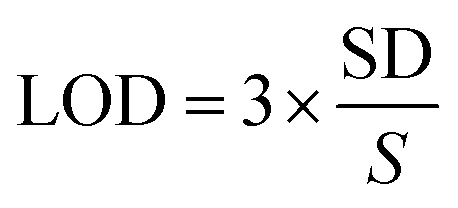DOI:
10.1039/D4NJ02548B
(Paper)
New J. Chem., 2024,
48, 15324-15337
Activity augmentation of a functionalized 2D conjugated polymer matrix with iron vanadate nano-bulbs for real-time detection of levofloxacin†
Received
5th June 2024
, Accepted 30th July 2024
First published on 8th August 2024
Abstract
In this study, we present a pioneering approach for the electrochemical detection of levofloxacin (LVO) in human blood serum utilising iron vanadate nano-bulbs FeVO4 (FVO) decorated on a two-dimensional conjugated polymer matrix g-C3N4 (CN). The iron vanadate nano-bulbs are synthesised via a simple hydrothermal approach and integrated into a 2D conjugated polymer matrix, providing a high surface area and numerous active sites for improved sensing of LVO with a detection limit of 0.06 μM, a broader linear range of 0.25–320 μM, and high sensitivity and selectivity towards LVO. The findings highlight the significant role of vanadium in electrocatalysis, unveiling the sensor's enhanced electrochemical properties and repeatability, reproducibility and stability. This work underscores the potential of FVO nano-bulbs in developing efficient and sensitive electrochemical sensors for pharmaceutical monitoring, providing a valuable tool for real-time drug toxicity analysis.
1. Introduction
Levofloxacin (LVO) is a chiral fluorinated carboxy quinolone developed as a broad-spectrum antibacterial medication with a chemical name of (S)-9-fluoro-2,3-dihydro-3-methyl-10-(4-methyl-1-piperazinyl)-7-oxo-7H-pyrido[1,2,3-de]-1,4-benzoxazine-6-carboxylicacid hemihydrate (C18H2FN3O4·1/2H2O). It is a fluoroquinolone type of compound that is administered for a wide range of ailments like chronic bronchitis, acute bacterial sinusitis, uncomplicated urinary tract infection, nosocomial pneumonia, acute bacterial prostatitis, acute pyelonephritis, skin or skin structure infections, and inhalational anthrax, and as prophylaxis.1 During the COVID-19 pandemic period, numerous worldwide reports have documented the use of LVO for COVID-19 management and treatment, often in response to unusual symptoms.2 The use of LVO carries a higher risk of serious side effects such as tendinitis, tendon rupture, peripheral neuropathy, CNS consequences, hepatotoxicity, and Stevens–Johnson syndrome (SJS), as well as primary side effects including photosensitivity, nausea, diarrhoea, headache, hyper/hypoglycaemia, and seizures.3 In addition, insufficient in vivo degradation of LVO causes 85% of the drug to discharge through emiction, causing the accumulation of LVO in the environment. This upsets the microecological equilibrium and ends up in the proliferation of bacterially resistant antibiotics in the environment.4,5 This also leads to indirect intake of LVO through the food chain by humans as well as animals, leading to the risk of excessive LVO residues in the meat, milk and other metabolic byproducts of animals.6 Considering these circumstances and exigency, it is essential to develop a simple, cost-effective, reliable and efficient transducer to perform trace-level analysis of the environment, food and biological samples. Other conventional techniques like chromatography, fluorometry, capillary electrophoresis, enzyme-linked immunosorbent assay, chemiluminescence and molecular imprinted polymers have some setbacks like pretreatment of the sample, simultaneous detection, time conception, expensive maintenance, requirement of harmful solvents, need for skilled professionals and setup, etc.7–9 Hence, electrochemical sensors are a valuable replacement owing to their high sensitivity, selectivity, rapid response time, cost-effectiveness, compatibility and portability. Moreover, being an electrochemically active substance, LVO can be determined through electrochemical analysis.
In the current study, we utilised a glassy carbon electrode (GCE) to detect LVO. Though the GCE has more advantageous features such as cost-effectiveness, versatility, customizability, portability, and compatibility, the bare GCE shows low sensitivity owing to its sluggish electron transfer kinetics at the interface.10–12 Hence, it needs to be modified with an appropriate electrode material to facilitate electron transfer at the electrode solution interface to sense the targeted analyte. Hence, we used a 2D conjugated polymer matrix – graphitic carbon nitride – CN, as a substrate. The repeated in-plane heptazine units in CN make up the layered two-dimensional arrangement, which stands out for its unique electronic configuration, remarkable stability, and conductivity, and simple pyrolysis can synthesize CN via one-step calcination of melamine. CN is a highly effective catalyst but stands back due to its low activity, and to improve the electrocatalytic activity of CN further, it was functionalised with bimetallic oxides of iron and vanadium. Unlike many other d-transition metals, vanadium is abundantly available and exhibits variable oxidation states ranging from 3 to 5, along with different coordination numbers. Fe is a naturally abundant and inexpensive element widely used across various fields. Its variable oxidation states (0 to 3) provide exceptional properties, particularly for applications involving redox reactions. Transition-metal vanadates, particularly those with the formula ABO4, where A consists of Ti, Fe, In, Sm, and Cr, have a diverse variety of applicability in many fields such as Li-ion batteries, sensors, photocatalysis, water splitting, and other optoelectronic applications.13 Among various metal vanadates, iron vanadate FVO stands out as a crucial candidate due to its exceptional catalytic and electrochemical properties.14 This is attributed to its mixed oxidation states, noncytotoxicity, accessible oxygen vacancies, and ease of band gap engineering.15 Iron and vanadium have an indirect band gap and are electrochemically active with improved redox reactions than other vanadates. Thus, it is a promising electrocatalyst.16,17 Electrocatalytic performance is optimized by interface engineering of the CN sheets with FVO. The significant interfacial interactions between two components of a heterostructure provide enormous electrochemically active sites, prominently boosting the electrocatalytic activity by accelerating the electron transfer process at the interface.18 The interaction between FVO and CN influences the redistribution of charges at the interface and regulates the electron transfer kinetics during the electrochemical process. This provides a possible pathway for achieving high electrocatalytic activity of the proposed electrocatalyst towards the detection of LVO. To our knowledge, electrocatalysts based on FVO/CN have not been previously reported for the voltammetric detection of fluoroquinolone antibiotics. Therefore, in this study, we created a new electrocatalyst with an iron vanadate-supported graphitic carbon nitride nanocomposite that was perfect for detecting trace amounts of LVO in the human blood sample.
2. Experimental procedure
2.1. Materials
Chemicals, reagents, solvents and analytes used in the synthesis procedure and analysis of the sensor were of analytical grade and used without any further purification unless mentioned. Phosphate buffer solution (PBS), which is prepared using monosodium hydrogen phosphate (NaHPO4) and disodium hydrogen phosphate (Na2HPO4) dissolved in Milli-Q water with a resistivity of 18.2 MΩ cm at a temperature of 25 ± 2 °C and [Fe(CN)6]3−/4− redox species was prepared with potassium ferrocyanide K4[Fe(CN)6], potassium ferricyanide K3[Fe(CN)6], potassium chloride (≥99% KCl) as a supporting electrolyte, and Milli-Q-water. The reagents and chemicals employed in the synthesis process include iron(III) nitrate nonahydrate (≥99%-Fe(NO3)3·9H2O), ammonium metavanadate (≥99%-NH4VO3), polyvinylpyrrolidone (≥99%-C6H9NO)n, and melamine (≥98.5%-C3H6N6). Analytes used were levofloxacin (≥99%-C18H20FN3O4·1/2H2O) and 5-fluorouracil (≥99%-C4H3FN2O2). Interferents like ascorbic acid (≥99%-C6H8O6), oxalic acid (≥99.5%-C2H2O4), uric acid (≥99%-C5H5N4O3), lactic acid (≥92% FISHER), sodium chloride (NaCl), magnesium chloride (MgCl2), and calcium chloride (CaCl2) were of analytical quality and were bought from FISHER, LOBA, Sigma-Aldrich, Alfa and SRL India.
2.2. Sample preparation and electrode fabrication
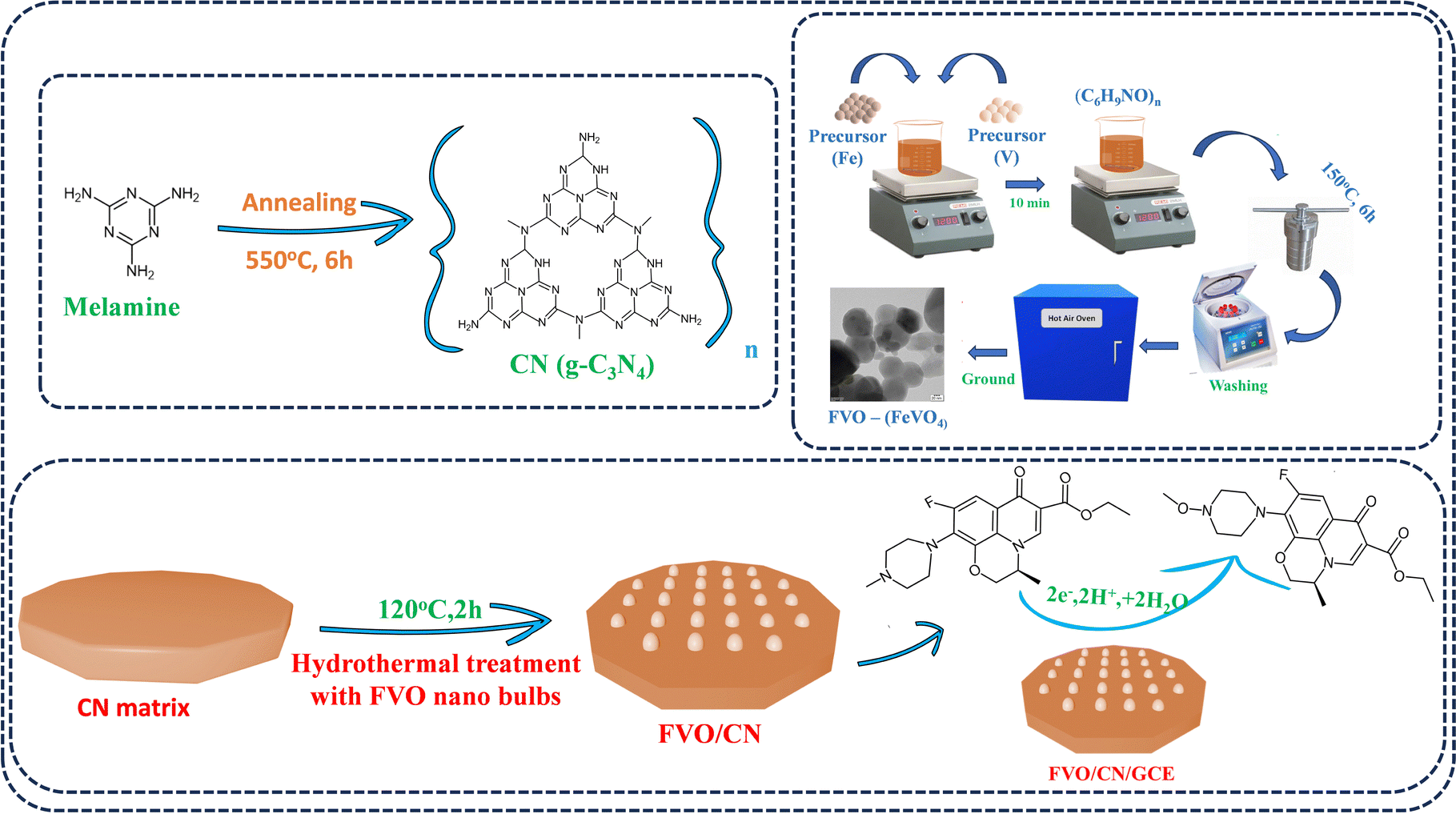
In this study, iron vanadate nano-bulbs were synthesized using a hydrothermal approach. The hydrothermal approach allows for controlled synthesis of FVO nano-bulbs in terms of size, shape, and structure. This approach enables us to tailor the properties of the nano-bulbs for specific applications and typically results in high-purity FVO nano-bulbs due to the homogeneous nucleation process taking place in a closed system under high temperature and pressure conditions. This method often yields materials with a suitable size and morphology, which is crucial for achieving consistent and predictable performance in various applications such as sensing. By adjusting the synthesis parameters, such as temperature, pressure, and reactant concentrations, we can optimize the properties of material nano-bulbs to meet specific requirements, such as enhanced catalytic activity or improved stability. Overall, the hydrothermal approach offers a versatile and effective method for synthesising FVO nano-bulbs with tailored properties, paving the way for various technological advancements and applications.19,20 Initially, 0.1 M Fe(NO3)3·9H2O was dissolved in 70 mL of deionized (DI) water. Then, 0.1 M NH4VO3 was added to the solution and stirred at 80 °C for 20 minutes. Following this, 500 mg of (C6H9NO)n was introduced into the solution, resulting in a bright yellow suspension. This suspension was then transferred to a 100 mL autoclave and heated at 150 °C for 6 hours. After cooling to room temperature, the resulting solution was thoroughly washed with ethanol and DI water multiple times to eliminate impurities. The yellow slurry was then dried and calcined at 600 °C for 3 hours in a muffle furnace and subsequently ground into a fine powder. The resulting material was designated as FVO. Carbon nitride (CN) was prepared through a one-step annealing process, wherein 5 g of melamine was heated in a tubular furnace at a ramp rate of 5 °C min−1 until reaching 550 °C and maintained at this temperature for 6 hours. The light-yellow product was ground into a fine powder using a mortar and pestle. A typical hydrothermal method was employed to synthesize a FVO/CN nanocomposite. CN was dispersed in distilled water using ultrasonication for 1 hour, followed by probe sonication for 30 minutes at an amplitude of 55% with a pulse ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 to exfoliate the CN sheets further. The prepared FVO was then added dropwise to the stirred CN dispersion in the desired ratio, and the mixture was stirred for a few minutes before being transferred to a Teflon-lined stainless-steel autoclave. This mixture was heated at 120 °C for 2 hours. After naturally cooling to room temperature, the contents were centrifuged at 6000 rpm and 15 °C. The residue was dried overnight at 60 °C and ground into a fine powder for further analysis. After polishing the GCE surface to a mirror finish with alumina slurry of 0.5 μm using a micro cloth pad, the electrode was ultrasonicated in ethanol and water with a low-frequency sonicator for 2 minutes to clean the surface residues. The sonicated GCE was rinsed with ethanol and left to dry, and 6 μL of material dispersion was drop casted on the electrode surface and allowed to dry in a hot air oven at 40 °C (Scheme 1). Real samples were prepared from human blood samples which were bought from SRM Medical Hospital and Research Centre. Samples were centrifuged at 10000 rpm for 10 minutes to discard the contaminants. With the help of a filtering membrane, the precipitate was filtered and the supernatant liquid was collected and diluted with 0.1 M PBS (pH = 7.4). The dilution is further used to detect LVO in the real sample analysis.
:2 to exfoliate the CN sheets further. The prepared FVO was then added dropwise to the stirred CN dispersion in the desired ratio, and the mixture was stirred for a few minutes before being transferred to a Teflon-lined stainless-steel autoclave. This mixture was heated at 120 °C for 2 hours. After naturally cooling to room temperature, the contents were centrifuged at 6000 rpm and 15 °C. The residue was dried overnight at 60 °C and ground into a fine powder for further analysis. After polishing the GCE surface to a mirror finish with alumina slurry of 0.5 μm using a micro cloth pad, the electrode was ultrasonicated in ethanol and water with a low-frequency sonicator for 2 minutes to clean the surface residues. The sonicated GCE was rinsed with ethanol and left to dry, and 6 μL of material dispersion was drop casted on the electrode surface and allowed to dry in a hot air oven at 40 °C (Scheme 1). Real samples were prepared from human blood samples which were bought from SRM Medical Hospital and Research Centre. Samples were centrifuged at 10000 rpm for 10 minutes to discard the contaminants. With the help of a filtering membrane, the precipitate was filtered and the supernatant liquid was collected and diluted with 0.1 M PBS (pH = 7.4). The dilution is further used to detect LVO in the real sample analysis.
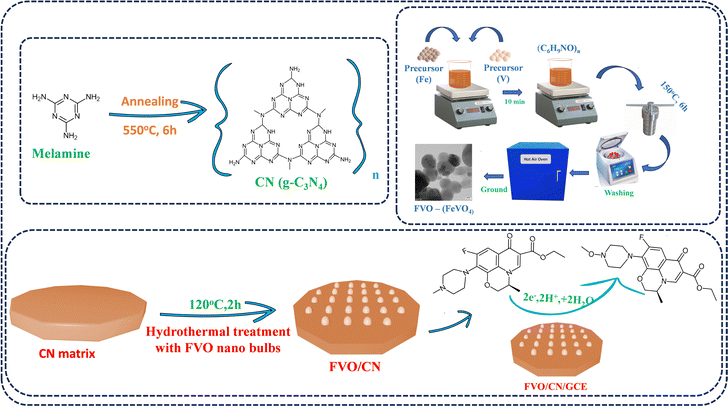 |
| | Scheme 1 Schematic representation demonstrating the synthesis of FVO/CN composite and its subsequent application in electrochemical sensing of LVO. | |
2.3. Characterization
Micromorphology analysis was done by using a transmission electron microscope (HRTEM – JEOL Japan, JEM-2100 Plus), and a scanning electron microscope (HRSEM – Thermo scientific Apreo S) equipped with an energy dispersive X-ray (EDX) spectrometer. The lateral crystalline structure and analytical properties of the prepared materials are analysed by using X-ray crystallography with (XRD – BRUKER USA D8 Advance, Davinci), within the range of 5–90° (2θ) and the chemical composition of the samples and specific chemical bonds were analysed with a FT-IR spectrometer (SHIMADZU, IRTRACER 100) in the wavenumber region of 4000–500 cm−1. Raman spectroscopy was performed using a HORIBA, LabRam HR Evolution spectrometer with a laser wavelength of 532 nm and a resolution of 0.4 cm−1. The oxidation state of the surface elements was investigated using X-ray photoelectron spectroscopy (XPS – Physical Electronics). pH studies were carried out by using Labman – LMPH 10 attached to a glass electrode. Electrochemical analysis was performed using an electrochemical workstation (CHI-760E) at 25 °C by purging nitrogen gas with Glassy carbon electrode as a working electrode, Ag/AgCl as a reference electrode and platinum as a counter electrode. For exfoliation and dispersion purposes, an ultrasonicator (Labman – LMCU – 9) with a frequency of 40 KHz and a power of 200 W was used. In addition to this, a probe sonicator (Labman – PRO 500) was employed with an amplitude of 55% and a pulse ratio of 3:2. The annealing process was done using a high-temperature tubular furnace (Ants. – 1200 SPT) with rated power – 3 kW, 230 V and 15 A. Centrifugation of the reaction mixtures was carried out by using a cooling centrifuge (Remi-C-24 plus) with a supply of 220 V, 50 Hz, 10 A.
3. Results and discussion
3.1. XRD and XPS
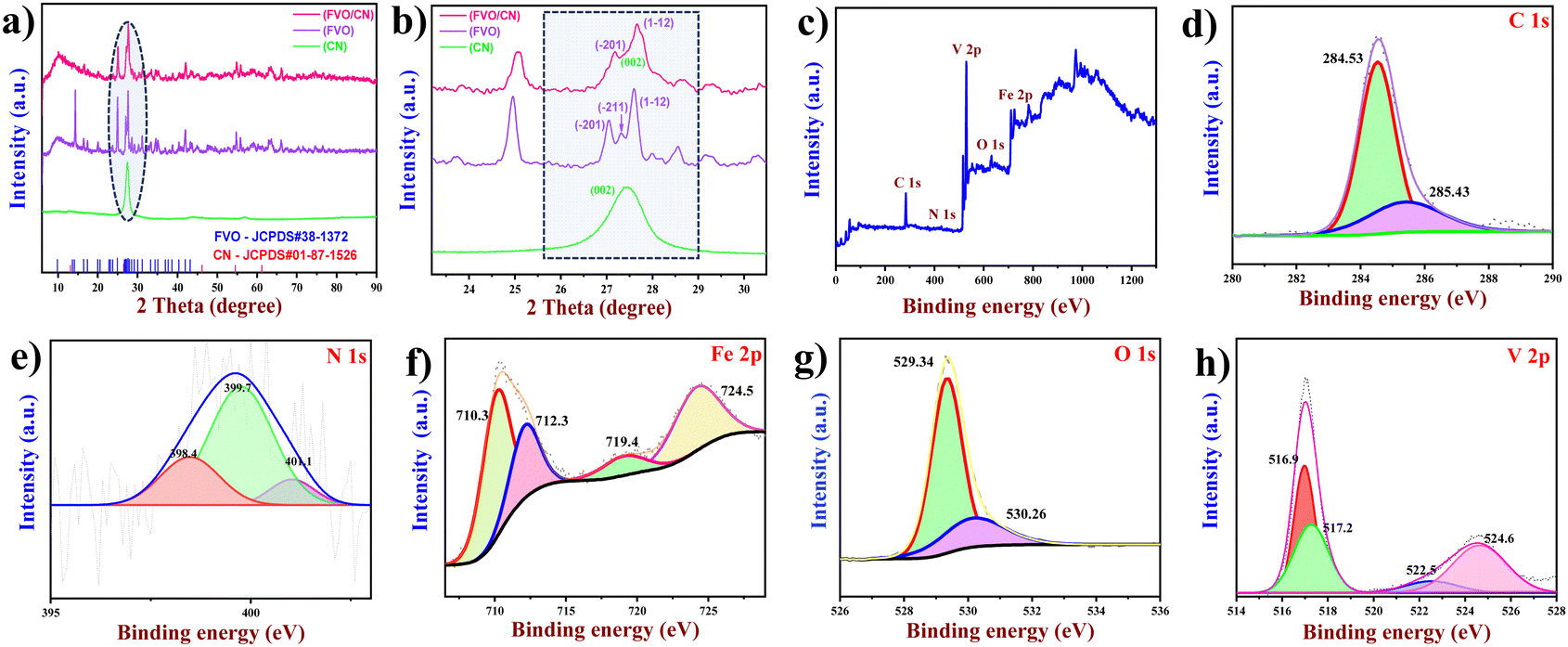
The crystalline structure, crystalline size, and phase purity of the FVO nanostructure, CN nanosheets, and FVO/CN nanocomposite were analysed using X-ray diffraction spectroscopy. In Fig. 1(a), well-defined diffraction peaks corresponding to FVO were observed at 2θ values of 16.45, 17.41, 20.05, 22.84, 23.29, 23.73, 24.96, 27.32, 27.60, 27.98, 28.19, 28.55, 29.15, 30.29, 30.61, 31.14, 31.53, 32.08, 33.35, 34.59, 35.11, and 42.03, corresponding to the diffraction planes (011), (![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 11), (110), (0
11), (110), (0![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) 1), (2), (12), (012), (01), (11), (12), (10), (200), (03), (120), (12), (3), (1), (21), (201), (03), (22), and (0
1), (2), (12), (012), (01), (11), (12), (10), (200), (03), (120), (12), (3), (1), (21), (201), (03), (22), and (0![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) 3), respectively. The obtained diffraction patterns match with the crystal database reference (JCPDS#38-1372) and confirm the single phase of the synthesized material.21 Diffraction peaks of FVO were distinct, well-defined, and more intense. This pattern closely aligns with FVO's anorthic (triclinic) crystalline phase. And CN (g-C3N4) shows reflections at 2θ values of 12.64° and 27. 37° corresponding to the graphitic planes of (100) and (002) due to the in-plane structural packing of repeated triazine units and aromatic rings stacked at the interlayer (JCPDS#01-87-1526).22 A broad peak around 57° persists in the refined crystallization pattern of the hexagonal space group with AB stacking of the triazine-based layers (Fig. 1(a)).23,24 For a clearer comparison, Fig. 1(b) displays an expanded XRD pattern of the prepared nanostructures spanning the range from 22.4 to 30°. Where the peak corresponding to the plane (11) of FVO has been overlayed by the (002) plane of CN, which confirms the formation of a nanocomposite; hence, it dominates the reflection attributed to FVO at ∼27° in the XRD pattern of FVO/CN. The average crystalline size of the FVO, CN and FVO/CN nanostructures was calculated to be 13.35, 19.49 and 10.38 nm using the Scherrer equation (eqn (1))
3), respectively. The obtained diffraction patterns match with the crystal database reference (JCPDS#38-1372) and confirm the single phase of the synthesized material.21 Diffraction peaks of FVO were distinct, well-defined, and more intense. This pattern closely aligns with FVO's anorthic (triclinic) crystalline phase. And CN (g-C3N4) shows reflections at 2θ values of 12.64° and 27. 37° corresponding to the graphitic planes of (100) and (002) due to the in-plane structural packing of repeated triazine units and aromatic rings stacked at the interlayer (JCPDS#01-87-1526).22 A broad peak around 57° persists in the refined crystallization pattern of the hexagonal space group with AB stacking of the triazine-based layers (Fig. 1(a)).23,24 For a clearer comparison, Fig. 1(b) displays an expanded XRD pattern of the prepared nanostructures spanning the range from 22.4 to 30°. Where the peak corresponding to the plane (11) of FVO has been overlayed by the (002) plane of CN, which confirms the formation of a nanocomposite; hence, it dominates the reflection attributed to FVO at ∼27° in the XRD pattern of FVO/CN. The average crystalline size of the FVO, CN and FVO/CN nanostructures was calculated to be 13.35, 19.49 and 10.38 nm using the Scherrer equation (eqn (1))| | | d = kλ/βcosθ | (1) |
where β indicates the FWHM (Full Width at half Maximum), K is the shape factor (0.9), λ is the wavelength of the X-ray source (0.154 nm), and θ refers to Bragg's angle. Mean crystalline size (d), dislocation density (∂), and microstrain percentage (ε) are shown in Table ST1 (ESI†).25
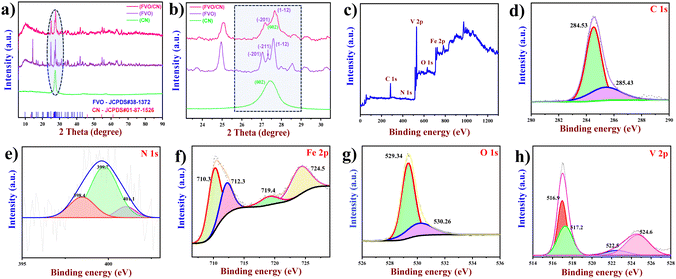 |
| | Fig. 1 (a) and (b) XRD patterns of CN, FVO and FVO/CN (c) XPS survey spectrum of FVO/CN, (d)–(h) deconvoluted spectrum of C 1s, N 1s, Fe 2p, and O 1s and the V 2p spectrum. | |
XPS was performed to study the electronic configuration of the prepared nanostructures’ surface elements, bonding states and chemical constituents. Fig. 1(c) shows the XPS survey spectrum of the FVO/CN nanocomposite, in which the elements including iron, vanadium, oxygen, carbon and nitrogen were confirmed to exist. The deconvoluted spectrum of C 1s of CN is plotted in Fig. 1(d), where two distinct peaks were observed, one at 284.4 eV representing the sp2 hybridization of the carbon in CN and another at 285 eV representing tertiary carbon present in N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N2.26 The N 1s spectrum in Fig. 1(e) confirms the existence of nitrogen with the presence of a peak at 398.4 eV due to the sp2 hybridization of triazine nitrogen present in CN–C, a peak at 399.7 eV represents the triennial nitrogen in –N(C) and a peak at 400.1 eV might correspond to the nitrogen bonded to the surface hydrogen atom (–N–H).27Fig. 1(f) depicts the inner shell level Fe 2p, doublet peaks observed at binding energies of 710.3 and 712.3 eV representing the Fe 2p3/2 energy level and a peak at 724.3 eV corresponding to the Fe 2p1/2 energy level, which confirm the existence of iron at +2 and +3 oxidation states. At 7l9.4 eV, the Fe3+ satellite peak was observed.28Fig. 1(g) illustrates the deconvoluted spectrum of the O 1s, where a prominent peak is observed at 529.3 eV and a distinct peak is observed at a binding energy of 530.3 eV, which corresponds to the hydroxylated oxygen atom present in the FVO.29 In Fig. 1(h), the peaks corresponding to V 2p3/2 were obtained at binding energies of 516.9 and 517.2 eV, confirming the presence of vanadium +4 and +5 oxidation states. For V 2p1/2, peaks at 522.5 and 522.4 eV correspond to +4 and +5 oxidation states.29 For V 2p1/2, a broadened peak was observed, this might be due to vibrational broadening, which is often referred to as Franck–Condon broadening due to the interactions of the substrate with the metal oxide in the composite formation.
N2.26 The N 1s spectrum in Fig. 1(e) confirms the existence of nitrogen with the presence of a peak at 398.4 eV due to the sp2 hybridization of triazine nitrogen present in CN–C, a peak at 399.7 eV represents the triennial nitrogen in –N(C) and a peak at 400.1 eV might correspond to the nitrogen bonded to the surface hydrogen atom (–N–H).27Fig. 1(f) depicts the inner shell level Fe 2p, doublet peaks observed at binding energies of 710.3 and 712.3 eV representing the Fe 2p3/2 energy level and a peak at 724.3 eV corresponding to the Fe 2p1/2 energy level, which confirm the existence of iron at +2 and +3 oxidation states. At 7l9.4 eV, the Fe3+ satellite peak was observed.28Fig. 1(g) illustrates the deconvoluted spectrum of the O 1s, where a prominent peak is observed at 529.3 eV and a distinct peak is observed at a binding energy of 530.3 eV, which corresponds to the hydroxylated oxygen atom present in the FVO.29 In Fig. 1(h), the peaks corresponding to V 2p3/2 were obtained at binding energies of 516.9 and 517.2 eV, confirming the presence of vanadium +4 and +5 oxidation states. For V 2p1/2, peaks at 522.5 and 522.4 eV correspond to +4 and +5 oxidation states.29 For V 2p1/2, a broadened peak was observed, this might be due to vibrational broadening, which is often referred to as Franck–Condon broadening due to the interactions of the substrate with the metal oxide in the composite formation.
3.2. FT-IR and Raman analysis
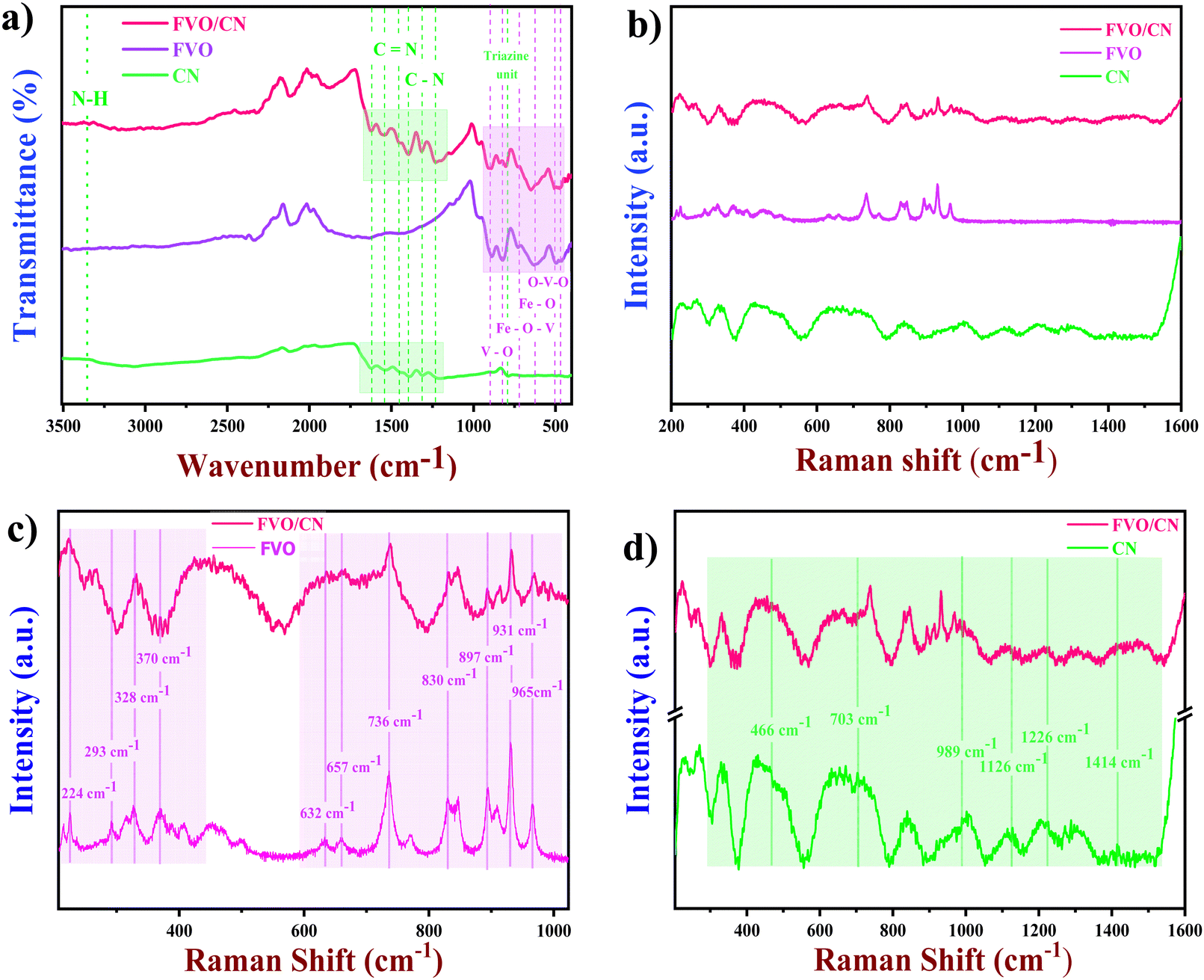
FT-IR spectroscopy was utilised as an analytical technique to elucidate the chemical composition of the samples and identify the specific chemical bonds within the as-synthesised nanostructures.30 As depicted in Fig. 2(a), the spectra acquired for FVO exhibited broad and less intense bands, indicating the successful formation of partial chemical bonds. Peaks corresponding to the stretching vibrations of V–O–V and Fe–O appeared at 503 cm−1. Mixed bridging of V–O–Fe and Fe–O stretching causes a peak at 625 cm−1. Symmetric and asymmetric stretching of V–O–O and VO causes peaks at 843 cm−1 and 900 cm−1.31,32 The surface functional groups on the prepared CN were also analysed, and a broad peak appeared at 3320 cm−1 owing to the stretching vibrations of NH2 and N–H. Peaks in the region 1200–1600 cm−1 correspond to the stretching of C–N and CN. A sharp but less intense peak appeared at 808 cm−1 attributed to the signature fingerprint stretching of triazine units of as-formed CN.33 Finally, the FT-IR spectrum of the composite has all the prominent peaks which were obtained in the individual spectrum, which confirms the existence of CN and FVO in the composite. For certain peaks, minor shifts are observed in Fig. 2(a) due to the combination mode of the stretching frequency of similar bonds in the molecules. Raman analysis was carried out to analyse the prepared nanostructures’ rotational or vibrational modes, chemical composition, and molecular structure. Through irradiating the material with laser light, the Raman spectrum obtained shows various vibrational modes due to the changes in the energies of the scattered photons. Fig. 2(b) illustrates the Raman spectrum of the freshly prepared nanostructures CN, FVO and FVO/CN exhibiting distinct peaks of varying intensity, revealing the formation of cohesive chemical bonds, and Fig. 2(c) portrays the spectrum of FVO where characteristic peaks obtained in the region of 224, 293, 328 and 370 cm−1 represent the stretching of Fe–O and V–O–O deformations.34 Mixed bridging of the stretches V–O⋯Fe and V⋯O⋯Fe causes a peak to arise at 632 cm−1, and bridging vibrational stretching of V–O⋯Fe was responsible for the peaks at 736 and 830 cm−1.35 V–O terminal stretching leads to the appearance of peaks at 897, 931 and and 965 cm−1.
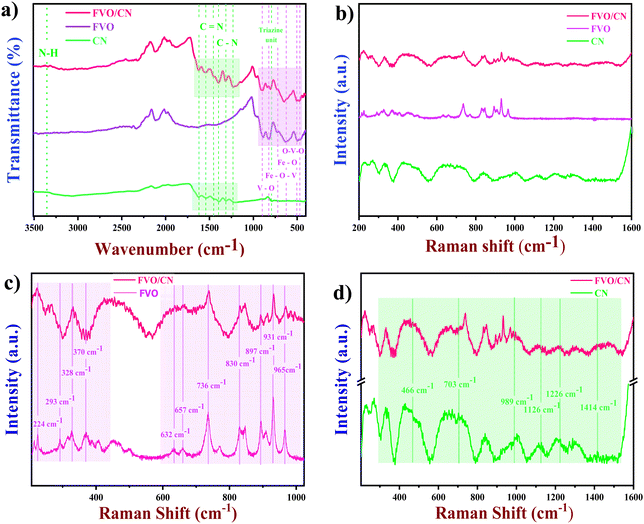 |
| | Fig. 2 (a) FT-IR spectra of CN, FVO and FVO/CN, (b) Raman spectra of CN, FVO and FVO/CN, (c) and (d) comparative Raman spectra of FVO/CN and FVO, and FVO/CN and CN. | |
Fig. 2(c) also confirms the presence of characteristic peaks of FVO in the FVO/CN spectrum.29Fig. 2(d) shows the Raman spectra of CN in detail; the characteristic peaks present at 466, 703, 989, 1126, 1226 and 1414 cm−1 in the spectrum correspond to the typical C–N (1200–1500 cm−1) and CN vibrational nodes in the heterocycles.36 This study validates the crystalline nature and purity of the FVO/CN composite. The FVO/CN hybrid enhances electron transfer by improving active sites through a significant coupling interaction between FVO and CN, leveraging the electrocatalyst's high electrical conductivity and large surface area. There exists a synergy between FVO and CN; when dispersed in water, CN acquires a slight negative charge owing to the deprotonation of the amine group, which causes a negative surface charge on the surface of the CN sheets.37ζ potentials were measured (Fig. S4, ESI†) to study the interaction of these two species, and it was found that CN has a ζ potential of −20.4 mV, whereas FVO's ζ potential was −16.5 mV and found to be more positive when compared to CN. The measured ζ potentials were lowered to −16.5 mV, which would promote interactions between particles rather than repulsion. This aligns with the stability of a colloidal system, where a ζ potential above +30 mV or below −30 mV is typically expected to facilitate maintaining stability.38 This causes an internal electrical field and a weak van der Waals interaction leads to the formation of a nanocomposite.39 The CN showcases a sheet-like morphology falling under the 2D category, offering a notably high surface area. By surface engineering these 2D sheets with iron vanadate nano-bulbs, the quantity of active sites is substantially increased. The distinctive contact interface created by the random dispersion of iron vanadate nano-bulbs on the sheet surface leads to a synergistic effect; facilitating rapid electron transfer between the components at the interface and enhancing the number of electrochemically active sites on the electrode surface. This improvement in electron transfer kinetics from the solution to the working electrode enhances the sensor's electrocatalytic activity, resulting in a more effective capture of the current response from electrooxidation reactions, ultimately influencing the sensor's performance.
3.3. Microstructure and morphology
To analyse the surface morphology of the prepared nanostructures, SEM and TEM micrographs were obtained, and a distinct nanostructure was observed in the micrographs of FVO, CN and the FVO/CN nanocomposite.37 In Fig. 3(a) and (b), smaller and elongated sphere-like nano-bulbs which are irregular in size and distributed non-homogenously in the micrograph of FVO (Fig. 3(b)) were observed, which shows the particle's agglomeration on a small scale.29Fig. 3(c) explains the exfoliated CN's flake/sheet-like morphology. A mixed morphology of nano-bulbs randomly decorated on the surface of the CN sheets was obtained. According to the experimental data obtained from the XRD analysis, the nanostructures have particle sizes less than 20 nm. This study also assessed the elemental composition of various materials through energy-dispersive X-ray analysis (EDX). The morphology and composition of FVO, CN, and the FVO/CN composite were investigated using HR-TEM. TEM images of FVO revealed the achievement of nanosphere formation (Fig. 3(e)). The inset of Fig. 3(e) displays the selected area electron diffraction (SAED) pattern of FVO. Additionally, Fig. 3(f) illustrates the TEM images of CN, showcasing the arrangement of nanosheets in stacked flat layers. Fig. 3(h) and (j) show the lattice fringes of the prepared FVO nano-bulb with a d-spacing value of d = 0.314 nm. It is intriguing that when FVO is combined with CN, it results in the formation of a FVO nano-bulb on the surface of the CN nanosheets, as illustrated in Fig. 3(d) and (g), and this along with Fig. 3(j) specify the composite formation and interlayer stacking of the FVO nanostructure on the CN sheets. The obtained EDX spectra Fig. 3(k) from different materials reveal the presence of diverse constituent elements (C, N, Fe, V and O) within the samples, confirming the formation of nanocomposites comprising CN and FVO. From an in-depth literature survey, it is found that the surface charge of the CN sheets is negative, whereas the presence of two metals in FVO provides a smear-positive charge, which creates a chance for the electrostatic interaction between them; this synergy forms a stable colloidal system adequate for sensing performance.37
 |
| | Fig. 3 SEM Micrographs of (a) and (b) FVO (FeVO4), (c) CN (g-C3N4), (d) FVO/CN,– TEM micrographs of (e)–(g) FVO, CN and FVO/CN, (h)–(j) lattice fringes of FVO, CN and FVO/CN, (k) EDX analysis of FVO/CN. | |
3.4. Electrochemical experiments
In this work, various electrochemical experiments were employed, including cyclic voltammetry (CV), an electrochemical technique employed to study the oxidation and reduction processes of any molecular species and chemical processes stimulated by the transport of electrons.40,41 CV was the preliminary electrochemical study employed throughout this work, and several electrochemical experiments were carried out for interpretation and optimization studies, including the catalyst ratio and loading optimization, the effect of the concentration of analyte (LVO), scan rates, pH of the electrolyte solution, and electrocatalytic activity of the materials. In the cyclic voltammogram, the x-axis represents the applied potential (E), and the y-axis represents the resulting current response (i); throughout this study IUPAC convention is used for reporting the CV studies. Electrochemical impedance spectroscopy (EIS) was performed to study the kinetic as well as mechanistic data of the electrochemical system, which is in the steady state confirmed using a sinusoidal signal observed for a wide range of frequencies and the corresponding sinusoidal response was monitored. In this work, EIS was employed to study the electrode/electrolyte interfaces, charge transfer processes, diffusion kinetics, and the overall performance of different modified electrodes.42,43 Differential pulse voltammetry (DPV), a powerful technique in electrochemical sensor studies, was performed to obtain qualitative and quantitative information about analyte behaviour and sensor performance. In this technique, a discrete pulse was applied to the working electrode in a controlled manner, and a potential sweep was applied between each pulse for a certain duration, where the corresponding current response was measured. Since the current response is measured between each pulse's start and end potentials, this differential measurement enhances the sensitivity and reduces the background current.44,45 The amperometry curve, also mentioned as the (i–t) curve, using which the current response was measured for the electrochemical reaction occurring at a constant applied potential through which several interpretations like the analyte concentration, selectivity, reaction kinetics, detection limits, and electrode performance were determined.46
Electrochemical impedance spectroscopy (EIS) was performed to study kinetic as well as mechanistic data of the electrochemical system in the steady state using a sinusoidal signal in a wide range of frequencies. In this work, EIS was employed to study the electrode/electrolyte interfaces, charge transfer processes, diffusion kinetics, and the overall performance of different modified electrodes.42 Impedance is measured by applying a small sinusoidal voltage perturbation superimposed on a potential that matches the redox reaction's standard electrode potential (Eo). Henceforth, we can also mention this as faradaic EIS, and the faradaic process is expressed as a general impedance (Zf), which includes both the kinetics of the redox process and the diffusion of the redox species [Fe(CN)6]4−/3− to the working electrode's surface. Zf can be broken down into two parts, (i) charge transfer resistance (Rct) – this pertains to the diverse electrochemical processes under the assumption that the redox species do not adhere to the electrode surface, (ii) Warburg impedance, (Zw) – this pertains to the obscurity in the mass transport of the redox species to the electrode surface reflecting a semi-linear diffusion. We can notice the fact that an equivalent circuit involving Cdl (double layer capacitance) and Rs (uncompensated solution resistance) along with the Rct and Zw was designated from the Randles circuit (Fig. 4(b)). In the same figure, we can notice a higher diameter of the semicircle and an excessive Rct value of 2581 Ω for the Bare GCE, 1665 Ω for CN/GCE, and 1479 Ω for FVO/GCE. In contrast, the FVO/CN/GCE nanocomposite showed a prominent drop in the Rct value to 535 Ω. This might be due to the higher surface area, conductivity, electron transfer kinetics and synergy of the nanocomposite, which augments the electrocatalytic activity of FVO/CN/GCE, facilitating effective sensing of the targeted analyte.
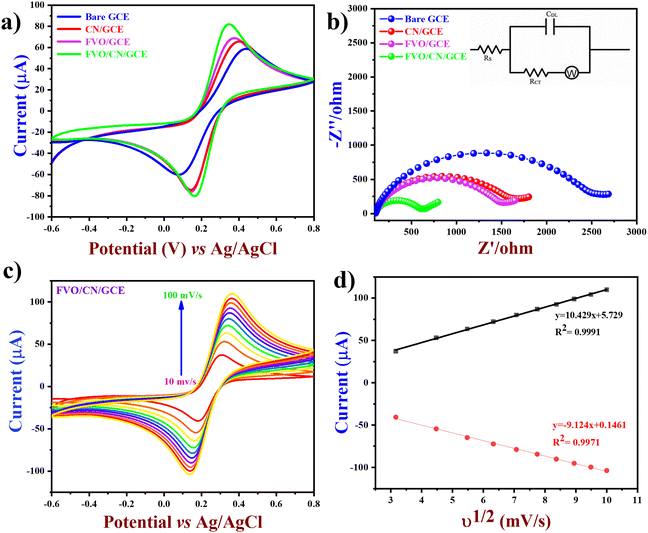 |
| | Fig. 4 (a) CV plots and (b) EIS plots of bare and different modified electrodes with 0.1 M KCl in 5 mM Fe(CN)63−/4−, (c) CV plots with increase in the scan rate from 10 to 100 mV s−1 and (d) linear plots of Ipa and Ipcvs. square roots of scan rates of FVO/CN/GCE with 0.1 M KCl in 5 mM Fe(CN)63−/4−. | |
The electron transfer ability of the bare and different modified electrodes was examined by engaging cyclic voltammetry (CV) in 0.1 M KCl with 5 mM Fe(CN)63−/4− redox species in the potential range of −0.6 to +0.8 V at a scan rate of 50 mV s−1 (Fig. 4(b)). The perceived electrochemical terms like anodic and cathodic potentials (Ea) and (Ec), oxidation and reduction peak currents (Ipa) and (Ipc), and potential difference (ΔEp) are plotted in Table ST2, ESI.† According to the results, the FVO/CN/GCE has lower (ΔEp) and prominent redox peak currents than the bare and other modified electrodes. The calibration plot of the square root of scan rate vs. redox peak current for bare GCE and different modified electrodes is provided in Fig. SF1, ESI† and FVO/CN/GCE was found to show linearity with the regression equations Ipa = 10.429x + 5.729 and Ipc = −9.124x − 0.146 with correlation coefficients R2 = 0.9991 and R2 = 0.9971. The electrochemically active surface area of bare and different modified electrodes (CN/GCE, FVO/GCE and FVO/CN/GCE) was calculated using the Randles–Sevcik equation (eqn (2))
| | | Ip = 2.6 × 105 × An3/2CD1/2ν1/2 | (2) |
where
Ip is the peak current, ‘
A’ is the surface area of the modified electrode, ‘
n’ is the number of electrons involved in the chemical reaction, ‘
C’ is the concentration of the [Fe(CN)
6]
4−/3− electrolyte in bulk solution (5 mM),
D1/2 is the square root of the diffusion coefficient, and
ν1/2 is the square root of the scan rate. Findings suggest that FVO/CN enhances the active sites on the electrode surface and facilitates the electron transfer rate at the interface. The active surface area was calculated to be 0.02, 0.043, 0.061, and 0.083 cm
2 for bare GCE, CN/GCE, FVO/GCE, and FVO/CN/GCE, respectively. The active surface area of the FVO/CN/GCE electrode increased 4 times compared to that of the bare GCE, which significantly increases the electrocatalytic activity of the modified electrode.
3.5. Effects of the catalyst loading and ratio
CV was performed in 0.1 M PBS (pH – 7.4) at a sweep rate of 50 mV s−1 for better understanding the electroanalytical performance of the bare and different modified electrodes. The catalyst concentration on the electrode is a vital factor for achieving superior sensitivity in electrodes.47 We used various quantities of the catalyst, ranging from 0.5 to 3 mg, and dispersed them in 1 mL of Milli-Q water to create a uniform suspension. 6 μL of each suspension was drop casted on the GCE and allowed to dry at 50 °C. A decline in the current spotted at 2 and 3 mg mL−1 dispersion might be attributable to the catalytic resistance on the electrode surface (Fig. 5(b)).48 Consequently, 6 μL of 1 mg mL−1 dispersion was used in all the upcoming weight ratio optimization. The FVO/CN nanocomposite was optimized at various weight ratios (w/w), and CN and FVO were tested at different weight ratios (1:1), (2:1) and (1:2). Fig. 5(a) displays the CV curves of FVO/CN in the presence of 120 μM of LVO, stipulating the oxidation peak current of LVO at various weight ratios, and among the weight ratios (1:1) exhibited higher oxidation peak current for LVO and optimum electrochemical performance. Henceforth, 6 μL of 1 mg mL−1 dispersion with a 1:1 weight ratio of FVO/CN was fixed as the optimal loading for the upcoming electrochemical studies. Under optimised conditions, the electrochemical responses of the bare GCE and the different modified electrodes were examined through CV, both with and without 120 μM LVO, as illustrated in Fig. 5(C). These electrochemical assessments were conducted in a 0.1 M PBS solution with a pH of 7.4, employing a sweep rate of 50 mV s−1. It is evident that the FVO/CN modified electrode has a higher peak current value and standard oxidation potential of LVO compared to other modified electrodes; hence, it was selected for further electrochemical experiments. In the absence of LVO, the bare GCE exhibited no noticeable peaks. However, in the presence of LVO, both the modified and unmodified GCEs displayed an oxidation peak. The bare GCE exhibited a weaker electrochemical response due to the slower electron transfer involved in LVO detection. Conversely, the FVO/CN-modified electrodes demonstrated a maximum peak current of 38.25 μA and a peak potential of +0.943 V, respectively. Incorporating FVO/CN on the electrode surface gave it an excellent electrochemical response owing to a larger specific surface area and improved electrocatalytic activity. The obtained oxidation peak current value for FVO/CN is significantly higher than that of the bare GCE in the presence of LVO.
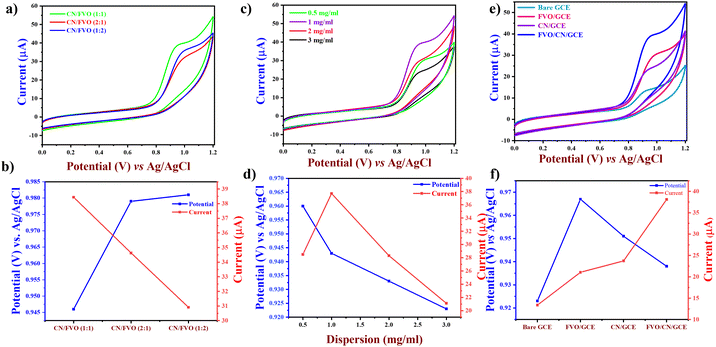 |
| | Fig. 5 (a) CV plots of FVO/CN/GCE with a different weight ratio of FVO and CN, (b) a nonlinear plot of oxidation peak potential and current vs. optimized weight ratios of FVO and CN, (c) CV plots of FVO/CN/GCE with various amounts of catalyst dispersion, (d) a nonlinear plot between oxidation peak current vs. various catalyst dispersion, (e) CV plots of bare and different modified electrodes in 0.1 M PBS (pH 7.4) in 120 μM of LVO, and (f) non-linear plot of oxidation peak potential and current for bare and different modified electrodes. | |
3.6. Effects of the concentration, pH and scan rate
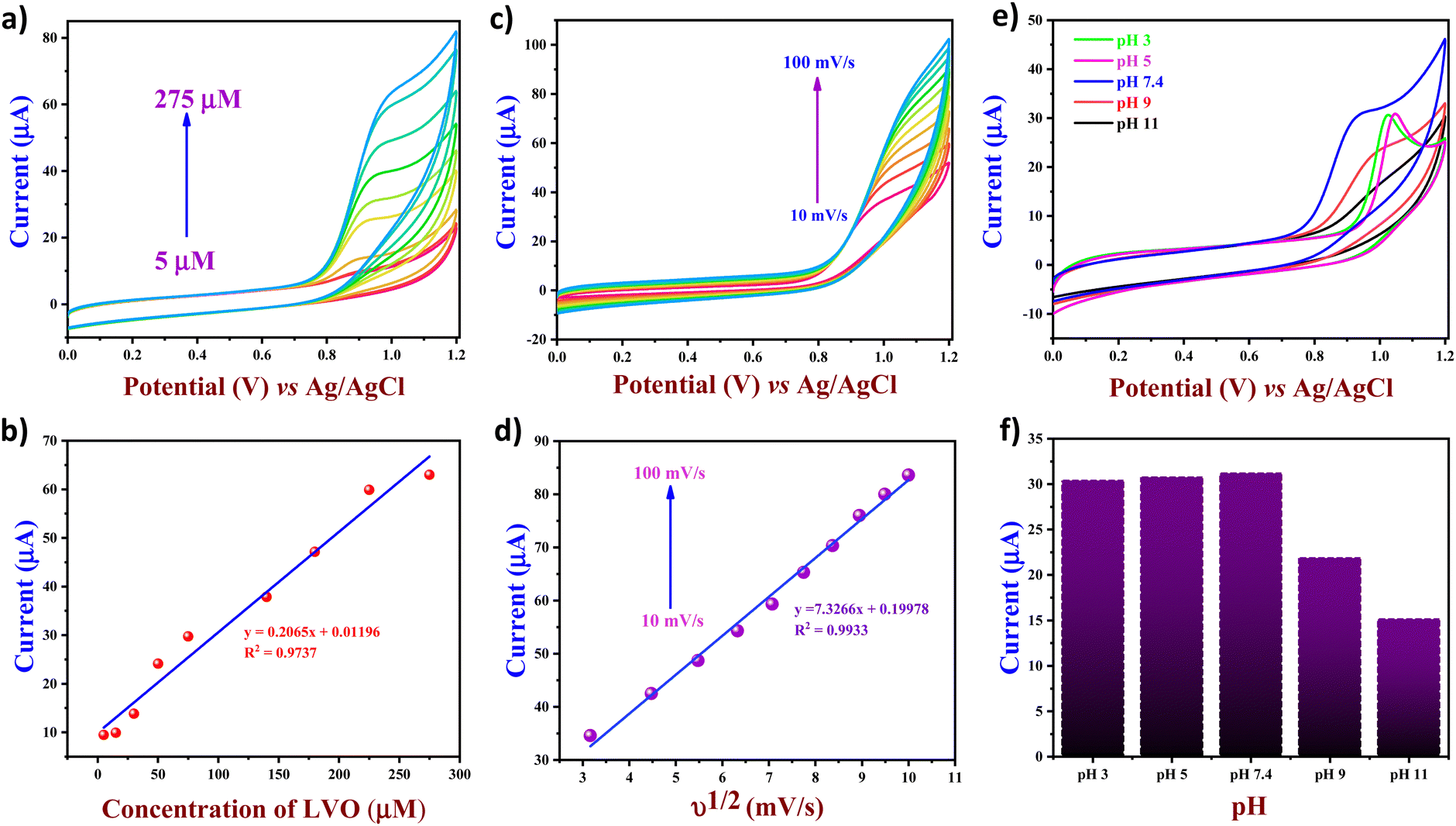
To evaluate the impact of the LVO concentration on the FVO/CN/GCE electrode, increasing amounts of LVO were tested in 0.1 M PBS (pH 7.4) at a sweep rate of 50 mV s−1. As shown in Fig. 6(a), the oxidation peak current was lower at 5 μM due to the lower concentration of the analyte, resulting in fewer deposition points. However, a gradual increase in current intensity was observed as the analyte concentration increased from 5 μM to 275 μM. The plot of peak current vs. concentration of the analyte is provided in Fig. 6(b), and the plot obtained was found to be linear (y = 0.2065x + 0.01196), with correlation coefficient R2 = 0.9737. The CV analysis was performed for the FVO/CN modified GCE in the presence of 250 μM LVO in the increasing scan rate from 10–100 mV s−1. From Fig. 6(c), it was observed that the peak potential shifted to a more positive direction with the increase in the scan rate due to modifications in the electrochemical reaction kinetics at the electrode interface. The change in peak intensity varies according to the sweep rate's linear regression equation confirming that the electrooxidation reaction on the electrode surface is a diffusion-controlled irreversible reaction.
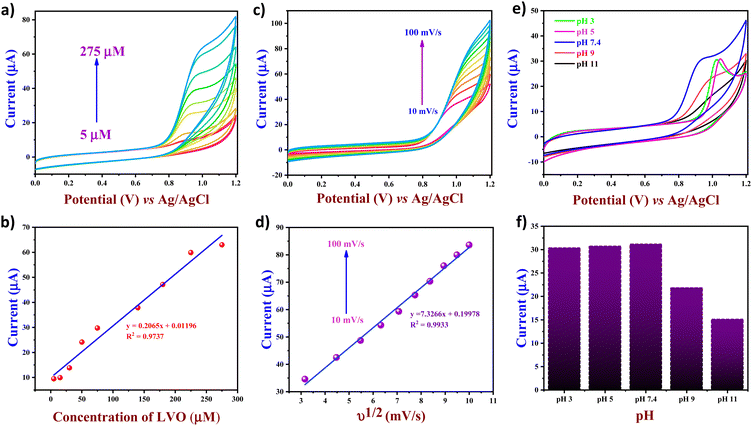 |
| | Fig. 6 (a) CV plots of FVO/CN/GCE with increasing concentration of LVO, (b) linear plot of peak current vs. the concentration of LVO, (c) CV plots of FVO/CN/GCE with increasing scan rates from 10 to 100 mV s−1 in the presence of 250 μM of LVO, (d) linear plot of peak current vs. square root of scan rates, (e) CV plots and (f) bar graph of current vs. pH of FVO/CN/GCE with various pH ranging from 3 to 11 with 120 μM of LVO. | |
According to the results obtained, the oxidation peak observed on the voltammogram corresponds to the two-step condensation of a piperazine moiety, emitting an electron. The initial stage involves the rate-determining step, where na = 1, resulting in forming a radical cation, followed by a subsequent rapid process leading to a cation. Subsequently, the hydrolysis and deprotonation facilitate the formation of LVO N-oxide through lateral pathways. Several electrochemical studies have looked into the changes in LVO oxidation and came up with a two-electron oxidation mechanism for the LVO molecule's piperazine part. The possible comprehensive mechanism elucidating the electron transfer process toward LVO is depicted in Scheme 1. Fig. 6(e) illustrates the influence of change in pH of 0.1 M PBS; as the pH varies a shift in the peak potential towards a positive value is observed, and a decrease in the peak current intensity from pH 3 to 7.4 is observed due to the production of the radical ions by the LVO during the electrooxidation. LVO exhibits zwitterionic properties around physiological pH levels due to the presence of carboxylic groups and piperazinyl groups, which act as proton-accepting functional groups. Moreover, pH of human blood is 7.4 and higher activity was observed at pH 7.4; thus, it was chosen as an optimum pH for further electrochemical studies.
3.7. Detection limit of LVO
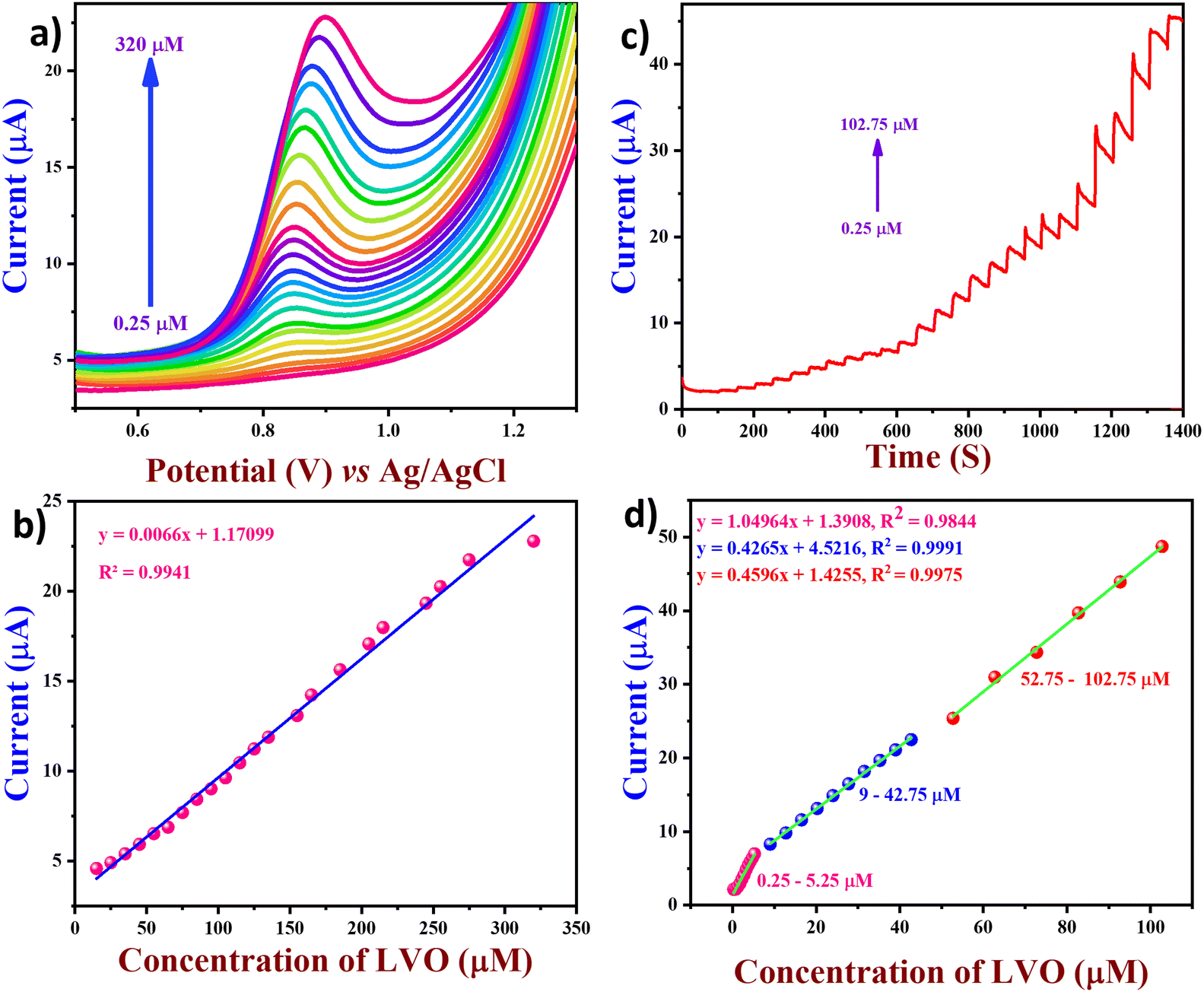
To analyse and quantify the electrochemical detection of the LVO through FVO/CN-modified GCE using differential pulse voltammetry (DPV) was performed. In this technique, higher sensitivity can be achieved due to the elimination of the non-faradaic and capacitive currents present in the cyclic voltammetry. The experiment was conducted with 0.1 M PBS in a N2 atmosphere at the optimum pH of 7.4. DPV conditions were fixed as follows: a scan rate of 50 mV s−1; an amplitude of 50 mV; a pulse width of 50 mS; a pulse period of 500 mS; and an increment of 0.004 V (Fig. 7(a)). Upon increasing the concentration of the LVO gradually from 0.25 to 320 μM, the peak current intensity also increased gradually linearly in accordance with the concentration of LVO. The correlation coefficient was obtained (R2 = 0.9941) from the linear regression equation (y = 0.0066x + 1.1709), and the calibration graph of the concentration of the analyte vs. the oxidation peak current is plotted in Fig. 7(b). The limit of detection was calculated to be 0.061 μM using eqn (3):| | 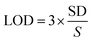 | (3) |
where ‘SD’ is the standard deviation of the blank, and ‘S’ is the slope of the calibration curve. In addition to DPV, amperometry was also performed to analyse the limit of detection, sensitivity, and selectivity of the FVO/CN-modified GCE. This experiment was carried out in 0.1 M PBS at pH 7.4 under dynamic conditions achieved by rotating a rice pellet at a rate of 1000 rpm with the addition of the analyte at regular intervals. The current response was monitored at a constant potential of +0.9 V, and in this study, the response time of the prepared sensor was also calculated to be (2.6 s). To investigate the electroanalytic performance of the FVO/CN modified GCE and quantify the concentration of the LVO, the amperometry technique was employed in 0.1 M PBS at pH of 7.4 with a constant applied potential of +0.9 V at a concentration of the LVO ranging from 0.25–102.1 μM. Throughout the study, a linear relationship existed between the current response and the concentration of the LVO upon every increment as shown in Fig. 7(d). A decline in the steady current was noted due to the saturation of the electrocatalytic active sites on the surface of the FVO/CN/GCE at a higher analyte concentration. The FVO/CN/GCE effectively captured the oxidation peak current of the LVO since a higher current response was obtained, owing to the synergistic effects of CN and FVO. The LOD and sensitivity of the FVO/CN/GCE were calculated to be 0.06 μM and 0.0834 μA μM−1 cm−2 towards the detection of LVO.
 |
| | Fig. 7 (a) DPV curves of FVO/CN/GCE in 0.1 M PBS for LVO in the concentration range of 0.25–320 μM, (b) linear plots of oxidation peak current vs. concentration of LVO, (c) (i–t) curve of FVO/CN/GCE was performed with the injection of different LVO concentrations (rotation rate = 1000 rpm and applied potential = +0.9 V) and (d) linear plots of current response vs. added LVO concentration. | |
3.8. Repeatability, reproducibility, selectivity, and stability
To assess the selectivity of the prepared electrode towards the detection of LVO, several electrochemically active substances as potential materials from a range of interferents in the biological fluids, and a few common structurally identical aromatic nitro compounds were chosen as interferents and tested using amperometry. Initially, 5 mM of LVO was added three times simultaneously at an interval of 50 s to 0.1 M PBS at pH 7.4, which is kept in a dynamic state at 1000 RPM, followed by the addition of interferents at 10-fold, higher concentrations including 5-fluorouracil, ciprofloxacin, acetaminophen, citric acid, uric acid, lactic acid, and often encountered metal ions comprising sodium, calcium, and magnesium ions. T here was a negligible current response noted for the addition of the interferents (Fig. SF2, ESI†), which might be due to the difference in oxidation potentials of the interferents with respect to LVO.
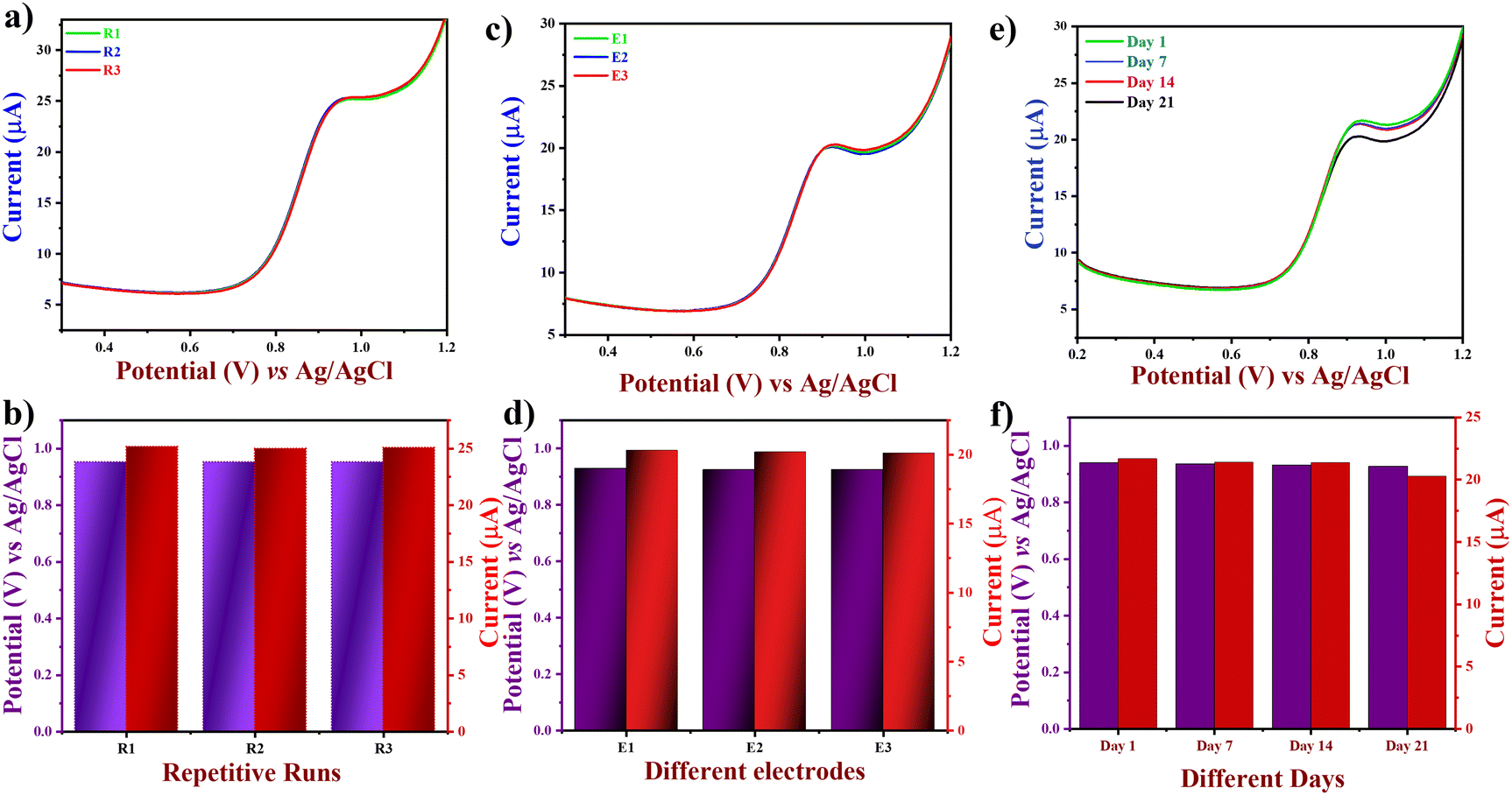
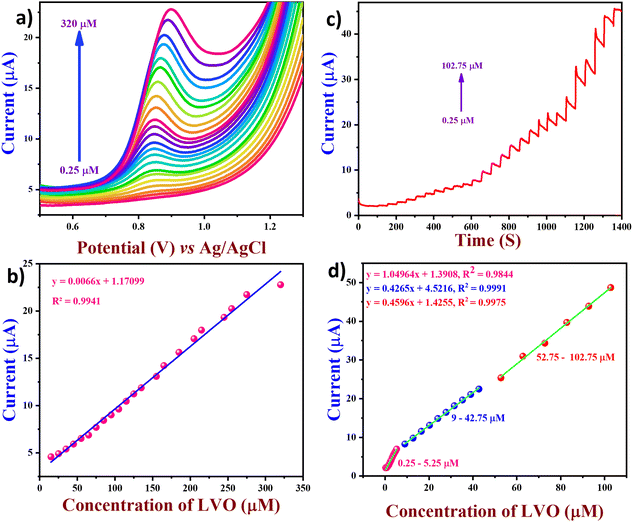
Hence, the relative error arising from the interferents was below 10% owing to their low adsorption capacity, indicating the remarkable selectivity of the suggested electrode. To assess the repeatability of the proposed electrode (Fig. 8(a)), the DPV technique was applied to three different electrodes on the same day, with equal time intervals between each measurement. Furthermore, the reproducibility of the proposed electrodes was tested by making FVO/CN-modified electrodes separately three times (Fig. 8(b)). In addition, the modified electrode's stability was tested by repeating the experiment with the same FVO/CN modified electrode at 1, 7, 14 and 21 days of storage (Fig. 8(c)). We subsequently assessed the long-range stability of the FVO/CN modified GCE through continuous observation using DPV in 0.1 M PBS, which revealed a decline of about 7% in the electrode's current response and a potential shift of 0.012 V was observed. The findings validated the outstanding repeatability, reproducibility, selectivity, and stability of the FVO/CN-modified electrode, even after numerous analyses and extended storage.
 |
| | Fig. 8 (a) DPV curves of LVO at FVO/CN/GCE with the same electrodes. (b) Bar graphs of potential and peak current vs. repeated runs. (c) DPV curves of LVO at FVO/CN/GCE with different electrodes. (d) Bar graphs of potential and peak current vs. different electrodes and (e) DPV curves of FVO/CN/GCE toward LVO for three weeks. (e) Bar graphs of potential and peak current vs. different days of storage. | |
3.9. Real sample analysis
Real-time monitoring of human serum samples is essential for analysing potential health hazards associated with pharmaceutical toxicity.49 Real samples were prepared from human blood samples bought through SRM Medical Hospital and Research Centre which ethical clearance by proceedings of the Institutional Ethics Committee (ID: 8521/IEC/2023). The blood serum sample was diluted with 0.1 M PBS (pH 7.4). Using the DPV technique, FVO/CN/GCE was analysed in 0.1 M PBS (pH 7.4) following the standard addition of LVO. Three blood serum samples with varying concentrations of LVO (90, 100 and 110 μM) were added to the resulting solutions, as illustrated in the ESI† (Fig. SF3). The recovery percentage of LVO was 97.5%. These findings showed that the FVO/CN/GCE can be utilized to analyse LVO in real samples.
4. Conclusion
In this study, we have successfully demonstrated an innovative and highly effective approach for the electrochemical detection of levofloxacin (LVO) in human blood serum using iron vanadate nano-bulbs (FVO) decorated on a two-dimensional conjugated polymer matrix (CN). The FVO nano-bulbs, synthesised through a straightforward hydrothermal method, were effectively integrated into the 2D conjugated polymer matrix, resulting in a sensor with numerous active sites. This configuration significantly improved the sensing capabilities for LVO, achieving a detection limit of 0.06 μM, a broad linear range of 0.02–320 μM, and high sensitivity and selectivity towards LVO. Our findings emphasize the critical role of vanadium in enhancing electrocatalytic performance and underscore the substantial potential of FVO nano-bulbs in developing efficient and sensitive electrochemical sensors for pharmaceutical monitoring. This research provides a valuable tool for detecting LVO in human blood serum, paving the way for advancements in electrochemical sensing and pharmaceutical safety monitoring.
Author contributions
B. R.: formal analysis, methodology, experimental operation, validation, data curation, visualization, writing – original draft, and review and editing; M. G.: data curation and writing review and editing; M. M.: review and editing; S. R.: review and editing, J. A. S.: guidance, visualization, supervision, and writing – review & editing the manuscript.
Data availability
The data supporting this article have been included in the main manuscript and the ESI,† and the data will be available upon request to the authors.
Conflicts of interest
The authors assert that they have no conflicting financial interests.
Acknowledgements
The authors acknowledge the Department of Chemistry, SRM Institute of Science and Technology, Tamil Nadu-603 203, India. We also acknowledge SCIF, the Nanotechnology Research Centre (NRC), SRMIST, and the MATERIAL CHARACTERIZATION FACILITY, Department of Physics and Nanotechnology, College of Engineering and Technology, SRMIST, for providing the instrumentation facilities.
References
-
V. Podder and N. M. Sadiq, Levofloxacin, StatPearls Publishing, Treasure Island (FL), 2023 Search PubMed
 .
.
- I. Karampela and M. Dalamaga, Could Respiratory Fluoroquinolones, Levofloxacin and Moxifloxacin, Prove to be Beneficial as an Adjunct Treatment in COVID-19?, Arch. Med. Res., 2020, 51, 741–742 CrossRef CAS PubMed .
- D. N. Fish, Fluoroquinolone Adverse Effects and Drug Interactions, Pharmacotherapy, 2001, 21, 253S–272S CAS .
- G. Li, X. Wan, Y. Xia, D. Tuo, X. Qi, T. Wang, M. Mehmandoust, N. Erk, Q. He and Q. Li, Lamellar α-Zirconium Phosphate Nanoparticles Supported on N-Doped Graphene Nanosheets as Electrocatalysts for the Detection of Levofloxacin, ACS Appl. Nano Mater., 2023, 6, 17040–17052 CrossRef CAS .
- M. K. Muthukumaran, M. Govindaraj, B. K. Raja and J. A. Selvi, In situ synthesis of polythiophene encapsulated 2D hexagonal boron nitride nanocomposite based electrochemical transducer for detection of 5-fluorouracil with high selectivity, RSC Adv., 2023, 13, 2780–2794 RSC .
- X. Yu, Q. Sui, S. Lyu, W. Zhao, J. Liu, Z. Cai, G. Yu and D. J. Es Barcelo, Municipal solid waste landfills: an underestimated source of pharmaceutical and personal care products in the water environment, Environ. Sci. Technol., 2020, 54, 9757–9768 CrossRef CAS PubMed .
- Y. Jin, G. Xu, X. Li, J. Ma, L. Yang, Y. Li, H. Zhang, Z. Zhang, D. Yao and D. J. Jo. A. Li, Compounds, Fast cathodic reduction electrodeposition of a binder-free cobalt-doped Ni-MOF film for directly sensing of levofloxacin, J. Alloys Compd., 2021, 851, 156823 CrossRef CAS .
- M. H. Ghanbari, P. Sharafi, S. Nayebossadr and Z. J. M. A. Norouzi, Utilizing a nanocomposite consisting of zinc ferrite, copper oxide, and gold nanoparticles in the fabrication of a metformin electrochemical sensor supported on a glassy carbon electrode, Microchim. Acta, 2020, 187, 1–11 CrossRef PubMed .
- M. Govindaraj, A. Srivastava, M. K. Muthukumaran, P. C. Tsai, Y. C. Lin, B. K. Raja, J. Rajendran, V. K. Ponnusamy and J. A. Selvi, Current advancements and prospects of enzymatic and non-enzymatic electrochemical glucose sensors, Int. J. Biol. Macromol., 2023, 253, 126680 CrossRef CAS PubMed .
- M. Ezzati, S. Shahrokhian and H. J. A. S. C. Hosseini, Engineering, In situ two-step preparation of 3D NiCo-BTC MOFs on a glassy carbon electrode and a graphitic screen printed electrode as nonenzymatic glucose-sensing platforms, ACS Sustainable Chem. Eng., 2020, 8, 14340–14352 CrossRef CAS .
- F. E. Galdino, J. P. Smith, S. I. Kwamou, D. K. Kampouris, J. Iniesta, G. C. Smith, J. A. Bonacin and C. E. J. Ac Banks, Graphite screen-printed electrodes applied for the accurate and reagentless sensing of pH, Anal. Chem., 2015, 87, 11666–11672 CrossRef CAS PubMed .
- M. K. Muthukumaran, M. Govindaraj, B. K. Raja and J. A. Selvi, Crystal plane-integrated strontium oxide/hexagonal boron nitride nanohybrids for rapid electrochemical sensing of anticancer drugs in human blood serum samples, Anal. Methods, 2023, 15, 5639–5654 RSC .
- C. E. Choong, C. M. Park, Y.-Y. Chang, J.-k Yang, J. R. Kim, S.-E. Oh, B.-H. Jeon, E. H. Choi, Y. Yoon and M. J. C. E. J. Jang, Interfacial coupling perovskite CeFeO3 on layered graphitic carbon nitride as a multifunctional Z-scheme photocatalyst for boosting nitrogen fixation and organic pollutants demineralization, Chem. Eng. J., 2022, 427, 131406 CrossRef CAS .
- S. Lakkepally, Y. Kalegowda, N. Ganganagappa and A. J. Jo. A. Siddaramanna, Compounds, A new and effective approach for Fe2V4O13 nanoparticles synthesis: evaluation of electrochemical performance as cathode for lithium secondary batteries, J. Alloys Compd., 2018, 737, 665–671 CrossRef CAS .
- S. Guo, A. Naeem, H. Fida, M. Hamayun, M. Muska, J. J. D. Chen and W. Treatment, Removal of Cu(II) from aqueous solution by iron vanadate: equilibrium and kinetics studies, Desalin. Water Treat., 2017, 75, 124–131 CrossRef CAS .
- K. Routray, W. Zhou, C. J. Kiely and I. E. J. A. C. Wachs, Catalysis science of methanol oxidation over iron vanadate catalysts: nature of the catalytic active sites, ACS Catal., 2011, 1, 54–66 CrossRef CAS .
- Z. Peng, Q. Wei, S. Tan, P. He, W. Luo, Q. An and L. J. Cc Mai, Novel layered iron vanadate cathode for high-capacity aqueous rechargeable zinc batteries, Chem. Commun., 2018, 54, 4041–4044 RSC .
- Y. P. Zhu, C. Guo, Y. Zheng and S.-Z. J. Aocr Qiao, Surface and interface engineering of noble-metal-free electrocatalysts for efficient energy conversion processes, Acc. Chem. Res., 2017, 50, 915–923 CrossRef CAS PubMed .
- N. Ding, S. Liu, X. Feng, H. Gao, X. Fang, J. Xu, W. Tremel, I. Lieberwirth and C. J. C. G. Chen, Design, Hydrothermal growth and characterization of nanostructured vanadium-based oxides, Cryst. Growth Des., 2009, 9, 1723–1728 CrossRef CAS .
- M. Zhang, Y. Fang, Y. F. Tay, Y. Liu, L. Wang, H. Jani, F. F. Abdi and L. H. J. A. A. E. M. Wong, Nanostructured iron vanadate photoanodes with enhanced visible absorption and charge separation, ACS Appl. Energy Mater., 2022, 5, 3409–3416 CrossRef CAS .
- R. Monsef, M. Ghiyasiyan-Arani, O. Amiri and M. Salavati-Niasari, Sonochemical synthesis, characterization and application of PrVO4 nanostructures as an effective photocatalyst for discoloration of organic dye contaminants in wastewater, Ultrason. Sonochem., 2020, 61, 104822 CrossRef PubMed .
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. J. Nm Antonietti, A metal-free polymeric photocatalyst for hydrogen production from water under visible light, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed .
- Y. Xiao, G. Tian, W. Li, Y. Xie, B. Jiang, C. Tian, D. Zhao and H. J. JotA. C. S. Fu, Molecule self-assembly synthesis of porous few-layer carbon nitride for highly efficient photoredox catalysis, J. Am. Chem. Soc., 2019, 141, 2508–2515 CrossRef CAS PubMed .
- T. Suter, V. Brázdová, K. McColl, T. S. Miller, H. Nagashima, E. Salvadori, A. Sella, C. A. Howard, C. W. Kay and F. J. T. Jo. P. C. C. Corà, Synthesis, structure and electronic properties of graphitic carbon nitride films, J. Phys. Chem. C, 2018, 122, 25183–25194 CrossRef CAS .
- M. Govindaraj, U. G. PK, M. K. Muthukumaran, K. Sekar, A. Maruthapillai and J. Arockia Selvi, Electrostatic Self Assembly of Metal-Free Hexagonal Boron Nitride/Protonated Carbon Nitride (h-BN/PCN) Nanohybrid: A Synergistically Upgraded 2D/2D Sustainable Electrocatalyst for Sulfamethazine Identification, ChemNanoMat, 2023, 9(11), e202300330 CrossRef CAS .
- M.-H. Vu, M. Sakar, C.-C. Nguyen and T.-O. Do, Chemically Bonded Ni Cocatalyst onto the S Doped g-C3N4 Nanosheets and Their Synergistic Enhancement in H2 Production under Sunlight Irradiation, ACS Sustainable Chem. Eng., 2018, 6, 4194–4203 CrossRef CAS .
- N. Murugan, M. B. Chan-Park and A. K. Sundramoorthy, Electrochemical Detection of Uric Acid on Exfoliated Nanosheets of Graphitic-Like Carbon Nitride (g-C3N4) Based Sensor, J. Electrochem. Soc., 2019, 166, B3163 CrossRef CAS .
- T.-C. Lin, G. Seshadri and J. A. Kelber, A consistent method for quantitative XPS peak analysis of thin oxide films on clean polycrystalline iron surfaces, Appl. Surf. Sci., 1997, 119, 83–92 CrossRef CAS .
- G. Kesavan, M. Pichumani and S.-M. J. A. A. N. M. Chen, Influence of crystalline, structural, and electrochemical properties of iron vanadate nanostructures on flutamide detection, ACS Appl. Nano Mater., 2021, 4, 5883–5894 CrossRef CAS .
-
Infrared Spectroscopy, in ACS Reagent Chemicals, American Chemical Society, 2017 Search PubMed.
- I. Khan and A. J. S. R. Qurashi, Shape controlled synthesis of copper vanadate platelet nanostructures, their optical band edges, and solar-driven water splitting properties, Sci. Rep., 2017, 7, 14370 CrossRef PubMed .
- M. Ghiyasiyan-Arani, M. Salavati-Niasari and S. J. Us Naseh, Enhanced photodegradation of dye in waste water using iron vanadate nanocomposite; ultrasound-assisted preparation and characterization, Ultrason. Sonochem., 2017, 39, 494–503 CrossRef CAS PubMed .
- S. Martha, A. Nashim and K. J. Jo. M. C. A. Parida, Facile synthesis of highly active gC 3 N 4 for efficient hydrogen production under visible light, J. Phys. Chem. A, 2013, 1, 7816–7824 CAS .
- M. M. Sajid, H. Zhai, N. A. Shad, M. Shafique, A. M. Afzal, Y. Javed, S. B. Khan, M. Ikram, N. Amin and Z. J. M. Si. S. P. Zhang, Photocatalytic performance of ferric vanadate (FeVO4) nanoparticles synthesized by hydrothermal method, Mater. Sci. Semicond. Process., 2021, 129, 105785 CrossRef CAS .
- A. Š. Vuk, B. Orel, G. Dražič, F. Decker and P. J. Jos-gs Colomban, UV-visible and IR spectroelectrochemical studies of FeVO 4 sol-gel films for electrochromic applications, J. Sol-Gel Sci. Technol., 2002, 23, 165–181 CrossRef CAS .
- D. Zeng, W.-J. Ong, H. Zheng, M. Wu, Y. Chen, D.-L. Peng and M.-Y. J. Jo. M. C. A. Han, Ni 12 P 5 nanoparticles embedded into porous gC 3 N 4 nanosheets as a noble-metal-free hetero-structure photocatalyst for efficient H 2 production under visible light, J. Phys. Chem. A, 2017, 5, 16171–16178 CAS .
- M. Govindaraj, J. Rajendran, U. G. PK, M. K. Muthukumaran, B. Jayaraman and J. Arockia Selvi, Graphitic carbon nitride nanosheets decorated with strontium tungstate nanospheres as an electrochemical transducer for sulfamethazine sensing, ACS Appl. Nano Mater., 2023, 6(2), 930–945 CrossRef CAS .
- G. V. Lowry, R. J. Hill, S. Harper, A. F. Rawle, C. O. Hendren, F. Klaessig, U. Nobbmann, P. Sayre and J. J. E. S. N. Rumble, Guidance to improve the scientific value of zeta-potential measurements in nanoEHS, Environ. Sci.: Nano, 2016, 3, 953–965 RSC .
- Y. Li, X. Li, S. Wang, S. Tan, Y. Xia, J. Shi, W. Chen, L. J. C. Gao, S. A. Physicochemical and E. Aspects, In-situ generated SrWO4/g-C3N4 heterojunction photocatalyst for enhanced visible light degradation activity of tetracycline, Colloids Surf., A, 2022, 643, 128806 CrossRef CAS .
- N. Elgrishi, K. J. Rountree, B. D. McCarthy, E. S. Rountree, T. T. Eisenhart and J. L. Dempsey, A Practical Beginner's Guide to Cyclic Voltammetry, J. Chem. Educ., 2018, 95, 197–206 CrossRef CAS .
- G. A. Mabbott, An introduction to cyclic voltammetry, J. Chem. Educ., 1983, 60, 697 CrossRef CAS .
- A. C. Lazanas and M. I. Prodromidis, Electrochemical Impedance Spectroscopy—A Tutorial, ACS Meas. Sci. Au, 2023, 3, 162–193 CrossRef CAS PubMed .
- B. K. Raja, A. Philips, S. Ravi, M. Ravi, A. S. Palakkal, R. S. Pillai, G. C. Senadi and J. A. Selvi, J, 3-Phenylquinazolin-4(3H)-one via a renewable approach as an efficient corrosion inhibitor for mild steel in acid media, Mater. Chem. Phys., 2023, 308, 128238 CrossRef CAS .
- J. Osteryoung, Pulse voltammetry, J. Chem. Educ., 1983, 60, 296 CrossRef CAS .
- A. Molina, C. Serna, J. A. Ortuño, J. Gonzalez, E. Torralba and A. Gil, Differential Pulse Voltammetry for Ion Transfer at Liquid Membranes with Two Polarized Interfaces, Anal. Chem., 2009, 81, 4220–4225 CrossRef CAS PubMed .
- Y. Lu, X. Li, D. Li and R. G. Compton, Amperometric Environmental Phosphate Sensors, ACS Sens., 2021, 6, 3284–3294 CrossRef CAS PubMed .
- K.-Y. Hwa and T. S. K. J. Sr Sharma, Nano assembly of NiFe spheres anchored on f-MWCNT for electrocatalytic reduction and sensing of nitrofurantoin in biological samples, Sci. Rep., 2020, 10, 12256 CrossRef CAS PubMed .
- J. Rajendran, T. S. Kannan, L. S. Dhanasekaran, P. Murugan, R. Atchudan, Z. A. ALOthman, M. Ouladsmane and A. K. J. C. Sundramoorthy, Preparation of 2D Graphene/MXene nanocomposite for the electrochemical determination of hazardous bisphenol A in plastic products, Chemosphere, 2022, 287, 132106 CrossRef CAS PubMed .
- T. Kokulnathan, E. A. Kumar, T.-J. Wang, I.-C. J. E. Cheng and E. Safety, Strontium tungstate-modified disposable strip for electrochemical detection of sulfadiazine in environmental samples, Ecotoxicol. Environ. Saf., 2021, 208, 111516 CrossRef CAS PubMed .
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ,
Muthukumar
Govindaraj
,
Muthukumar
Govindaraj