
Bubbling resilient 3D free-standing nanoporous graphene with an encapsulated multicomponent nano-alloy for enhanced electrocatalysis†
Linshan
Zhu‡
a,
Qingqing
Li‡
ac,
Yixuan
Hu‡
b,
Xin
Wu
a,
Kolan Madhav
Reddy
 *b,
Kaikai
Li
*a,
Guoqiang
Xie
a,
Xingjun
Liu
a and
Hua-Jun
Qiu
*ad
*b,
Kaikai
Li
*a,
Guoqiang
Xie
a,
Xingjun
Liu
a and
Hua-Jun
Qiu
*ad
aSchool of Materials Science and Engineering, Harbin Institute of Technology (Shenzhen), Shenzhen, China. E-mail: qiuhuajun@hit.edu.cn; likaikai@hit.edu.cn
bFrontier Research Center for Materials Structure, School of Materials Science and Engineering, Shanghai Jiao Tong University, Shanghai 200240, China. E-mail: kmreddy@sjtu.edu.cn
cZhejiang Laboratory, Hangzhou 311100, China
dShenzhen Key Laboratory of Advanced Functional Carbon Materials Research and Comprehensive Application, Harbin Institute of Technology, Shenzhen, 518055, China
First published on 18th June 2024
Abstract
The design and synthesis of highly durable and active electrocatalysts are crucial for improving the hydrogen evolution reaction (HER) and oxygen reduction reaction (ORR) in proton exchange membrane fuel cells (PEMFCs). In this work, we present a novel dealloyed nanoporous PtCuNiCoMn multicomponent alloy with ligaments/pores ranging from 2–3 nm, which is in situ encapsulated in a three-dimensional, free-standing nanoporous nanotubular graphene network featuring a pore/tube diameter of ∼200 to 300 nm. This method allows precise control over the noble metal loading and alloy composition while preventing noble metal loss throughout the preparation process. The innovative bimodal nanoporous graphene/alloy structure, coupled with an open 3D spongy morphology, and optimized surface Pt electronic structure through multicomponent interaction, significantly enhances the activity for the HER/ORR, outperforming commercial Pt/C. Moreover, this design addresses the issues of Pt nanoparticle aggregation and detachment from carbon supports that typically exist in Pt/C-type catalysts, thereby substantially improving the catalytic durability, even under intense gas bubbling conditions.
New conceptsIn this work, we introduce a novel synthesis strategy for a unique type of nanoporous graphene/nanoalloy composite with nanoporous Pt-based multicomponent alloys in situ encapsulated in a freestanding 3D nanoporous nanotubular graphene network. By effectively bypassing the common problems of Pt NP aggregation and the loss of contact to the carbon support involved in the traditional Pt/C-type supported catalyst powders, this new type of freestanding 3D nanotubular graphene/nanoporous alloy composite electrocatalyst exhibits remarkably enhanced activity and especially durability for both the ORR and HER even under large current density/strong gas bubbling conditions when compared with Pt/C. Moreover, the developed strategy is scalable, highly controllable in terms of the Pt loading amount (by controlling the Pt content in the precursor alloy), completely avoids any waste of Pt during the whole synthesis process, and can be extended to other noble metal-based alloy systems. The unique bimodal 3D nanoporous structure of this freestanding nanocomposite makes it a promising candidate for applications in bubbling involved electrochemical processes. |
Proton exchange membrane fuel cells (PEMFCs) have attracted considerable research interests owing to their great potential in automobile and portable electronics.1,2 The development of PEMFCs, however, is hampered by challenges such as slow cathodic oxygen reduction kinetics and insufficient pure hydrogen production. Therefore, it is imperative to find effective catalysts for the oxygen reduction reaction (ORR) and hydrogen evolution reaction (HER) in PEMFCs.3–5 Although significant progress has been made for developing Pt-free catalysts, Pt-based electrocatalysts remain the most effective ones for the ORR and HER in acidic media.6,7 The current gold standard involves commercial Pt/C catalysts, where fine Pt nanoparticles (∼3 to 4 nm) are anchored on the carbon powders. However, these catalysts fall short in catalytic durability and the specific activity.8 Specifically, owing to the weak interaction between the Pt nanoparticles (NPs) and carbon supports alongside the carbon support corrosion during working conditions, Pt NPs usually undergo aggregation, Ostwald ripening as well as detachment from the carbon supports, which would induce enormous loss of the electrochemical active surface area and subsequent performance degradation.9–16 This issue is exacerbated under conditions with strong gas bubbling, such as in the HER.
To design electrocatalysts better than Pt/C-type supported catalysts,17–19 recently supportless Pt nanocatalysts are attracting much attention due to their interesting properties, such as enhanced electrochemical durability.20–22 For example, Jin et al.20 reported anisotropic mesoporous Pt@Pt-skin Pt3Ni core–shell framework nanowires for efficient electrocatalysis. Sievers et al.21 prepared a support-free nanostructured Pt–Cu electrocatalyst by alternating sputtering, which exhibits a higher ORR activity compared to supported nanoparticles. However, the decline of performance during long-term potential cycling is still obvious.22–24 Thus, it is highly important and still challenging to develop new electrocatalysts with greatly enhanced durability and catalytic activity for practical applications.
Alloying Pt with transition metals has proven effective for enhancing the inherent catalytic activity of Pt-based catalysts by tuning the electronic structure and leveraging the ligand/strain effects.25–34 Recent studies have also shown that the inherent activity and catalytic durability could be further improved by fabricating Pt-based intermetallic nanostructures or designing multicomponent high-entropy nanoalloys.35–47 In particular, the multicomponent alloy design allows theoretically numberless combinations to widely tune the surface electronic structure/strain for catalytic activity optimization.
In this study, we introduce a novel fabrication of a unique type of nanoporous Pt-based multicomponent alloy in situ encapsulated in a free-standing 3D nanoporous nanotubular graphene network. This innovative approach addresses the issues associated with Pt NP aggregation and loss of contact with the carbon support commonly found in the traditional Pt/C-type supported catalyst powders. Our free-standing 3D nanotubular graphene/nanoporous Pt-based alloy composite electrocatalyst demonstrates remarkably enhanced activity and especially durability for both the ORR and HER, even under conditions of large current density/strong gas bubbling when compared with traditional Pt/C catalysts. Furthermore, the developed strategy is scalable, allows precise control over Pt loading and completely avoids any waste of Pt during the whole fabrication process.
The fabrication process is schematically explained in Fig. 1a. Firstly, a nanoporous multicomponent alloy (MnNiCuCoPt) with nanopore sizes of ∼3 to 5 nm was obtained by dealloying a designed Mn75Ni10Cu10Co4.5Pt0.5 precursor alloy. It was then used as a 3D nanoporous template/catalyst for the growth of nanoporous graphene by chemical vapor deposition (CVD). The high-temperature CVD process would cause the alloy ligament/pore coarsening of the nanoporous MnNiCuCoPt template from 3–5 nm to 200–300 nm accompanied by graphene coating. The 3D coarsened porous MnNiCuCoPt with graphene coating was then further dealloyed in 8.0 M HNO3 solution, leading to the formation of an ultrafine nanoporous Pt-based alloy with ligament/pore sizes of 2–3 nm encapsulated in free-standing nanotubular graphene with nanotubular/nanopore sizes of ∼200 to 300 nm.
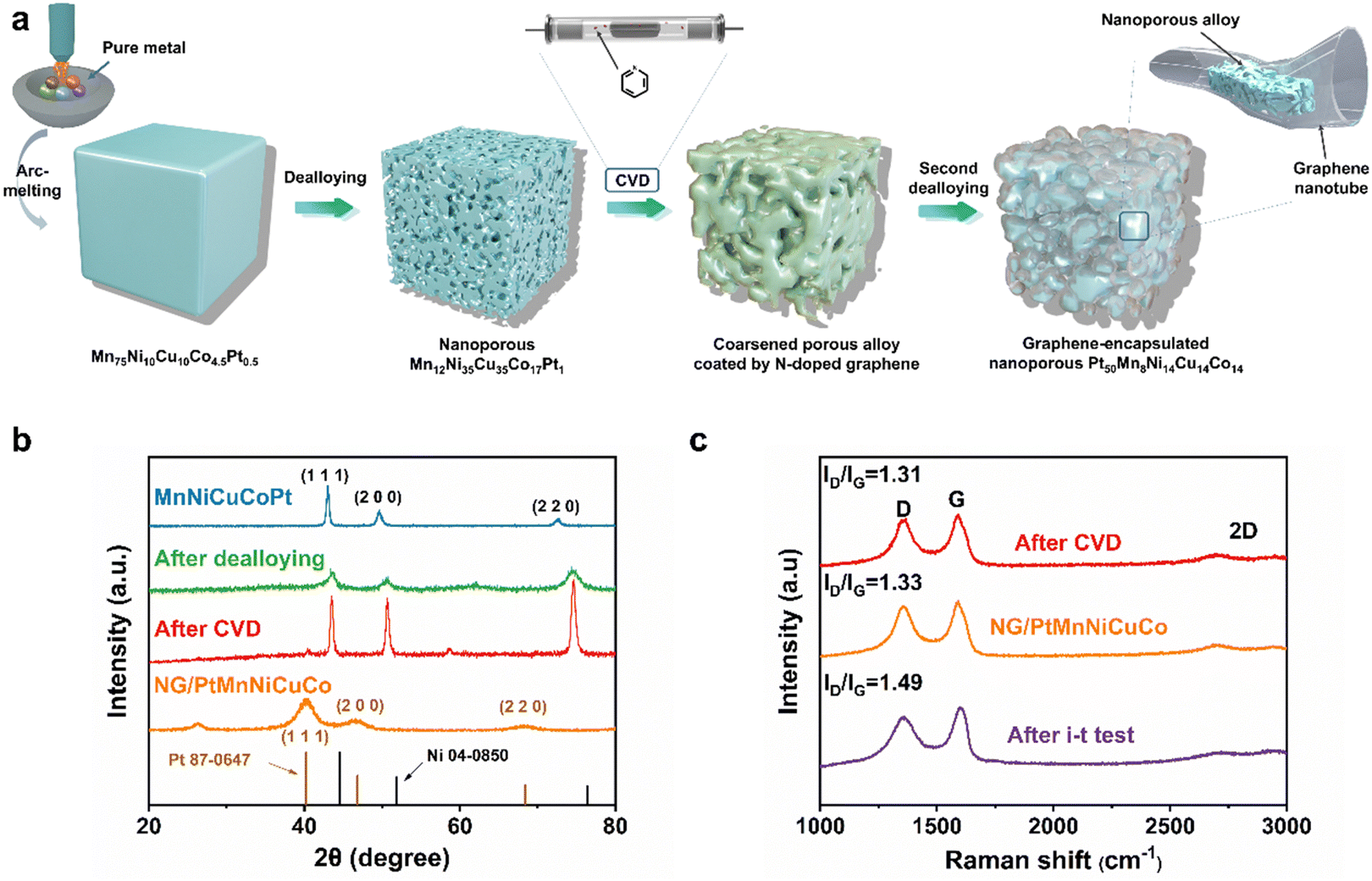 | ||
| Fig. 1 (a) Schematic illustration of the synthesis process of the bimodal nanoporous graphene/alloy composite. (b) XRD patterns of the precursor alloy, after dealloying, after CVD growth of graphene and after the removal of the templates by HNO3 (NG/PtMnNiCuCo). (c) Raman spectra of the prepared samples at different stages. | ||
X-ray diffraction (XRD) analysis shows that the designed Mn75Ni10Cu10Co4.5Pt0.5 precursor alloy is a single-phase face-centered cubic (fcc) alloy (Fig. 1b). The first dealloying in 1.0 M (NH4)2SO4 aqueous solution would selectively remove most Mn and result in a nanoporous MnNiCuCoPt alloy template with an ultrafine ligament/pore size of ∼3 to 5 nm (Fig. S1a, ESI†).48 This process is confirmed by the disappearance of the original XRD peaks and the appearance of a new set of broader fcc peaks (Fig. 1b). These new peaks also shift obviously to high angles due to the removal of larger Mn atoms. After the growth of N-doped graphene by CVD, the XRD diffraction peaks of the nanoporous alloy template become sharp again, indicating the crystal growth (coarsening) of the nanoporous template (Fig. 1b). We then carried out a second dealloying in a diluted HNO3 solution to remove most of these non-noble metal components (Mn, Ni, Cu, and Co), and generate a nanoporous Pt-rich multicomponent alloy, which is encapsulated in the 3D nanoporous nanotubular N-doped graphene network (named NG/PtMnNiCuCo). As shown in Fig. 1b, the NG/PtMnNiCuCo exhibits a broad peak at about 25°, which is attributed to the C (002) diffraction. Other diffraction peaks can be assigned to the (111), (200) and (220) diffraction of Pt-based alloys, suggesting the crystal nature of the dealloyed alloy. The removal of smaller Ni, Cu, Co and Mn also caused the fcc diffraction peaks to shift to smaller angles when compared with those of the nanoporous template. In comparison, the pure N-doped nanoporous graphene (NG) shows only one XRD peak at around 25° (Fig. S2a, ESI†). The composition change of the multicomponent alloy after each step is analyzed by energy dispersive spectra (EDS) and shown in Table S1 (ESI†).
The Raman spectrum in Fig. 1c shows that after CVD, the intensity ratios of the D and G bands (ID/IG) of NG were 1.31. After the second dealloying, there was almost no significant change in ID/IG value (1.33), indicating that the graphene defects did not increase significantly by dealloying. The appearance of 2D bands and the I2D/IG intensity ratio of ∼0.43 suggest the synthesized high-quality multilayered graphene.49,50
After the CVD growth of graphene, the scanning electron microscopy (SEM) image shows the coarsened nanoporous structure with greatly increased alloy ligament/pore sizes to ∼200 to 300 nm (Fig. S1, ESI†). The thin graphene, which is closely coated on the ligament surface, cannot be observed by SEM at this stage. After the second dealloying, the nanoporous nanotubular graphene network can be clearly observed by SEM (Fig. 2a). Dealloyed alloy fragments (by the second dealloying) are uniformly encapsulated in the free-standing 3D graphene. The formation of separated alloy fragments is mainly due to the low content of noble Pt in the coarsened porous MnNiCuCoPt template. It is expected that the large-scale removal of non-noble elements (Mn, Ni, Cu, and Co) by the second dealloying would cause severe volume shrinkage and breakage of the coarsened ligaments. As shown by the red dotted line in Fig. 2b and h, the shrunken alloy profile matches the shape of the graphene pipe, indicating that the shrinkage scale is relatively uniform. Both transmission electron microscopy (TEM, Fig. 2c) and high-resolution high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM, Fig. 2d and e) confirm the ultrafine 3D nanoporous ligament-pore structure (2–3 nm) in these alloy fragments. From the high-resolution TEM (HRTEM) image in Fig. 2c, the atomic interplanar spacing is ∼0.23 nm, corresponding well to the (111) plane of an fcc Pt alloy (Fig. 2c). Thus, a bimodal nanoporous structure was obtained with a first level graphene-based coarsened porous structure and second level ultrafine nanoporous alloy structure.
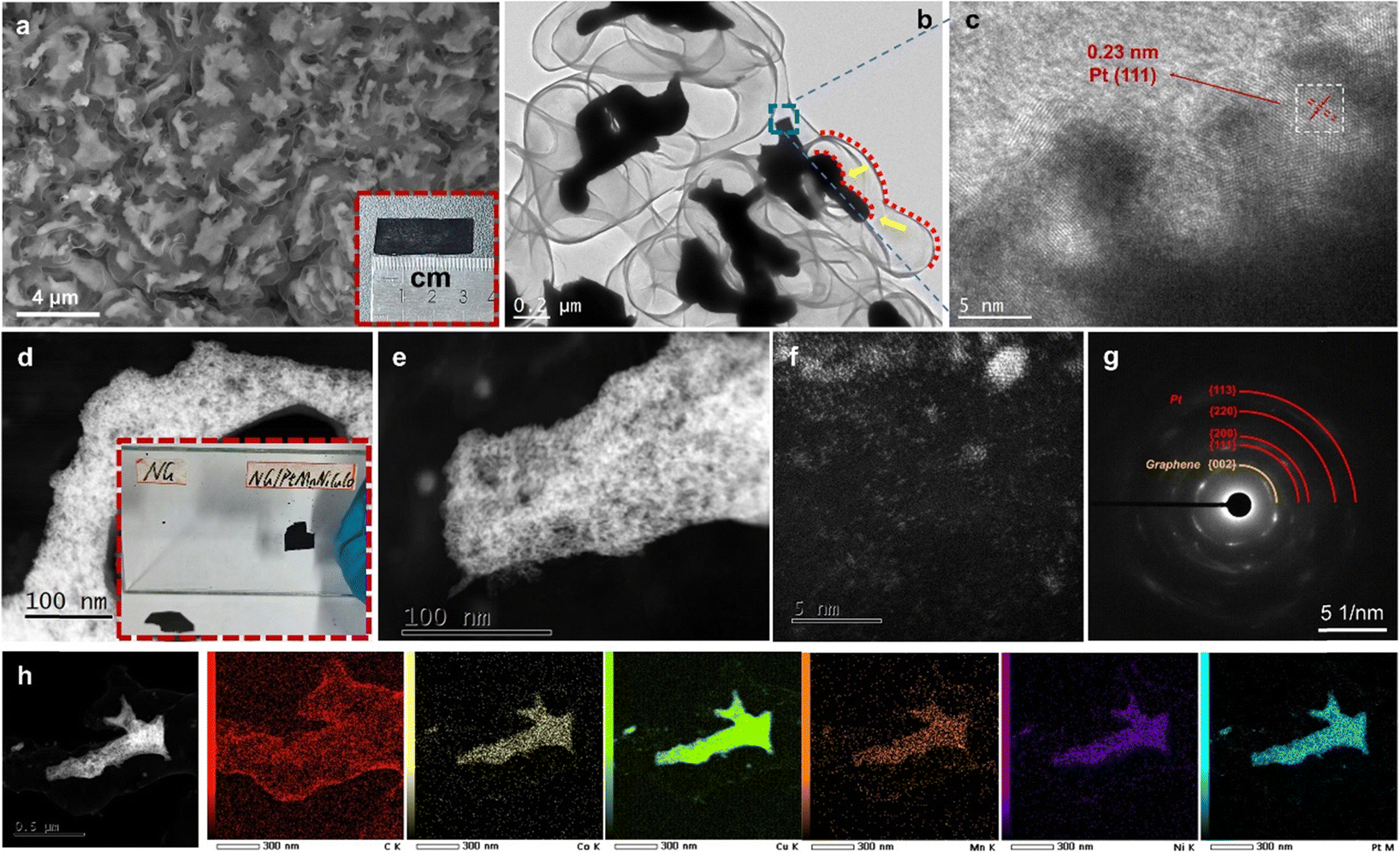 | ||
| Fig. 2 (a) Plane-view SEM image and photograph (inset in (a)), (b) TEM image, (c) HRTEM image, (d)–(f) HAADF-STEM images, (g) SAED pattern, and (h) STEM-EDS mapping of the NG/PtMnNiCuCo. (inset in (d)) Photograph of NG and NG/PtMnNiCuCo, which are pasted on the glass and then dried. | ||
Besides the nanoporous fragments, some metal clusters and single atoms are also observed on the graphene surface as revealed by the HAADF-STEM image (Fig. 2f). The selected area electron diffraction (SAED) pattern discloses mixed circles, which are confirmed as graphene and polycrystalline fcc Pt alloy (Fig. 2g). The STEM-EDS elemental mapping reveals the homogeneous distribution of the five metal elements in the encapsulated nanoporous alloy fragments and the carbon-based graphene nanotubular network (Fig. 2h). The EDS result of the alloy fragment elemental composition is shown in Table S2 (ESI†). Due to the interference from the copper grid in TEM, the content of Cu is not shown. It can be seen that the content of Pt is about 57 at%. Mn is about 9 at%. Ni and Co each account for about 17 at%. In contrast, the pure N-doped nanoporous graphene (NG) has a similar coarsened nanoporous morphology, but without the encapsulated alloy fragments (Fig. S2b–d, ESI†). It is worth noting that NG/PtMnNiCuCo has a stronger adsorption capacity than NG. As shown in Fig. 2d inset and Video S1 (ESI†), 99.7% ethanol is used to paste the two samples on a glass plate. After the complete vaporization of ethanol by illumination, the NG/PtMnNiCuCo was strongly adsorbed onto the glass plate, while the NG fell off easily. This should be due to the existence of the multi-component nanoporous alloy with high surface energy. The presence of the nanoporous PtMnNiCuCo inside of the graphene tube also accelerates the charge transfer/conductivity of NG as shown in the Nyquist plots (Fig. S3, ESI†).
X-ray photoelectron spectroscopy (XPS) analysis further reveals the N-doped graphene/Pt-rich alloy composite structure (Fig. S4, ESI†). The C 1s spectrum shows one main peak at 284.8 eV, which corresponds to the sp2 carbon from the conjugated honeycomb lattice. Low amounts of functional groups such as hydroxyl and carboxyl can also be detected. From the N 1s spectrum (Fig. S4, ESI†), we found the peaks of oxidized N (403.3 eV), graphitic N (401.1 eV) and pyridinic N (398.9 eV) in the graphene network. On the basis of the XPS analysis, the atomic percentage of nitrogen in NG/PtMnNiCuCo is estimated to be 3.61 at%, which is higher than that of NG grown on nanoporous Ni (∼3 at%).51 Moreover, the highest peak is from the pyridinic N, which is different from the grown N-doped graphene on the nanoporous Ni template (the highest peak is from graphitic N).52 This difference suggests that other metal elements in the nanoporous multicomponent alloy template would affect the doping states of N during the CVD process. Although the exact affecting mechanism is still unclear, the high amount of pyridinic N ratio has been demonstrated to be beneficial for the electrochemical ORR.53,54 The analysis of other elements by XPS is shown in the ESI.†
The electrocatalytic activity of the NG/PtMnNiCuCo towards the ORR was evaluated in O2-saturated 0.1 M HClO4 solution (Fig. 3a). The nanoporous pure NG and commercial Pt/C are also studied for comparison. We find that NG/PtMnNiCuCo exhibits the highest activity with a half-wave potential (E1/2) of 0.9 V, which is higher than that of Pt/C (∼0.85 V). When compared with literature data about acidic ORR catalysts, the performance of our free-standing catalyst is comparable with the best reported catalysts (Table S3, ESI†). The inset in Fig. 3b shows the ORR polarization curves of the NG/PtMnNiCuCo at different rotating speeds. The corresponding Koutecky–Levich plots in Fig. 3b show a good linear relationship. The calculated electron transfer number (n) ≈ 3.97 indicates that O2 is reduced to H2O with negligible side reactions.
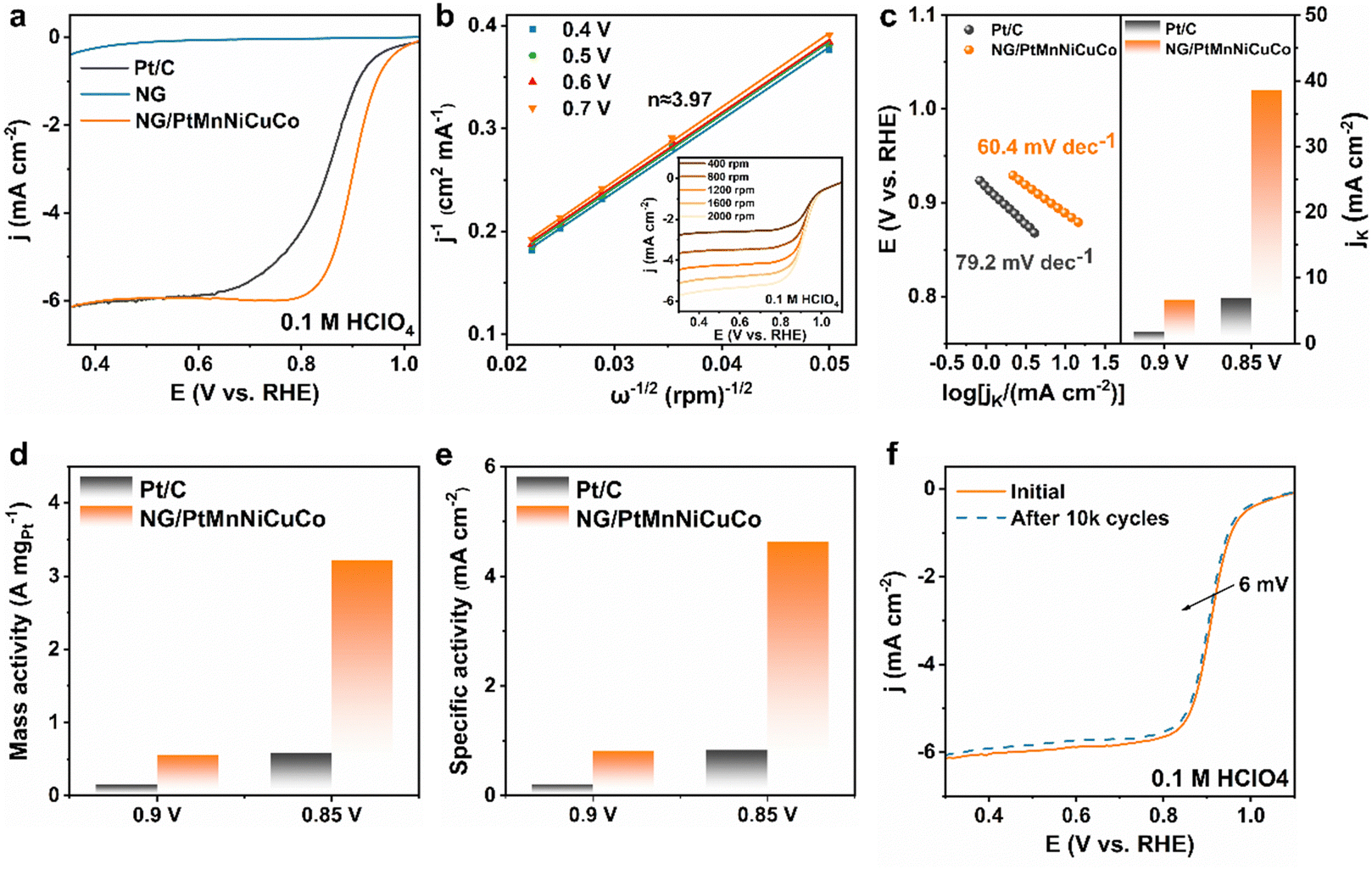 | ||
| Fig. 3 (a) ORR polarization curves. (b) The K–L plots of the NG/PtMnNiCuCo at different potentials, with the inset showing the LSV curves at various rotating speeds. (c) Tafel plots and Jk for the NG/PtMnNiCuCo and commercial Pt/C catalysts. (d) Mass activity and (e) specific activity of NG/PtMnNiCuCo and Pt/C at 0.85 and 0.9 V for the ORR. (f) ORR polarization curves of NG/PtMnNiCuCo before and after 10 k cycles. | ||
Fig. 3c shows that NG/PtMnNiCuCo had a lower Tafel slope of 60.4 mV dec−1 than Pt/C (79.2 mV dec−1), suggesting a faster ORR kinetics on NG/PtMnNiCuCo. In addition, the kinetic current densities (Jk) of NG/PtMnNiCuCo at 0.9 and 0.85 V are 6.67 and 38.63 mA cm−2, respectively, which are about 3.71 and 5.56 times those of Pt/C, indicating a favorable kinetics in line with the Tafel slope results. Furthermore, the mass activity of NG/PtMnNiCuCo at 0.90 V (vs. RHE) is calculated to be 0.56 A mgPt−1 (Fig. 3d), which is 3.73 times that of the Pt/C catalyst (0.15 A mgPt−1). At 0.85 V (vs. RHE), its mass activity (3.22 A mgPt−1) was 5.55 times that of Pt/C (0.58 A mgPt−1). For the calculation of specific surface activities, the electrochemical active surface area (ECSA) of the catalyst was first tested following an H-adsorption approach (Fig. S5, ESI†). In Fig. 3e, the specific surface activities of NG/PtMnNiCuCo at 0.9 V and 0.85 V are 0.81 and 4.64 mA cm2, respectively, which are 3.86 and 5.59 times that of Pt/C (0.21 mA cm2 at 0.90 V, and 0.83 mA cm2 at 0.85 V). As shown in Fig. 3f, the activity of NG/PtMnNiCuCo is well maintained during continuous potential cycling, and a loss of only about 6 mV in the half-wave potential was observed after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles.
000 cycles.
To further test the catalytic activity and especially durability under strong gas bubbling conditions, we carried out a HER test in 0.5 M H2SO4 electrolyte. According to Fig. 4a, NG/PtMnNiCuCo exhibits an excellent HER activity, requiring an overpotential of only 23 mV to reach 10 mA cm2, which is even better than Pt/C (32 mV) and comparable to other reported Pt-based HER catalysts (Table S4, ESI†). The similar Tafel slope values for both NG/PtMnNiCuCo and Pt/C (inset in Fig. 4a) suggest that the Tafel mechanism should be mainly responsible for the HER process.
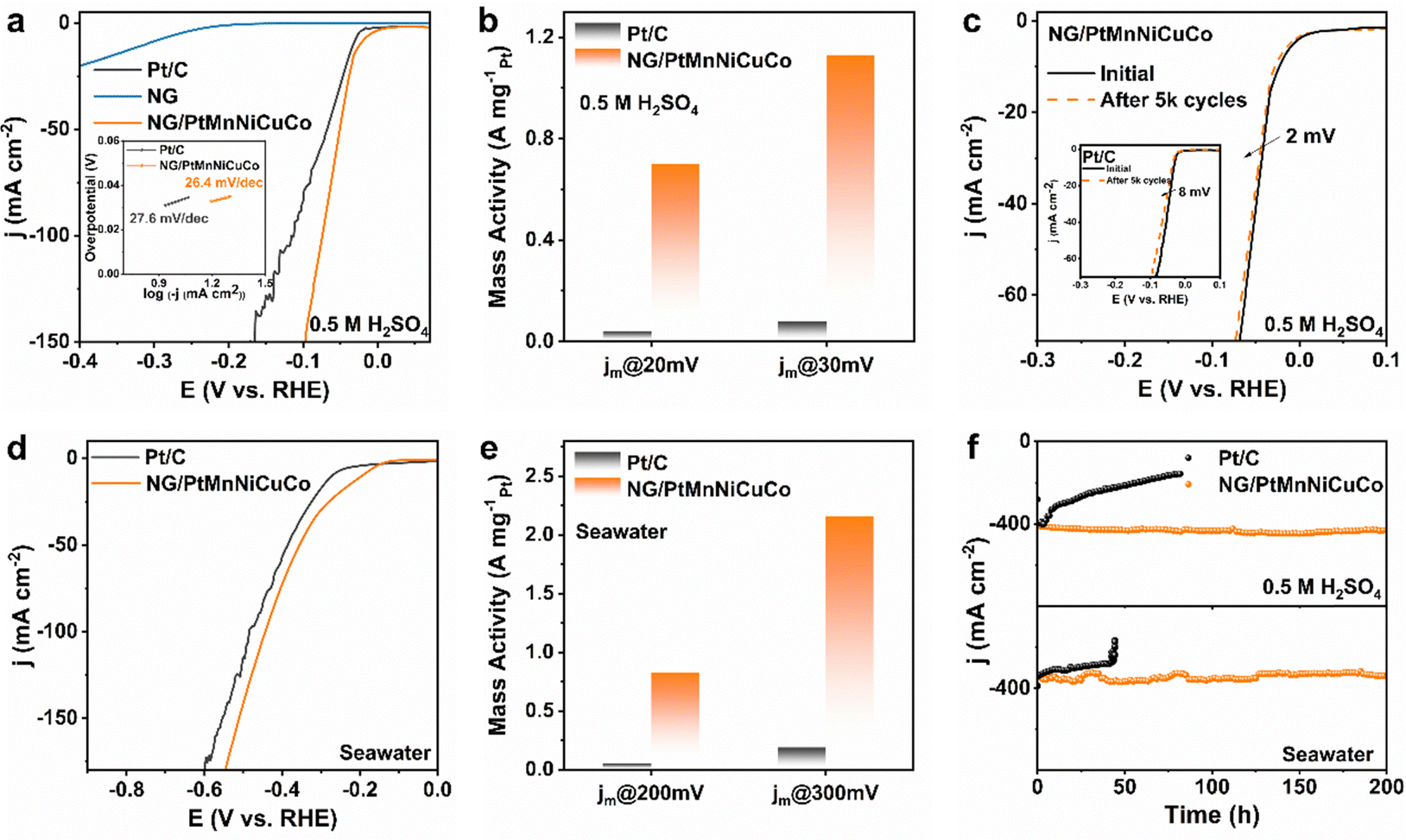 | ||
| Fig. 4 (a) HER polarization curves and (inset) Tafel plots. (b) Mass activity of NG/PtMnNiCuCo and Pt/C at 20 and 30 mV. (c) Polarization curves of NG/PtMnNiCuCo and Pt/C (inset) before and after 5 k cycles. (d) LSV curves in the seawater. (e) Mass activity of NG/PtMnNiCuCo and Pt/C at 200 and 300 mV in the seawater. (f) Long-term HER durability tests. | ||
Due to the relatively low loading amount and the alloy nature of the nanoporous catalyst, the mass activities of NG/PtMnNiCuCo are 0.7 and 1.12 A mgPt−1 at 20 and 30 mV, respectively (Fig. 4b), which are about 14 times higher than that of Pt/C. For the stability test, NG/PtMnNiCuCo shows almost overlapped LSV curves before and after 5000 cycles. The overpotential at 10 mA cm−2 only increased by 2 mV, while that of Pt/C increased by 8 mV (Fig. 4c). These results indicate the better HER activity and stability of NG/PtMnNiCuCo in acidic medium.
For the HER in seawater, our NG/PtMnNiCuCo also exhibits a better performance than Pt/C (Fig. 4d) with an overpotential of 194 mV at 10 mA cm2. When LSV is normalized by Pt loading amount (Fig. S6, ESI†), the mass activity of NG/PtMnNiCuCo was about 16.6 and 11.4 times that of Pt/C at 200 and 300 mV overpotential, respectively (Fig. 4e). Finally, long-term stability under large current densities (strong H2 gas bubbling) was assessed by chronoamperometry measurements. It was found that NG/PtMnNiCuCo could work stably under ∼400 mA cm−2 and a long period of 200 h in both acidic medium and seawater, while Pt/C has obvious performance degradation under such operations (Fig. 4f). Besides the inherent activity degradation, the drastic current density decrease may be due to the detachment of Pt/C from the current collector surface owing to the strong bubbling. In comparison, the physical entrapment can effectively avoid the detachment of the nanoporous catalyst, resulting in high durability even under strong gas bubbling conditions. After the long-term i–t test in 0.5 M H2SO4 solution, SEM and XRD analysis suggest that the structural integrity and elemental composition of NG/PtMnNiCuCo does not obviously change (Fig. S7 and Table S1, ESI†). Raman analysis shows the slightly increased ID/IG value to 1.49 from 1.33 (Fig. 1c), indicating more graphene structural defects caused by the large current HER.
Density functional theory (DFT) calculations were conducted to gain insightful understanding on the enhanced ORR/HER activities of the multicomponent alloy. Based on XRD/HRTEM characterizations and the acidic working media, we used the (1 1 1) facet of PtMnNiCuCo with a pure Pt surface and sublayer doped with Mn, Ni, Cu, and Co as the study surface (Fig. 5a). Fig. 5b shows electron difference contour maps of pure Pt and PtMnNiCuCo, in which blue and red regions correspond to the electron depletion and accumulation, respectively. It is clear that the Pt surface in PtMnNiCuCo has more charge exchange interaction than that of pure Pt, resulting in more electron deficient surface Pt sites. This is in good agreement with the XPS results. Projected density of states (PDOS) analysis shows that both Pt and PtMnNiCuCo have a high level of occupied electron states at the Fermi level (Fig. 5c), suggesting their metallic character with high electronic conductivity. The occupied electron states of PtMnNiCuCo are higher than that of Pt, which may explain the enhanced ORR activity of the multicomponent alloy. To gain further insight, we analyze their d-band center (εd), as the binding energy of adsorbates is intimately associated with the d-band center of catalysts. As shown in Fig. 5d, the εd of Pt and PtMnNiCuCo is −2.097 eV and −2.000 eV, respectively. It is believed that the upward shift of εd would strengthen the bonding between the catalyst and adsorbate, which will facilitate the reaction on PtMnNiCuCo. We finally sought to evaluate the free energy profiles along the reaction coordinates of the ORR/HER. For the ORR, the rate-determining step on both Pt and PtMnNiCuCo is the formation of OOH* (Fig. 5e). By adding these non-noble elements in the sublayer, the ΔG of the rate-determining step on PtMnNiCuCo becomes lower, which illustrates the more active surface of PtMnNiCuCo for the ORR. For the HER, the HER kinetics and activity rely greatly on the Gibbs free energy of H* adsorption (ΔGH*). As shown in Fig. 5f, the ΔGH* is negative, and the smaller |ΔGH*| value on the multicomponent alloy reveals that the desorption of H2 from PtMnNiCuCo is facilitated, leading to enhanced HER activity. It is worth mentioning that these residual single atom metal sites should also be active for the ORR/HER. Considering the ratios of the single atoms and nanoporous alloys, we believe that the encapsulated nanoporous alloy is mainly responsible for the tested high activity.
 | ||
| Fig. 5 (a) The structure of Pt and PtMnNiCuCo, (b) the charge density distribution, (c) the projected density of states, and (d) the d-band center of Pt and PtMnNiCuCo. Free energy diagrams of the (e) ORR and (f) HER on both Pt and PtMnNiCuCo. | ||
In summary, by effectively bypassing the common problems of Pt NP aggregation and detachment from the carbon support involved in Pt/C-type supported catalysts, the free-standing composite with encapsulation design has significantly enhanced catalytic durability even under strong gas bubbling conditions. Moreover, the multicomponent alloy design significantly optimized the electronic structure of Pt, resulting in enhanced inherent catalytic activities for both the ORR and HER. This work provides a promising approach for designing/constructing next-generation multicomponent encapsulated catalysts for water splitting and fuel cells.
Author contributions
H.-J. Qiu conceived and designed the work. L. Zhu prepared the materials and tested the catalytic performance. Q. Li and K. Li did the DFT calculations. Y. Hu and K. Reddy did the TEM characterizations. L. Zhu, H.-J. Qiu, K. Reddy and Y. Hu wrote the manuscript. All authors discussed the results and revised the manuscript.Data availability
All data generated or used during the study appear in the submitted article. Raw data is available from the authors on request.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors are thankful for the financial support from Shenzhen Key Laboratory of Advanced Functional Carbon Materials Research and Comprehensive Application (Grant No. ZDSYS20220527171407017).References
- R. Kavanagh, X. Cao, W. Lin, C. Hardacre and P. Hu, Angew. Chem., Int. Ed., 2012, 51, 1572–1575 CrossRef CAS PubMed.
- K. Jiao, J. Xuan, Q. Du, Z. Bao, B. Xie, B. Wang, Y. Zhao, L. Fan, H. Wang, Z. Hou, S. Huo, N. P. Brandon, Y. Yin and M. D. Guiver, Nature, 2021, 595, 361–369 CrossRef CAS PubMed.
- L. Liang, H. Jin, H. Zhou, B. Liu, C. Hu, D. Chen, Z. Wang, Z. Hu, Y. Zhao, H.-W. Li, D. He and S. Mu, Nano Energy, 2021, 88, 106221 CrossRef CAS.
- J. Gao, X. Zhou, Y. Wang, Y. Chen, Z. Xu, Y. Qiu, Q. Yuan, X. Lin and H. Qiu, Small, 2022, 18, 2202071 CrossRef CAS PubMed.
- L. Gong, J. Zhu, F. Xia, Y. Zhang, W. Shi, L. Chen, J. Yu, J. Wu and S. Mu, ACS Catal., 2023, 13, 4012–4020 CrossRef CAS.
- X. Tian, X. Zhao, Y. Q. Su, L. Wang, H. Wang, D. Dang, B. Chi, H. Liu, E. Hensen, X. W. Lou and B. Y. Xia, Science, 2019, 366, 850–856 CrossRef CAS PubMed.
- X. Liu, S. Hao, G. Zheng, Z. Su, Y. Wang, Q. Wang, L. Lei, Y. He and X. Zhang, J. Energy Chem., 2022, 64, 315–322 CrossRef CAS.
- K. Qiu, G. Chai, C. Jiang, M. Ling, J. Tang and Z. Guo, ACS Catal., 2016, 6, 3558–3568 CrossRef CAS.
- J. Zhang, Y. Yuan, L. Gao, G. Zeng, M. Li and H. Huang, Adv. Mater., 2021, 33, 2006494 CrossRef CAS PubMed.
- J. C. Meier, C. Galeano, I. Katsounaros, A. A. Topalov, A. Kostka, F. Schüth and K. J. J. Mayrhofer, ACS Catal., 2012, 2, 832–843 CrossRef CAS.
- K. Paperzh, A. Alekseenko, I. Pankov and V. Guterman, J. Electroanal. Chem., 2024, 952, 117972 CrossRef CAS.
- S. Sui, X. Wang, X. Zhou, Y. Su, S. Riffat and C. Liu, J. Mater. Chem. A, 2017, 5, 1808–1825 RSC.
- L. Bu, N. Zhang, S. Guo, X. Zhang, J. Li, J. Yao, T. Wu, G. Lu, J.-Y. Ma, D. Su and X. Huang, Science, 2016, 354, 1410–1414 CrossRef CAS PubMed.
- E. Pahon, D. Bouquain, D. Hissel, A. Rouet and C. Vacquier, J. Power Sources, 2021, 510, 230385 CrossRef CAS.
- E. Guilminot, A. Corcella, F. Charlot, F. Maillard and M. Chatenet, J. Electrochem. Soc., 2007, 154, B96 CrossRef CAS.
- R. M. Darling and J. P. Meyers, J. Electrochem. Soc., 2004, 152, A242 CrossRef.
- R. Wang, D. Li, S. Maurya, Y. S. Kim, Y. Wu, Y. Liu, D. Strmcnik, N. M. Markovic and V. R. Stamenkovic, Nanoscale Horiz., 2020, 5, 316–324 RSC.
- C. Ma, C. Yang, H. Zhuo, C. Chen, K. Lu, F. Wang, Z. Shi, H. Xiao, M. Song and G. Jiang, J. Am. Chem. Soc., 2023, 145, 10890–10898 CrossRef CAS PubMed.
- S. Xu, Y. Kim, J. Park, D. Higgins, S.-J. Shen, P. Schindler, D. Thian, J. Provine, J. Torgersen, T. Graf, T. D. Schladt, M. Orazov, B. H. Liu, T. F. Jaramillo and F. B. Prinz, Nat. Catal., 2018, 1, 624–630 CrossRef CAS.
- H. Jin, Z. Xu, Z.-Y. Hu, Z. Yin, Z. Wang, Z. Deng, P. Wei, S. Feng, S. Dong, J. Liu, S. Luo, Z. Qiu, L. Zhou, L. Mai, B.-L. Su, D. Zhao and Y. Liu, Nat. Commun., 2023, 14, 1518 CrossRef CAS PubMed.
- G. W. Sievers, J. R. Bowen, V. Brüser and M. Arenz, J. Power Sources, 2019, 413, 432–440 CrossRef CAS.
- X. Zhu, L. Huang, M. Wei, P. Tsiakaras and P. K. Shen, Appl. Catal., B, 2021, 281, 119460 CrossRef CAS.
- A. Kongkanand and M. F. Mathias, J. Phys. Chem. Lett., 2016, 7, 1127–1137 CrossRef CAS PubMed.
- K. Kodama, T. Nagai, A. Kuwaki, R. Jinnouchi and Y. Morimoto, Nat. Nanotechnol., 2021, 16, 140–147 CrossRef CAS PubMed.
- Y. Da, R. Jiang, Z. Tian, G. Chen, Y. Xiao, J. Zhang, S. Xi, Y. Deng, W. Chen, X. Han and W. Hu, Adv. Energy Mater., 2023, 13, 2300127 CrossRef CAS.
- H. Yang, Y. Ko, W. Lee, A. Züttel and W. Kim, Mater. Today Energy, 2019, 13, 374–381 CrossRef.
- L. Ren, D. Yang and J.-H. Yang, Int. J. Hydrogen Energy, 2022, 47, 13876–13886 CrossRef CAS.
- S. Zhu, L. Yang, J. Bai, Y. Chu, J. Liu, Z. Jin, C. Liu, J. Ge and W. Xing, Nano Res., 2023, 16, 2035–2040 CrossRef CAS.
- N. Seselj, S. M. Alfaro, E. Bompolaki, L. N. Cleemann, T. Torres and K. Azizi, Adv. Mater., 2023, 35, 2302207 CrossRef CAS PubMed.
- H. Wang, R. Wang, S. Sui, T. Sun, Y. Yan and S. Du, Automot. Innov., 2021, 4, 144–164 CrossRef.
- A. O. Elnabawy, E. A. Murray and M. Mavrikakis, J. Phys. Chem. C, 2022, 126, 4374–4390 CrossRef CAS.
- Y. Wang, H.-Z. Yu, J. Ying, G. Tian, Y. Liu, W. Geng, J. Hu, Y. Lu, G.-G. Chang, K. I. Ozoemena, C. Janiak and X.-Y. Yang, Chem. – Eur. J., 2021, 27, 9124–9128 CrossRef CAS PubMed.
- I. Jang, S. Lee, E. Lee, D.-W. Lee, H.-Y. Park, B. B. Choi, H. C. Ham and S. J. Yoo, Nano Today, 2021, 41, 101316 CrossRef CAS.
- C. Lim, A. R. Fairhurst, B. J. Ransom, D. Haering and V. R. Stamenkovic, ACS Catal., 2023, 13, 14874–14893 CrossRef CAS PubMed.
- Q. Zhang, T. Shen, M. Song, S. Wang, J. Zhang, X. Huang, S. Lu and D. Wang, J. Energy Chem., 2023, 86, 158–166 CrossRef CAS.
- X. Zhang, Y. Yang, Y. Liu, Z. Jia, Q. Wang, L. Sun, L. Zhang, J. J. Kruzic, J. Lu and B. Shen, Adv. Mater., 2023, 35, 2303439 CrossRef CAS PubMed.
- Z.-X. Cai, H. Goou, Y. Ito, T. Tokunaga, M. Miyauchi, H. Abe and T. Fujita, Chem. Sci., 2021, 12, 11306–11315 RSC.
- H.-J. Qiu, G. Fang, J. Gao, Y. Wen, J. Lv, H. Li, G. Xie, X. Liu and S. Sun, ACS Mater. Lett., 2019, 1, 526–533 CrossRef CAS.
- W. Zhang, X. Feng, Z. X. Mao, J. Li and Z. Wei, Adv. Funct. Mater., 2022, 32, 2204110 CrossRef CAS.
- S. Li, X. Tang, H. Jia, H. Li, G. Xie, X. Liu, X. Lin and H.-J. Qiu, J. Catal., 2020, 383, 164–171 CrossRef CAS.
- Y. Wang, Z. Wang, J. Zhang, C. Zhang, H. Gao, J. Niu and Z. Zhang, Nanoscale, 2018, 10, 17070–17079 RSC.
- Z. Jin, X. Zhou, Y. Hu, X. Tang, K. Hu, K. M. Reddy, X. Lin and H.-J. Qiu, Chem. Sci., 2022, 13, 12056–12064 RSC.
- X. Lin, Y. Hu, K. Hu, X. Lin, G. Xie, X. Liu, K. M. Reddy and H.-J. Qiu, ACS Mater. Lett., 2022, 4, 978–986 CrossRef CAS.
- T. Yu, Y. Zhang, Y. Hu, K. Hu, X. Lin, G. Xie, X. Liu, K. M. Reddy, Y. Ito and H.-J. Qiu, ACS Mater. Lett., 2022, 4, 181–189 CrossRef CAS.
- Q. Mao, X. Mu, K. Deng, H. Yu, Z. Wang, Y. Xu, X. Li, L. Wang and H. Wang, Adv. Funct. Mater., 2023, 33, 2304963 CrossRef CAS.
- M. Wei, Y. Sun, J. Zhang, F. Ai, S. Xi and J. Wang, Energy Environ. Sci., 2023, 16, 4009–4019 RSC.
- H. Xu, L. Yang, K. Wang, L. Jin, Y. Liu, G. He and H. Chen, Inorg. Chem., 2023, 62, 11271–11277 CrossRef CAS PubMed.
- H.-J. Qiu, J. L. Kang, P. Liu, A. Hirata, T. Fujita and M. W. Chen, J. Power Sources, 2014, 247, 896–905 CrossRef CAS.
- Z. Chen, W. Ren, L. Gao, B. Liu, S. Pei and H.-M. Cheng, Nat. Mater., 2011, 10, 424–428 CrossRef CAS PubMed.
- Y. Zhang, L. Zhang and C. Zhou, Acc. Chem. Res., 2013, 46, 2329–2339 CrossRef CAS PubMed.
- H.-J. Qiu, Y. Ito, W. Cong, Y. Tan, P. Liu, A. Hirata, T. Fujita, Z. Tang and M. Chen, Angew. Chem., Int. Ed., 2015, 127, 14237–14241 CrossRef.
- H. Qiu, P. Du, K. Hu, J. Gao, H. Li, P. Liu, T. Ina, K. Ohara, Y. Ito and M. Chen, Adv. Mater., 2019, 31, 1900843 CrossRef PubMed.
- R. Ma, Y. Zhou, C. Hu, M. Yang, F. Wang, K. Yan, Q. Liu and J. Wang, Energy Storage Mater., 2018, 13, 142–150 CrossRef.
- L. Zhu, D. Zheng, Z. Wang, X. Zheng, P. Fang, J. Zhu, M. Yu, Y. Tong and X. Lu, Adv. Mater., 2018, 30, 1805268 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Comparison of NG/PtMnNiCuCo and NG absorbability (MP4). Experimental details, SEM, XRD, electrochemical analysis, and comparison tables (PDF). See DOI: https://doi.org/10.1039/d4nh00190g |
| ‡ L. Zhu, Q. Li, and Y. Hu contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |