
Zwitterionic dioxidovanadium(V) complexes containing fluorinated triphenylphosphonium ligands: structure and biomacromolecule studies†
Francisco Mainardi
Martins
 a,
Daniele Cocco
Durigon
bc,
Otávio Augusto
Chaves
d,
Rosely Aparecida
Peralta
c,
Davi Fernando
Back
*a and
Hernán
Terenzi
*b
a,
Daniele Cocco
Durigon
bc,
Otávio Augusto
Chaves
d,
Rosely Aparecida
Peralta
c,
Davi Fernando
Back
*a and
Hernán
Terenzi
*b
aLaboratory of Inorganic Materials, Department of Chemistry, CCNE, Federal University of Santa Maria, Santa Maria, RS 97105-900, Brazil. E-mail: davi.f.back@ufsm.br
bLaboratory of Structural Molecular Biology, Department of Biochemistry, Federal University of Santa Catarina, Florianópolis, SC 88040-900, Brazil. E-mail: hernan.terenzi@ufsc.br
cLaboratory of Bioinorganic and Crystallography, Department of Chemistry, Federal University of Santa Catarina, Florianópolis, SC 88040-900, Brazil
dCQC-IMS, Department of Chemistry, University of Coimbra, Coimbra, 3004-535, Portugal
First published on 26th September 2024
Abstract
Research on metallodrugs with biological properties remains at the forefront, focusing on the development of compounds that interact non-covalently with deoxyribonucleic acid (DNA) and possess the ability to cleave the double helix strands of this biomacromolecule. In this context, vanadium(V) imine complexes featuring a fluorinated triphenylphosphonium group, (3-formyl-4-hydroxybenzyl)tris(4-fluorophenyl)phosphonium chloride ([AF]Cl), were studied for their targeting and accumulation in mitochondria, in addition to their interactions and ability to cleave DNA. The solid-state structures of complexes C1–C3 were elucidated using single-crystal X-ray diffraction and were characterized using vibrational techniques and elemental analysis, along with extensive characterization in solution. These studies revealed that the complexes contain cis-dioxidovanadium(V) species and are zwitterionic species. It was shown that C1–C3 can interact with and are capable of cleaving plasmid DNA through oxidative mechanisms without the need for photoinduction. When potential interactions with bovine serum albumin were analyzed, it was revealed that interactions in the order of 104 M−1 (Stern–Volmer quenching constant, KSV) were observed. Additionally, in silico molecular docking studies showed that C1–C3 can preferentially interact with the minor grooves of DNA and with domain IB (site III) of bovine and human serum albumins.
Introduction
Interactions between small molecules and deoxyribonucleic acid (DNA) can be irreversible or reversible. In the first case, covalent bonds are formed, for example, when using platinum-based antitumor drugs.1 The employment of these Pt-complexes, particularly cisplatin, leads to different and severe side effects2,3 and also cases of chemoresistance.4,5 The formation of covalent bonds with DNA represents the main drawback for the use of cisplatin.6 Reversible recognition of nucleic acids involves non-covalent interactions and usually can be defined as intercalative, major/minor groove binding, and/or electrostatic interactions.1 The development of compounds that interact in non-covalent and reversible ways with DNA targets is at the forefront of the synthesis of new antineoplastic metallodrugs.6–8The cleavage of both strands of the DNA double helix is medically significant because it causes damage to cells, resulting in cell death.9 Therefore, the study of complexes capable of cleaving the DNA chain at specific sites has garnered interest due to their potential contributions to genomics and their applications in photodynamic therapy and chemotherapy against neoplasms.10,11
Mechanistically, two major types of cleavage can occur, namely hydrolytic or induced by reactive oxygen species (ROS) and the last one being categorized into oxidative or photocleavage. Generally speaking, synthetic metallonucleases that cleave DNA through hydrolytic mechanisms typically feature metal ions with inert redox properties and strong Lewis acidity via a reversible reaction, whereas those with ROS-induced mechanisms usually occur via irreversible reactions involving redox-active metal ions or photosensitizers.9,12,13 The interaction and cleavage of DNA by redox-active metal complexes are crucial for developing antineoplastic therapeutic compounds.14 In this context, certain cations have garnered increased attention, such as manganese(III),15 iron(III),15 cobalt(II),16 ruthenium(II),17,18 and vanadium(IV/V).19,20
Vanadium is a redox-active transition metal abundantly dissolved in marine waters,21 exhibiting ions in variable oxidation states,22 with III, IV, and V being the most common and biologically relevant.23,24 When tetra- or pentavalent, vanadium metal centers are often stabilized in their oxido ([VO]2+ or [VO]3+) or cis-dioxidovanadium (cis-[VO2]+) forms due to their high oxophilicities and Lewis acidity in these oxidation states.25,26 Vanadium ions perform various biological functions,27–29 such as constituting nitrogenase enzymes30,31 and haloperoxidases.32,33
Several types of vanadium-containing compounds have been studied for their potential therapeutic properties,34–36 including insulin-enhancing properties37 (notably, complexes bis(maltolate)oxidovanadium(IV), BMOV, and bis(ethylmaltolate)oxidovanadium(IV), BEOV),38,39 antibacterial,40,41 antiviral,42–44 antiparasitic,45,46 and antineoplastic activities47–49 (e.g., bis(4,7-dimethyl-1,10-phenanthroline)sulfateoxidovanadium(IV), metvan).50 In recent years, several vanadium(V) complexes containing different imine ligands have been synthesized and evaluated for their interactions with biomacromolecules, including serum albumins and DNA,51–53 cytotoxic potential,35,54–56 and/or photoinduced DNA cleavage.57–59
Recently, the focus on vanadium complexes has extended to specifically target mitochondria,60 offering a potential strategy for effectively inducing tumor cell death. Mitochondria play key roles in cellular energy production and in the execution of cell death through apoptotic mechanisms. Therefore, the production of reactive oxygen species, mitochondrial changes, upregulation of anti-apoptotic factors, and the acquired inability of the tumor suppressor protein p53 to activate the pro-apoptotic proteins Bak and Bax connect this organelle to the formation and growth of neoplasms.61
For mitochondrial targeting of a compound of interest, derivatization with triphenylphosphonium groups (+TPP) has been a strategy employed by the pharmaceutical industry for years,62 as these groups preferentially accumulate in the anionic mitochondrial matrix due to their cationic charge.61 These groups, widely regarded as the “gold standard” for mitochondrial vectors, also exhibit high lipophilicity, exceptional stability under physiological conditions, and facile conjugation with the compound of interest.63 Consequently, the potential for derivatizing +TPP groups with 4-fluoro substituents is assessed.
Fluorination of molecules is a prevalent step in modern medicinal chemistry aimed at developing compounds with enhanced metabolic stability, bioavailability, and protein interactions. Fluorine atoms can modulate lipophilicity and facilitate weak hydrogen bonds and electrostatic interactions in organofluorine compounds.64 The unique physical and chemical characteristics of fluorinated compounds arise from the small atomic radius and high electronegativity of fluorine.65 Significant advances in fluorinated drug development with varying properties have occurred in recent years, including antifungals and antivirals, as well as in antineoplastic therapies.66
This study reports the synthesis of (3-formyl-4-hydroxybenzyl)tris(4-fluorophenyl)phosphonium chloride ([AF]Cl), along with its use as a building block in the synthesis, characterization, and biological applications of three new zwitterionic complexes of cis-dioxidovanadium(v) (C1–C3) containing imine hydrazone ligands ([H2L1]Cl–[H3L3]Cl). The structures of complexes C1–C3 were elucidated by single-crystal X-ray diffraction and characterized by various techniques in solution and in the solid state. Concerning biologically relevant applications, these complexes were evaluated for their interactions and cleavage potentials with DNA and bovine serum albumin through gel electrophoresis assays, fluorescence and UV-vis spectroscopies, and also molecular docking studies.
Experimental
General characterization and instrumentation
All the spectra of the techniques used to characterize the chemical structure of the compounds under study are presented in Fig. S1–S48 in the ESI.†The nuclear magnetic resonance (NMR) spectra of nucleus 1H (600 MHz), 13C (151 MHz), 19F (565 MHz), 31P (243 MHz), and 51V (158 MHz) were recorded using a Bruker AVANCE III 600 MHz spectrometer at 25 °C. Dimethyl sulfoxide (DMSO-d6) and chloroform (CDCl3) were used as deuterated solvents. Tetramethylsilane (TMS, Me4Si) was used as the internal reference, while trichlorideoxidovanadium(V) in deuterated benzene (90% VOCl3/C6D6 solution), triphenylphosphane (PPh3), and trifluorotoluene (CF3Ph) were used as external references. Chemical shifts were reported in parts per million (δ, ppm) and were referenced to residual DMSO-d6 or TMS (CDCl3) peaks. The multiplicities were expressed as follows: s, singlet; d, doublet; t, triplet, and m, multiplet.
The Fourier-transform infrared (FT-IR) spectra in transmission mode were recorded using a Bruker Vertex 70 spectrometer equipped with an ATR diamond accessory in the 4000–30 cm−1 region with 64 scans and 4 cm−1 resolution. On the other hand, the Fourier-transform Raman (FT-Raman) spectra of the complexes (crystalline materials) in absorbance mode were recorded using a Bruker SENTERRA confocal Raman microscope. The scattered beam was collected using an Olympus 20× objective and the spectra were recorded in the 3500–50 cm−1 range with a laser line of 785 nm. The spectra were recorded at a power of 25 mW with 6 coadditions of 10 seconds each.
The mass spectra of the inorganic complexes were obtained using an Amazon ion trap mass spectrometer via electrospray ionization (ESI-MS). The analysis was carried out in an ultrapure acetonitrile solution at a concentration of 500 ppb at 180 μL min−1. The capillary temperature was maintained at 180 to 200 °C and a voltage of −400 to −500 V. The simulated spectra were calculated using the mMass software.67
Finally, the electronic spectra in the UV-Vis region were obtained using a Varian Cary 50 BIO UV-vis spectrometer coupled to a thermostatic bath at 25 ± 1 °C, using N,N-dimethylformamide (DMF) as the solvent and a cuvette with 1.0 cm optical path.
Crystallography
Data were collected using a Bruker D8 Venture Photon 100 diffractometer equipped with an Incoatec IμS high brilliance Mo-Kα X-ray tube with two-dimensional Montel micro-focusing optics. Measurements were made at low temperature using a Cryostream 800 unit from Oxford Cryosystems. The structures were initially solved by the intrinsic phasing method using the XT/SHELXT program and refined with XL/SHELXL with anisotropic displacement factors for non-hydrogen atoms.68 All refinements were made by full-matrix least-squares on F2 with anisotropic displacement parameters for all non-hydrogen atoms. The hydrogen atoms were included in the refinement in calculated positions but the atoms (of hydrogens) that are performing special bonds were located in the Fourier map. Drawings were done using ORTEP-3 2020.1 for Windows.69,70 Crystal data and more details of the data collection/refinements of C1–C3 are presented in Table S1 in the ESI.†DNA binding and cleavage studies
In a typical experiment, the cleavage of plasmid DNA (pBSK-II) was initiated with 2 μL (330 ng DNA), and 2 μL of various buffers (10 mM) alongside 5 μL of the complexes, where concentrations ranged from 0 to 500 μM. To reach a total volume of 20 μL, 11 μL of ultra-pure water was added in an Eppendorf microtube. The reactions were conducted for various durations (8 or 16 hours) at an incubation temperature of 50 °C, shielded from light exposure. To stop the reactions, 5 μL of the sample buffer (0.25 M EDTA, pH 8.0, 50% glycerol, and 0.01% bromophenol blue) was introduced. The samples were subsequently stored at 4 °C until analysis via 1% agarose gel electrophoresis, which contained ethidium bromide (0.3 μg mL−1) for 100 minutes at 90 V in TBE buffer (44.5 mM TRIS, 44.5 mM boric acid, and 1.00 mM EDTA, pH 8.0).The gels were imaged and quantitatively analysed by densitometry using the ImageJ software. Given that ethidium bromide intercalates to a lesser extent with the supercoiled form of the plasmid DNA, a correction factor of 1.47 was applied when assessing this specific conformation of plasmid DNA.71
To determine the DNA interaction and cleavage preferences by the complexes, external agents were incorporated into the reaction mixtures. This included lithium perchlorate (LiClO4, to augment the ionic strength of the reaction media) and minor-groove binders (4′,6-diamidino-2-phenylindole, DAPI, and netropsin, NET; 50 μM) along with a DNA major-groove binder (methyl green; 50 μM).
Studies on the interactions between the complexes and calf thymus DNA (ct-DNA, stock solution 1.0 mM) were conducted using a Varian Cary 50 Bio UV-vis spectrophotometer connected to a thermostatic bath maintained at 25 ± 1 °C. Complexes C1–C3 were examined in a DMF/buffer (TRIS 0.5 mM) solution (5% v/v) at pH 7.40 and an ionic strength (I) of 0.5 mM (NaCl).
Protein binding and cleavage studies
The binding affinities of the complexes for proteins were investigated through their interactions with bovine serum albumin (BSA) using fluorescence spectroscopy. Tryptophan fluorescence quenching assays of BSA (2 μM) were performed in a solution (0.1 M TRIS–HCl buffer–0.1 M NaCl, pH 7.5) at room temperature. The emission spectra were monitored at 290–550 nm with excitation at 280 nm. Upon the incremental addition of complexes, fluorescence spectra were acquired after 5-minute incubation for each addition, utilizing a Cary Varian Eclipse spectrofluorometer at ambient temperature.Molecular docking procedures
The crystallographic structure of DNA, BSA, and human serum albumin (HSA) was obtained from the Protein Data Bank with access codes of 1BNA,724F5S,73 and 3JRY,74 respectively. The chemical structure of compounds C1–C3 was obtained from the experimental X-ray data described in this work. Molecular docking calculations were performed with the GOLD 2022.3 software (Cambridge Crystallographic Data Centre, Cambridge, CB2 1EZ, UK). Hydrogen atoms were added to the biomacromolecules following tautomeric states and ionization data inferred using the GOLD 2022.3 software at pH 7.4.For the DNA structure, an 8 Å radius around the two main possible binding sites (major and minor grooves) was explored,75 while for BSA and HSA, an 8 Å radius around the three main binding pockets (subdomains IIA, IIIA and IB – known as sites I, II, and III, respectively)76,77 was defined and explored. For all biomacromolecules, the standard ChemPLP was used as the scoring function. Protein–ligand interaction profiler (PLIP) webserver (https://plip-tool.biotec.tu-dresden.de/plip-web/plip/index)78 was used for the identification of protein–ligand interactions and the 3D-figures were generated using the PyMOL Molecular Graphics System 1.0 level software (Delano Scientific LLC software, Schrödinger, New York, NY, USA).79
Synthetic procedures
The complexes were synthesized through one-pot in situ (metal template) reactions. All the synthetic methodologies (Scheme 1) and spectroscopic data for starting materials, ligands [H2L1]Cl–[H3L3]Cl, and complexes C1–C3 are detailed in Section S1 in the ESI.†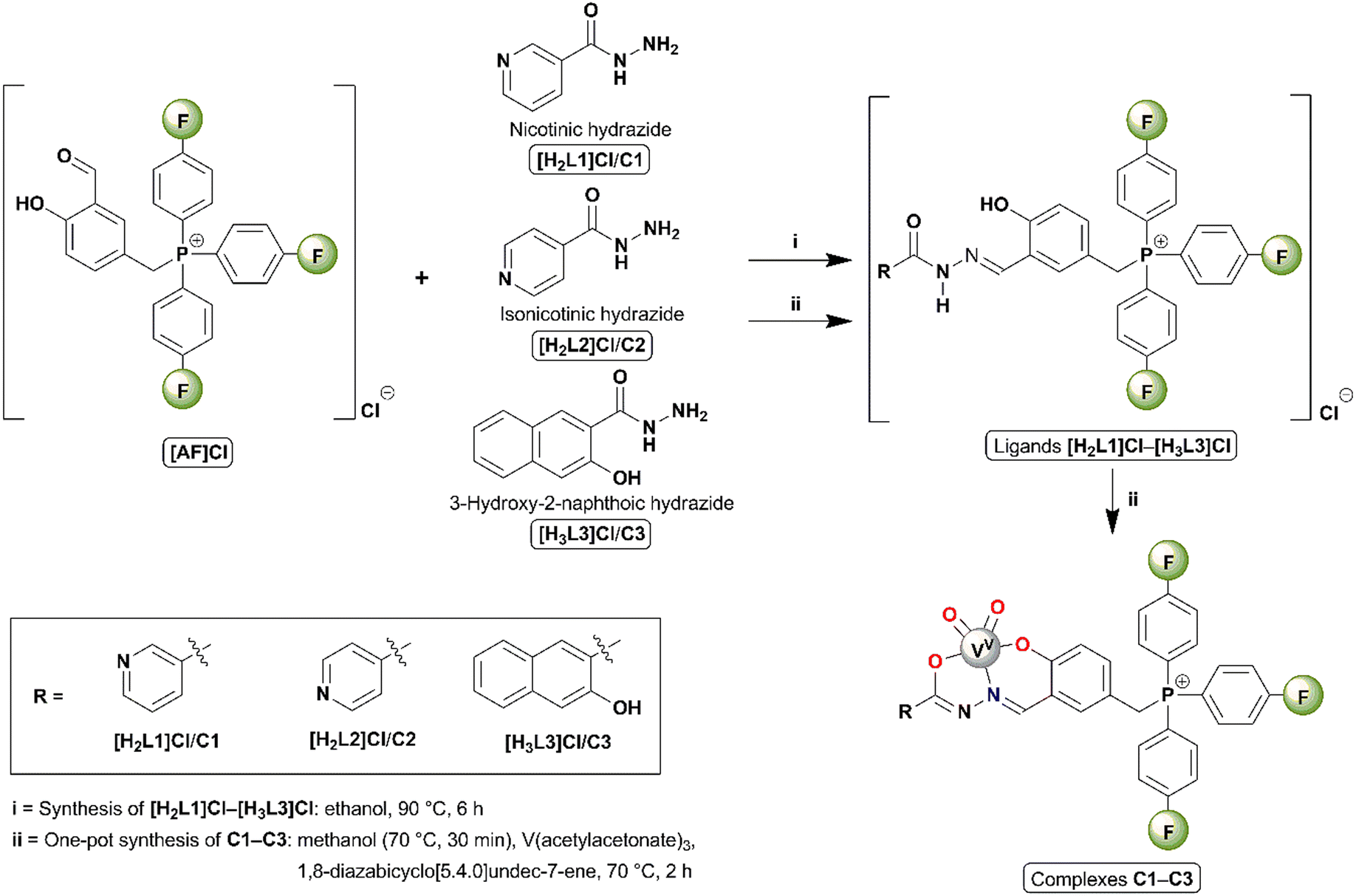 | ||
| Scheme 1 General synthesis of ligands [H2L1]Cl–[H3L3]Cl and complexes C1–C3. | ||
Results and discussion
Solid-state structures
The solid-state structures of the C1–C3 complexes were elucidated by means of single-crystal X-ray diffraction, indicating their same coordination sphere; consequently, the solid-state structures of the complexes will be discussed collectively in a generalized manner. The ORTEP-3 crystallographic structural projections of complexes C1–C3 in the solid state are depicted in Fig. 1. The values of selected bond lengths and angles for C1–C3, alongside similar complexes from the literature, are summarized in Tables S2 and S3 in the ESI.†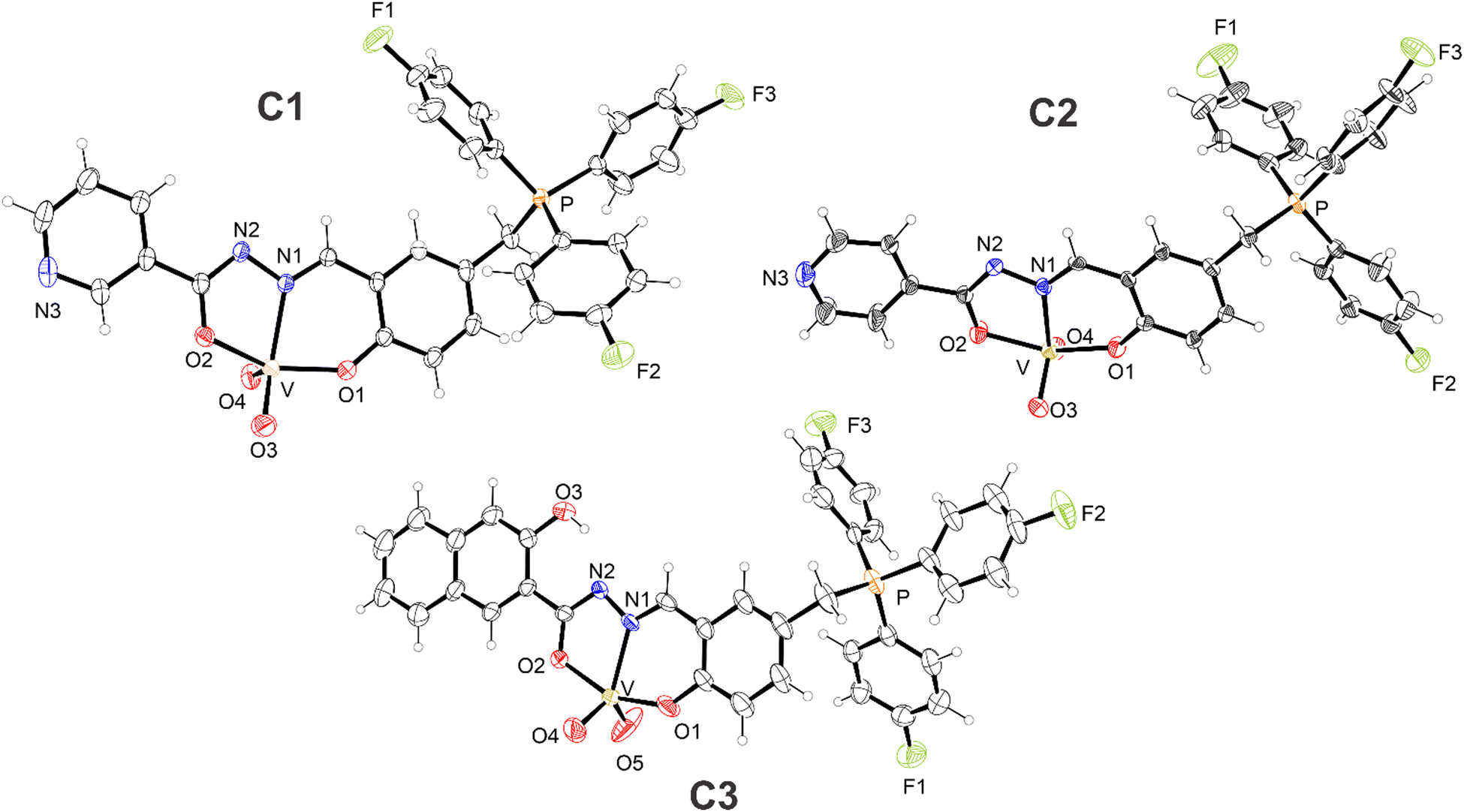 | ||
| Fig. 1 ORTEP-3 crystallographic structural projections of complexes C1, C2, and C3 in the solid state. Thermal ellipsoids are calculated with a 50% probability level. Crystallization solvates are omitted for better visualization. | ||
The complexes feature an iminic hydrazone ligand unity arising from the condensation of (3-formyl-4-hydroxybenzyl)tris(4-fluorophenyl) chloride and the respective hydrazide, resulting in a tridentate chelating cavity that comprises phenol and carbonyl oxygens, and an iminic nitrogen as potential donor atoms for coordination.80 However, the vanadium metal centers in C1–C3 (mononuclear complexes) are coordinated to the respective ligands through the anionic forms of such oxygen atoms. One is in the phenolate form (ranging from 1.898(5) to 1.915(2) Å) and the other is in the enolate form (ranging from 1.973(5) to 1.982(4) Å).
The enolate-form oxygen atom is generated by deprotonation of the amide nitrogen of the hydrazone moiety, as indicated by the bond lengths of C–N(amide) (ranging from 1.292(7) to 1.311(3) Å) and C–O(enolate) (ranging from 1.288(6) to 1.305(3) Å), and the C–N(amide)–N(imine) bond angles81 (ranging from 108.1(2)° to 109.0(5)°) in C1–C3. Moreover, a neutral iminic nitrogen (ranging from 2.138(5) to 2.156(2) Å) acts as a third site in ligands characterized by an O,N,O tridentate dianionic motif. These values for C1–C3, along with the bond length values of their coordination spheres, bear similarities to those of selected complexes in the literature (Tables S2 and S3 in the ESI†).82,83
In addition to the ligand donor atoms, the vanadium metal centers in C1–C3 coordinate with two oxido ligands (ranging from 1.588(6) to 1.646(2) Å), adopting the form of cis-dioxidovanadium (cis-[VO2]+), a distinctive species of vanadium(V). Consequently, C1–C3 exhibit pentacoordinate coordination spheres. The geometries of C1–C3 have been determined based on the values of the structural index parameter τ, calculated as (β − α)/60, with β and α being, respectively, the largest and second largest bond angle values of the coordination spheres. The values of this parameter range from 0, representing a perfect square-based pyramid, to 1, denoting a perfect trigonal bipyramid. Non-integer values within this range indicate distorted geometries.84 The β and α angles (°), calculated τ values (dimensionless), and coordination geometries of C1–C3 are summarized in Table 1, with comparisons to some dioxidovanadium(V) complexes from the literature presented in Table S4 in the ESI.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 82,83
82,83
| Complex | β anglea (°) | α angleb (°) | τ | Coordination geometriesd |
|---|---|---|---|---|
| a Highest bond angle value. b Second highest bond angle value. c Structural index parameter τ (dimensionless) calculated as τ = (β − α)/60. d SBP = square based pyramidal geometry and TB = trigonal bipyramidal geometry. e Two independent units of the complex in the asymmetric unit. | ||||
| C1 | 147.59(8) | 145.77(11) | 0.03 | Slightly distorted SBP |
| C2 | 155.46(17)/153.50(18) | 125.6(2)/136.2(2) | 0.50/0.29 | Distorted between SBP and TB/distorted SBP |
| C3 | 154.9(2) | 129.8(3) | 0.42 | Distorted between SBP and TB |
Furthermore, given the sum of cationic and anionic charges, alongside the absence of counterions, these three complexes are classified as neutral zwitterionic complexes,85 and this is due to the presence of the delocalized cationic charge intrinsic to the ligand of the tris(4-fluorophenyl)phosphonium group. This characteristic distinguishes them from the complexes reported by Maurya et al.86,87 and Dash et al.,57 wherein protonation and coordination of the pyridinic nitrogen atoms occur.
Spectroscopic characterization
The electronic UV-vis spectra of the dioxidovanadium complexes C1–C3 (Fig. S45 of the ESI†) were recorded in DMF solution. For all three compounds, transition bands at 280–410 nm were revealed. The bands around 410 nm are best described as ligand-to-metal charge transfer (LMCT) from the phenolate ligand to the metal, while the other remaining and more energetic bands at 260–350 nm are attributed to intraligand transitions (π → π* and n → π*).88–90The stability of C1–C3 in DMF/buffer – (TRIS 0.5 mM) 5% v/v; pH 7.40, I = 0.5 mM (NaCl) at 25 °C (Fig. S46–S48 of the ESI†) – was examined over 24 h using UV-vis absorption spectroscopy. The absorption spectra of all three compounds remained unchanged, indicating their stability and structural integrity during the investigated period.
DNA binding and cleavage studies
The agarose gel electrophoresis technique is employed to assess the DNA interaction properties of complexes using the plasmid DNA. The ability of vanadium(V) compounds to alter the supercoiled conformation of the pBSK-II plasmid DNA is observed through their electrophoretic mobility on agarose gels. This technique allows for the observation of two distinct DNA forms: the supercoiled circular form I and the open, singly nicked, circular form II.Initially, the impact of pH on the reaction medium was assessed (Fig. 2). For all three complexes, a notable increase in DNA cleavage was observed at pH 6.0 and 7.0, after 8 hours of reaction at a complex concentration of 500 μM. Additionally, we noted that C1–C3 exhibited similar activity. Consequently, pH 7.0 was selected for further assays.
 | ||
| Fig. 2 pH effects on plasmid DNA cleavage. Experimental conditions: [DNA] = 330 ng, ≈25 μM; [buffer] = 10 mM, MES (6.0); HEPES (pH 7.0); HEPES (8.0); CHES (9.00); complexes concentration = 500 mM; temperature = 50 °C; incubation time = 8 hours sheltered from light. | ||
Notably, unlike similar compounds described by Dash and coworkers,58,59 which involve mononuclear dioxovanadium(V) complexes containing aroylhydrazone derivative ligands and have shown DNA cleavage activity through photoinduced reactions, the compounds described herein, C1–C3, can cleave DNA in the absence of light and under mild pH conditions, specifically at pH 7.0.
The effect of complex concentrations was also investigated. For C1–C3, as shown in Fig. S52–S54 in the ESI,† it was possible to observe that the complexes exhibit a capacity to cleave DNA in a concentration-dependent manner at 500–30 μM. This demonstrates that increasing the concentration of the compound also increases its DNase activity.
The cleavage reaction was carried out in both aerobic and anaerobic environments, in a glovebox using argon as an inert gas. As shown in Fig. S55 in ESI,† the absence of oxygen significantly decreased the DNase activity of compounds C1–C3. Therefore, it can be inferred that the cleavage activity is dependent on oxidative processes. To obtain information on the preference for cleavage sites, the cleavage was investigated using DNA groove blockers, and the results of which are shown in Fig. S56 in the ESI.† Generally, complexes C1–C3 showed an increase in DNA cleavage activity in the presence of minor groove binders, DAPI and NET, suggesting higher affinities of the vanadium complexes with the minor groove. Conversely, methyl green, a major groove binder, did not affect the DNase activity.
To comprehend the forms of interaction between the complexes and DNA, the impact of the ionic strength within the reaction medium was initially explored. We found that as the concentration of lithium perchlorate increased from 0 to 50 mM for all three complexes (Fig. 3 for C1 and Fig. S57 and S58 in the ESI† for C2 and C3, respectively); the cleavage of DNA by the complexes also exhibited a gradual decrease for all three complexes. Considering the negative charge of the DNA molecule at pH 7.0, this finding suggests that electrostatic interactions, probably the positive charge centered on the tris(4-fluorophenyl)phosphonium group, between the complex and DNA might play a role in their binding and subsequent cleavage.
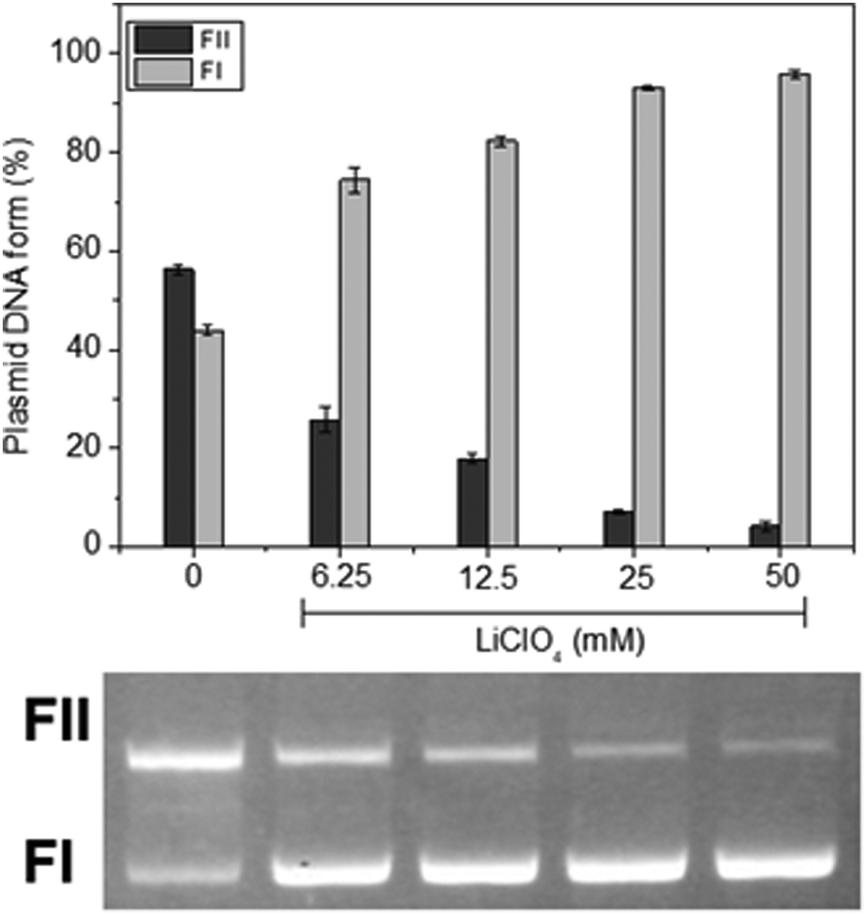 | ||
| Fig. 3 Ionic strength effects on plasmid DNA cleavage for complex C1. Experimental conditions: [DNA] = 330 ng, ≈25 μM; [buffer] = 10 mM, HEPES (pH 7.0); complex concentration = 500 μM; temperature = 50 °C; incubation time = 16 hours sheltered from light. | ||
The nature of the binding interaction between complexes C1–C3 and ct-DNA was also examined using the UV-Vis absorption titration method, observing changes in absorbance and shifts in the wavelength of the complex spectra with increasing amounts of ct-DNA. The results are presented in Fig. 4 for C3 and Fig. S59 and S60 in the ESI† for C1 and C2, respectively. As a result, significant hypochromic changes were observed for all three compounds, along with slight bathochromic shifts.
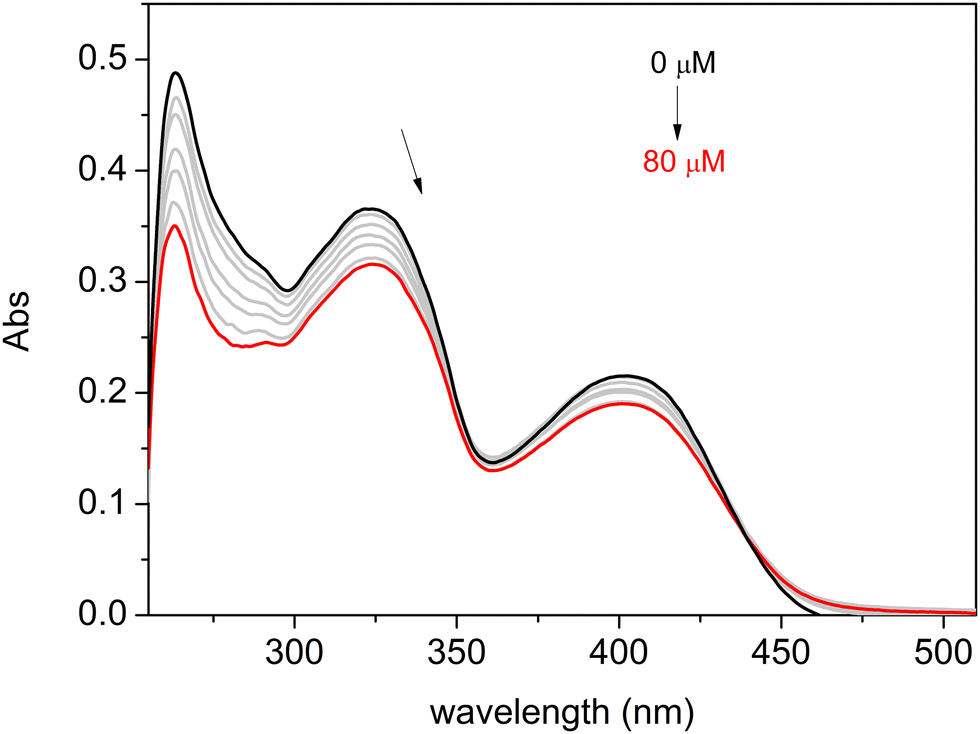 | ||
| Fig. 4 Absorption spectra of complex C3 on the titration of 0–80 μM ct-DNA in DMF/buffer (TRIS 0.5 mM) 5% v/v; pH 7.40, I = 0.5 mM (NaCl); 25 °C. An arrow indicates the changes in absorbance with the increasing concentration of ct-DNA. | ||
In the absorption spectrum of a compound in the presence of DNA, changes such as hypochromism or hyperchromism and a wavelength shift (red or blue) frequently occur. Hypochromism and bathochromism (red shift) in UV-vis spectra are typically linked to the binding of a metal complex to DNA through intercalation involving π-stacking, van der Waals, and hydrophobic interactions between the metal complex and DNA.91
Protein binding studies
Serum albumin is the most abundant protein in the blood, transporting nutrients and drugs throughout the body.92 More precisely, BSA is one of the most extensively studied protein models, with over 75% sequence similarity to human serum albumin and comparable drug-binding affinities.93 Interaction studies between complexes C1–C3 and BSA were conducted through steady-state fluorescence quenching assays at room temperature.BSA fluorescence is primarily governed by the aromatic amino acid residues, namely tyrosine, phenylalanine, and tryptophan (W134 and W213), with excitation at 280 nm. It is well-known that changes in the fluorescence behavior of albumin result from the interactions and/or changes in the microenvironment of these fluorophores caused by the quencher.93
Fig. 5 depicts the protein's maximum emission band at approximately 340 nm when excited at 280 nm. Observations from the steady-state fluorescence spectra revealed that an increase in the complexes’ concentration (Fig. 5 for C1 and C3, and Fig. S61 in the ESI† for C2) led to a significant decrease in the albumin's fluorescence intensity. This evidence suggests the binding between the complexes and the fluorophores of BSA.
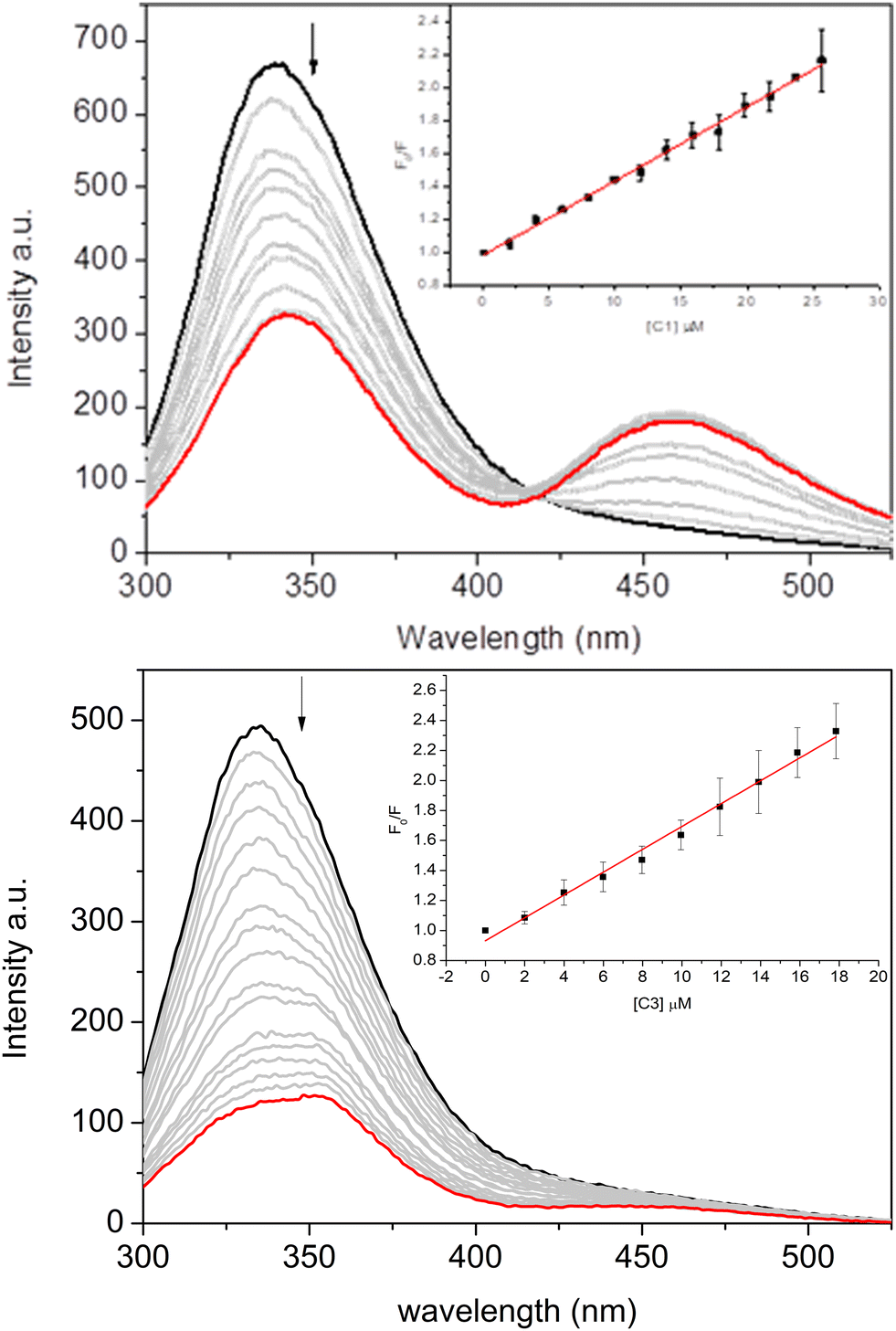 | ||
| Fig. 5 Fluorescence spectra of BSA (2 μM) quenched by complexes C1 (up) and C3 (down) in the concentration range of 0–20 μM. Inset: The Stern–Volmer plot. | ||
An isosbestic point was also observed along with a new fluorescence band, indicating an increased fluorescence intensity in the 425–525 nm range. This phenomenon suggests the formation of an exciplex with BSA, which can be considered further evidence of more complex interactions between albumin and vanadium complexes.94,95 This phenomenon was more evident for C1 and C2 than for C3, indicating the impact of the nicotinic and isonicotinic moieties on the spectroscopic signature of the binding of the inorganic complexes under study. Rio et al.95 previously reported the capacity of albumin in forming exciplex in the presence of heterocyclic compounds (e.g., 1,2,4-triazolo-3-thiones), highlighting the impact of the sulfide and sulfoxide groups.
To further explore these interactions, the Stern–Volmer quenching (KSV) and binding (Kb) constants of the complexes were calculated and are summarized in Table 2. The Stern–Volmer equation is F0/F = 1 + KSV[Q] = 1 + kqτ0[Q], where F0 and F represent the fluorescence intensity without and with the complexes, respectively, KSV is the Stern–Volmer quenching constant, [Q] is the concentration of the complexes, kq is the bimolecular quenching rate, and τ0 is the fluorescence lifetime of BSA (6.06 × 10−9 s).96 This calculation used corrected fluorescence data to account for the effect of dilution. The Kb values and the number of binding sites (n) were obtained from the Scatchard equation using double logarithm plots through the equation log(F0 − F/F) = nlog[Q] + logKb.
| Complex | K SV × 104 (M−1) | k q × 1012 (s−1 M−1) | K b × 104 (M−1) | n | ΔGe (kJ mol−1) |
|---|---|---|---|---|---|
| a Stern–Volmer quenching constant. b Bimolecular quenching rate constant with BSA (τ = 6.06 ns). c Double logarithm binding constant. d Number of binding sites. e Gibbs free-energy values (R = 8.314 J mol−1 and T = 298.15 K). | |||||
| C1 | 4.48 ± 0.09 | 7.34 | 6.03 ± 0.13 | 1.03 ± 0.03 | −27.28 |
| C2 | 3.88 ± 0.07 | 6.36 | 7.76 ± 0.12 | 1.06 ± 0.02 | −27.91 |
| C3 | 7.62 ± 0.26 | 12.49 | 36.31 ± 0.18 | 1.22 ± 0.04 | −31.73 |
In Table 2, one can observe that the number of interaction sites for C1–C3 is preferably one. The results (both KSV and Kb) demonstrated that the binding constants of C1 and C2 are very similar, which is reasonable given their structural similarities. Conversely, compound C3 exhibited the highest binding constant among the three complexes, reflecting the structural effects of incorporating naphthalenol as a pendant arm on the ligand. The values observed are in the same range or higher than those for similar [VVO2]-complexes reported in the literature, indicating that C1–C3 might be distributed in the human bloodstream by the interaction with albumin.26,52,59,97
The bimolecular quenching rate constant (kq) of all the complexes are larger than the diffusion rate constant in water (kdiff ≈ 7.40 × 109 M−1 s−1 at 298 K, following the Smoluchowski–Stokes–Einstein theory98), indicating a ground-state association.99 The negative sign for the Gibb's free energy (ΔG), calculated based on the Van’t Hoff equation (ΔG = −RTlnKb, where R is the gas constant that is equal to 8.314 J mol−1 and T is the temperature in Kelvin), indicates that the binding process is spontaneous at 298.15 K.100
Molecular docking calculations for DNA and proteins
To offer a molecular point of view on the interaction between the [VVO2]-complexes and DNA/BSA/HSA, in silico calculations by the molecular docking approach were carried out at pH 7.4. Table 3 summarizes the docking score value (dimensionless) into the main binding pockets of DNA and serum albumins. Since each pose obtained using the GOLD 2022.3 software is considered as the negative value of the sum of energy terms from the mechanical-molecular type component, and more positive the docking score value indicates better interaction.| Complex | DNA | Bovine serum albumin | Human serum albumin | |||||
|---|---|---|---|---|---|---|---|---|
| Major groove | Minor groove | Site I | Site II | Site III | Site I | Site II | Site III | |
| C1 | 26.1 | 63.1 | 44.9 | 40.5 | 88.6 | 60.4 | 11.9 | 84.2 |
| C2 | 32.1 | 65.1 | 41.4 | 25.2 | 84.9 | 59.0 | 7.76 | 87.8 |
| C3 | 25.6 | 92.0 | 47.8 | 16.8 | 91.1 | 57.0 | 10.1 | 98.7 |
Thus, according to the computational results, for DNA which has two main binding sites (minor and major grooves), the [VVO2]-complexes fit preferentially into minor grooves than into major grooves (Table 3), agreeing with the experimental competitive binding assays. The same trend was previously in silico reported for the interaction between DNA and VV-complexes derived from pyridoxal/salicylaldehyde.52 On the other hand, BSA has three main binding pockets for both endogenous and exogenous compounds: subdomain IIA (site I) located in a hydrophobic binding pocket, subdomain IIIA (site II) also located in a hydrophobic binding pocket, and subdomain IB (site III) located on the surface of the protein.101,102
All binding sites were able to accommodate the [VVO2]-complexes; however, site III is the main pocket (Table 3) with docking score values close to 2-fold higher than for sites I and II. Interestingly, subdomain IB was not reported as the main binding site to VV-complexes derived from pyridoxal or salicylaldehyde,52,103 indicating dependence on the chemical nature of the ligands complexed with vanadium(V) species.
To verify if C1–C3 will also have the same binding trend obtained from bovine to the human analogue, in silico calculations for HSA were also carried out and the results are summarized in Table 3. In this case, subdomain IB was also identified as the main binding site, reinforcing site III as the feasible cavity to the interaction of the [VVO2]-complexes. However, after a deep analysis of the docking score values, it is possible to note that BSA and HSA had practically the same score in site III, with differences in the other binding sites. This was expected due to the highest superposition of the 3D structure of the two proteins into subdomain IB compared to the other subdomains IIA and IIIA (Fig. 7A) with a global root mean square deviation (RMSD) value of 1.634 dimensionless. Interestingly, albumin-C3 had the highest docking score value than albumin-C1 or albumin-C2, following the same experimental binding trend.
In the case of DNA, molecular docking results suggested van der Waals interactions as the main intermolecular force responsible for the interaction with [VVO2]-complexes C1–C3 (Table S5 in the ESI†), and despite the high steric volume of the inorganic complexes under study, they had good fit capacity in the double helix of DNA (Fig. 6) with the positive charge centered in the tris(4-fluorophenyl)phosphonium group of C1–C3 close to the negative charged phosphate moieties of the DNA structure, agreeing with the experimental trend.
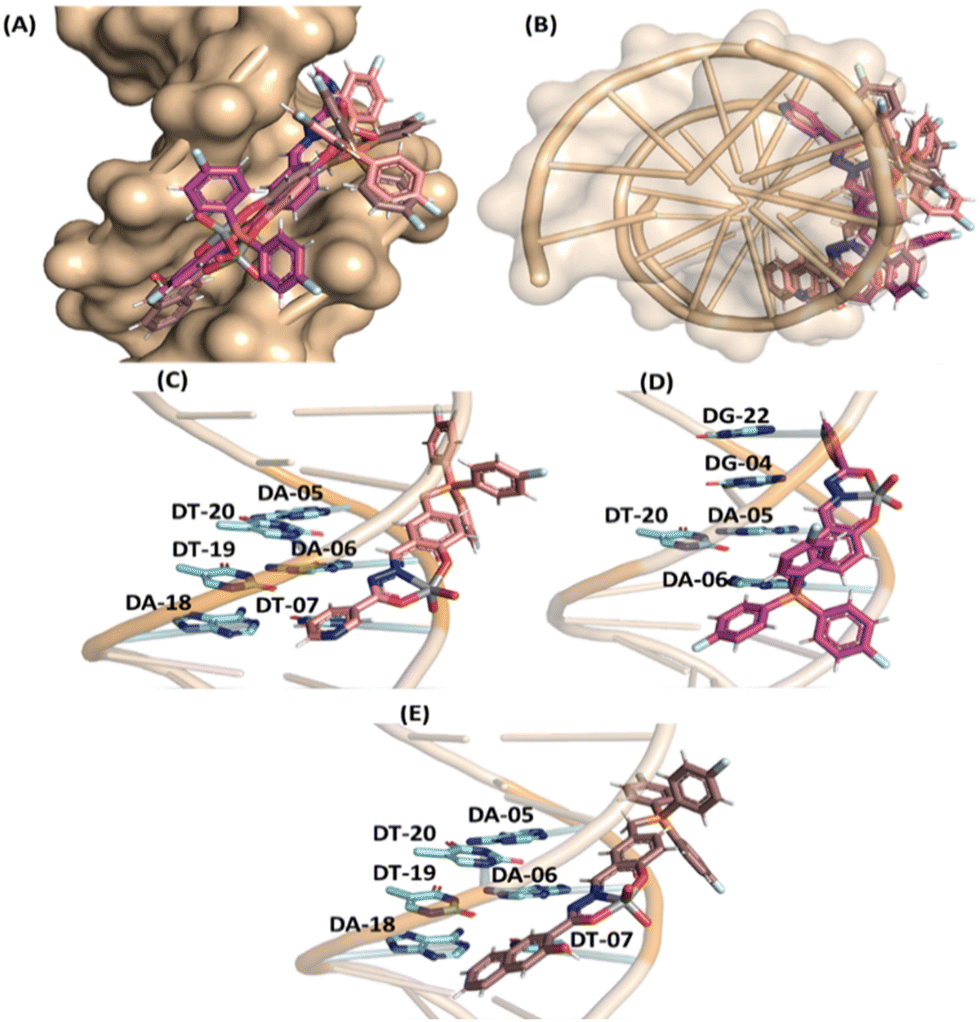 | ||
| Fig. 6 Superposition of the best docking pose for the interaction DNA:[VVO2]–complexes in the minor groove as the (A) front view and (B) top view. A zoom representation for the interaction: (C) DNA:C1, (D) DNA:C2, and (E) DNA:C3. Selected nucleobases, C1, C2, and C3, are in stick representation in light blue, beige, pink, and brown, respectively. Elements’ color: hydrogen, oxygen, nitrogen, fluorine, phosphorus, and vanadium(V) are in white, red, dark blue, cyan, orange, and gray, respectively. For better interpretation, the hydrogen atoms from nucleobases were omitted. | ||
On the other hand, for BSA, interactions via hydrophobic, π-cation, and hydrogen bonding were suggested as the main binding forces, while for HSA hydrophobic interaction was detected and hydrogen bonding with π-stacking interaction was detected only for HSA-C3 (Tables S6, S7 in the ESI† and Fig. 7). Interestingly, Dias et al.104 reported that the interaction of the mixed species cis-[VO(carrier)2-(HSA)] (carrier is picolinato, maltolato, or 3-hydroxy-1,2-dimethyl-4-pyridinone) involves hydrogen bonding and/or hydrophobic interactions with the protein surface, corroborating with the data described above. Finally, in silico calculations suggested that the naphthalenol moiety of C3 is more buried into the protein cavity than the nicotinic and isonicotinic moieties of C1 and C2, respectively, reinforcing the obtained highest experimental binding capacity for C3 to albumin.
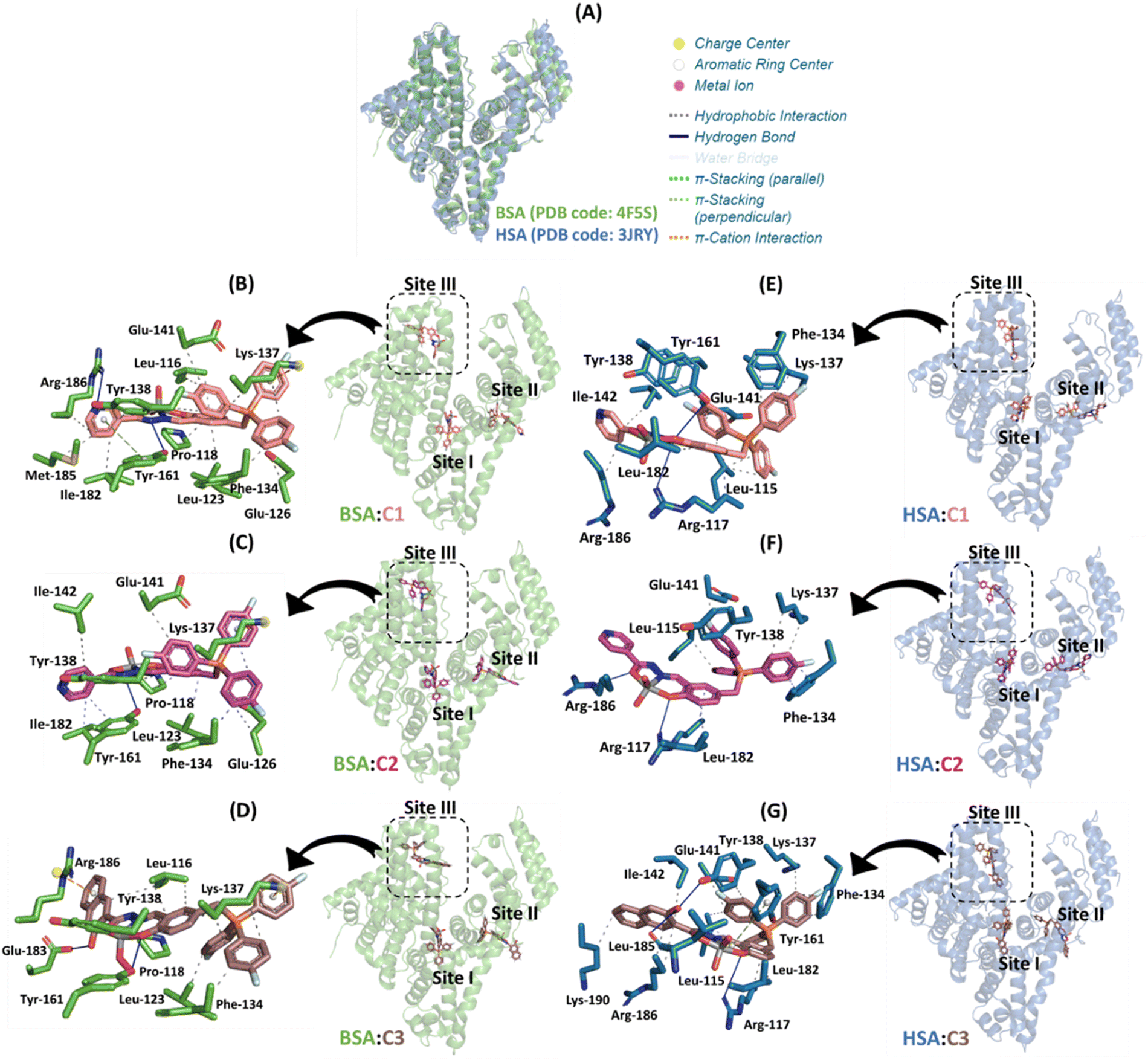 | ||
| Fig. 7 (A) Superposition of the 3D structures of BSA and HSA (PDB codes 4F5S and 3JRY, respectively). The best docking pose for the interaction: (B) BSA:C1, (C) BSA:C2, (D) BSA:C3, (E) HSA:C1, (F) HSA:C2, and (G) HSA:C3 in the three main binding sites with a zoom representation for subdomain IB. Selected amino acid residues for BSA and HSA, C1, C2, and C3, are in stick representation in green, duck blue, beige, pink, and brown, respectively. Elements’ color: oxygen, nitrogen, fluorine, phosphorus, and vanadium(V) are in red, dark blue, cyan, orange, and gray, respectively. For better interpretation, all hydrogen atoms were omitted. | ||
Both experimental and in silico results indicated that the chemical nature of the pendant arms impacts not only the binding capacity to the biomacromolecules but also the steady-state fluorescence signal. For example, for C1, the isosbestic point detected an exciplex formation in the presence of albumin (Fig. 5). Molecular docking calculations suggested that it occurs due to the capacity of the nicotinic moiety from C1 to interact with the fluorophore Tyr-161 via π-stacking force (Fig. 7B). In other words, since albumin was excited at 280 nm, the amino acid residue Tyr in its excited state might transfer energy to the nicotinic moiety of C1. In the case of C2 and C3, a weak and non-exciplex formation was detected, respectively (Fig. S61 and Fig. 5B, respectively). The in silico results suggested that the isonicotinic and naphthalenol moieties from C2 and C3, respectively, interact differently in the subdomain IB of albumin, i.e., the isonicotinic moiety interacts with Tyr-161 via hydrophobic forces (Fig. 7C). In contrast, naphthalenol does not interact with aromatic amino acid residues (Fig. 7D).
Finally, despite the number of connecting points for BSA-C3 is lower than that for BSA-C1 and BSA-C2 (Fig. 7), the inorganic complex C3 presented the highest binding affinity (around 6- and 4.7-fold higher than C1 and C2, respectively, Table 2) probably due to the capacity of the naphthalenol moiety in interaction via π-cation forces with the charge center of Arg-186 driven by the hydrogen bond between the hydroxyl group of the naphthalenol moiety and Glu-183.
Conclusions
In this work, we successfully synthetized three novel vanadium complexes (C1–C3) derived from three iminic ligands ([H2L1]Cl–[H3L3]Cl) containing a tris(4-fluorophenyl)phosphonium ([AF]Cl) moiety. The complexes, obtained as single-crystals, had their solid-state structure elucidated by the single-crystal X-ray diffraction technique, revealing C1–C3 as cis-dioxidovanadium(V) zwitterionic complexes. The structures of the complexes were also corroborated by several solid-state and solution characterization techniques.In vitro tests with DNA revealed that complexes C1–C3 are capable of interacting with and cleaving DNA. Competitive binding studies combined with in silico modeling indicated a preference for all the complexes to bind to the minor groove of DNA, significant for the specificity of DNA cleavage. All the new vanadium(V) compounds exhibited significant interactions with the protein, as demonstrated by the binding constant values from emission spectroscopy, indicating a substantial interaction between BSA and the complexes.
Experimental and molecular docking studies showed that compound C3, which incorporates naphthalenol as a pendant arm on the ligand, interacts more strongly with the protein than C1 and C2. This heightened interaction is attributed to π-stacking, enhancing the attraction between the amino acid (the naphthalenol moiety of C3 is more buried into the protein cavity than the nicotinic and isonicotinic moieties of C1 and C2, respectively) residues and the compound.
Author contributions
Francisco Mainardi Martins: methodology, formal analysis, investigation, writing – original draft, writing – review and editing, and visualization. Daniele Cocco Durigon: methodology, formal analysis, investigation, writing – original draft, writing – review and editing, and visualization. Otávio Augusto Chaves: software, validation, investigation, writing – original draft, writing – review and editing, and visualization. Rosely Aparecida Peralta: formal analysis, investigation, writing – review and editing, and visualization. Davi Fernando Back: methodology, validation, formal analysis, investigation, writing – original draft, writing – review and editing, visualization, supervision, and funding acquisition. Hernán Terenzi: methodology, validation, formal analysis, investigation, writing – original draft, writing – review and editing, visualization, supervision, project administration, and funding acquisition.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.† CCDC 2367124, 2367125, and 2367126 contain the supplementary crystallographic data for complexes C1–C3, respectively.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Laboratório de Biologia Molecular Estrutural (LABIME) from Universidade Federal de Santa Catarina (UFSC) for ESI-MS analysis and would like to thank the financial agencies: Brazilian Research Councils: CNPq – Edital PQ 308411/2022-6 and CAPES-PROEX – Finance Code 001. We would also like to thank Atlas Assessoria Linguística for support with the English version of this manuscript. O. A. C. also thanks Fundação para a Ciência e Tecnologia (FCT, Portuguese agency for scientific research) for his PhD fellowship 2020.07504.BD (https://doi.org/10.54499/2020.07504.BD).Notes and references
- C. Metcalfe and J. A. Thomas, Chem. Soc. Rev., 2003, 32, 215 RSC.
- R. Oun, Y. E. Moussa and N. J. Wheate, Dalton Trans., 2018, 47, 6645–6653 RSC.
- A. Callejo, L. Sedó-Cabezón, I. Juan and J. Llorens, Toxics, 2015, 3, 268–293 CrossRef CAS PubMed.
- L. Galluzzi, I. Vitale, J. Michels, C. Brenner, G. Szabadkai, A. Harel-Bellan, M. Castedo and G. Kroemer, Cell Death Dis., 2014, 5, e1257 CrossRef CAS PubMed.
- L. Galluzzi, L. Senovilla, I. Vitale, J. Michels, I. Martins, O. Kepp, M. Castedo and G. Kroemer, Oncogene, 2012, 31, 1869–1883 CrossRef CAS PubMed.
- B. K. Biswas, N. Biswas, S. Saha, A. Rahaman, D. P. Mandal, S. Bhattacharjee, N. Sepay, E. Zangrando, E. Garribba and C. Roy Choudhury, J. Inorg. Biochem., 2022, 237, 111980 CrossRef CAS PubMed.
- A. Kellett, Z. Molphy, C. Slator, V. McKee and N. P. Farrell, Chem. Soc. Rev., 2019, 48, 971–988 RSC.
- B. J. Pages, D. L. Ang, E. P. Wright and J. R. Aldrich-Wright, Dalton Trans., 2015, 44, 3505–3526 RSC.
- C. Wende, C. Lüdtke and N. Kulak, Eur. J. Inorg. Chem., 2014, 2597–2612 CrossRef CAS.
- E. Palmajumder, S. R. Dash, J. Mitra and K. K. Mukherjea, ChemistrySelect, 2018, 3, 7429–7438 CrossRef CAS.
- E. Palmajumder, N. Sepay and K. K. Mukherjea, J. Biomol. Struct. Dyn., 2018, 36, 919–927 CrossRef CAS PubMed.
- Z. Yu and J. Cowan, Curr. Opin. Chem. Biol., 2018, 43, 37–42 CrossRef CAS PubMed.
- M. Anjomshoa and B. Amirheidari, Coord. Chem. Rev., 2022, 458, 214417 CrossRef CAS PubMed.
- I. Correia, S. Roy, C. P. Matos, S. Borovic, N. Butenko, I. Cavaco, F. Marques, J. Lorenzo, A. Rodríguez, V. Moreno and J. C. Pessoa, J. Inorg. Biochem., 2015, 147, 134–146 CrossRef CAS PubMed.
- L. Shi, Y.-Y. Jiang, T. Jiang, W. Yin, J.-P. Yang, M.-L. Cao, Y.-Q. Fang and H.-Y. Liu, Molecules, 2017, 22, 1084 CrossRef PubMed.
- D. Li, N. Zhang, Z. Yang and Z. Tao, Appl. Organomet. Chem., 2017, 31, e3548 CrossRef.
- M. Ganeshpandian, M. Palaniandavar, A. Muruganantham, S. K. Ghosh, A. Riyasdeen and M. A. Akbarsha, Appl. Organomet. Chem., 2018, 32, e4154 CrossRef.
- S. Akhter, M. Usman, F. Arjmand and S. Tabassum, Polyhedron, 2022, 213, 115618 CrossRef CAS.
- P. R. Inamdar and A. Sheela, Int. J. Biol. Macromol., 2015, 76, 269–278 CrossRef CAS PubMed.
- R. Paulpandiyan and N. Raman, Bioorg. Chem., 2017, 73, 100–108 CrossRef CAS PubMed.
- E. K. Moore, J. Hao, S. J. Spielman and N. Yee, Geobiology, 2020, 18, 127–138 CrossRef CAS PubMed.
- K. Kustin, J. Inorg. Biochem., 2015, 147, 32–38 CrossRef CAS PubMed.
- L. Hernández, M. L. Araujo, W. Madden, E. Del Carpio, V. Lubes and G. Lubes, J. Inorg. Biochem., 2022, 229, 111712 CrossRef PubMed.
- S. M. Shaheen, D. S. Alessi, F. M. G. Tack, Y. S. Ok, K.-H. Kim, J. P. Gustafsson, D. L. Sparks and J. Rinklebe, Adv. Colloid Interface Sci., 2019, 265, 1–13 CrossRef CAS PubMed.
- M. Sutradhar, J. A. L. Da Silva and A. J. L. Pombeiro, in Vanadium Catalysis, ed. M. Sutradhar, A. J. L. Pombeiro and J. A. L. Da Silva, The Royal Society of Chemistry, 2020, pp. 1–11 Search PubMed.
- G. Sahu, A. Banerjee, R. Samanta, M. Mohanty, S. Lima, E. R. T. Tiekink and R. Dinda, Inorg. Chem., 2021, 60, 15291–15309 CrossRef CAS PubMed.
- D. Rehder, Metallomics, 2015, 7, 730–742 CrossRef CAS PubMed.
- D. Rehder, Inorg. Chim. Acta, 2023, 549, 121387 CrossRef CAS.
- J. Costa Pessoa, E. Garribba, M. F. A. Santos and T. Santos-Silva, Coord. Chem. Rev., 2015, 301–302, 49–86 CrossRef CAS.
- L. C. Seefeldt, Z.-Y. Yang, D. A. Lukoyanov, D. F. Harris, D. R. Dean, S. Raugei and B. M. Hoffman, Chem. Rev., 2020, 120, 5082–5106 CrossRef CAS PubMed.
- N. S. Sickerman, Y. Hu and M. W. Ribbe, Chem. - Asian J., 2017, 12, 1985–1996 CrossRef CAS PubMed.
- Z. Chen, Coord. Chem. Rev., 2022, 457, 214404 CrossRef CAS.
- C. C. McLauchlan, H. A. Murakami, C. A. Wallace and D. C. Crans, J. Inorg. Biochem., 2018, 186, 267–279 CrossRef CAS PubMed.
- A. A. Sharfalddin, I. M. Al-Younis, H. A. Mohammed, M. Dhahri, F. Mouffouk, H. Abu Ali, Md. J. Anwar, K. A. Qureshi, M. A. Hussien, M. Alghrably, M. Jaremko, N. Alasmael, J. I. Lachowicz and A.-H. Emwas, Inorganics, 2022, 10, 244 CrossRef CAS.
- G. Sahu, E. R. T. Tiekink and R. Dinda, Inorganics, 2021, 9, 66 CrossRef CAS.
- J. C. Pessoa, S. Etcheverry and D. Gambino, Coord. Chem. Rev., 2015, 301–302, 24–48 CrossRef CAS PubMed.
- S. Treviño and A. Diaz, J. Inorg. Biochem., 2020, 208, 111094 CrossRef PubMed.
- J. H. McNeill, V. G. Yuen, H. R. Hoveyda and C. Orvig, J. Med. Chem., 1992, 35, 1489–1491 CrossRef CAS PubMed.
- K. H. Thompson and C. Orvig, J. Inorg. Biochem., 2006, 100, 1925–1935 CrossRef CAS PubMed.
- A. Sinha, R. Chaudhary, D. S. Reddy, M. Kongot, M. M. Kurjogi and A. Kumar, Heliyon, 2022, 8, e10125 CrossRef CAS PubMed.
- N. Samart, Z. Arhouma, S. Kumar, H. A. Murakami, D. C. Crick and D. C. Crans, Front. Chem., 2018, 6, 519 CrossRef CAS PubMed.
- A. Zahirović, B. Tüzün, S. Hadžalić, I. Osmanković, S. Roca, S. Begić and M. Fočak, J. Mol. Struct., 2023, 1294, 136564 CrossRef.
- S. Semiz, J. Trace Elem. Med. Biol., 2022, 69, 126887 CrossRef CAS PubMed.
- S.-Y. Wong, R. Wai-Yin Sun, N. P.-Y. Chung, C.-L. Lin and C.-M. Che, Chem. Commun., 2005, 3544 RSC.
- M. F. Mosquillo, P. Smircich, A. Lima, S. A. Gehrke, G. Scalese, I. Machado, D. Gambino, B. Garat and L. Pérez-Díaz, Bioinorg. Chem. Appl., 2020, 2020, 1–10 CrossRef PubMed.
- B. Casarrubias-Tabarez, N. Rivera-Fernández, M. Rojas-Lemus, N. López-Valdez and T. I. Fortoul, Toxicol. Rep., 2020, 7, 1001–1007 CrossRef CAS PubMed.
- I. Leon, J. Cadavid-Vargas, A. Di Virgilio and S. Etcheverry, Curr. Med. Chem., 2017, 24, 112–148 CrossRef CAS PubMed.
- S. Kumar, S. Kumari, R. Karan, A. Kumar, R. K. Rawal and P. Kumar Gupta, Inorg. Chem. Commun., 2024, 161, 112014 CrossRef CAS.
- C. Amante, A. L. De Sousa-Coelho and M. Aureliano, Metals, 2021, 11, 828 CrossRef CAS.
- O. J. D’Cruz and F. M. Uckun, Expert Opin. Invest. Drugs, 2002, 11, 1829–1836 CrossRef PubMed.
- S. P. Dash, A. K. Panda, S. Dhaka, S. Pasayat, A. Biswas, M. R. Maurya, P. K. Majhi, A. Crochet and R. Dinda, Dalton Trans., 2016, 45, 18292–18307 RSC.
- L. P. Fioravanço, J. B. Pôrto, F. M. Martins, J. D. Siqueira, B. A. Iglesias, B. M. Rodrigues, O. A. Chaves and D. F. Back, J. Inorg. Biochem., 2023, 239, 112070 CrossRef PubMed.
- D. Patra, N. Biswas, B. Kumari, P. Das, N. Sepay, S. Chatterjee, M. G. B. Drew and T. Ghosh, RSC Adv., 2015, 5, 92456–92472 RSC.
- S. Saswati, P. Adão, S. Majumder, S. P. Dash, S. Roy, M. L. Kuznetsov, J. Costa Pessoa, C. S. B. Gomes, M. R. Hardikar, E. R. T. Tiekink and R. Dinda, Dalton Trans., 2018, 47, 11358–11374 RSC.
- L. Mato-López, A. Sar-Rañó, M. R. Fernández, M. L. Díaz-Prado, A. Gil, Á. Sánchez-González, N. Fernández-Bertólez, J. Méndez, V. Valdiglesias and F. Avecilla, J. Inorg. Biochem., 2022, 235, 111937 CrossRef PubMed.
- G. Sahu, S. A. Patra, P. D. Pattanayak, W. Kaminsky and R. Dinda, Inorg. Chem., 2023, 62, 6722–6739 CrossRef CAS PubMed.
- S. P. Dash, A. K. Panda, S. Pasayat, R. Dinda, A. Biswas, E. R. T. Tiekink, S. Mukhopadhyay, S. K. Bhutia, W. Kaminsky and E. Sinn, RSC Adv., 2015, 5, 51852–51867 RSC.
- S. P. Dash, A. K. Panda, S. Pasayat, R. Dinda, A. Biswas, E. R. T. Tiekink, Y. P. Patil, M. Nethaji, W. Kaminsky, S. Mukhopadhyay and S. K. Bhutia, Dalton Trans., 2014, 43, 10139 RSC.
- S. P. Dash, A. K. Panda, S. Pasayat, S. Majumder, A. Biswas, W. Kaminsky, S. Mukhopadhyay, S. K. Bhutia and R. Dinda, J. Inorg. Biochem., 2015, 144, 1–12 CrossRef CAS PubMed.
- A. Kumar, I. Pant, A. Dixit, S. Banerjee, B. Banik, R. Saha, P. Kondaiah and A. R. Chakravarty, J. Inorg. Biochem., 2017, 174, 45–54 CrossRef CAS PubMed.
- P. Lu, B. J. Bruno, M. Rabenau and C. S. Lim, J. Controlled Release, 2016, 240, 38–51 CrossRef CAS PubMed.
- W. Xu, Z. Zeng, J. Jiang, Y. Chang and L. Yuan, Angew. Chem., Int. Ed., 2016, 55, 13658–13699 CrossRef CAS PubMed.
- G. Schanne, L. Henry, H. C. Ong, A. Somogyi, K. Medjoubi, N. Delsuc, C. Policar, F. García and H. C. Bertrand, Inorg. Chem. Front., 2021, 8, 3905–3915 RSC.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- R. Guo and J. Chen, RSC Adv., 2018, 8, 17110–17120 RSC.
- E. A. Ilardi, E. Vitaku and J. T. Njardarson, J. Med. Chem., 2014, 57, 2832–2842 CrossRef CAS PubMed.
- M. Strohalm, D. Kavan, P. Novák, M. Volný and V. Havlíček, Anal. Chem., 2010, 82, 4648–4651 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- Y. Jin, M. A. Lewis, N. H. Gokhale, E. C. Long and J. A. Cowan, J. Am. Chem. Soc., 2007, 129, 8353–8361 CrossRef CAS PubMed.
- H. R. Drew, R. M. Wing, T. Takano, C. Broka, S. Tanaka, K. Itakura and R. E. Dickerson, Proc. Natl. Acad. Sci. U. S. A., 1981, 78, 2179–2183 CrossRef CAS PubMed.
- A. Bujacz, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2012, 68, 1278–1289 CrossRef CAS PubMed.
- K. L. Hein, U. Kragh-Hansen, J. P. Morth, M. D. Jeppesen, D. Otzen, J. V. Møller and P. Nissen, J. Struct. Biol., 2010, 171, 353–360 CrossRef CAS PubMed.
- T. Bessega, O. A. Chaves, F. M. Martins, T. V. Acunha, D. F. Back, B. A. Iglesias and G. M. De Oliveira, Inorg. Chim. Acta, 2019, 496, 119049 CrossRef CAS.
- O. A. Chaves, M. R. De Lima Santos, M. C. C. De Oliveira, C. M. R. Sant’Anna, R. C. Ferreira, A. Echevarria and J. C. Netto-Ferreira, J. Mol. Liq., 2018, 254, 280–290 CrossRef CAS.
- O. A. Chaves, D. Cesarin-Sobrinho, C. M. R. Sant’Anna, M. G. De Carvalho, L. R. Suzart, F. E. A. Catunda-Junior, J. C. Netto-Ferreira and A. B. B. Ferreira, J. Photochem. Photobiol., A, 2017, 336, 32–41 CrossRef CAS.
- M. F. Adasme, K. L. Linnemann, S. N. Bolz, F. Kaiser, S. Salentin, V. J. Haupt and M. Schroeder, Nucleic Acids Res., 2021, 49, W530–W534 CrossRef CAS PubMed.
- S. Yuan, H. C. S. Chan and Z. Hu, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2017, 7, e1298 Search PubMed.
- S. A. Aboafia, S. A. Elsayed, A. K. A. El-Sayed and A. M. El-Hendawy, J. Mol. Struct., 2018, 1158, 39–50 CrossRef CAS.
- W. Henderson, L. L. Koh, J. D. Ranford, W. T. Robinson, J. O. Svensson, J. J. Vittal, Y. M. Wang and Y. Xu, J. Chem. Soc., Dalton Trans., 1999, 3341–3343 RSC.
- S. D. Kurbah, M. Asthana, I. Syiemlieh, A. A. Lywait, M. Longchar and R. A. Lal, J. Organomet. Chem., 2018, 876, 10–16 CrossRef CAS.
- M. Mohanty, S. K. Maurya, A. Banerjee, S. A. Patra, M. R. Maurya, A. Crochet, K. Brzezinski and R. Dinda, New J. Chem., 2019, 43, 17680–17695 RSC.
- A. W. Addison, T. N. Rao, J. Reedijk, J. Van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- M. Sutradhar and A. J. L. Pombeiro, Coord. Chem. Rev., 2014, 265, 89–124 CrossRef CAS.
- M. R. Maurya, C. Haldar, A. Kumar, M. L. Kuznetsov, F. Avecilla and J. Costa Pessoa, Dalton Trans., 2013, 42, 11941 RSC.
- M. R. Maurya, N. Jangra, F. Avecilla, N. Ribeiro and I. Correia, ChemistrySelect, 2019, 4, 12743–12756 CrossRef CAS.
- R. Borah, S. Lahkar, N. Deori and S. Brahma, RSC Adv., 2022, 12, 13740–13748 RSC.
- R. Borah, N. Deori, S. Lahkar, S. Paul and S. Brahma, J. Mol. Struct., 2023, 1293, 136224 CrossRef CAS.
- S. Thakur, S. Wahedur, R. M. Gomila, A. Frontera and S. Chattopadhyay, Polyhedron, 2023, 235, 116335 CrossRef CAS.
- M. Sirajuddin, S. Ali and A. Badshah, J. Photochem. Photobiol., B, 2013, 124, 1–19 CrossRef CAS PubMed.
- F. Kratz, J. Controlled Release, 2008, 132, 171–183 CrossRef CAS PubMed.
- S. Ketrat, D. Japrung and P. Pongprayoon, J. Mol. Graphics Modell., 2020, 98, 107601 CrossRef CAS PubMed.
- N. J. Turro, V. Ramamurthy and J. C. Scaiano, Principles of molecular photochemistry: an introduction, University Science Books, Sausalito, Calif, 1st edn, 2009 Search PubMed.
- G. F. Rio, L. H. E. Castro, G. S. R. Souza, O. Augusto Chaves, M. Edilson Freire De Lima, D. Cesarin-Sobrinho and C. M. R. Sant’Anna, J. Mol. Liq., 2024, 407, 125247 CrossRef CAS.
- O. A. Chaves, R. J. S. Loureiro, C. Serpa, P. F. Cruz, A. B. B. Ferreira and J. C. Netto-Ferreira, Int. J. Biol. Macromol., 2024, 263, 130279 CrossRef CAS PubMed.
- F. M. Martins, B. A. Iglesias, O. A. Chaves, J. L. Gutknecht Da Silva, D. B. R. Leal and D. F. Back, Dalton Trans., 2024, 53, 8315–8327 RSC.
- M. Montalti, A. Credi, L. Prodi and M. T. Gandolfi, Handbook of Photochemistry, CRC Press, Boca Raton, FL, USA, 3rd edn, 2006 Search PubMed.
- M. A. G. Soares, F. Souza-Silva, C. R. Alves, L. Vazquez, T. S. De Araujo, C. Serpa and O. A. Chaves, Sci. Pharm., 2024, 92, 32 CrossRef CAS.
- T. V. Acunha, B. M. Rodrigues, J. A. Da Silva, D. D. M. Galindo, O. A. Chaves, V. N. Da Rocha, P. C. Piquini, M. H. Köhler, L. De Boni and B. A. Iglesias, J. Mol. Liq., 2021, 340, 117223 CrossRef CAS.
- O. A. Chaves, C. S. H. Jesus, P. F. Cruz, C. M. R. Sant’Anna, R. M. M. Brito and C. Serpa, Spectrochim. Acta, Part A, 2016, 169, 175–181 CrossRef CAS PubMed.
- V. S. Câmara, O. A. Chaves, B. B. De Araújo, P. F. B. Gonçalves, B. A. Iglesias, M. A. Ceschi and F. S. Rodembusch, J. Mol. Liq., 2022, 349, 118084 CrossRef.
- O. A. Chaves, M. C. C. De Oliveira, C. M. C. De Salles, F. M. Martins, B. A. Iglesias and D. F. Back, J. Inorg. Biochem., 2019, 200, 110800 CrossRef CAS PubMed.
- D. M. Dias, J. P. G. L. M. Rodrigues, N. S. Domingues, A. M. J. J. Bonvin and M. M. C. A. Castro, Eur. J. Inorg. Chem., 2013, 4619–4627 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2367124–2367126 for C1–C3. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4nj03087g |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |