM (HRTEM) image (Fig. 3d) and selected area electron diffraction (SAED) patterns (Fig. S12, ESI†) of Cu–Sn alloy exhibit the lattice spacings of 0.238, 0.216, and 0.208 nm, corresponding to the (100), (002), and (101) planes of Cu3Sn.
Fig. 3e–h display the structures of Cu41Sn11/CNFs fabricated by the calcination of electrospun Cu shell, Sn core coaxial NFs by full reduction. We observed the hollow-structured CNFs by the existence of SAN in the core of electrospun nanofibers which decomposes to gas products during calcination. In the electrospun NFs, PAN in the shell would undergo carbonization to form synthesis of hollow CNFs. We found the precipitated Cu–Sn alloy nanoparticles, formed outside of the hollow CNFs (Fig. 3e and f) The distribution of Cu, Sn, and C distributions in the nanoparticles and NFs were verified using TEM and EDS elemental mapping (Fig. 3g). In Fig. 3h, the lattice spacings of 0.212 nm correspond to the Cu41Sn11 (660) plane.
Fig. 3i–l reveal the structures of Cu6Sn5/CNFs fabricated by the high vacuum calcination of electrospun Sn shell, Cu core coaxial NFs. SEM and TEM images and EDS elemental mapping show that Cu6Sn5 nanoparticles are located at the surface of hollow CNFs, which shows the similar structures with that of Cu41Sn11 (Fig. 3i–k). The HRTEM image in Fig. 3l shows (![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 13) and (132) planes of Cu6Sn5 with a lattice spacing of 0.297 nm and 0.210 nm, respectively.
13) and (132) planes of Cu6Sn5 with a lattice spacing of 0.297 nm and 0.210 nm, respectively.
Effect of Cu–Sn phase control on CO2RR
CO2RR performances of Cu–Sn alloys/CNFs were evaluated using a flow cell with the electrolyte of 1 M KOH (Fig. S13, ESI†). Fig. 4 and Table S3–S7 (ESI†) show the CO2RR product distributions of CuO–SnO2 NFs, Cu–SnO2 NFs, Cu3Sn/CNFs, Cu41Sn11/CNFs, and Cu6Sn5/CNFs according to the potentials. In the CO2RR of CuO–SnO2 NFs, CO and HCOO− were main products with FEs of 26.9% and 59.7% at the −0.54 V (vs. RHE). Despite the potential increased to −0.99 V (vs. RHE), the FEs of CO and HCOO− maintained with the FE of 26.9% and 65.3% respectively (Fig. 4a). In the CO2RR of Cu–SnO2 NFs, CO and HCOO− were the main products with FE of 24.9% and 66.9% at the −0.58 V (vs. RHE) (Fig. 4b). As the potential increases to −0.89 V (vs. RHE), the FE of CO increased to 59.1% and the FE of HCOO− decreased to 37.3%. Interestingly, CuO–SnO2 NFs and Cu–SnO2 NFs exhibit similar selectivity at low potential, −0.54 V and −0.58 V (vs. RHE), while, as the potential increases, CuO–SnO2 NFs maintains the HCOO−/CO FE ratio over 1.8, whereas Cu–SnO2 NFs gradually decreases the HCOO−/CO FE to 0.63, despite the same phases as Cu and Sn under the CO2RR process (Fig. 4c). XRD patterns after CO2RR show that Cu in CuO–SnO2 derived Cu–Sn exhibits lower intensity than Cu–SnO2 derived Cu–Sn (Fig. S7b, ESI†). This result implies that CuO–SnO2 derived Cu–Sn composed of high ratio of Sn–Sn phase and shows high HCOO− selectivity. In contrast, Cu–SnO2 derived Cu–Sn composed of high ratio of Cu–Cu phase and exhibits high CO selectivity.29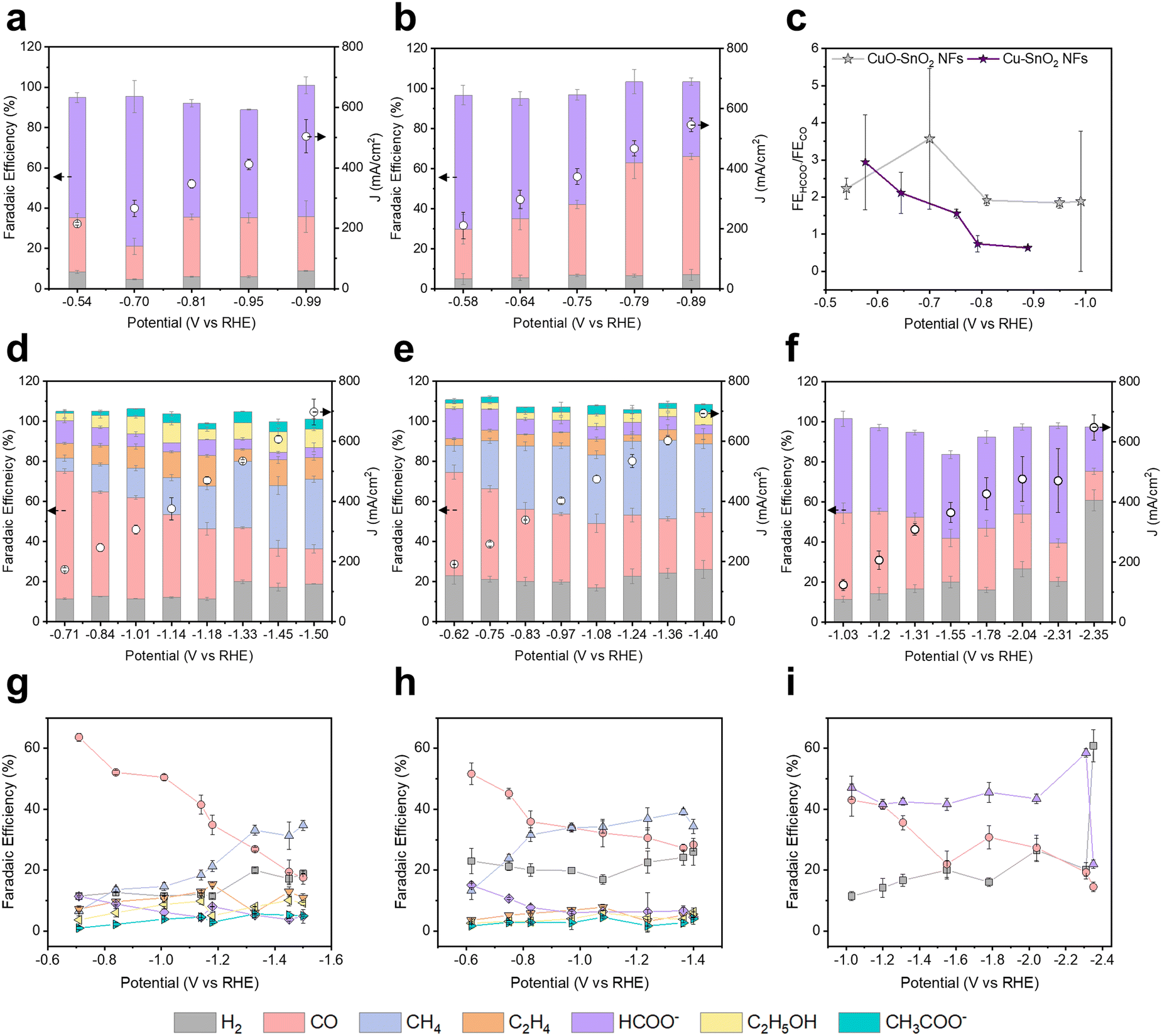 | ||
| Fig. 4 Comparison of electrochemical CO2RR performances in terms of product distribution according to the potential. (a) CuO–SnO2 NFs, (b) Cu–SnO2 NFs, (c) FE ratio of HCOO− and CO in CuO–SnO2 NFs and Cu–SnO2 NFs. (d) and (g) Cu3Sn/CNFs, (e) and (h) Cu41Sn11/CNFs, and (f) and (i) Cu6Sn5/CNFs. CO2RR of each catalyst was evaluated in the flow cell with the electrolyte of 1 M KOH. | ||
In the CO2RR of Cu3Sn/CNFs, CO was the main product with the FE of 63.7% at the −0.71 V (vs. RHE) (Fig. 4d and g). As the potential increased to −1.50 V (vs. RHE), the FE of CO decreased to 17.6%. On the contrary, we found the gradual increase in the CH4 selectivity from 6.57% to 34.7% as the potential increased −0.71 V to −1.50 V (vs. RHE) due to the *CO protonation.30,31 This result indicates that the main CO2RR product is the CH4 in Cu3Sn alloy/CNFs.
In the CO2RR of Cu41Sn11/CNFs, which have a higher Cu ratio in Cu–Sn alloy than that of Cu3Sn/CNFs, we found similar trends of CO2RR products from CO to CH4 according to the potential (Fig. 4e and h). At the potential of −0.62 V (vs. RHE), CO FE was 51.7% and CH4 FE was 13.3%. As the potential increased, we found the similar trend with that of Cu3Sn/CNFs, where the FE of CO decreased and the FE of CH4 increased with *CO protonation. When the potential increased to −1.36 V (vs. RHE), the FE of CO was 27.3%. Interestingly, the FE of CH4 was increased from 13.3% to 39.1%. However, at −1.40 V (vs. RHE) the CH4 selectivity decreased to 34.4% due to the degradation of the catalyst at high over potential. Compared to the CH4 production of Cu3Sn/CNFs (FE of 6.57% and 34.7% at −0.71 V and −1.50 V (vs. RHE)) and Cu41Sn11/CNFs (FE of 13.3% and 39.1% at −0.62 V and −1.36 V (vs. RHE)), at the low potential range, Cu41Sn11 exhibit high CH4 selectivity compared to Cu3Sn and at the high potential range, similar selectivity was shown between Cu3Sn and Cu41Sn11.
When the Sn ratio increased from Cu3Sn to Cu6Sn5 (Cu6Sn5/CNFs), the major CO2RR product was changed from CH4 to HCOO− (Fig. 4f and i). At the −1.03 V (vs. RHE), we found the production of HCOO−, CO, and H2 with the FE of 47.1%, 43.0%, and 11.4%. CO FE decrease accordance with the potential increase. However, the major product was HCOO− in the CO2RR of Cu6Sn5/CNFs. This resulted in the promoted HCOO− production with the FE of 41.7% and 58.6% at the potentials of −1.55 V and −2.31 V (vs. RHE) respectively. When the potential exceeds −2.31 V (vs. RHE), we observed the increased H2 FE and decreased HCOO− FE. This reveals the tunable CO2RR selectivity by modulating the Cu–Sn bimetallic catalyst phases based on the Ellingham diagram and binary phase diagram. To confirm the stability of Cu–Sn bimetallic catalyst in the electrochemical CO2RR process, we conducted a long-term stability with 1 M KOH electrolyte in a flow cell (Fig. S14, ESI†).
Theoretical understanding of steering CO2RR pathway by Cu–Sn alloy
We observed that either CO or CH4 was the main product of the Cu41Sn11 and Cu3Sn catalysts and HCOO− was the main product of the Cu6Sn5 catalyst. To understand the difference in product selectivity between Cu-rich and Sn-rich Cu–Sn alloy catalysts, we conducted density functional theory (DFT) calculations of the free energy changes in CO2RR to *CO and *HCOOH on the surface of Cu41Sn11 and Cu6Sn5 (Fig. 5a–d). The facet of each catalyst to model was determined based on the HRTEM and XRD analysis results. The XRD pattern for Cu41Sn11 catalyst showed one major peak corresponding to (660) plane and the XRD pattern for Cu6Sn5 catalyst shows two major peaks corresponding to the (132) and (13) plane (Fig. 2a and 3h, l). From these results, the (660) plane of Cu41Sn11 and the (132) and (13) planes of Cu6Sn5 were modeled (Fig. S15, ESI†). In the unit cell for DFT calculation, several water molecules and the K atom were included to incorporate solvation effect32 and simulate the charged surface of the catalyst.33,34
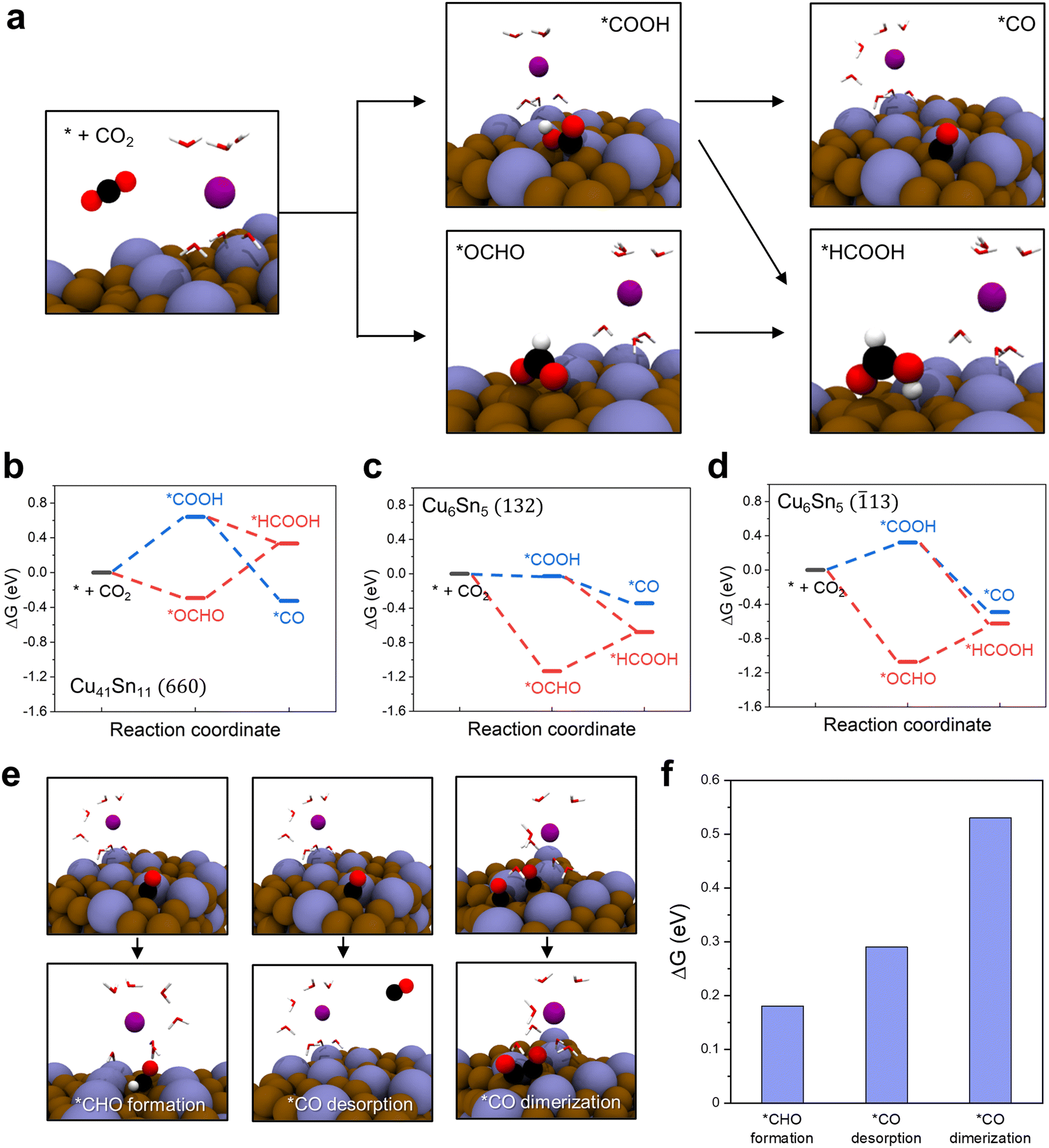 | ||
| Fig. 5 DFT calculation of CO2RR pathways. (a) CO2RR pathways to *CO and *HCOOH on Cu41Sn11 (660) surface. Energy diagram of CO2RR to *CO (blue path) and *HCOOH (red path) on (b) Cu41Sn11 (660), (c) Cu6Sn5 (132), and (d) Cu6Sn5 (13) surfaces. (e) and (f) Atomic structure and energy changes for possible reactions after *CO formation on Cu41Sn11 (660) surface. Brown, blue, black, red, white, and purple spheres represent Cu, Sn, C, O, H, and K atoms, respectively. H2O molecules are shown as lines for brevity. | ||
The reaction pathways of CO2RR to *CO and to *HCOOH diverge at the early stage of CO2RR. When the CO2 is adsorbed on the catalyst surface, CO2 can be reduced to *COOH or *OCHO. Then *COOH is reduced to *CO or *HCOOH and *OCHO is reduced to *HCOOH.35Fig. 5a shows the atomic structure changes in these processes on the Cu41Sn11 (660) surface. On the other two surfaces, Cu6Sn5 (132) and Cu6Sn5 (13), the same process was carried out (Fig. S16, ESI†). The free energies of each step in the reaction pathways on the Cu41Sn11 (660), Cu6Sn5 (132), and Cu6Sn5 (13) surfaces were calculated (Fig. 5b–d). On the Cu41Sn11 (660) surface, *CO has lower energy by 0.66 eV than *HCOOH, whereas *HCOOH is more stable in energy at 0.34 eV and 0.13 eV than *CO on the Cu6Sn5 (132) and Cu6Sn5 (13) surfaces. This means that *CO formation is more favorable than *HCOOH formation on the Cu41Sn11 (660) surface and *HCOOH formation is more favorable than *CO formation on the Cu6Sn5 (132) and Cu6Sn5 (13) surfaces. This may explain the preference of the Cu6Sn5 catalyst for HCOO− production. However, this result is not sufficient to explain the preference of the Cu41Sn11 catalysts for CO or CH4 products because *CO can be converted into various products through diverse reaction pathways.
To explain the preference for CO or CH4 products of the Cu41Sn11 catalyst, we calculated free energy changes in possible reactions after *CO formation on the Cu41Sn11 (660) surface (Fig. 5e). The reactions shown in Fig. 5e are *CHO formation for CH4 production (left), *CO desorption for CO production (middle), and *CO dimerization for the production of other hydrocarbons containing two or more carbons (right). The free energy change of *CHO formation and *CO desorption is 0.18 eV and 0.29 eV, lower than that of *CO dimerization at 0.53 eV (Fig. 5f). From this result, the preference of the Cu41Sn11 catalyst for CO or CH4 products over other products can be explained. Therefore, the high CO or CH4 product selectivity of Cu-rich Cu–Sn alloy catalysts and the high HCOO− product selectivity of Sn-rich Cu–Sn alloy catalysts were elucidated by DFT calculations.
Conclusion
We studied the thermodynamic based phase control of Cu–Sn alloy catalysts for the efficient steering of CO2RR pathway toward selective CO2 conversion. We designed the processing window to modulate the Cu–Sn bimetallic phases by designing the electrospinning (uniaxial and coaxial) and calcination (full oxidation, selective reduction, and full reduction). CuO–SnO2 NFs, and Cu–SnO2 NFs were fabricated by the calcination of CuSn uniaxial NFs at full oxidation and selective reduction. In the CuSn coaxial NFs, Cu–Sn alloy phase was controlled by modulating the Cu and Sn precursor ratio based on the Cu–Sn binary phase diagram. This was enabled by the calcination at full reduction of CuSn uniaxial NFs (Cu:Sn = 1:1), Cu shell Sn core NFs (Cu:Sn = 2:1), and Sn shell Cu core NFs (Cu:Sn = 1:2). CuSn uniaxial NFs, Cu shell Sn core NFs, and Sn shell Cu core NFs calcined under full reduction formed Cu3Sn/CNFs, Cu41Sn11/CNFs, and Cu6Sn5/CNFs respectively. CuO–SnO2 NFs exhibited high HCOO− selectivity at high potential (FE of 65.3% at −0.99 V vs. (RHE)), whereas Cu–SnO2 NFs showed high CO selectivity at high potential (FE of 59.1% at −0.89 V vs. (RHE)). The CO2RR of Cu–Sn alloy/CNFs exhibited the CH4 formation in Cu rich Cu3Sn/CNFs and Cu41Sn11/CNFs, while Sn rich Cu6Sn5 showed the HCOO− formation. Especially in the low potential range, Cu41Sn11 shows high CH4 selectivity compared to Cu3Sn (FE of 13.3% at −0.62 V (vs. RHE) and FE of 6.57% at −0.71 V (vs. RHE)). On the other hand, at the high potential range, Cu41Sn11 and Cu3Sn exhibit similarly high CH4 selectivity (FE of 39.1% at −1.36 V (vs. RHE) and FE of 34.7% at −1.50 V (vs. RHE)). Theoretical study about the effect of Cu–Sn active site design on CO2RR confirmed that the Cu41Sn11 and Cu6Sn5 can steer the CO2RR pathways between CH4 and HCOO−. We confirm that in a bimetallic system, the oxidation state of metals and alloy phases can modulate the CO2RR product selectivity. We expect that our methodology can provide an insight to control the phase of bimetallic and alloy catalysts elaborately and optimize their catalytic activity and selectivity.
Experimental
Thermodynamic calculations
The Ellingham diagram and phase diagram were established by a thermochemical database program (FactsageTM software). The FACT pure substances database (FactPS) and Fact light metal database (FTlite) were used for thermodynamic calculations of Cu, Sn and Cu–Sn intermetallic compounds. To predict Cu–Sn intermetallic compounds, a binary phase diagram was calculated in the phase diagram tab of program. For Ellingham diagram, the formation Gibbs free energies according to Sn partial pressure were calculated in the reaction tab of program.Fabrication of Cu–Sn bimetallic catalysts via electrospinning and calcination
For uniaxial electrospinning, the metal precursor and polymer solutions were prepared by dissolving (1) 3 mmol SnCl2 and 3 mmol Cu(CH3COO)2 (Cu(Ac)2) in 6 mL DMF, and (2) 1.2 g PAN in 10 mL DMF. They were stirred on a hot plate (120 °C) for 2 h. After dissolution of the solutes, the metal precursor solution and PAN solution were mixed by stirring at room temperature during overnight. For electrospinning, the solution was loaded into a 12 mL Luer lock syringe. The distance between the tip and collector was 15 cm. While applying 17 kV from a voltage source to the metal needle tip, the solution-loaded syringe was pressed by a syringe pump at a rate of 1 ml h−1.For coaxial electrospinning, the shell solution (Cu(Ac)2 or SnCl2) was prepared with 3 mmol metal precursor and 0.56 g PAN in 5 mL DMF. The core solution (SnCl2 or Cu(Ac)2) was prepared with 3 mmol metal precursor and 1.66 g SAN in 5 mL DMF. Each core and shell contained 3 mmol metal precursor (Cu(Ac)2 or SnCl2). The solution was electrospun with an the voltage of 20 kV, pressed by 0.5 mL h−1 and 0.25 mL h−1, and formed a fibrous mat on a collector of aluminum foil. The distance between the needle and collector was 15 cm.
After electrospinning, we conducted the calcination of the electrospun nanofibers at a temperature of 700 °C for 4 h, with a ramping rate of 4 °C min−1. Full oxidation region was built in the air atmosphere. Selective reduction region was built in co-flowing the O2 gas 5 sccm and Ar gas 20 sccm with rotary pump operation. Full reduction region was built in high vacuum condition modulated under pressure (1.0 × 10−5 torr) using a rotary pump and a turbo molecular pump. After calcination, cooling from 700 °C to room temperature took over 3 h.
Materials characterization
The microstructures of Cu–Sn alloys/CNFs were analyzed using a field emission scanning electron microscope (FE-SEM, S-4800, Hitachi) and a transmission electron microscope (TEM, Tecnai G2 F20 TWIN TMP, FEI) with an energy dispersive spectroscope (EDS). The phase of Cu–Sn alloys/CNFs was analyzed using an X-ray diffractometer (XRD, Miniflex 600 Mini, Horiba). The chemical states of the Cu–Sn alloys/CNFs were investigated by XPS (ESCALAB 250Xi, Thermo Fisher Scientific). The quantitative analysis of Cu and Sn in Cu–Sn alloys/CNFs was analyzed by inductively coupled plasma optical emission spectroscopy (ICP-OES, Thermo Scientific/iCAP7400DUO). The XAS measurement was conducted at the 10C beamline of Pohang Accelerator Laboratory, Pohang, Republic of Korea.Electrochemical CO2RR performance measurement
The electrochemical CO2RR performances of Cu–Sn alloys/CNFs were measured in flow cell with the electrolyte of 1 M KOH. In the flow cell, catholyte and anolyte were circulated independently, and CO2 gas was supplied directly from the backside of the gas diffusion electrode (GDE) with a constant 50 sccm flow rate. The anolyte and catholyte were separated by the anion exchange membrane (AEM, Fumasep). Cathode for CO2RR was prepared by dispersing 2 ml of isopropanol (IPA, Sigma-Aldrich), 40 μl of Nafion binder (5 wt%, Ew 1000), and 20 mg of Cu–Sn catalyst by spray-coating the catalyst ink on the carbon paper-based gas diffusion layer (GDL, Carbon paper, Sigracet 39BB) with the amount of 1.25 mg cm−2. Ag/AgCl (3 M KCl) was used as a reference electrode and Ni foam was used as the counter electrode for OER. Ag/AgCl potential was transformed to a reversible hydrogen electrode (RHE) scale considering iR compensation (80%). The RHE transform formula is:| ERHE = EAg/Agcl + 0.197 + 0.059 × pH + 0.8 × iR |
The electrochemical CO2RR performance analysis was conducted during 1 h. The gas products of CO2RR were collected in a gas tight syringe and injected three times (5 min, 30 min, and 55 min) to gas chromatography (Micro GC Fusion, INFICON Inc.) for the analysis. The liquid products were obtained from catholyte after 1 h electrolysis and analyzed by nuclear magnetic resonance spectrometer (NMR, Bruker). A mixture of catholyte, D2O, and dimethyl sulfoxide (DMSO) as an internal standard was collected in an NMR tube. The FE for each product was calculated using the following equation:
| FE (%) = (z·n·F)/Q |
DFT calculation
DFT calculations were performed with the Vienna ab initio simulation package (VASP)36,37 using the generalized gradient approximation (GGA) of the Perdew–Burke–Ernzerhof (PBE) functional.38 We used the plane-wave basis with energy cut-off of 450 eV. The Brillouin zone was sampled using a 3 × 3 × 1 Monkhorst–Pack mesh. Structural optimization was performed until the force on each atom was less than 0.01 eV Å−1. The unit cells on the Cu41Sn11 (660), Cu6Sn5 (132), and Cu6Sn5 (13) surfaces contained a 15 Å-thick vacuum layer to avoid interaction with periodic images. To describe the van der Waals (vdW) interactions, the DFT-D3 method in Grimme's scheme was applied.39 For electrochemical reactions involving the proton-coupled electron transfer step, free energies of the reaction were calculated based on a computational hydrogen electrode model.40
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (NRF-2021R1C1C1013784 and 2021M3D1A2047041). Experiments at PLS-II were supported in part by MSIT and POSTECH.References
- B. Zhang, F. Kang, J.-M. Tarascon and J.-K. Kim, Prog. Mater. Sci., 2016, 76, 319–380 CrossRef CAS.
- N. Uddin, H. Zhang, Y. Du, G. Jia, S. Wang and Z. Yin, Adv. Mater., 2020, 32, 1905739 CrossRef CAS PubMed.
- S. Furukawa and T. Komatsu, ACS Catal., 2017, 7, 735–765 CrossRef CAS.
- S. Zhang, S. E. Saji, Z. Yin, H. Zhang, Y. Du and C.-H. Yan, Adv. Mater., 2021, 33, 2005988 CrossRef CAS PubMed.
- P. Strasser, S. Koh, T. Anniyev, J. Greeley, K. More, C. Yu, Z. Liu, S. Kaya, D. Nordlund, H. Ogasawara, M. F. Toney and A. Nilsson, Nat. Chem., 2010, 2, 454–460 CrossRef CAS PubMed.
- J. Wang, Y. Ji, Q. Shao, R. Yin, J. Guo, Y. Li and X. Huang, Nano Energy, 2019, 59, 138–145 CrossRef CAS.
- H. Li, X. Yue, Y. Qiu, Z. Xiao, X. Yu, C. Xue and J. Xiang, Mater. Today Energy, 2021, 21, 100797 CrossRef CAS.
- J.-Y. Kim, H. S. Ahn, I. Kim, D. Hong, T. Lee, J. Jo, H. Kim, M. K. Kwak, H. G. Kim, G. Kang, S. Go, W. H. Ryu, G.-D. Lee, M. Kim, D.-H. Nam, E. S. Park and Y.-C. Joo, Nat. Synth., 2024, 3, 452–465 CrossRef.
- D. Li, L. Huang, Y. Tian, T. Liu, L. Zhen and Y. Feng, Appl. Catal., B, 2021, 292, 120119 CrossRef CAS.
- Y. P. Zhu, T. Y. Ma, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2017, 56, 1324–1328 CrossRef CAS PubMed.
- Y. Wu, X. Liu, D. Han, X. Song, L. Shi, Y. Song, S. Niu, Y. Xie, J. Cai, S. Wu, J. Kang, J. Zhou, Z. Chen, X. Zheng, X. Xiao and G. Wang, Nat. Commun., 2018, 9, 1425 CrossRef PubMed.
- M. Luo, Y. Sun, X. Zhang, Y. Qin, M. Li, Y. Li, C. Li, Y. Yang, L. Wang, P. Gao, G. Lu and S. Guo, Adv. Mater., 2018, 30, 1705515 CrossRef PubMed.
- H. C. Shin, J. Dong and M. Liu, Adv. Mater., 2003, 15, 1610–1614 CrossRef CAS.
- J. Wang, J. Zou, X. Hu, S. Ning, X. Wang, X. Kang and S. Chen, J. Mater. Chem. A, 2019, 7, 27514–27521 RSC.
- B. Zhang, Y. Jiang, M. Gao, T. Ma, W. Sun and H. Pan, Nano Energy, 2021, 80, 105504 CrossRef CAS.
- Z.-Z. Wu, F.-Y. Gao and M.-R. Gao, Energy Environ. Sci., 2021, 14, 1121–1139 RSC.
- W. J. Dong, J. W. Lim, D. M. Hong, J. Y. Park, W. S. Cho, S. Baek, C. J. Yoo, W. Kim and J.-L. Lee, ACS Appl. Energy Mater., 2020, 3, 10568–10577 CrossRef CAS.
- L. Bu, S. Guo, X. Zhang, X. Shen, D. Su, G. Lu, X. Zhu, J. Yao, J. Guo and X. Huang, Nat. Commun., 2016, 7, 11850 CrossRef CAS PubMed.
- S. Bai, L. Bu, Q. Shao, X. Zhu and X. Huang, J. Am. Chem. Soc., 2018, 140, 8384–8387 CrossRef CAS PubMed.
- Y. Ge, X. Duan, M. Zhang, L. Mei, J. Hu, W. Hu and X. Duan, J. Am. Chem. Soc., 2018, 140, 193–199 CrossRef CAS PubMed.
- M. Li, Z. Zhao, T. Cheng, A. Fortunelli, C.-Y. Chen, R. Yu, Q. Zhang, L. Gu, B. V. Merinov, Z. Lin, E. Zhu, T. Yu, Q. Jia, J. Guo, L. Zhang, W. A. Goddard, Y. Huang and X. Duan, Science, 2016, 354, 1414–1419 CrossRef CAS PubMed.
- K. Huan, Y. Li, D. Deng, H. Wang, D. Wang, M. Li and L. Luo, Appl. Surf. Sci., 2022, 573, 151528 CrossRef CAS.
- H. Guan, Y. Zhao, J. Zhang, Y. Liu, S. Yuan and B. Zhang, Sens. Actuators, B, 2018, 261, 354–363 CrossRef CAS.
- D.-H. Nam, J. W. Kim, J.-H. Lee, S.-Y. Lee, H.-A. S. Shin, S.-H. Lee and Y.-C. Joo, J. Mater. Chem. A, 2015, 3, 11021–11030 RSC.
- S. Zeng, S. Shan, A. Lu, S. Wang, D. T. Caracciolo, R. J. Robinson, G. Shang, L. Xue, Y. Zhao, A. Zhang, Y. Liu, S. Liu, Z. Liu, F. Bai, J. Wu, H. Wang and C.-J. Zhong, Catal. Sci. Technol., 2021, 11, 5712–5733 RSC.
- M. Morimoto, Y. Takatsuji, R. Yamasaki, H. Hashimoto, I. Nakata, T. Sakakura and T. Haruyama, Electrocatalysis, 2018, 9, 323–332 CrossRef CAS.
- M. G. Kim, S. Sim and J. Cho, Adv. Mater., 2010, 22, 5154–5158 CrossRef CAS PubMed.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095–14107 CrossRef CAS.
- G. A. El-Nagar, F. Yang, S. Stojkovikj, S. Mebs, S. Gupta, I. Y. Ahmet, H. Dau and M. T. Mayer, ACS Catal., 2022, 12, 15576–15589 CrossRef CAS PubMed.
- R. Kortlever, J. Shen, K. J. P. Schouten, F. Calle-Vallejo and M. T. Koper, J. Phys. Chem. Lett., 2015, 6, 4073–4082 CrossRef CAS PubMed.
- H. Pan and C. J. Barile, Energy Environ. Sci., 2020, 13, 3567–3578 RSC.
- F. Calle-Vallejo, R. F. de Morais, F. Illas, D. Loffreda and P. Sautet, J. Phys. Chem. C, 2019, 123, 5578–5582 CrossRef CAS.
- E. Skúlason, G. S. Karlberg, J. Rossmeisl, T. Bligaard, J. Greeley, H. Jónsson and J. K. Nørskov, Phys. Chem. Chem. Phys., 2007, 9, 3241–3250 RSC.
- J. Rossmeisl, E. Skúlason, M. E. Björketun, V. Tripkovic and J. K. Nørskov, Chem. Phys. Lett., 2008, 466, 68–71 CrossRef CAS.
- L. Shang, X. Lv, L. Zhong, S. Li and G. Zheng, Small Methods, 2022, 6, 2101334 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nh00393d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |