DOI:
10.1039/D4NH00252K
(Communication)
Nanoscale Horiz., 2024,
9, 1582-1586
Enhancing DNA-based nanodevices activation through cationic peptide acceleration of strand displacement†
Received
4th June 2024
, Accepted 12th July 2024
First published on 13th July 2024
Abstract
Dynamic DNA-based nanodevices offer versatile molecular-level operations, but the majority of them suffer from sluggish kinetics, impeding the advancement of device complexity. In this work, we present the self-assembly of a cationic peptide with DNA to expedite toehold-mediated DNA strand displacement (TMSD) reactions, a fundamental mechanism enabling the dynamic control and actuation of DNA nanostructures. The target DNA is modified with a fluorophore and a quencher, so that the TMSD process can be monitored by recording the time-dependent fluorescence changes. The boosting effect of the peptides is found to be dependent on the peptide/DNA N/P ratio, the toehold/invader binding affinity, and the ionic strength with stronger effects observed at lower ionic strengths, suggesting that electrostatic interactions play a key role. Furthermore, we demonstrate that the cationic peptide enhances the responsiveness and robustness of DNA machinery tweezers or logic circuits (AND and OR) involving multiple strand displacement reactions in parallel and cascade, highlighting its broad utility across DNA-based systems of varying complexity. This work offers a versatile approach to enhance the efficiency of toehold-mediated DNA nanodevices, facilitating flexible design and broader applications.
New concepts
This work demonstrates the novel concept of using cationic peptides, specifically oligoarginine, to accelerate toehold-mediated strand displacement (TMSD) reactions in DNA nanotechnology. Unlike previous methods that rely on complex modifications or harsh conditions to accelerate TMSD, this approach utilizes simple, bio-inspired peptides. The cationic peptides act as DNA chaperones, enhancing the interaction between the target and invading strands through electrostatic interactions. This not only accelerates the strand displacement process but also enables the operation of DNA nanodevices at lower ionic strengths, a condition not easily achievable with existing methods. This work provides new insights into the role of electrostatic interactions in DNA nanotechnology. It demonstrates that simple peptides can be used to manipulate and control DNA interactions, opening up new possibilities for the design and development of dynamic DNA nanodevices. The ability to operate these devices at lower ionic strengths could also expand their potential applications in biological and medical settings.
|
Introduction
DNA nanotechnology has emerged as a powerful tool for the development of intricate nanodevices with diverse applications.1–3 Central to the functionality of these devices is the toehold-mediated strand displacement (TMSD) reaction, a fundamental mechanism in DNA nanotechnology, enabling the dynamic control and actuation of DNA nanostructures.4–6 However, the inherent slow kinetics of TMSD impedes the realization of complex DNA nanodevices with rapid operation and robust performance, essential for applications such as biosensing,7 diagnostics,8,9 and targeted drug delivery.10 To address this challenge, various approaches have been explored to accelerate TMSD as well as the TMSD-driven operation of DNA nanodevices. Toehold length and sequence optimization are frequently used to enhance the initial binding rate of the invading strand to the target strand, thus accelerating the reaction.11–14 Longer toeholds can lead to unwanted leak reactions, particularly in complex DNA networks involving cross talks or cascaded events. Other approaches developed in recent years include biological acceleration (e.g. through biocatalytic or protein binding pathway),15,16 freeze–thaw cycling,17 manipulation of nanointerfaces,18 use of polar organic solvents,19 introduction of block copolymers,20,21 supramolecular polymer templating,22 and host–guest interactions.23 They also raise concerns, such as the stability of the DNA nanostructures, complex synthetic procedures and accompanied costs, operational complexity, or unintended side reactions, hindering their applicability to complex DNA devices.
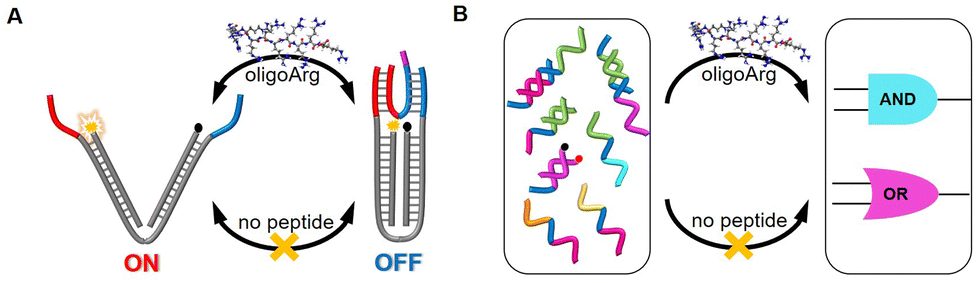
Biological systems can exert exquisite control over genetic information, as evidenced by the dynamic interplay between DNA and enzymes. Enzymes such as DNA polymerases, helicases, and ligases catalyze a myriad of DNA transactions, ranging from replication and repair to recombination and transcription, with precision reminiscent of a finely tuned mechanism. The highly conserved amino acid residues in the active sites of these enzymes play crucial roles in catalyzing and modulating DNA transactions.24,25 Inspired by the importance of the DNA–peptide interactions observed in these native enzymatic systems, we seek to design synthetic peptide catalysts capable of enhancing the efficiency and versatility of DNA operations. In this work, we reported the employment of a positively charged peptide, oligoarginine (oligoArg) of varying lengths, acting like a DNA chaperone26–28 to manipulate the toehold-mediated strand displacement reactions. These peptides bind target and invading DNA strands via noncovalent interactions, such as electrostatic forces, facilitating proximity between the toehold domains and invading DNA, thereby accelerating subsequent strand displacement. Furthermore, the presence of the oligoArg peptide can cause the promotion of the DNA mechanical and computing operations, particularly pronounced at low ionic strengths (Fig. 1). Compared to the literature (ref. 11–23), our approach is distinguished by its ease of operation, such as the time-saving synthesis of peptides and the self-assembly of DNA/peptides, along with the lack of spatial limitations or DNA sequence constraints.
 |
| | Fig. 1 Schematic illustration of the cationic peptide-promoted activation of (A) DNA mechanical and (B) computing devices. | |
Results and discussion
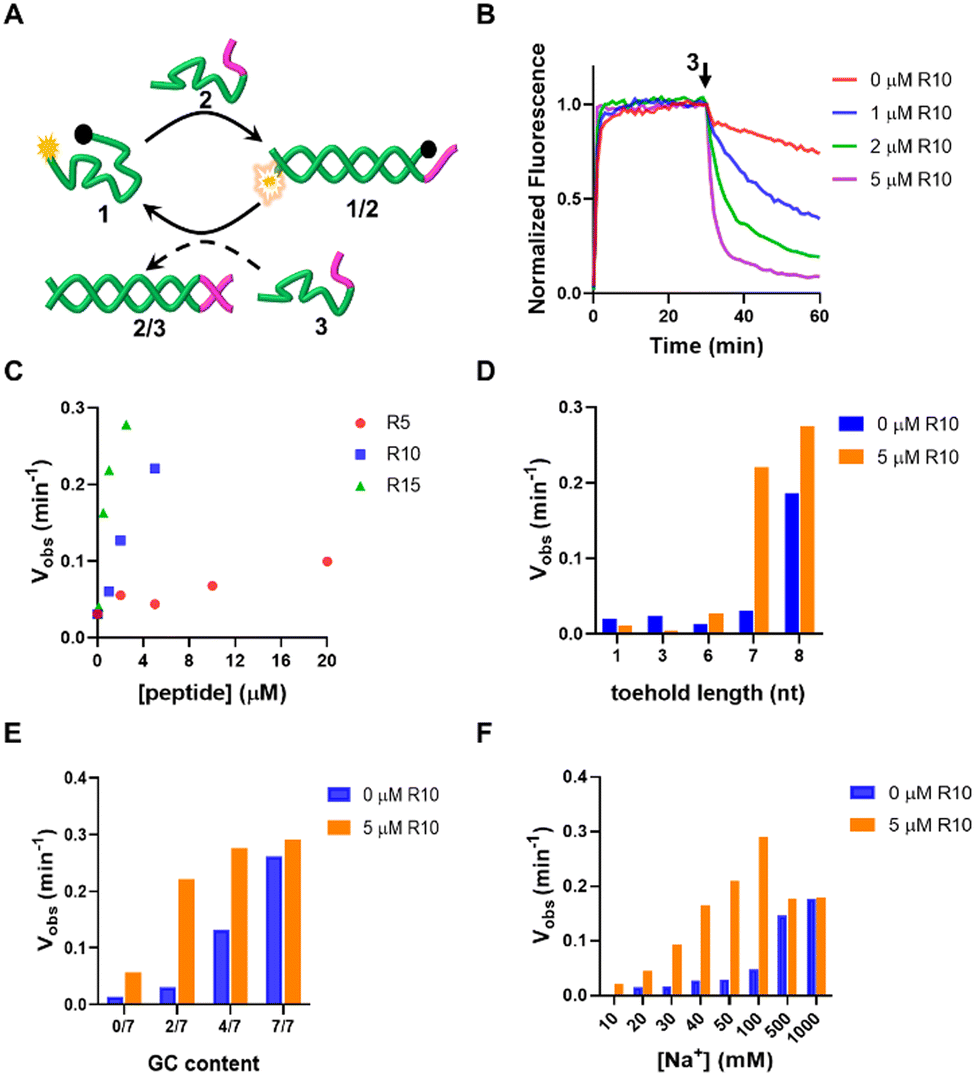
First, we investigated the effect of oligoArg on the strand displacement reactions of the DNA, with 1/2 as the targets and 3 as the invader strand. 1 is labeled with the fluorophore Cy5 at the 5′-end and the Black Hole quencher 2 (BHQ2) at the 3′-end. Upon hybridization of 1 to the target, Cy5 and BHQ2 separated, resulting in a low-FRET state and high fluorescence of Cy5. In the presence of 3 that is fully complementary to 2, 2 is displaced due to toehold-mediated strand displacement reactions with 1/2. This causes 1 to transform into a randomly coiled state, resulting in a high-FRET state and low fluorescence of Cy5 (Fig. 2A). It can be observed that when 1/2 has a toehold length of 7 nt (for the sequences used in this work, see Table S1, ESI†), the strand displacement was not evident. When R10 containing 10 Arg residues (N/P: 0.56) was added, the reaction rate and yield were significantly accelerated (Fig. 2B), which was dependent on the length of oligoArg and the concentrations (Fig. 2C and Fig. S1, ESI†). R5 with N/P ratio 0.28 to 1/2 caused a similar strand displacement rate to R10 with N/P ratio 0.11, and R15 with N/P ratio 0.02 to 1/2, which may be attributed to the weaker interaction of R5 with DNA than R10 and R15 (Fig. S2, ESI†). The cationic side chain group and the amide backbone can form interactions with DNA through charged, hydrogen bonding or van der Waals forces, and may both contribute to the enhancement of the strand displacement reactions. Therefore, we introduced a cationic polymer (polyethylenimine, PEI with Mw 800) with no amide or a neutral oligoGly peptide (six residues, Gly6) with no side chain group, instead of oligoArg. We found that the addition of PEI caused the acceleration of the strand displacement, while the Gly6 did not (Fig. S3, ESI†), confirming the importance of the electrostatic interactions.
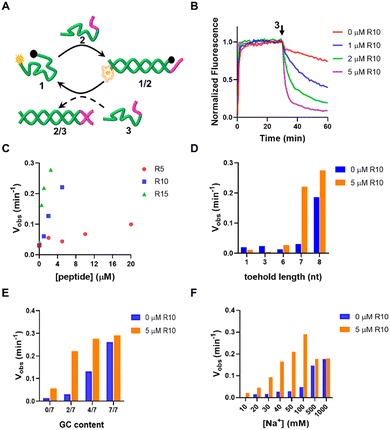 |
| | Fig. 2 (A) Schematic diagram of the strand displacement process. The yellow exploding and black circle represent the Cy5 fluorophore and BHQ2 quencher, respectively. (B) Effects of different concentrations of R10 on time-dependent fluorescence changes following DNA strand displacement reaction. [DNA] = 1 μM. (C) Effect of the lengths and concentrations of the oligoArg peptides on the Vobs (taken from the normalized fluorescence changes during the initial three minutes). Effect of oligoArg R10 on the Vobs of the strand displacement reaction with different (D) toehold length, (E) GC content in the toehold sequence and (F) in the presence of different concentrations of Na+. | |
We then explored the enhancement of the strand displacement reactions by R10 in the target DNA with varying toehold lengths (Fig. 2D and Fig. S4, ESI†). When the toehold consisted of 1 nt (DNA 4) or 3 nt (DNA 6), we observed no strand displacement after adding the invading strands 5 and 7, even after the addition of R10. However, with a 6 nt (DNA 8) or 8 nt (DNA 10) toehold length, there was only a slight increase in the rate of the strand displacement in the presence of the invaders 9 and 11, after the addition of R10. In a parallel investigation, we changed the GC content in the toehold sequence (Fig. 2E and Fig. S5, ESI†). It was observed that when the toehold comprised only A and T bases or G and C bases, the strand displacement reactions cannot occur or proceeded very fast. R10 enhanced the strand displacement less significantly compared to the scenario involving the 1/2 target. The toehold length or the GC content in the toehold both influenced the rate of strand displacement through affecting the affinity of the invading strand to the target duplex. These results indicate that the positive oligoArg peptide can only accelerate toehold-mediated strand displacement reactions with an appropriate rate. It's worth noting that the accelerating effect of the R10 peptide was particularly pronounced at lower Na+ concentrations. At 1.0 M Na+, the strand displacement occurred rapidly, and the addition of R10 didn't further enhance this process (Fig. 2F and Fig. S6, ESI†). This is likely because the high concentration of Na+ shields the charges of DNA, hindering the electrostatic interactions between the DNA and the peptides. However, when Na+ was only 10 mM, the formation of the 1/2 duplex was notably slow, and the strand displacement reactions between 1/2 and 3 couldn't be observed. R10 significantly accelerated both events, underscoring the importance of DNA/peptide charged interactions between phosphate and guanidinium groups, as shown in the theoretical structural model (Fig. S7, ESI†), in promoting the strand displacement reactions. As the reaction temperature was elevated, the acceleration effect was still observed, although less significant (Fig. S8, ESI†), indicating that the R10/DNA complexes were robust. In addition to R10, the other charged peptides, such as oligolysine (K10), the block copolymer (e.g. R5K5) or the alternating copolymer (RK)5, can also accelerate the strand displacement (Fig. S9, ESI†). The allowed operation under low ionic strengths may expand the applications of the DNA-based dynamic systems. In the following, we will examine the application of this strategy in the DNA systems of greater complexity, including DNA tweezer or logic gates.
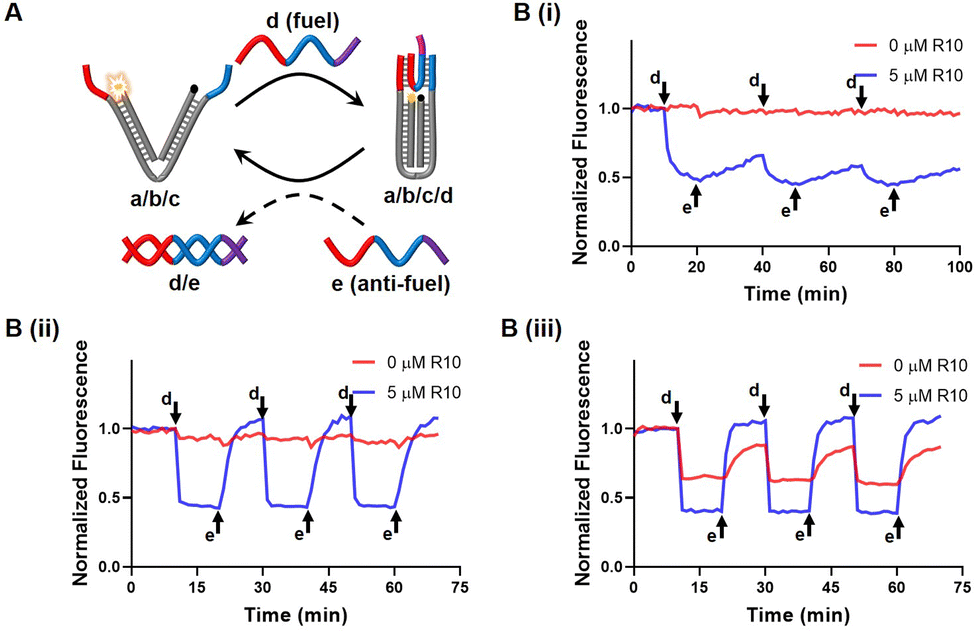
We designed a DNA nanotweezer, which consists of two nucleic acid arms (a and b) bridged by a “reporter” nucleic acid (c) that includes a Cy5/BHQ2 pair at its ends (Fig. 3A). A fuel strand d hybridized with the dangling ends of the tweezer, leaving a 7 nt toehold, and generating the closed configuration of the tweezer. This can be demonstrated by quenching of the Cy5 fluorescence. An anti-fuel strand e, which is fully complementary to the fuel strand, was introduced to release d from the tweezer, resulting in the opening of the tweezer, as indicated by the fluorescence recovery of Cy5. By cyclically adding fuel and anti-fuel strands, the DNA tweezer could be closed and opened reversibly, switching the fluorescence between an Off and On state. Fig. 3B illustrated that the efficiency of these processes was influenced by the Na+ concentration, impacting the hybridization yield (Fig. 3B). Effective operation of the DNA tweezer was observed only when the Na+ concentration exceeded 70 mM. The introduction of R10 successfully triggered activation of the tweezer at 70 mM Na+, albeit only moderately accelerating the configuration transformation at 100 mM Na+.
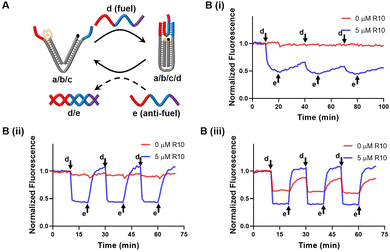 |
| | Fig. 3 (A) Working principle of the DNA nanotweezer. (B) Cyclic operation of the DNA nanotweezer accelerated by R10, in the presence of different concentrations of Na+, (i) [Na+] = 30 mM, (ii) [Na+] = 70 mM, (iii) [Na+] = 100 mM. | |
The AND and OR logic gates were constructed using multiple strand displacement reactions in parallel and cascade, comprising a threshold, a fuel, a reporter (reporter B), two gates (gate A and gate B), and two inputs (A and B) (Fig. 4A). Simultaneous introduction of inputs A and B (1 ∧ 1) to the AND gate led to a notable rise in fluorescence, and either input A (1 ∧ 0) and input B (0 ∧ 1), or no input (0 ∧ 0) resulted in a slight fluorescence enhancement. In the case of the OR gate, inputs of (1 ∨ 0), (0 ∨ 1), or (1 ∨ 1) produced increases in the output signal, while no significant increase in output signal was observed for (0 ∨ 0), indicating the accurate functioning of the OR gate. These gates can be operated when exposed to a concentration of 1 M Na+ or 12 mM Mg2+, with no impact from R10 (Fig. S10, ESI†). However, when the concentration of Na+ was 0.1 M, the addition of R10 significantly improved the signal change over time (Fig. 4B–E). When R10 was absent, the AND gate reacted a 1 ∧ 1 input at a rate of 0.0026 min−1, whereas in the presence of R10, it responded at a rate of 0.0207 min−1 (Fig. 4F). For the OR gate, in the absence of R10, the rate values for input 1 ∨ 1, 1 ∨ 0 and 0 ∨ 1 were 0.0071 min−1, 0.0045 min−1 and 0.0034 min−1, respectively, and the corresponding values in the presence of R10 were 0.0288 min−1, 0.0246 min−1, and 0.0273 min−1, respectively (Fig. 4G). It is noted that the fluorescence changes were also observed in the absence of the inputs (i.e. leak in the strand displacement) (Fig. S11, ESI†). It can be concluded that oligoArg can drastically promote the rates of logic operations at low Na+ concentrations.
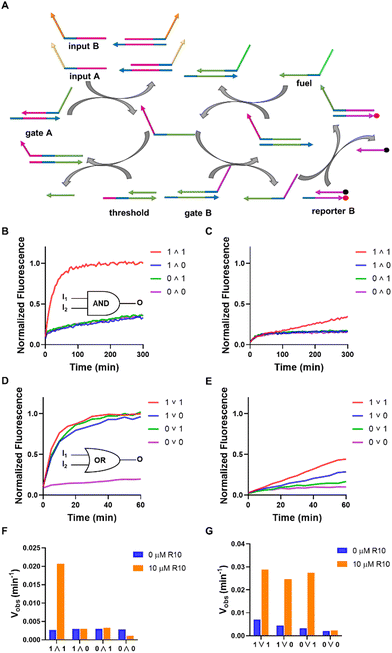 |
| | Fig. 4 (A) Schematic illustration of AND or OR gates based on seesaw gates. The 3′ ends of the DNA strands are indicated by arrowheads. The colors in the strands indicate complementary sequences. The red and black circles represent Cy5 and BHQ2, respectively. (B)–(E) Time-dependent fluorescence signal changes for the (B) AND gate and (D) OR gate in the presence of R10, and for the (C) AND gate and (E) OR gate in the absence of R10. The concentrations of gate A, gate B, fuel, and reporter B were 2×, 1×, 1×, 1.5×, respectively, with 1× equal to 0.5 × 10−6 M. The concentrations of the input strands were 0.1 × (0, logic off) or 0.9 × (1, logic on). Vobs (taken from the initial thirty minutes) of the (F) AND and (G) OR gate in the presence or absence of R1. | |
Conclusions
In summary, we have reported employment of positive-charged oligoArg peptides to accelerate the toehold-mediated strand displacement reactions of DNA. The accelerating effect was dependent on the N/P ratio, the toehold length and sequence, as well as the ionic strength. We additionally demonstrate that the introduction of a minute quantity of the peptide boosted the operations of a DNA machine, as well as two logic gates (AND and OR), particularly notable under low ionic strengths, thereby affirming the significance of electrostatic interactions in strand displacement reactions. The approach presented by this work may be applicable to the operations of a variety of complex DNA nanodevices, and can contribute to the development of dynamic DNA nanotechnology.
Experimental
Materials
The oligonucleotides were purchased from HIPPO BIOTECHNOLOGY and purified with HPLC. The peptides were purchased from Shanghai ZiYu Biotechnology Co. Ltd and SciLight Biotechnology, and the purity was above 98%. Sequences of oligonucleotides and peptides are listed in Tables S1 and S2, respectively, ESI.† All freeze-dried DNA and peptides were stored at −20 °C upon receipt. The DNA strands were dissolved in Milli-Q water to prepare a 100 μM stock solution. The peptides were dissolved in Milli-Q water to prepare a 2 mM stock solution, shaken for 30 min, and then left at room temperature for one day before use. Na2HPO4/NaH2PO4 salts, NaCl, and dimethyl sulfoxide (DMSO) were purchased from Aladdin (purity of all chemicals was over 99%). PEI (MW = 800) was purchased from MREDA. Gly6 was purchased from Macklin, and the purity was above 98%.
Fluorescence analysis of the strand displacement reaction
The fluorescence was measured using a Cary Eclipse fluorescence spectrophotometer (Agilent Technologies, Australia). Excitation and emission wavelengths were set at 648 nm and 670 nm, respectively, with slit widths at 5 nm. All kinetic experiments were conducted in phosphate buffer at pH 7.0. Initially, the peptide was mixed with 1 in the buffer to measure the fluorescence intensity at 0 minutes. Immediately afterward, 2 was added to the mixture, and fluorescence intensity was recorded every minute. After 30 minutes, 3 was introduced, and the fluorescence intensity was monitored until the end of the experiment. The peptide was added to the DNA solution at various N/P ratios, representing the ratio of positively charged amino groups of the peptide to negatively charged phosphate groups of the DNA. The fluorescence data were normalized as follows:| | | Normalized fluorescence = Ft/F∞ | (1) |
where Ft is the fluorescence intensity at a given time t, and F∞ is the maximum fluorescence intensity in the reaction.
CD spectra
The CD spectra were measured in a Jasco J-815 spectropolarimeter in nitrogen at a certain flow rate and the following parameters were used during the measurement: optical length: 0.5 cm, scanning speed: 100 nm min−1, bandwidth: 10 nm, wavelength range: 200–350 nm.
Fluorescence analysis of the DNA nanotweezer
First, R10 was mixed with a/b/c in phosphate buffer (pH 7) containing different concentrations of Na+. The fuel/anti-fuel strands were sequentially injected to monitor the fluorescence changes after the fluorescence intensity was stable. a/b/c is thermally annealed at 1 μM concentration in phosphate buffer (pH 7). The thermal annealing was carried out by slowly decreasing the temperature of the solution from 95 °C to 4 °C over a period of ∼2 h in a thermal cycler.
Fluorescence analysis of AND/OR logic gates
All the experiments were performed in phosphate buffer (pH 7.0). In AND or OR gates, input A (0.45 × 10−6 M or 0.05 × 10−6 M) and/or input B (0.45 × 10−6 M or 0.05 × 10−6 M) were added to gate A (1 × 10−6 M), gate B (0.5 × 10−6 M), fuel (0.5 × 10−6 M), reporter B (0.75 × 10−6 M), and threshold (0.6 × 10−6 M for AND gate, 0.3 × 10−6 M for OR gate) in the absence or the presence of R10.
Molecular dynamics simulations
The DNA models were built using the NAB tool in Ambertools, and the R10 model was built using Avogadro, with all amino homogenized protons. All molecular dynamic simulations used Gromacs 2020.6 software using amber14sb_parmbsc1 force fields and TIP3P models. During the simulation, Na+ was introduced into the system to equilibrate the charge in solution. At the same time, the conjugate gradient method was used to minimize the energy of the system in steps of 0.01 nm. Prior to formal dynamic simulations, we performed 100 ps restricted molecular dynamics simulations on DNA and R10 molecules. V-rescale and Parrinello–Rahman methods were used to maintain the temperature and pressure of the system at 295.15 K and 1 standard atmosphere in steps of 2 fs. The neighbor list was generated by the Verlet method, and PME and a cut-off were used to treat the remote electrostatic interaction (rcoulomb = 1.0 nm) and van der Waals interaction (rvwd = 1.0 nm) under periodic boundary conditions, respectively, and to perform long-range correction of van der Waals effects. The first 5 ns were removed, and the Gromos clustering method was used to perform cluster analysis on all subsequent frames (cutoff = 0.3 nm).
Author contributions
Z. G. W. conceived, designed, and supervised the experiments. X. Z. designed and performed the experiments. Z. G. W. and X. Z. collected and analyzed the data. S. X. performed the simulations. R. D., S. X. and X. W. provided suggestions on the work. Z. G. W. and X. Z. wrote the manuscript. All authors have given approval to the final version of the manuscript.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors are grateful for the National Science Foundation China (52173194), Beijing Natural Science Foundation (2232017), and Fundamental Research Funds for the Central Universities (buctrc201902).
Notes and references
- S. Ranallo, C. Prevost-Tremblay, A. Idili, A. Vallee-Belisle and F. Ricci, Nat. Commun., 2017, 8, 15150 CrossRef CAS
 .
.
- S. W. Oh, A. Pereira, T. Zhang, T. Li, A. Lane and J. Fu, Angew. Chem., Int. Ed., 2018, 57, 13086–13090 CrossRef CAS .
- J. Zhu, L. Zhang, T. Li, S. Dong and E. Wang, Adv. Mater., 2013, 25, 2440–2444 CrossRef CAS PubMed .
- S. Ranallo, D. Sorrentino and F. Ricci, Nat. Commun., 2019, 10, 5509 CrossRef CAS PubMed .
- A. Peil, L. Xin, S. Both, L. Shen, Y. Ke, T. Weiss, P. Zhan and N. Liu, ACS Nano, 2022, 16, 5284–5291 CrossRef CAS .
- L. N. Green, H. K. K. Subramanian, V. Mardanlou, J. Kim, R. F. Hariadi and E. Franco, Nat. Chem., 2019, 11, 510–520 CrossRef CAS .
- X. Xu, L. Wang, K. Li, Q. Huang and W. Jiang, Anal. Chem., 2018, 90, 3521–3530 CrossRef CAS PubMed .
- X. Gong, W. Zhou, D. Li, Y. Chai, Y. Xiang and R. Yuan, Biosens. Bioelectron., 2015, 71, 98–102 CrossRef CAS .
- M. You, G. Zhu, T. Chen, M. J. Donovan and W. Tan, J. Am. Chem. Soc., 2015, 137, 667–674 CrossRef CAS PubMed .
- M. Xiao, W. Lai, F. Wang, L. Li, C. Fan and H. Pei, J. Am. Chem. Soc., 2019, 141, 20354–20364 CrossRef CAS .
- D. Y. Zhang and E. Winfree, J. Am. Chem. Soc., 2009, 131, 17303–17314 CrossRef CAS PubMed .
- A. J. Genot, D. Y. Zhang, J. Bath and A. J. Turberfield, J. Am. Chem. Soc., 2011, 133, 2177–2182 CrossRef CAS PubMed .
- D. Lysne, T. Hachigian, C. Thachuk, J. Lee and E. Graugnard, J. Am. Chem. Soc., 2023, 145, 16691–16703 CrossRef CAS PubMed .
- X. Yang, Y. Tang, S. M. Traynor and F. Li, J. Am. Chem. Soc., 2016, 138, 14076–14082 CrossRef CAS .
- N. J. Bonde, Z. J. Romero, S. Chitteni-Pattu and M. M. Cox, Nucleic Acids Res., 2022, 50, 2201–2210 CrossRef CAS PubMed .
- F. Li, Y. Tang, S. M. Traynor, X. F. Li and X. C. Le, Anal. Chem., 2016, 88, 8152–8157 CrossRef CAS .
- Y. Zhu, X. Xiong, M. Cao, L. Li, C. Fan and H. Pei, Sci. Adv., 2023, 9, eaax7983 CrossRef CAS PubMed .
- M. Oishi and K. Saito, ACS Nano, 2020, 14, 3477–3489 CrossRef CAS .
- T. Zhang, C. Shang, R. Duan, A. Hakeem, Z. Zhang, X. Lou and F. Xia, Analyst, 2015, 140, 2023–2028 RSC .
- J. Wang, H. Raito, N. Shimada and A. Maruyama, Small, 2023, 19, 2304091 CrossRef CAS .
- N. Shimada, K. Saito, T. Miyata, H. Sato, S. Kobayashi and A. Maruyama, Adv. Funct. Mater., 2018, 28, 1707406 CrossRef .
- W. Engelen, S. P. W. Wijnands and M. Merkx, J. Am. Chem. Soc., 2018, 140, 9758–9767 CrossRef CAS .
- D. Kankanamalage, J. H. T. Tran, N. Beltrami, K. Meng, X. Zhou, P. Pathak, L. Isaacs, A. L. Burin, M. F. Ali and J. Jayawickramarajah, J. Am. Chem. Soc., 2022, 144, 16502–16511 CrossRef CAS PubMed .
- P. Soultanas and D. B. Wigley, Nucleic Acids Res., 2002, 30, 4051–4060 CrossRef CAS .
- G. Ziegelin, T. Niedenzu, R. Lurz, W. Saenger and E. Lanka, Nucleic Acids Res., 2003, 31, 5917–5929 CrossRef CAS .
- M. S. Kwak, W. J. Rhee, Y. J. Lee, H. S. Kim, Y. H. Kim, M. K. Kwon and J. S. Shin, Redox Biol., 2021, 40, 101858 CrossRef CAS PubMed .
- R. Moriyama, N. Shimada, A. Kano and A. Maruyama, Biomaterials, 2011, 32, 7671–7676 CrossRef CAS .
- D. Kashiwagi, S. Sim, T. Niwa, H. Taguchi and T. Aida, J. Am. Chem. Soc., 2018, 140, 26–29 CrossRef CAS PubMed .
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *
*