
Insight into mechanism for remarkable photocatalytic hydrogen evolution of Cu/Pr dual atom co-modified TiO2†
Hongshun
Zheng
ab,
Baoye
Zi
a,
Tong
Zhou
a,
Guoyang
Qiu
a,
Zhongge
Luo
a,
Qingjie
Lu
a,
Alain Rafael Puente
Santiago
 cd,
Yumin
Zhang
a,
Jianhong
Zhao
a,
Jin
Zhang
a,
Tianwei
He
*a and
Qingju
Liu
*ab
cd,
Yumin
Zhang
a,
Jianhong
Zhao
a,
Jin
Zhang
a,
Tianwei
He
*a and
Qingju
Liu
*ab
aNational Center for International Research on Photoelectric and Energy Materials, Yunnan Key Laboratory for Micro/nano Materials & Technology, School of Materials Science and Engineering, Yunnan University, Kunming 650091, China. E-mail: he.tianwei@ynu.edu.cn; qjliu@ynu.edu.cn
bSouthwest United Graduate School, Kunming 650091, China
cWalker Department of Mechanical Engineering, University of Texas at Austin, Austin, TX 78712, USA
dFlorida International University (FIU), Department of Chemistry and Biochemistry, Miami, FL, USA
First published on 8th July 2024
Abstract
The development of high-activity photocatalysts is crucial for the current large-scale development of photocatalytic hydrogen applications. Herein, we have developed a strategy to significantly enhance the hydrogen photocatalytic activity of Cu/Pr di-atom co-modified TiO2 architectures by selectively anchoring Cu single atoms on the oxygen vacancies of the TiO2 surface and replacing a trace of Ti atoms in the bulk with rare earth Pr atoms. Calculation results demonstrated that the synergistic effect between Cu single atoms and Pr atoms regulates the electronic structure of Cu/Pr–TiO2, thus promoting the separation of photogenerated carriers and their directional migration to Cu single atoms for the photocatalytic reaction. Furthermore, the d-band center of Cu/Pr–TiO2, which is located at −4.70 eV, optimizes the adsorption and desorption behavior of H*. Compared to TiO2, Pr–TiO2, and Cu/TiO2, Cu/Pr–TiO2 displays the best H* adsorption Gibbs free energy (−0.047 eV). Furthermore, experimental results confirmed that the photogenerated carrier lifetime of Cu/Pr–TiO2 is not only the longest (2.45 ns), but its hydrogen production rate (34.90 mmol g−1 h−1) also significantly surpasses those of Cu/TiO2 (13.39 mmol g−1 h−1) and Pr–TiO2 (0.89 mmol g−1 h−1). These findings open up a novel atomic perspective for the development of optimal hydrogen activity in dual-atom-modified TiO2 photocatalysts.
New conceptsThe development of high-performance photocatalysts is crucial for the current large-scale development of photocatalytic hydrogen applications. In this work, we have developed a strategy to significantly enhance the hydrogen photocatalytic activity of Cu/Pr di-atom co-modified TiO2 architectures by selectively anchoring Cu single atoms (SAs) on the oxygen vacancies of the TiO2 surface and replacing a small amount of Ti atoms in the bulk with rare earth Pr atoms. The mechanism of synergistic effects between Cu SAs and Pr atoms was systematically investigated and confirmed by density function theory (DFT) calculations and experimental methods. Specifically, the synergistic effect between Cu SAs and Pr atoms can transform the Cu sites into efficient electron-concentration and photocatalytically reactive sites, promoting more photogenerated carriers from the bulk and surface to participate in the photocatalytic reaction. Additionally, the adsorption and desorption behaviors of H atoms on the TiO2 surface are optimized to dramatically reduce the HER reaction energy barrier, so that the Cu/Pr–TiO2 photocatalyst exhibits an excellent ΔGH* of −0.047 eV. Furthermore, the experimental results confirmed that the superior hydrogen production rate (34.90 mmol g−1 h−1) of Cu/Pr–TiO2 significantly outperforms those of state-of-the-art hydrogen photocatalysts. This work opens a novel atomic perspective for the development of optimal hydrogen activity in dual-atom-modified TiO2 photocatalysts. |
1. Introduction
With the vigorous development of industrialization and urbanization, a large amount of non-renewable fossil energy has been drastically consumed, provoking a series of global energy and environmental crises. Hydrogen production through photocatalytic water splitting, which is capable of converting low-density sunlight energy into high-density chemical energy, provides a better solution for meeting the increasing energy needs in modern society.1–3 In this case, the selection of suitable materials is crucial for the development of photocatalytic water splitting for hydrogen production.4–6 In 1972, when Fujishima and Honda first reported that TiO2 single-crystal electrodes could split water to produce hydrogen under xenon lamp irradiation conditions, research on the TiO2 photocatalytic splitting of water to produce hydrogen was officially unveiled.7 Since then, TiO2 has attracted more and more attention by virtue of its remarkable advantages, such as green friendliness, abundant sources, better chemical stability, suitable redox potential and low cost.7–9 It has promoted the depth and breadth of TiO2 applications in photocatalysis. As a result, TiO2 has not only become a classical photocatalyst carrier, but is also one of the most studied and widely used semiconductor materials in the field of photocatalytic water splitting for hydrogen production.10,11However, the large bandgap value of TiO2 leads to its poor response to visible light, and the low photogenerated carrier separation efficiency limits the quantum efficiency of TiO2, which are among the principal limitations that need to be overcome for the large-scale application of TiO2.9 Therefore, it is of great significance to develop and fabricate efficient TiO2 photocatalysts.11–15
Metal-atom-doped TiO2 photocatalysts have attracted widespread attention, especially the surface doping of metal single atoms (SAs) and the doping of metal atoms into the bulk. According to a large number of experimental and density functional theory (DFT) computational studies, it is found that metal atom doping usually generates new absorption band edges by introducing impurity energy levels into the bandgap, which not only expands the spectral response range, but also improves the redox properties of photocatalysts.16–18 It can be seen that the number of active sites created by SAs are considerably higher than those of nanoparticles under the same loading amount, which boost the catalytic activity of SA photocatalysts and effectively improve utilization of the material.19–21 Currently, almost all metal-doped TiO2-based catalysts use noble metals, but the scarce earthly reserves and high prices of noble metals limit their scaling-up. In contrast, the transition metal Cu, with its advantages of high activity, low cost and good stability, has become an ideal non-noble metal co-catalyst material in the photocatalytic decomposition of water to produce hydrogen.22 Cheng et al.23 successfully doped Cu SAs into the TiO2 surface to form an SA photocatalyst (Cu/TiO2), which demonstrated that doping with Cu SAs can simultaneously enhance the light absorption and photocatalytic performance of TiO2. Zhang et al.24 successfully prepared surface-doped Cu SAs for TiO2 photocatalysts and ensured the dispersion of Cu atoms with higher loadings (∼1.5 wt%). It was found that the photocatalytic hydrogen evolution rate of Cu/TiO2 reached 101.70 mmol g−1 h−1, which was slightly better than that of the noble metal Pt SAs-TiO2. Notably, the combination of Cu SAs and TiO2 changes the intrinsic electronic structure of the photocatalyst, which further improves the performance of the photocatalytic hydrogen evolution reaction (HER). Up to now, to the best of our knowledge, all the SA-photocatalytic systems reported in the literature contain one type of metal active site, which makes it difficult to break the linear relationship that exists in the actual catalytic reaction processes, and also fails to completely utilize the advantages of SAs. In addition, doping of metal atoms into the bulk of photocatalysts to enhance catalytic activity has been considered a modification method with high feasibility and has attracted a lot of attention. Out of the numerous doped metal elements, the rare earth (RE) elements25–29 are attractive because even extremely minor amounts of an RE element can significantly alter the properties of photocatalysts.30–34 Moreover, due to the relatively weak shielding of the f electrons of RE elements, the interactions with the outer electrons are stronger, thus exhibiting unique chemical and electronic interactions.31,35–37 In particular, RE elements optimize the electronic structure with distinct splitting and overlapping of the 4f electronic states owing to interactions between electrons and crystal field effects.38 It is worth mentioning that recent studies have found that RE atoms, with their unique valence electron structure and large atomic radius, are able to form higher coordination numbers, which in turn produces a stronger substrate anchoring effect with a wider range of electron delocalization effects.39 It is undeniable that the surface of a photocatalyst is the principal part where the photocatalytic reaction takes place. Additionally, the doping of RE metal atoms into the bulk has a limited effect on improving photocatalytic activity, so the advantages of RE metal atoms still cannot be fully utilized. That is, the optimization of both the surface microstructure and the electronic structure of photocatalysts is essential to improve the effect of RE metal atoms in the bulk.
Surprisingly, we found that the combination of bulk RE atoms and surface Cu SAs using a surface doping strategy exhibits incomparable advantages for hydrogen evolution photocatalysis. That is, the doping of RE atoms into the bulk can well regulate the coordination environment of Cu SAs and optimize their reactivity and stability on the surface. Moreover, their coupling with each other will play a key role in the separation and transport of photocatalytic carriers both in the bulk and on the surface. Remarkably, there is a great possibility that the catalytic activity of TiO2 photocatalysts can be improved by several orders of magnitude through the synergistic interactions between bulk-doped RE atoms and Cu SAs.
It has been reported that the sol–gel preparation method can effectively dope Pr atoms into the TiO2 bulk.40 The doping of Pr atoms induces significant changes in TiO2, particularly enhancing its utilization of visible light and improving the separation and migration of photogenerated carriers. Additionally, the introduction of Pr atoms into the lattice not only enhances photocatalytic activity but also improves the selectivity and structural stability of TiO2.38–41 Currently, Pr-atom-doped TiO2 has demonstrated the successful degradation of organic pollutants under visible light.40 Therefore, there is great potential for utilizing rare earth Pr atoms to optimize the electronic structure of TiO2 bulk. Herein, we have chosen to use Pr atoms and Cu SAs to develop a new family of Cu/Pr–TiO2 photocatalysts. An elegant combination of DFT and experimental techniques has allowed us to investigate the intrinsic mechanism of the synergistic effect of Cu SAs and Pr doping on the photocatalytic activity of TiO2. In order to better balance the effect of trace Pr atom doping and computational efficiency, we set the Pr doping ratio at 1%, that is, replacing 1 Ti atom in the bulk. For Cu SAs loading, it has been found that Cu SAs can be anchored to the O vacancies and Ti vacancies on the Ti surface, respectively.42 We selectively anchor Cu SAs to the O vacancies on the TiO2 surface. To systematically investigate the effect of the synergistic interaction between Cu SAs and Pr atoms on the photocatalytic activity of TiO2, the microstructures, electronic structures and H* adsorption Gibbs free energies (ΔGH*) of TiO2, Pr–TiO2, Cu/TiO2, and Cu/Pr–TiO2 were analyzed in depth. The results illustrate that the synergistic mechanism between Cu SAs and Pr atoms not only well tailors the coordination environment of Cu SAs on the surface, but also effectively regulates the TiO2 electronic structure to provide more photogenerated carriers to participate in the surface reaction. Meanwhile, the adsorption and desorption behaviors of surface H* were optimized, resulting in Cu/Pr–TiO2 exhibiting a satisfactory ΔGH* (−0.047 eV). Further, the synergistic mechanism between them was confirmed by DFT calculations combined with partial experimental characterization. Clearly, the photogenerated carrier separation efficiency, lifetime and hydrogen production rate of different photocatalysts have demonstrated that the HER activity of TiO2 photocatalysts can be dramatically boosted. This work will certainly provide experimental and theoretical guidance for the development of more efficient TiO2 photocatalysts in the future.
2. Experimental
2.1. Computational methods and details
All calculations for periodic material structures are based on a DFT framework performed in the Vienna ab initio simulation package (VASP)43–46 with the projector-augmented wave (PAW)47 method. The exchange–correlation function is handled using the generalized gradient approximation (GGA) formulated by the Perdew–Burke–Ernzerhof (PBE) functional.48 The interaction between the atomic core and electrons is described by the PAW method to improve the accuracy and efficiency of DFT calculations. The strong electron correlation is suitably described by the GGA+U approach in Grimme's scheme, in which the on-site Coulombic interaction is included. It is worth mentioning that GGA+U is currently the most chosen approach to research the large system of strongly correlated d and f orbital electrons, which provides a good balance between computational efficiency and accuracy. Here we apply the GGA+U scheme with U = 4.2, 6.0 and 6.0 eV for the Ti-3d,49–51 Cu-3d52,53 and Pr-4f54,55 electrons, respectively. It is well known that slab models with only a few atomic layers are one of the best choices for DFT atomic simulations in surface science. In this way, the anatase TiO2 (101) surface56 is modelled with six O–Ti–O layers, and the lattice length in the z direction is set to 35 Å to ensure adequate vacuum thickness between the slabs. Among them, the Cu/TiO2 surface model is built by anchoring Cu SAs on a stabilized oxygen vacancy of the TiO2 (101) surface (Fig. S1, ESI†), while Pr–TiO2 is constructed by adding a Pr atom to the bulk in place of a Ti atom. After that, the Cu/Pr–TiO2 surface model is modeled by anchoring Cu SAs to the oxygen vacancy on the TiO2 surface and a Pr atom to the Ti atom position in the bulk (Fig. S2, ESI†). The plane-wave basis set energy cutoff is set to 500 eV. The Brillouin zone is sampled with gamma (Γ)-centered Monkhorst–Pack mesh sampling (3 × 3 × 1 for the surface structures of TiO2, Pr–TiO2, Cu/TiO2 and Cu/Pr–TiO2) for geometric relaxation. In order to obtain more reliable results for calculation of the electronic structure, the sampling of the Monkhorst–Pack grid with gamma (Γ) as the center is increased to 5 × 4 × 1. The energy and gradient in geometric optimization are converged to <1 × 10−6 eV and 0.01 eV Å−1 per atom, respectively.The corresponding Gibbs free energy has been calculated to study the effect of active sites on the hydrogen evolution reaction (HER) performance of the TiO2 photocatalyst. The corresponding calculations are based on the computational hydrogen electrode (CHE) proposed by Nørskov et al.57 According to this method, the Gibbs free energy of the proton and electron pair (H+ + e−) is equal to half of the free energy of gaseous hydrogen (H2) under standard reaction conditions. Moreover, the change in Gibbs free energy (ΔG) of each species can be obtained from the following equation:
| ΔG = ΔEDFT + ΔEZPE − TΔS | (1) |
2.2. Synthesis of Pr–TiO2 and Cu/Pr–TiO2
Preparation of Pr–TiO2 photocatalytic catalysts: Pr–TiO2 was synthesized by doping Pr atoms into the bulk of TiO2 using a proprietary sol–gel method previously developed by our group. Specifically, 76 mg of Pr(NO3)3·6H2O was dissolved in 75 mL of deionized water and stirred thoroughly. Subsequently, 3 mL of hydrolysis inhibitors and 5 mL of tetrabutyl titanate (Ti(OBu)4) were added to the solution while stirring. After stirring for 10 minutes, HNO3 was added to adjust the pH level to 2–3. The resulting mixture underwent a water bath at 60 °C for 15 h, leading to the formation of a clear and translucent sol–gel. Upon aging and transforming into xerogel form, the powders were calcined in air at a temperature of 400 °C for a duration of 2 h. Consequently, we successfully obtained Pr doped into the bulk of TiO2, which is referred to as PrX-TiO2 (X = 1) in this study. In contrast, TiO2 was synthesized using an identical method but without adding Pr(NO3)3·6H2O.Preparation of Cu/Pr–TiO2 photocatalytic catalysts: Cu SAs were anchored on Pr–TiO2 by the incipient wetness impregnation method using cupric chloride as the Cu source. In detail, 0.2 g of Pr–TiO2 powers were dispersed into a solution of cupric chloride using ultrasonic waves for 5 h at 40 °C. Afterwards, the resulting samples of Cu SAs supported on Pr–TiO2 were collected by rinsing with ultrapure water and subsequently dried in ambient air at 60 °C. These samples were labelled Cu/Pr–TiO2. It is worth noting that the final loading amount of Cu was 0.47 wt%. Similarly, the Cu/TiO2 catalyst was prepared using an identical method but with TiO2 powder.
3. Results and discussion
3.1. Microstructural features of the photocatalysts
The microstructural features of TiO2, Pr–TiO2, Cu/TiO2 and Cu/Pr–TiO2 photocatalytic systems are shown in Fig. 1. The TiO2 microstructure is characterized as follows: the outermost atomic layer consists of 2-coordinated O atoms, 3-coordinated O atoms and 5-coordinated Ti atoms; both the subatomic layer and the third layer consist of 3-coordinated O atoms and 6-coordinated Ti atoms. The reduced coordination number of outermost layer atoms (including some O atoms and Ti atoms) is concomitant with the appearance of dangling bonds. In terms of layer spacing, compared with the initial value of 1.17 Å, variation in the spacing of layers is almost negligible. With doping of Pr atoms into the TiO2 bulk to replace Ti atoms, the structure of the Pr–TiO2 surface can be characterized as: the outermost atomic layer consists of 2-coordinated O atoms, 3-coordinated O atoms and 5-coordinated Ti atoms; the subatomic layer consists of 3-coordinated O atoms and 6-coordinated Ti atoms; the third layer consists of 3-coordinated O atoms, 6-coordinated Ti atoms and 6-coordinated Pr atoms. In terms of layer spacing, compared with the initial value of 1.17 Å, the shrinkage of layer spacing from the first to the second layer and from the second to the third layer is relatively obvious, and the maximum changes are 0.11 and 0.05 Å, respectively. This is attributed to the fact that the substitution of Ti atoms by Pr atoms with a larger atomic radius leads to distortion of lattice, thus reducing the distance between the layers. When Cu SAs are anchored at the oxygen vacancy location on the surface, the Cu/TiO2 surface structure can be characterized as: the outermost atomic layer consists of 2-coordinated O atoms, 3-coordinated O atoms, 5-coordinated Ti atoms and 2-coordinated Cu atoms; both the subatomic layer and the third layer consist of 3-coordinated O atoms and 6-coordinated Ti atoms. From this, Cu SAs, Ti atoms and some O atoms on the outermost surface exhibit dangling bonds. In terms of layer spacing, compared with the initial value of 1.17 Å, shrinkage of layer spacing from the first to second layer is relatively obvious, and the maximum change is 0.40 Å. This is attributed to the fact that the substitution of Ti atoms by Pr atoms with a larger atomic radius leads to distortion of the lattice, thus decreasing the spacing between all layers. This means that Cu SAs are anchored to the outermost surface, giving rise to a shrinkage of the first atomic layer in the bulk direction, which increases the atomic interactions between the first and second layers. As Cu SAs are anchored to the surface and Pr atoms are doped into the bulk, the structure of the Cu/Pr–TiO2 surface can be characterized as: the outermost atomic layer consists of 2-coordinated O atoms, 3-coordinated O atoms, 5-coordinated Ti atoms and 3-coordinated Cu atoms; the subatomic layer consists of 3-coordinated O atoms and 6-coordinated Ti atoms; the third layer consists of 3-coordinated O atoms, 6-coordinated Ti atoms and 6-coordinated Pr atoms. Similarly, dangling bonds appear on the outermost surface at Cu SAs, Ti atoms and some O atoms. Compared with the initial value of 1.17 Å, the lattice is more distorted due to the synergic effect of Cu SAs and Pr atoms, which leads to a more pronounced contraction of the interlayer spacing from the first to the second and the second to third layers, with maximum changes of 0.65 Å and 0.13 Å, respectively.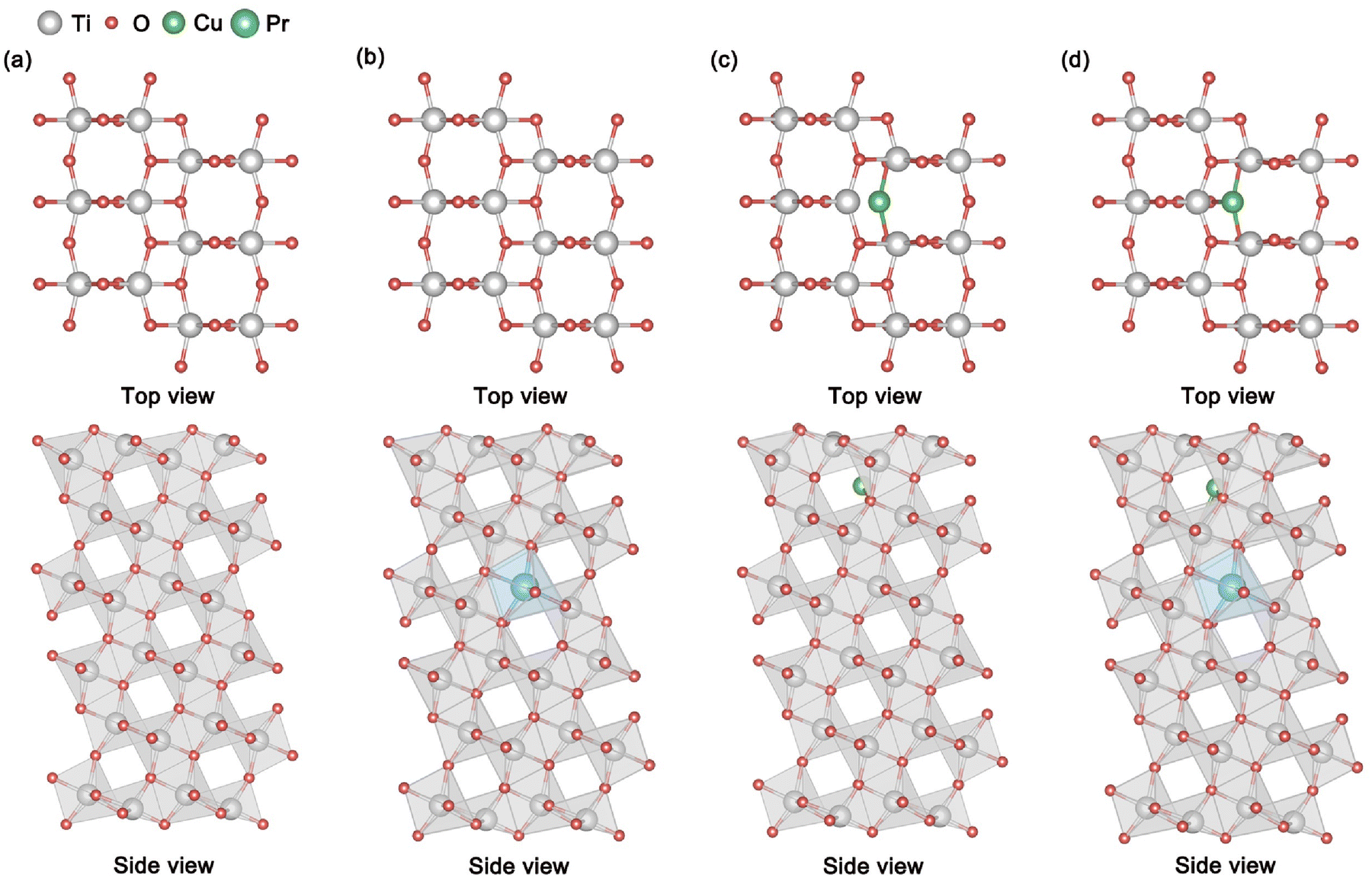 | ||
| Fig. 1 The structure of the (101) surface: (a) TiO2, (b) Pr–TiO2, (c) Cu/TiO2 and (d) Cu/Pr–TiO2. | ||
In addition, the formation energies (Ef) of TiO2, Pr–TiO2, Cu/TiO2 and Cu/Pr–TiO2 systems under oxidizing and reducing conditions were calculated. The formation energy, Ef, is calculated using the following equation,
| Ef = Esystem − Epure − nμCu + nμO − mμPr + mμTi | (2) |
3.2. Synergistic interaction and Gibbs free energy reaction
In order to investigate in depth the effect of the synergistic mechanism between Cu SAs and Pr atoms on the photocatalytic activity of TiO2, the electronic structures of TiO2, Pr–TiO2, Cu/TiO2, and Cu/Pr–TiO2 systems were analyzed in depth in a systematic and comparative manner by visualizing the results of the corresponding layer-resolved localized density of states (LDOS) and the outermost layer projected density of states (PDOS), as shown in Fig. 2 and 3. For the relaxed TiO2 (101) surface, it is clear that the changes were pronounced due to the presence of dangling bonds between Ti and O atoms located on the outermost surface. Compared with other layers, the characteristics of delocalization are more obvious. Combined with PDOS for the outermost surface, the bandgap value is 2.64 eV, and the valence band maximum (VBM) and conduction band minimum (CBM) are mainly contributed by O-2p and Ti-3d states (Fig. 2a and 3a).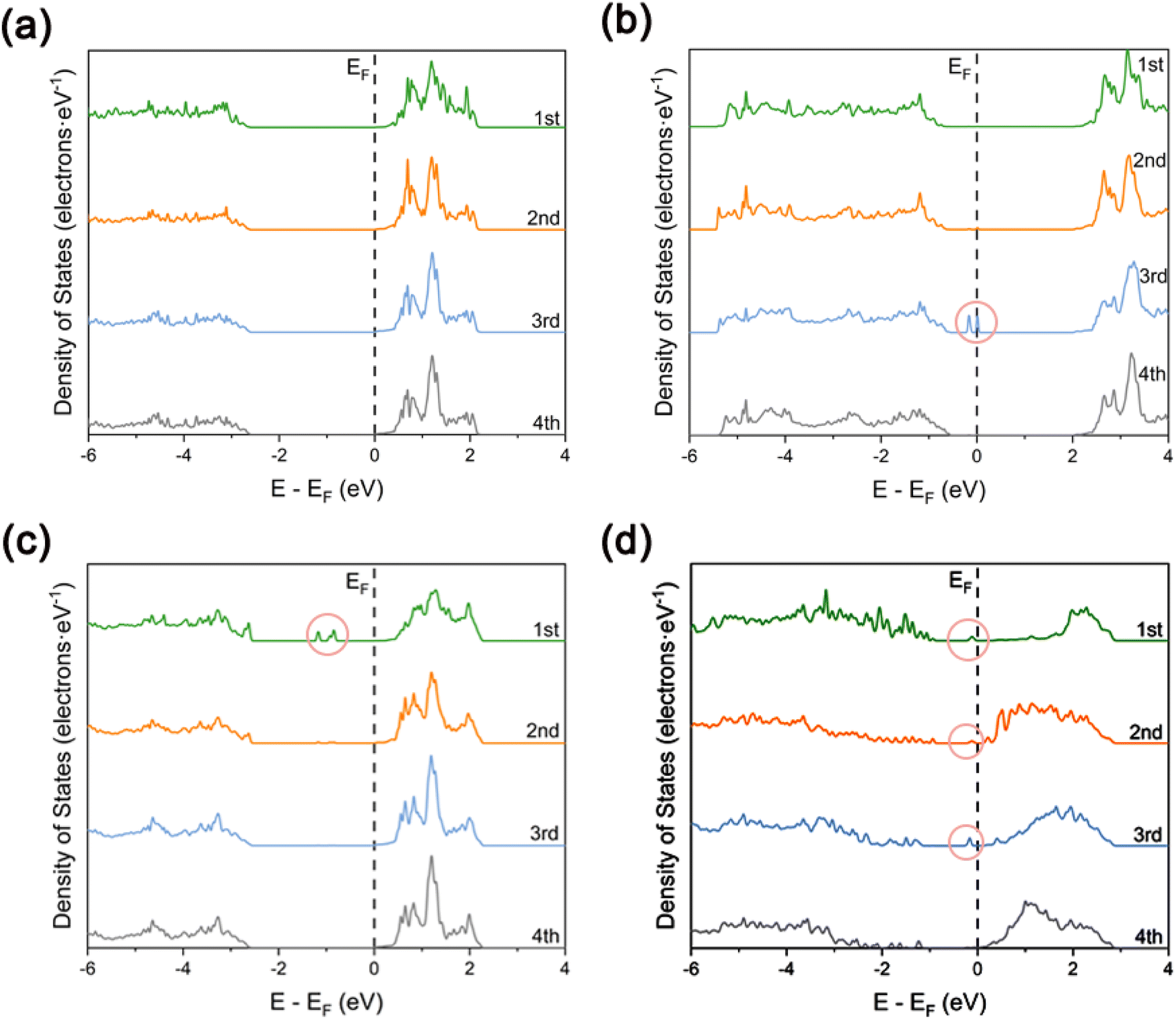 | ||
| Fig. 2 The calculated layered local density of states for the relaxation (101) surface: (a) TiO2, (b) Pr–TiO2, (c) Cu/TiO2 and (d) Cu/Pr–TiO2. (The pink circles mark the density of states peaks formed by impurity energy levels in the forbidden band). | ||
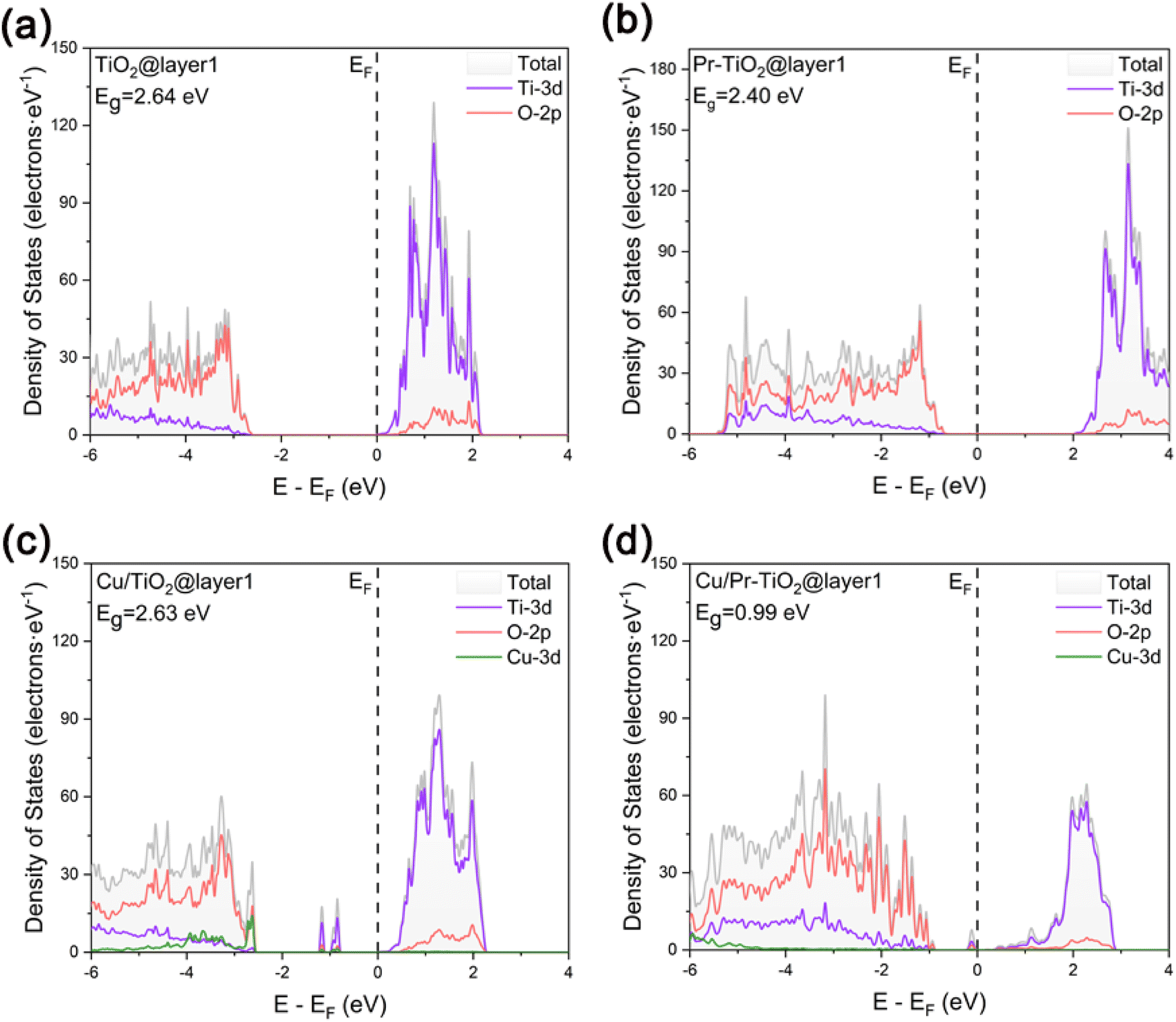 | ||
| Fig. 3 The calculated projected density of states for the relaxed (101) surface: (a) TiO2, (b) Pr–TiO2, (c) Cu/TiO2 and (d) Cu/Pr–TiO2. | ||
With the substitution of Ti atoms by Pr atoms at the surface of the third layer, the valence and conduction bands are shifted to higher energies as a whole. Moreover, new density of states peaks are generated due to impurity energy levels in the forbidden band at the surface of the third layer. The presence of these localization characteristic peaks near the VBM facilitates inhibition of the recombination of photogenerated carriers in the bulk of TiO2. In other words, more internal photogenerated carriers will migrate to the surface, which can improve the photocatalytic activity. Combined with PDOS for the outermost surface of Pr–TiO2, the bandgap value is 2.4 eV, and the VBM and CBM are likewise mainly contributed by O-2p and Ti-3d states (Fig. 2b and 3b). Compared with PDOS for the outermost surface of TiO2, the VBM is increased from −2.61 to −0.64 eV, and the CBM is likewise increased from 0.033 to 1.99 eV. In conclusion, the doping of Pr atoms has a much greater effect on the electronic structure of TiO2 bulk than on the electronic structure of its outermost surface.
When Cu SAs are anchored to the outermost surface of TiO2, this causes a significant alteration in the originally existing dangling bonds. The outermost layer shows a more pronounced delocalization characteristic compared to the other layers. This is conducive to promoting the migration of photogenerated carriers on the surface. However, new density of states peaks with localization characteristics are generated in the forbidden band of the outermost surface. These peaks can act as recombination centers, making the carriers on the surface more prone to inactivation due to the occurrence of recombination during migration, thus inhibiting the photocatalytic activity of the system. Combined with PDOS for the outermost surface of Cu/TiO2, the bandgap value is 2.63 eV. The VBM is mainly contributed by the hybridization of Cu-3d and O-2p states, the CBM is mainly contributed by Ti-3d states, and the density of states peaks present in the forbidden band are constructed by the hybridization of Ti-3d, O-2p and Cu-3d states. Compared with PDOS for the outermost surface of TiO2, the VBM is increased from −2.61 to −2.54 eV, and the CBM is decreased from 0.033 to 0.025 eV (Fig. 2c and 3c). Therefore, the anchoring of Cu SAs has a much greater effect on the electronic structure of TiO2 outermost surface than on the electronic structure of its bulk.
In contrast, Cu SAs are anchored on the outermost surface of Cu/Pr–TiO2, while Pr atoms replace Ti atoms in its bulk. In this way, Cu/Pr–TiO2 not only exhibits the characteristics of both Pr–TiO2 and Cu/TiO2, but also the synergistic interaction between Cu SAs and Pr atoms can significantly regulate the electronic structure of TiO2, which boosts the photocatalytic activity. Specifically, Cu/Pr–TiO2 shows the most pronounced delocalization characteristics of LDOS from the outermost surface to the third layer surface compared to TiO2, Pr–TiO2, and Cu/TiO2 systems. Besides, new state density peaks with localization characteristics were generated in the forbidden band near the CBM, regardless of the outermost surface or the third surface. This indicates that the synergistic effect between Cu SAs and Pr atoms in the photocatalyst can provide a directional transfer channel for the photogenerated carriers, inhibit recombination of photogenerated carriers in the bulk and on the surface, and promote the migration of more photogenerated carriers to the active sites on the surface to participate in the photocatalytic reaction. Combined with PDOS for the outermost surface of Cu/Pr–TiO2, it can be noted that both the VBM and CBM are contributed by the hybridization of Ti-3d, O-2p and Cu-3d states. Furthermore, the density of states peaks located in the forbidden band and close to the CBM are likewise contributed by the hybridization of Ti-3d, O-2p and Cu-3d states. Compared with PDOS for the outermost surface of TiO2, both the VBM and the CBM are shifted toward the Fermi energy level, the VBM is increased from −2.61 to −0.87 eV, and the CBM is decreased from 0.033 to 0.12 eV. It is worth noting that the value of the outermost surface of Cu/Pr–TiO2 is 0.99 eV, which is much smaller than that of TiO2, Pr–TiO2 or Cu/TiO2 (Fig. 2d and 3d). This is mainly attributed to the fact that the synergistic interaction between Cu SAs and Pr atoms significantly affects the surface states, resulting in a significant decrease in the bandgap value. In other words, the reduction in the bandgap value can effectively broaden the optical response range of Cu/Pr–TiO2. In summary, the synergistic interaction of Cu SAs and Pr atoms can fully utilize the advantages of Cu SA photocatalysts42,58 and rare earth metal Pr doping. On the one hand, the synergistic effect of Cu SAs and Pr atoms can promote the separation and migration of photogenerated carriers in both the bulk and the surface of TiO2, and prolong the lifetime of the photogenerated carriers; on the other hand, it can reduce the bandgap value, broaden the photo-responsive range of Cu/Pr–TiO2, and increase the electron density near the Fermi energy level, resulting in a significant enhancement in the photocatalytic activity of TiO2.
Subsequently, we analyzed the differential charge densities of the Pr–TiO2, Cu/TiO2 and Cu/Pr–TiO2 systems, as shown in Fig. 4. The differential charge density, δρ, is calculated using the following equation,
| δρ = ρAB − ρA − ρB | (3) |
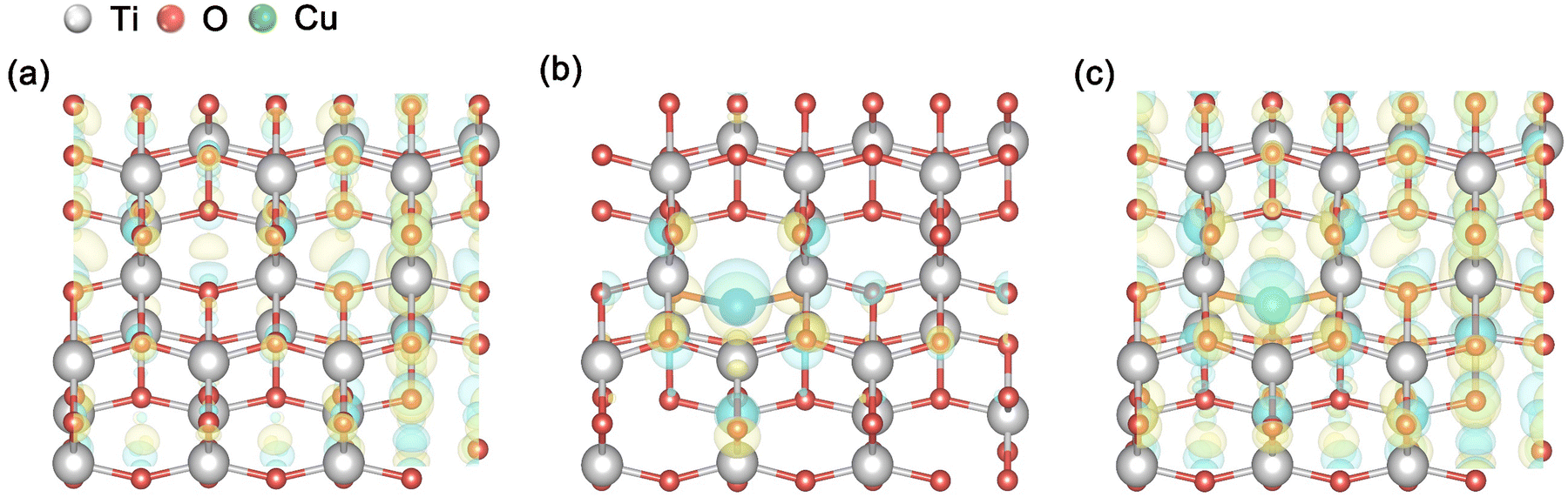 | ||
| Fig. 4 Charge density difference maps from the top view of different photocatalysts: (a) Pr–TiO2, (b) Cu/TiO2 and (c) Cu/Pr–TiO2. (Blue stands for the outflow and yellow for the inflow of electrons). | ||
To reveal more systematically the effect of the synergistic interaction between Cu SAs and Pr atoms on the photocatalytic HER activity, the ΔGH* at different active sites (O and Cu sites) of TiO2, Pr–TiO2, Cu/TiO2, and Cu/Pr–TiO2 were calculated (Fig. S3 and S4, ESI†). The ΔGH* of TiO2, Pr–TiO2, Cu/TiO2, and Cu/Pr–TiO2 are −0.24, −0.43, −0.15, and −0.34 eV, respectively, when H* is adsorbed at the O site on the surface. In this sense, the best activity for HER is displayed at the O site of the Cu/TiO2 surface, since the ΔGH* of Cu/TiO2 is the closest to 0 eV (Fig. 5a). It is worth pointing out that the variation in ΔGH* at the O site for TiO2, Pr–TiO2, Cu/TiO2, and Cu/Pr–TiO2 can be properly understood by the alteration of the p-band center. With the p-band center gradually moving away from the Fermi energy level, the corresponding reaction energy barriers become lower, and the HER reaction is performed more easily. As shown in Fig. 5b, the value of the p-band center of Pr–TiO2 (−1.76 eV) is the closest to the Fermi energy level and ΔGH* is −0.43 eV; whereas the value of p-band center of Cu/TiO2 (−3.70 eV) is the furthest away from the Fermi energy level and ΔGH* is −0.15 eV. Combined with the PDOS results, it is evident that the internal Pr-4f orbital electrons cause an upshift in the p-band center on the outermost Pr–TiO2 surface. From a thermodynamic point of view, the ability of the O site on the Pr–TiO2 surface to bind H* is the strongest compared to other systems, making it difficult to desorb H*, leading to easy inactivation of the O site.
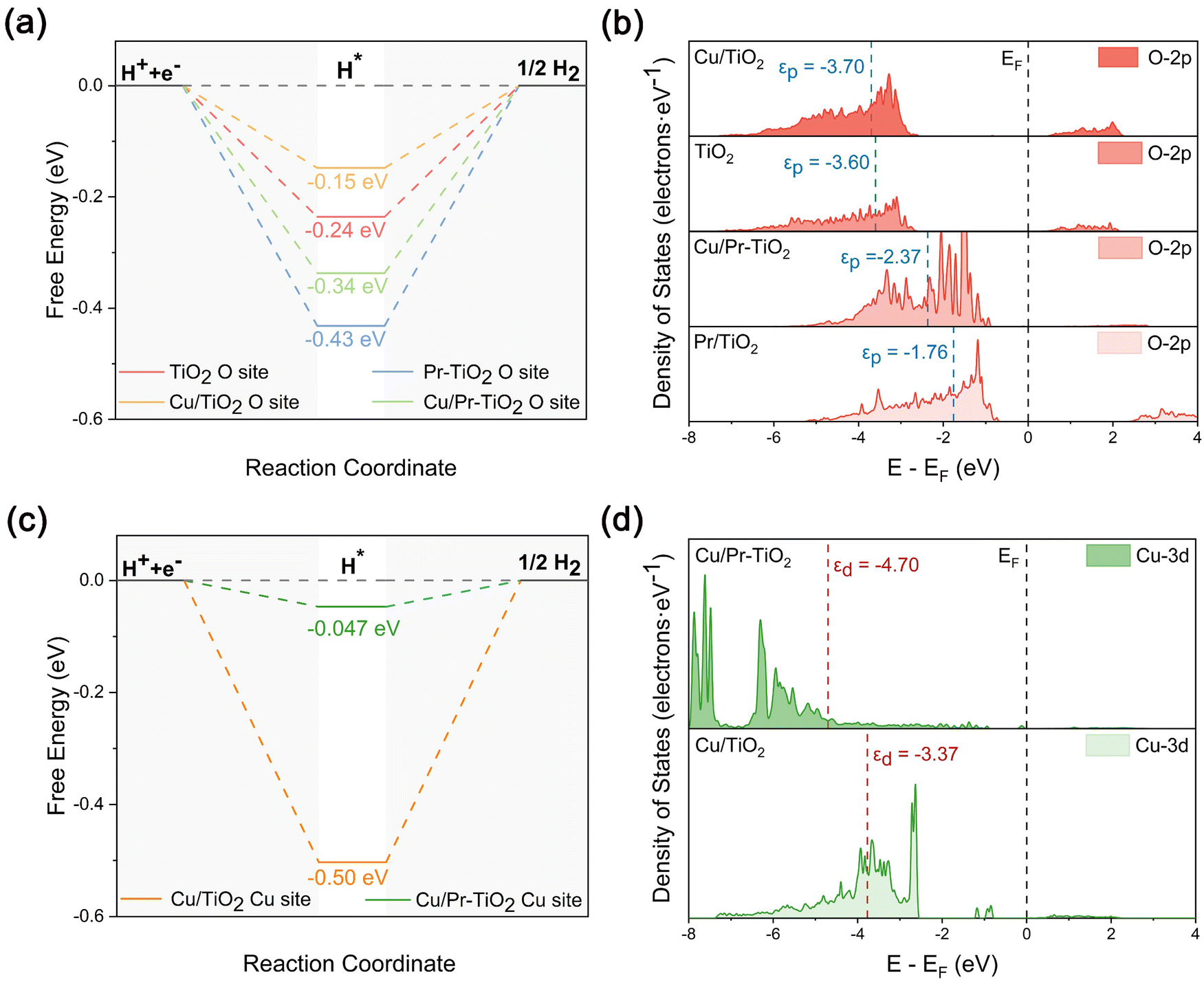 | ||
| Fig. 5 (a) The Gibbs free energy of H* adsorption at O sites of TiO2, Pr–TiO2, Cu/TiO2 and Cu/Pr–TiO2 photocatalysts. (b) The corresponding p-band center position of TiO2, Pr–TiO2, Cu/TiO2 and Cu/Pr–TiO2 photocatalysts. (c) The Gibbs free energy of H* adsorption at Cu sites of Cu/TiO2 and Cu/Pr–TiO2 photocatalysts. (d) The corresponding d-band center position of Cu/TiO2 and Cu/Pr–TiO2 photocatalysts. | ||
In contrast, when Cu SA is used as the surface HER active site, the ΔGH* of Cu/TiO2 and Cu/Pr–TiO2 are −0.50 and −0.047 eV, respectively. Clearly, the ΔGH* results at different active sites (O and Cu sites) are evidence that not only is the HER activity of Cu/Pr–TiO2 the best among all the systems, but that Cu SAs on the surface of Cu/Pr–TiO2 are also the main active sites for the HER (Fig. 5c). In addition, ΔGH* can be equally well characterized by modification of the d-band center. The reaction energy barrier of HER decreases when the d-band center shifts away from the Fermi energy level. Specifically, the d-band center of Cu/TiO2 is located at −3.37 eV, corresponding to a more negative ΔGH* (−0.50 eV) facilitating the promotion of H* adsorption on the surface Cu SAs sites. However, a lot of energy from outside is necessary to overcome the energy barrier to disengage H* on the Cu SAs sites on the Cu/TiO2 surface. For Cu/Pr–TiO2, the d-band center is located at −4.70 eV, which is far away from the Fermi energy level (Fig. 5d). Unlike Cu/TiO2, the synergistic interaction between Cu SAs and Pr atoms in Cu/Pr–TiO2 adjusted the position of the d-band center, thus significantly reducing the reaction energy barriers that have to be overcome for the HER at the Cu SA site. Therefore, the above results once again demonstrate that Cu SAs and O atoms on the Cu/TiO2 surface act as electron aggregation sites and HER active sites, respectively, which implies that the separation of electron aggregation sites and active sites significantly inhibits photocatalytic HER activity. By contrast, the synergistic interaction between Cu SAs and Pr atoms results in Cu SAs on the Cu/Pr–TiO2 surface being both electron aggregation sites and HER sites. This can effectively reduce the surface reaction energy barriers to significantly boost the photocatalytic HER activity of TiO2. That is, the synergistic interaction between Cu SAs and Pr atoms is the essential reason for the optimal photocatalytic HER activity of Cu/Pr–TiO2.
3.3. Photocatalytic hydrogen performance
To verify the reliability of the computational findings, we experimentally prepared anatase TiO2, Pr–TiO2, Cu/TiO2, and Cu/Pr–TiO2 photocatalysts. The experimental route for the Cu/Pr–TiO2 photocatalyst is shown in Fig. 6a. Sequentially, Pr atoms are doped into the TiO2 bulk and Cu SAs are anchored on the TiO2 surface by sol–gel and impregnation methods. The characterization results of Cu/Pr–TiO2 are shown in Fig. S5–S7 (ESI†). The above DFT calculations show that, on the one hand, the synergistic interaction between Cu SAs and Pr atoms can effectively promote photogenerated carrier separation and transport in the bulk and on the surface of TiO2. On the other hand, it can significantly reduce the energy barrier of the surface HER reaction. It is well known that light excites the electrons of a photocatalyst to shift from the valence band to the conduction band, and generates holes in the valence band. The recombination of electrons and holes leads to photoluminescence (PL), which generates the PL spectra. Researchers test the efficiency of photogenerated carrier separation in photocatalysts by comparing the fluorescence intensity of PL peaks of different photocatalysts. As shown in Fig. 6b, the order of fluorescence intensity of the PL peaks of the photocatalysts is TiO2 > Pr–TiO2 > Cu/TiO2 > Cu/Pr–TiO2. The results of the PL spectra clearly illustrate that the photogenerated carrier separation efficiency of TiO2 is the lowest among the four photocatalysts, owing to TiO2 producing the largest number of photons after the photogenerated electrons and holes are recombined; whereas the synergistic effect between the Cu SAs and Pr atoms make the photogenerated carrier separation efficiency of Cu/Pr–TiO2 higher than that of TiO2, Pr–TiO2, or Cu/TiO2. Time-resolved photoluminescence (TRPL) spectra are performed to characterize the lifetimes of photogenerated carriers in different photocatalysts. It can be seen that Cu/Pr–TiO2 (2.45 ns) has the longest average lifetime of charge carriers compared to Cu/TiO2 (2.23 ns), Pr–TiO2 (2.04 ns) or TiO2 (1.89 ns) in Fig. 6c. Therefore, the enhancement in photogenerated carrier separation efficiency and transport by the synergistic interaction of Cu SAs and Pr atoms was further demonstrated by the fluorescence intensity in the PL and TRPL spectra.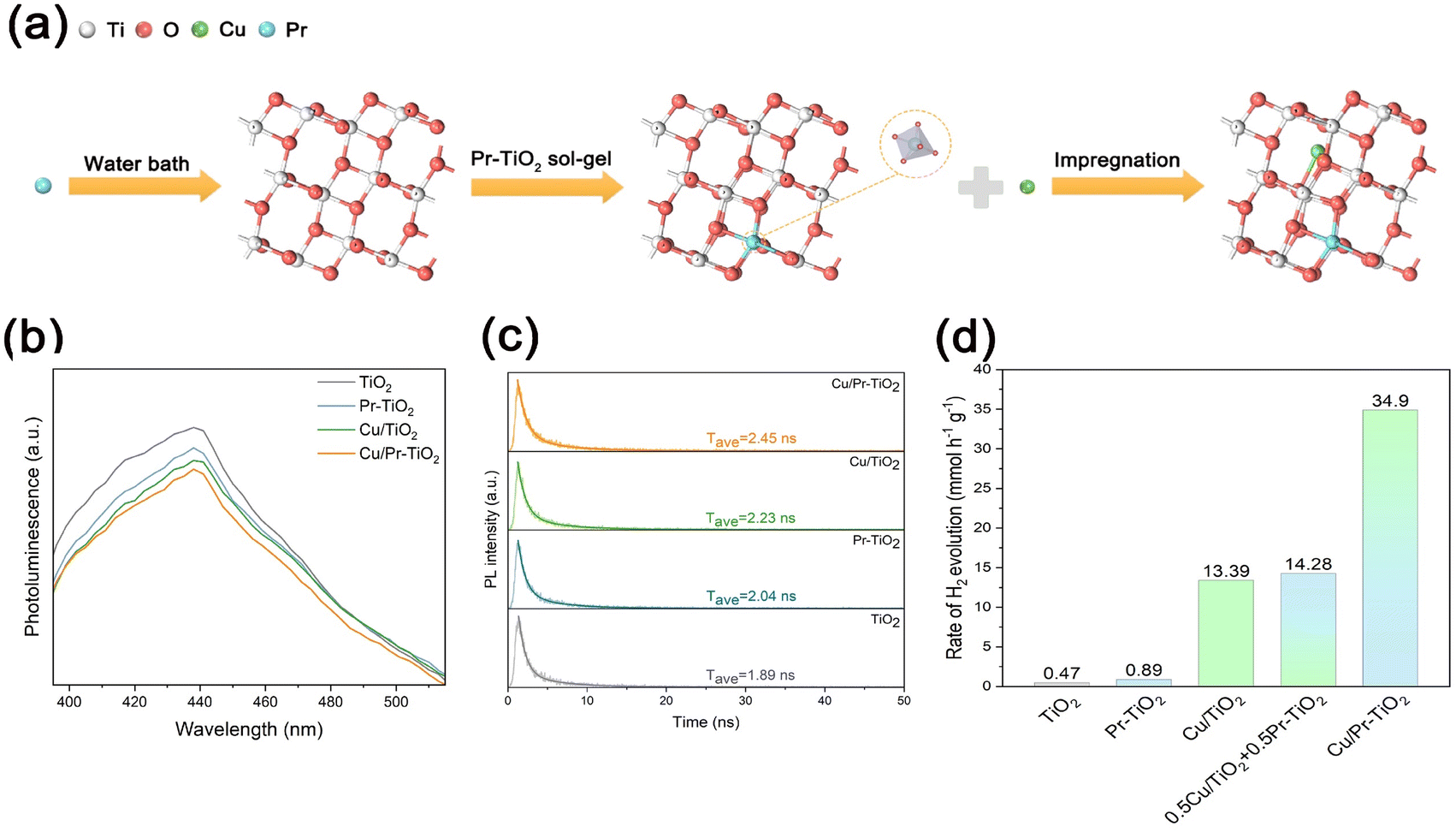 | ||
| Fig. 6 (a) Schematic illustration for the fabrication of the Cu/Pr–TiO2 photocatalyst. (b) PL spectra and (c) TRPL spectra of TiO2, Pr–TiO2, Cu/TiO2 and Cu/Pr–TiO2 photocatalysts. (d) Photocatalytic H2 evaluation rates of TiO2, Pr–TiO2, Cu/TiO2, mixed Pr–TiO2 and Cu/TiO2, and Cu/Pr–TiO2 photocatalysts. | ||
In addition, the HER activities of TiO2, Pr–TiO2, Cu/TiO2 and Cu/Pr–TiO2 photocatalysts under the same reaction conditions were evaluated, as illustrated in Fig. 6d. The Cu/Pr–TiO2 photocatalyst shows the highest HER activity with a hydrogen production rate of 34.90 mmol g−1 h−1, which is considerably higher than that of TiO2 (0.47 mmol g−1 h−1), Pr–TiO2 (0.89 mmol g−1 h−1) or Cu/TiO2 (13.39 mmol g−1 h−1). In order to demonstrate that the boost in the HER activity of Cu/Pr–TiO2 photocatalyst is due to the existence of a synergistic interaction between Cu SAs and Pr atoms rather than superposition of the effects of Cu SAs and Pr atoms, the hydrogen production efficiency of a mixture of Cu/TiO2 and Pr–TiO2 is also tested under the same reaction conditions. The test results show that the hydrogen production efficiency of Cu/Pr–TiO2 is much higher than that of the mixture of Cu/TiO2 and Pr–TiO2, which confirms the synergistic effect between Cu SAs and Pr atoms in the Cu/Pr–TiO2 photocatalyst to enhance the photocatalytic HER activity. Taken together, these results strongly illustrate that the enhanced photocatalytic activity can be attributed to the atomic synergistic interaction between Cu SAs and Pr atoms, which is in good agreement with the DFT calculation analysis.
4. Conclusions
In this work, the effect of synergistic interactions between Cu single atoms and Pr atoms on the HER reactivity of Cu/Pr–TiO2 photocatalysts was systematically investigated by a combination of DFT calculations and experiments. The microstructures, electronic structures and H* adsorption Gibbs free energies of TiO2, Pr–TiO2, Cu/TiO2, and Cu/Pr–TiO2 were compared by DFT calculation. The results indicate that the synergistic effect between Cu single atoms and Pr atoms can make the structure of Cu/Pr–TiO2 more stable than that of Cu/TiO2. This not only enables the existence of doping energy levels in Cu/Pr–TiO2 that are conducive to promoting the separation of photogenerated carriers, but also regulates the electronic structure to promote the directional migration of photogenerated carriers to the Cu single atoms for the photocatalytic hydrogen evolution reaction. Specifically, the synergistic effect between Cu single atoms and Pr atoms can transform the Cu single atom sites into efficient electron-concentration and photocatalytically reactive sites. This means that more photogenerated carriers from the bulk and surface can be aggregated to the Cu site on the Cu/Pr–TiO2 surface to participate in the photocatalytic reaction. Compared with the position of the d-band center of Cu/TiO2 (−3.37 eV), the position of the d-band center of Cu/Pr–TiO2 (−4.70 eV) is farther away from the Fermi energy level, which suggests that the synergistic effect obviously optimizes the adsorption and desorption behaviors of H atoms on the TiO2 surface. Therefore, the Cu/Pr–TiO2 photocatalyst exhibits an excellent ΔGH* of −0.047 eV, which dramatically reduces the HER reaction energy barrier to notably optimize its HER activity. Findings from the PL, TRPL spectra and hydrogen production rate not only demonstrate that Cu single atoms and Pr atoms can establish a synergistic interaction at the atomic level that surpasses the photocatalytic HER activity of both pristine TiO2 and monometallically-modified TiO2, but also strongly supports the reliability of the DFT calculations. Undoubtedly, the Cu/Pr–TiO2 photocatalyst paves the way towards the development of more green, economical and efficient active sites for photocatalytic hydrogen production.Data availability
The data supporting this article have been included as part of the ESI.† The authors will supply the relevant data in response to reasonable requests.Author contributions
Hongshun Zheng: conceptualization, methodology, formal analysis, writing – original draft. Baoye Zi: formal analysis, investigation, validation. Tong Zhou: formal analysis, visualization. Guoyang Qiu: visualization. Zhongge Luo: software. Qingjie Lu: software. Alain Rafael Puente Santiago: formal analysis. Yumin Zhang: resources. Jianhong Zhao: resources. Jin Zhang: project administration. Tianwei He: conceptualization, methodology, supervision, writing – review & editing. Qingju Liu: conceptualization, methodology, supervision, funding acquisition, writing – review & editing.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was funded by the National Key Research and Development Program of China (2022YFB3803600), the National Natural Science Foundation of China (22378346, 22368050), the Yunnan Fundamental Research Projects for funding under project 202401CF070185, the Key Research and Development Program of Yunnan Province (202302AF080002). Authors thank the Electron Microscopy Center, the Advanced Computing Center, the Advanced Analysis and Measurement Center of Yunnan University for the sample testing and computations service.References
- L. Cremonese, G. K. Mbungu and R. Quitzow, Int. J. Hydrogen Energy, 2023, 48, 19422–19436 CrossRef CAS.
- S. Wang, W. Huo, H. Feng, Z. Xie, J. K. Shang, E. V. Formo, P. H. C. Camargo, F. Fang and J. Jiang, Adv. Mater., 2023, 35, 2304494 CrossRef CAS PubMed.
- F. Xu and B. Weng, J. Mater. Chem. A, 2023, 11, 4473–4486 RSC.
- Y. Liu, Y. Sun, E. Zhao, W. Yang, J. Lin, Q. Zhong, H. Qi, A. Deng, S. Yang, H. Zhang, H. He, S. Liu, Z. Chen and S. Wang, Adv. Funct. Mater., 2023, 33, 2301840 CrossRef CAS.
- Y.-P. Zhu, J. Yin, E. Abou-Hamad, X. Liu, W. Chen, T. Yao, O. F. Mohammed and H. N. Alshareef, Adv. Mater., 2020, 32, 1906368 CrossRef CAS PubMed.
- L. Lin, T. Hisatomi, S. Chen, T. Takata and K. Domen, Trends Chem., 2020, 2, 813–824 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- A. Fujishima, K. Hashimoto and T. Watanabe, Russ. J. Electrochem., 1999, 35, 1137–1138 Search PubMed.
- Z. Li, S. Wang, J. Wu and W. Zhou, Renewable Sustainable Energy Rev., 2022, 156, 111980 CrossRef.
- Y. Wang, S. Qin, N. Denisov, H. Kim, Z. Bad'ura, B. B. Sarma and P. Schmuki, Adv. Mater., 2023, 35, 2211814 CrossRef CAS PubMed.
- Q. Guo, C. Zhou, Z. Ma and X. Yang, Adv. Mater., 2019, 31, 1901997 CrossRef CAS PubMed.
- J. Ran, H. Zhang, S. Fu, M. Jaroniec, J. Shan, B. Xia, Y. Qu, J. Qu, S. Chen, L. Song, J. M. Cairney, L. Jing and S.-Z. Qiao, Nat. Commun., 2022, 13, 4600 CrossRef CAS PubMed.
- B. Xia, B. He, J. Zhang, L. Li, Y. Zhang, J. Yu, J. Ran and S.-Z. Qiao, Adv. Energy Mater., 2022, 12, 2201449 CrossRef CAS.
- Y. Chen, D. Yang, X. Xin, Z. Yang, Y. Gao, Y. Shi, Z. Zhao, K. An, W. Wang, J. Tan and Z. Jiang, J. Mater. Chem. A, 2022, 10, 9717–9725 RSC.
- G. Shu, Y. Wang, Y. Li, S. Zhang, J.-X. Jiang and F. Wang, J. Mater. Chem. A, 2020, 8, 18292–18301 RSC.
- V. J. Babu, S. Vempati, T. Uyar and S. Ramakrishna, Phys. Chem. Chem. Phys., 2015, 17, 2960–2986 RSC.
- V. Kumaravel, S. Mathew, J. Bartlett and S. C. Pillai, Appl. Catal., B, 2019, 244, 1021–1064 CrossRef CAS.
- Q.-F. Liu, Q. Zhang, B.-R. Liu, S. Li and J.-J. Ma, Chin. J. Catal., 2018, 39, 542–548 CrossRef CAS.
- C. Tang, L. Chen, H. Li, L. Li, Y. Jiao, Y. Zheng, H. Xu, K. Davey and S. Z. Qiao, J. Am. Chem. Soc., 2021, 143, 7819–7827 CrossRef CAS PubMed.
- Q. Xu, C. Guo, B. Li, Z. Zhang, Y. Qiu, S. Tian, L. Zheng, L. Gu, W. Yan, D. Wang and J. Zhang, J. Am. Chem. Soc., 2022, 144, 4321–4326 CrossRef CAS.
- T. He, A. R. Puente-Santiago, S. Xia, M. A. Ahsan, G. Xu and R. Luque, Adv. Energy Mater., 2022, 12, 2200493 CrossRef CAS.
- S. Luo, H. Song, F. Ichihara, M. Oshikiri, W. Lu, D.-M. Tang, S. Li, Y. Li, Y. Li, P. Davin, T. Kako, H. Lin and J. Ye, J. Am. Chem. Soc., 2023, 145, 20530–20538 CrossRef CAS PubMed.
- C. Cheng, W.-H. Fang, R. Long and O. V. Prezhdo, JACS Au, 2021, 1, 550–559 CrossRef CAS PubMed.
- Y. Zhang, J. Zhao, H. Wang, B. Xiao, W. Zhang, X. Zhao, T. Lv, M. Thangamuthu, J. Zhang, Y. Guo, J. Ma, L. Lin, J. Tang, R. Huang and Q. Liu, Nat. Commun., 2022, 13, 58 CrossRef CAS.
- J. Xu, X. Chen, Y. Xu, Y. Du and C. Yan, Adv. Mater., 2020, 32, 1806461 CrossRef CAS.
- F. Donati, M. Pivetta, C. Wolf, A. Singha, C. Wäckerlin, R. Baltic, E. Fernandes, J.-G. de Groot, S. L. Ahmed, L. Persichetti, C. Nistor, J. Dreiser, A. Barla, P. Gambardella, H. Brune and S. Rusponi, Nano Lett., 2021, 21, 8266–8273 CrossRef CAS PubMed.
- J. Liu, W. Chen, J. Li and C. Cui, ACS Catal., 2018, 8, 2230–2235 CrossRef CAS.
- F.-X. Zhang, P.-Q. Hu, Z.-M. Zhang, J.-H. Gong and D.-H. Wang, Rare Met., 2023, 42, 606–613 CrossRef CAS.
- X.-J. Pan, Z.-Q. Yang, Y. Xu, M. Wang, X.-W. Huang, Z.-Y. Feng, Q. Zhong and X.-L. Peng, Rare Met., 2023, 42, 2725–2735 CrossRef CAS.
- S. Bingham and W. A. Daoud, J. Mater. Chem., 2011, 21, 2041–2050 RSC.
- B. Zheng, J. Fan, B. Chen, X. Qin, J. Wang, F. Wang, R. Deng and X. Liu, Chem. Rev., 2022, 122, 5519–5603 CrossRef CAS PubMed.
- S. Huang, N. Zhu, Z. Lou, L. Gu, C. Miao, H. Yuan and A. Shan, Nanoscale, 2014, 6, 1362–1368 RSC.
- J. Wang, H. Wang, S. Zuo, X. Jin, B. Zheng, R. Deng, W. Liu and J. Wang, Environ. Sci.: Nano, 2020, 7, 3333–3342 RSC.
- F. Sordello, I. Berruti, C. Gionco, M. C. Paganini, P. Calza and C. Minero, Appl. Catal., B, 2019, 245, 159–166 CrossRef CAS.
- J.-C. G. Bünzli, Chem. Rev., 2010, 110, 2729–2755 CrossRef PubMed.
- S. Stojadinović, N. Radić, B. Grbić, S. Maletić, P. Stefanov, A. Pačevski and R. Vasilić, Appl. Surf. Sci., 2016, 370, 218–228 CrossRef.
- J. Liu, X. Kong, L. Zheng, X. Guo, X. Liu and J. Shui, ACS Nano, 2020, 14, 1093–1101 CrossRef CAS PubMed.
- Z. Zhan, Z. Sun, Z. Wei, Y. Li, W. Chen, S. Li and S. Pang, Nano Res., 2024, 17, 3493–3515 CrossRef CAS.
- X. Wang, Y. Zhu, H. Li, J.-M. Lee, Y. Tang and G. Fu, Small Methods, 2022, 6, 2200413 CrossRef CAS.
- M. T. Colomer, K. J. Duarte, A. L. Ortiz, D. F. Mercado and L. M. Ballesteros-Rueda, Mater. Charact., 2021, 182, 111536 CrossRef CAS.
- A. Mikolajczyk, E. Wyrzykowska, P. Mazierski, T. Grzyb, Z. Wei, E. Kowalska, P. N. A. Caicedo, A. Zaleska-Medynska, T. Puzyn and J. Nadolna, Appl. Catal., B, 2024, 346, 123744 CrossRef CAS.
- B.-H. Lee, S. Park, M. Kim, A. K. Sinha, S. C. Lee, E. Jung, W. J. Chang, K.-S. Lee, J. H. Kim, S.-P. Cho, H. Kim, K. T. Nam and T. Hyeon, Nat. Mater., 2019, 18, 620–626 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 13115–13118 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- Y. Li, H. Zhou, S. Cai, D. Prabhakaran, W. Niu, A. Large, G. Held, R. A. Taylor, X.-P. Wu and S. C. E. Tsang, Nat. Catal., 2024, 7, 77–88 CrossRef CAS.
- G. Ren, M. Zhou and H. Wang, J. Am. Chem. Soc., 2024, 146, 6084–6093 CrossRef CAS.
- J. Zhang, C. Peng, H. Wang and P. Hu, ACS Catal., 2017, 7, 2374–2380 CrossRef CAS.
- L. Y. Isseroff and E. A. Carter, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 235142 CrossRef.
- H. H. Pham, G. T. Barkema and L.-W. Wang, Phys. Chem. Chem. Phys., 2015, 17, 26270–26276 RSC.
- F. Tran, J. Schweifer, P. Blaha, K. Schwarz and P. Novák, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 085123 CrossRef.
- K. B. Vural, Ç. Kaderoğlu and Ş. Ellialtıoğlu, Appl. Surf. Sci., 2023, 613, 156042 CrossRef CAS.
- H. G. Yang, C. H. Sun, S. Z. Qiao, J. Zou, G. Liu, S. C. Smith, H. M. Cheng and G. Q. Lu, Nature, 2008, 453, 638–641 CrossRef CAS PubMed.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, Energy Environ. Sci., 2010, 3, 1311–1315 RSC.
- B. Xiao, C. Shen, Z. Luo, D. Li, X. Kuang, D. Wang, B. Zi, R. Yan, T. Lv, T. Zhou, J. Zhang and Q. Liu, Chem. Eng. J., 2023, 468, 143650 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nh00196f |
| This journal is © The Royal Society of Chemistry 2024 |