
Accelerated size-focusing light activated synthesis of atomically precise fluorescent Au22(Lys–Cys–Lys)16 clusters†
Parimah
Aminfar
a,
Travis
Ferguson
a,
Emily
Steele
a,
Emerson M.
MacNeil
a,
María Francisca
Matus
 b,
Sami
Malola
b,
Hannu
Häkkinen
b,
Paul N.
Duchesne
a,
Hans-Peter
Loock
c and
Kevin G.
Stamplecoskie
*a
b,
Sami
Malola
b,
Hannu
Häkkinen
b,
Paul N.
Duchesne
a,
Hans-Peter
Loock
c and
Kevin G.
Stamplecoskie
*a
aDepartment of Chemistry, Queen's University, Kingston, Ontario K7L 3N6, Canada. E-mail: kevin.stamplecoskie@queensu.ca
bDepartments of Physics and Chemistry, Nanoscience Center, University of Jyväskylä, FI-40014 Jyväskylä, Finland
cDepartment of Chemistry, University of Victoria, Victoria, British Columbia V8N 5C2, Canada
First published on 1st December 2023
Abstract
Atomically precise metal nanoclusters are promising candidates for various biomedical applications, including their use as photosensitizers in photodynamic therapy (PDT). However, typical synthetic routes of clusters often result in complex mixtures, where isolating and characterizing pure samples becomes challenging. In this work, a new Au22(Lys–Cys–Lys)16 cluster is synthesized using photochemistry, followed by a new type of light activated, accelerated size-focusing. Fluorescence excitation–emission matrix spectroscopy (EEM) and parallel factor (PARAFAC) analysis have been applied to track the formation of fluorescent species, and to assess optical purity of the final product. Furthermore, excited state reactivity of Au22(Lys–Cys–Lys)16 clusters is studied, and formation of type-I reactive oxygen species (ROS) from the excited state of the clusters is observed. The proposed size-focusing procedure in this work can be easily adapted to conventional cluster synthetic methods, such as borohydride reduction, to provide atomically precise clusters.
Atomically precise gold and silver nanoclusters (NCs) have been extensively developed in recent years, due to their remarkable potential in many areas, from catalysis to biomedical applications.1–8 A primary motivation for using pure atomically-precise samples is the structural dependence of their properties. This is particularly important in their biological applications, where purity and optical/electronic characterization become crucial in their utility.9 In addition, a deep insight into ground state and excited state behavior of clusters is required. Finally, tracking of NCs in the body, and understand how they interact with biological environments is critical.
Synthesis of clusters of defined size is a significant challenge, and the most easily obtained solutions of metal clusters are often complex mixtures of different sizes of clusters (different exact numbers of metal atoms and ligands).10,11 Despite remarkable progress in separation of clusters with different sizes from crude solutions,12–14 separation techniques are often time-consuming and need to be well-optimized for new clusters. Furthermore, most of the purification techniques have focused on just a few specific metal cores and ligands such as Au25 and glutathione (GSH).15 Size-focussing to obtain pure samples can take a week or more when one requires pure clusters. The structure/properties of individual clusters can vary greatly, with major electronic changes occurring with the exchange of even a single metal atom, or ligand. For more than a decade, studies of clusters have included mixtures of similar sizes of cluster and numbers of coordinating ligands. Especially in the light of biological and medicinal applications, exhaustive efforts are required to size-focus and purify.
Selecting appropriate characterization methods of metal NCs is as important as optimization of their synthesis. While absorbance, emission, and mass spectroscopy are widely used, fluorescence excitation–emission matrix spectroscopy (EEM) supported by parallel factor (PARAFAC) analysis can also be applied as a powerful, non-destructive, in situ technique to assess the optical purity of metal clusters.16
Considering that synthesis using typical reducing agents (i.e., NaBH4) often yields an unavoidable polydispersity of clusters, light-activated synthesis was previously proposed as an alternative. This photochemical route provides unique control over the most challenging aspects of metal cluster synthesis (Scheme 1).17,18 While the Brust–Schiffrin method is known to provide mixture of clusters, this new photochemical approach along with the size focusing can be used to provide different results and isolate individual clusters rather than mixtures. Using a Norrish type I reaction, alpha-hydroxy radicals are generated in situ as a reducing agent from a photochemical initiator. In this work, Omnirad 2959 is used as a photochemical reducing agent to synthesize emissive Au clusters in the presence of Lysine–Cysteine–Lysine (Lys–Cys–Lys) tripeptide as a ligand. A new, rapid size-focusing step is added to the synthesis to isolate the most stable structure, Au22(Lys–Cys–Lys)16. The formation of Au22(Lys–Cys–Lys)16 clusters was monitored during the synthesis using, both, absorbance spectroscopy and fluorescence EEM spectroscopy. PARAFAC analysis was used to determine the number of intermediates and track their formation during the synthesis. Subsequently, an accelerated size-focusing step was performed by irradiating the crude mixture with a LED at 620 nm for 24 h. The red-light irradiation yields atomically precise Au22(Lys–Cys–Lys)16 clusters (Scheme 1).
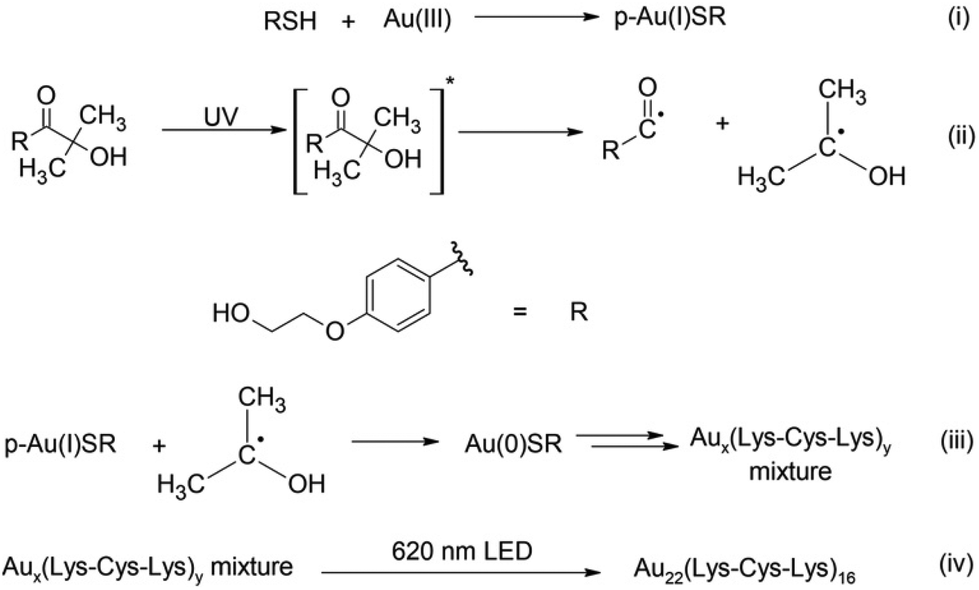 | ||
| Scheme 1 Formation of Au22(Lys–Cys–Lys)16 clusters using Omnirad-2959 as a photochemical initiator to generate reducing alpha-hydroxy radicals, followed by an accelerated size-focusing of the crude mixture. | ||
The Lys–Cys–Lys motif has been previously used to synthesize highly luminescent Ag nanoclusters.19 This cationic tripeptide is highly water soluble and facilitates cellular uptake due to the positively charged Lys unit under neutral pH – a property favourable for many biomedical applications. Using Lys–Cys–Lys ligand, Huang et al. have reported a mixture of red-fluorescent Au nanoclusters that can stain to nucleoli for bio-imaging.20 The work herein is focused more on atomic precision for clusters using this ligand; critical to further studies and advancing to real applications in medicine.
Fluorescence EEM spectroscopy and PARAFAC analysis were used to support the formation of emissive clusters during the synthesis and optical purity of the final product. The assignment of Au22(Lys–Cys–Lys)16 was supported by high-resolution electrospray ionization mass spectrometry (ESI-MS). The excited state reactivity and energy transfer capability of Au22(Lys–Cys–Lys)16 clusters were then studied using chemical probes specific for type-I and type-II reactive oxygen species (ROS) generation.
The Au22(Lys–Cys–Lys)16 synthesis was performed similarly to a method previously reported for Au25 and Ag18 clusters.17,18 9 mM aqueous solution of Lys–Cys–Lys was first reacted with aqueous HAuCl4 (3 mM), in the presence of 9 mM Omnirad 2959 as a photo-initiator. 1 mM NaOH was added to adjust the pH of clear solution to 11. To remove oxygen, the solution was subsequently purged with nitrogen gas for 15 minutes in a quartz cuvette. The solution was then irradiated with 5 UVA lamps (250 W m−2). Absorbance and fluorescence EEM spectra of the solution were monitored during irradiation (ESI 1†). After 12 minutes of irradiation, a mixture of Aux(Lys–Cys–Lys)y NCs was formed with an absorbance feature at 600 nm and emission at 760 nm (Fig. 1). Fluorescence EEM spectroscopy followed by PARAFAC analysis revealed the formation of three emissive clusters while UVA irradiation (Fig. 1). Excitation scan of the solution did not match its absorbance spectrum, indicating poor material purity (Fig. 1). The light activated synthesized mixture was kept at 4 °C and the absorbance and fluorescence EEM of the sample was monitored over the course of two weeks (ESI 2†). Interestingly, the absorbance and fluorescence EEM spectrum of the mixture changed, suggesting the crude mixture was going through a slow size-focusing process towards the formation of the most stable structure. This observation motivated the accelerated size-focusing process, where we use intense LED light to accelerate the process of generating atomically precise clusters.
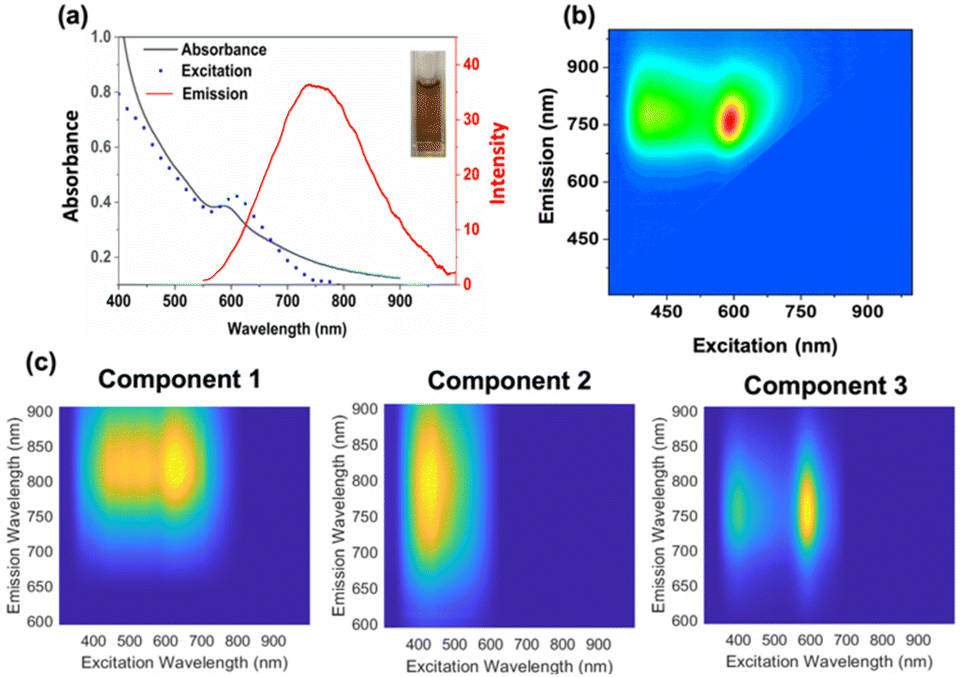 | ||
| Fig. 1 (a) Absorbance spectrum of Aux(Lys–Cys–Lys)y mixture of clusters isolated after 12 minutes of irradiation with UVA light, along with its emission spectrum (λexc = 600 nm) and excitation spectrum (λem = 760 nm); inset: appearance of the product, (b) fluorescence EEM plot of the Aux(Lys–Cys–Lys)y mixture, and (c) fluorescence EEM plot of components obtained from PARAFAC analysis. | ||
To accelerate size-focusing, the solution mixture was irradiated with a 620 nm LED (Luzchem; power: 1.5 mW) and the absorbance spectrum was monitored during irradiation (Fig. 2). After 24 h, the most stable cluster, Au22(Lys–Cys–Lys)16, was identified by fluorescence EEM through its characteristic absorbance feature at 500 nm and strong emission at 790 nm (Fig. 2). The work-up of Au22(Lys–Cys–Lys)16 clusters was performed by concentrating and purifying samples by centrifugation using centrifugal filters with a 3 kDa cut-off. Au22(L)16 clusters were then stored in a refrigerator where they were stable for more than a month at 4 °C.
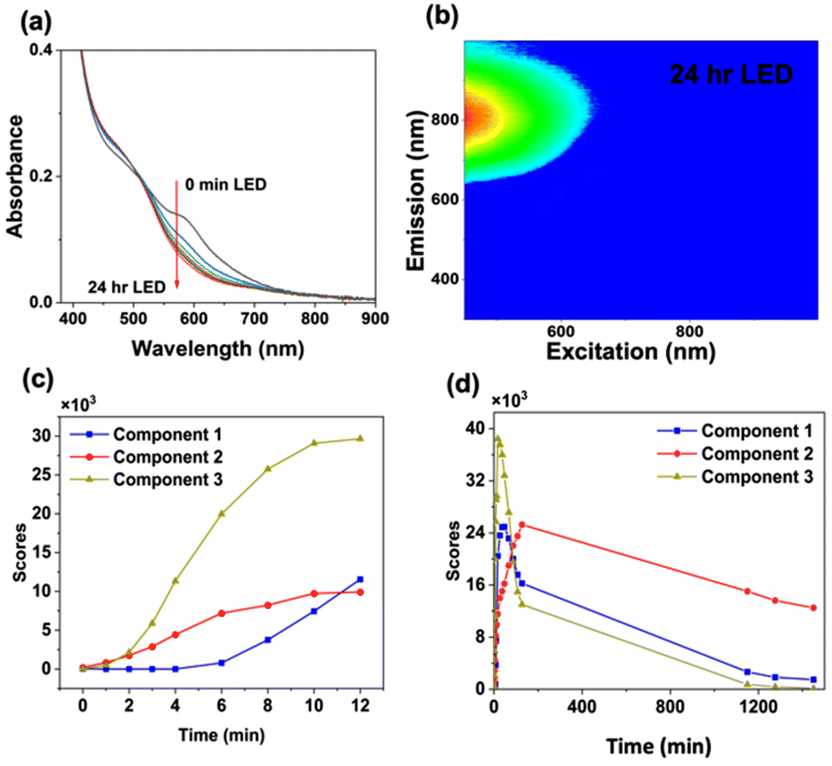 | ||
| Fig. 2 (a) Absorbance spectra of Aux(Lys–Cys–Lys)y mixture over 24 h LED irradiation for size-focusing, (b) fluorescence EEM plot of size-focused Au22(Lys–Cys–Lys)16 clusters after 24 h LED irradiation, and score plot of the 3-component model obtained from PARAFAC analysis during (c) 12 min of UVA irradiation and (d), 12 min of UVA irradiation followed by 24 h size-focusing. | ||
In the first step of synthesis of Au22(L)16 clusters, thiol containing tripeptide solution reduces Au(III) to Au(I) to form thiol coordinated intermediate Au(I)–SR complexes.17 The second step of the synthesis involves formation of alpha-hydroxy radicals upon UVA irradiation and reduction of Au(I)–SR species to form the metal cores (Scheme 1, eqn (i)). In the final step, size-focusing is performed using LED irradiation, providing the most stable cluster for 24 h (Scheme 1, eqn (ii)). During the synthesis, the fluorescence EEM spectrum of reaction solution was monitored every 30 seconds.
PARAFAC analysis is a powerful multivariate analysis technique that is well suited to monitor reaction kinetic in simple mixtures of fluorophores.21,22 PARAFAC analysis was performed on fifteen EEM spectra to qualitatively indicate the number of emissive components in EEM data. Before performing PARAFAC analysis, the scattering contribution was removed from each EEM spectra of the data set using EZspectr software, and each EEM was subsequently normalized using MATLAB. Prior to mathematical decomposition, the number of components that best describe the dataset must be approximated. PARAFAC analysis was then performed on the EEM scans using drEEM Toolbox for MATLAB.23 The best fit to the dataset was obtained using three significant components, when constraining the components to be non-negative in all dimensions (Fig. 1). The fluorescence EEM spectra generated by PARAFAC were found to explain 99.1% of the data when fit to this three-component model. The components were then used to calculate the scores for the components, where the score can be used as a proxy for relative concentration multiplied with the quantum yield. The fluorescence EEM spectra of the three components overlap considerably making multivariate analysis an important tool; it would not have been possible to determine concentrations using just peak positions and intensities in the EEM spectra (see Fig. 1).
Score values obtained from PARAFAC model are proportional to the concentration of each component. From the score plot, it can be observed that after performing accelerated size-focusing (620 nm LED irradiation overnight), component 2 is the most abundant emitter in the solution (Fig. 2). Interestingly, we found that the cluster purity is highest when using shorter UVA irradiation time of the crude mixture (12 min) rather than longer irradiation time (30 min UVA) (ESI 3 and 4†).
The concentrations of precursors and the pH of the solution can influence which material is isolated in light activated synthesis of metal clusters.18 The concentration of reducing agent (Omnirad-2959) played an important role in formation of the Aux(Lys–Cys–Lys)y mixture. A concentration of reducing agent that was three times higher than the initial concentration of HAuCl4 salts, gave the highest yield, as expected when reducing Au(III) ions to Au(0). Important to note, with lower concentrations of Omnirad-2959 no species emitting at wavelengths longer than 600 nm formed even after 1 h of UVA irradiation (ESI 5†). Higher concentrations of reducing agent cause inner-filter effects which slow the reduction step (necessary for formation of clusters rather than nanoparticles) by lowering the steady state concentration of reducing radicals, thus forming near-infrared emitting clusters. As the reduction of Au(I)–SR intermediates is pH-dependent (Scheme 1, eqn (i)), the pH of Au(I)–SR containing solution was adjusted to four different values (8–12) prior to UVA irradiation. The highest yield for the product was obtained at pH 11; determined from absorbance after 12 min of UVA irradiation (ESI 6†).
The effect of non-linear processes was also investigated. Should only the photon flux (concentration) be important, we would expect no difference in yield if the photons were provided continuously or in short, intense pulses. The Au(I)–SR reaction solutions were irradiated with 340 nm pulsed laser light (∼1 nJ per pulse, with 250 fs FWHM). After 11 h of laser irradiation, the total deposited energy is identical to that of continuous excitation under UVA lamps for 1 min. We found that the absorbance of the product was similar to the absorbance profile of clusters formed after 1 min of UVA irradiation (ESI 7†). Non-linear effects therefore do not appear to be important.
In order to assess the thermal effects of irradiation, a thermal reduction method was studied with the Lys–Cys–Lys ligand.24 The Au(I)–SR intermediates were heated to 55 °C and after 24 h blue emitting clusters were formed. They were similar to the products obtained from light activated synthesis when low concentration of the photochemical reducing agent was used (ESI 8†) as opposed to Au22(Lys–Cys–Lys)16.
The emission quantum yield of Au22(Lys–Cys–Lys)16 was found to be 5%, measured with a Hamamatsu absolute quantum yield spectrometer. TEM imaging of Au22(Lys–Cys–Lys)16 solution confirmed the presence of particles that were significantly smaller than 2 nm (∼1.66 nm) (ESI 9†).
The assignment of Au22(Lys–Cys–Lys)16 using mass spectrum of atomically precise clusters was obtained in positive ion mode in ESI-MS (Fig. 3). The most intense peak at 1547.37 m/z is well matched with Au22(C12H22NS)16H8 (z = +5). As a result of ligand fragmentation, only a portion of the ligand remains attached to the metal core. Similar fragmentation patterns have been observed for captopril and glutathione protected Au18 clusters.25 Exhaustive efforts have been done to characterize the other components (ESI 10†). While the major component was characterized as Au22(Lys–Cys–Lys)16, the others do not appear to survive the ionization. Future characteristic studies and identification of the species in the mixture are of great importance since the size distribution range of the crude product has an impact on the final product.26
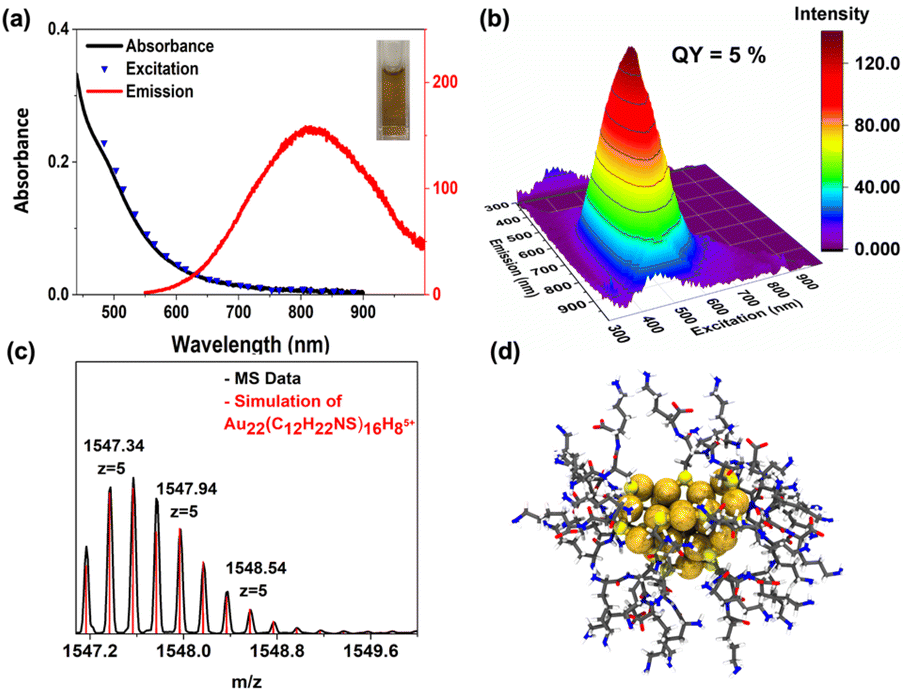 | ||
| Fig. 3 (a) Absorbance spectrum of Au22(Lys–Cys–Lys)16 cluster, along with its emission spectrum (λexc = 500 nm) and excitation spectrum (λem = 790 nm); inset: appearance of the product, (b) fluorescence EEM spectrum of Au22(Lys–Cys–Lys)16 clusters, (c) ESI-MS of the Au22(Lys–Cys–Lys)16 clusters obtained using a spray voltage of 4 kV in a positive ion mode, and (d) proposed structure for Au22(Lys–Cys–Lys)16 clusters obtained with DFT. | ||
A three-dimensional, non-spherical model has been proposed for the structure of Au22(Lys–Cys–Lys)16 clusters (Fig. 3) using density functional theory (DFT) as implemented in software GPAW27 and Perdew–Burke–Ernzerhof (PBE) exchange correlation functional.28 The created model structure is based on the published crystal structure from Li et al., reporting a metastable cluster of the same molecular composition Au22L16 but using adamantanethiolates (SAdm) thiolates as surface ligands L.29 Here, we took the starting structure for the metal core and Au–S interface structure from the crystal and the original SAdm ligands were replaced with new Lys–Cys–Lys ligands. Bond angles and directions at the metal ligand interface were modified only if it was necessary to avoid overlaps between neighbouring ligands. The initial structures were first optimized by the ligand layer only to avoid artifacts due to the non-optimal initial arrangement of the organic part. At next the full structure was optimized by letting all atoms be free. We solved the electronic structure of the model cluster with three charge states +2, neutral and −2 which gave HOMO–LUMO gaps of 0.02 eV, 1.27 eV and 0.83 eV respectively. The electronic structure analysis shows that for neutral cluster there are two 1P symmetric occupied delocalized metal core states and one 2S unoccupied state located close to the Fermi energy. The significant splitting is seen for the third 1P state due to flat oblate shape of the cluster model, which makes the realisation of the gap at 6 superatom electrons reasonable. 1P and 2S states are visualized in ESI 15.† We decided to use the neutral charge structure in further modelling because it exhibited the best electronic stability. Calculated absorption spectrum of the neutral system (ESI 15†) and is in reasonable agreement with the experimentally measured spectrum. Both show a monotonous slow increase of absorption intensity below 900 nm until the change to higher probability transitions with an onset around 600 nm. Calculated spectrum shows four identifiable but features at 675 nm (low extinction), 505 nm, 450 nm and 390 nm. Hence, based on UV-Vis characterization the model structure is consistent with and explains the observed spectra. The first absorption peak at 675 nm originates from 1P to 2S type of transitions that determine the optical gap as visualized in ESI 15.†
In addition, the stability of the proposed model in explicit solvent at room temperature was confirmed via molecular dynamics (MD) simulations (see the ESI† for more details). During 500 ns of production MD, it is observed that the Au22(Lys–Cys–Lys)16 nanocluster adopts a stable conformation indicated by the minimal fluctuation of the atomic positions and compactness of the structure, as described by the root-mean-square deviation (RMSD) and radius of gyration (Rg), respectively (ESI 17†). The dynamics of the ligand layer is governed by the flexibility of the side chains of lysine, while the metal–ligand interface and the proposed Au22 core geometry remains the same (a ESI video is also provided as ESI†). Unstable Au22(SAdm)16 have previously been studied by other models (Ring model) to understand their decomposition and to propose possible isomers.30 However, the clusters isolated herein have absorbance spectra and stability well described by the superatom model. While the present work focusses on synthesis and isolation of Au22(Lys–Cys–Lys)16 this data adds to understanding and theoretical investigations of Au22(SR)16 and its possible isomers.
X-ray absorption spectroscopy (XAS) was used to further characterize the Au22(Lys–Cys–Lys)16 clusters by probing the local structure of the Au atoms in the cluster. Au L3-edge extended X-ray absorption fine structure (EXAFS) data was collected in transmission mode at the Sector 20-BM beamline of the Advanced Photon Source (Argonne National Laboratory in Argonne, Illinois) with an Au foil reference being measured simultaneously with all scans. Samples were analyzed in the aqueous phase at room temperature. Background subtraction and scan averaging were performed using Athena, part of the DEMETER package for EXAFS refinement.31 EXAFS fitting and EXAFS simulation were performed using WINXAS 4.0.2 software.32 The scattering paths and extended X-ray absorption fine structure spectrum of the DFT-calculated structure were simulated using FEFF8 computational software and compared to the experimental Au22(Lys–Cys–Lys)16 EXAFS data (ESI 12†).33 To account for thermal disorder in the simulated EXAFS data of the DFT-calculated structure, an empirical Debye–Waller factor was obtained by fitting the experimental spectrum. This fitting was performed using an Au–S scattering path from the DFT-calculated structure to quantitatively examine the experimental data (ESI 12†); the results are presented in Table S2.† The Au–S coordination number of 2.19, obtained from EXAFS fitting, is greater than the theoretical coordination number of 1.5 from the DFT-calculated structure. This suggests the significant presence of residual Au(I)–thiolate oligomer structures; these oligomers have a higher Au–S coordination number than the cluster, thereby artificially increasing the coordination number value obtained from the fit. The Au–S bond length of 2.295 Å from the EXAFS fitting (Table S2†) is also in good agreement with both the DFT-calculated structure and other Au(I)–thiolate nanoclusters with similar semi-ring core structures.34,35 Although the Au–Au scattering representing Au core structures were not of high enough intensity to reliably fit, the longer-range scattering peaks observed in the experimental EXAFS spectrum show good agreement with those of the DFT-calculated structure EXAFS spectrum.
To elucidate the excited state behaviour of Au22(Lys–Cys–Lys)16, femtosecond transient absorbance spectroscopy (commonly also called pump–probe spectroscopy) was used. The technique provides valuable information in assessing excited state lifetimes and properties related to activity as a photosensitizer. It uses two laser pulses: the first, the pump pulse, is used to excite the sample at a single wavelength and the second, the probe pulse (a white light pulse), is delayed in time and probes changes in absorbance of the sample in the excited state. The difference spectrum (excited state absorbance minus ground state absorbance) is recorded in time, allowing for dynamics of the excited state and relaxation to the ground state to be monitored. Herein, an aqueous solution (pH = 7) of Au22(Lys–Cys–Lys)16 clusters was excited with 250 fs FWHM pump centred at 340 nm and the transient absorption was recorded up to 5 ns after excitation. The results are shown in Fig. S11† and measured relaxation rates (lifetimes) are tabulated in (Table S1†). These clusters behave similarly to other peptide stabilized aqueous clusters,25,36 which exhibit three relaxation components; (1) thermalization of hot electrons, (2) dynamics on the ∼1000 ps timescale that we attribute to formation of a reactive/emissive excited state, and (3) nanosecond to microsecond emission or reactivity from the lowest lying excited state. A Jablonski diagram is shown in Fig. 4 to illustrate the excited state behaviour of the clusters and possible reaction pathways with oxygen.
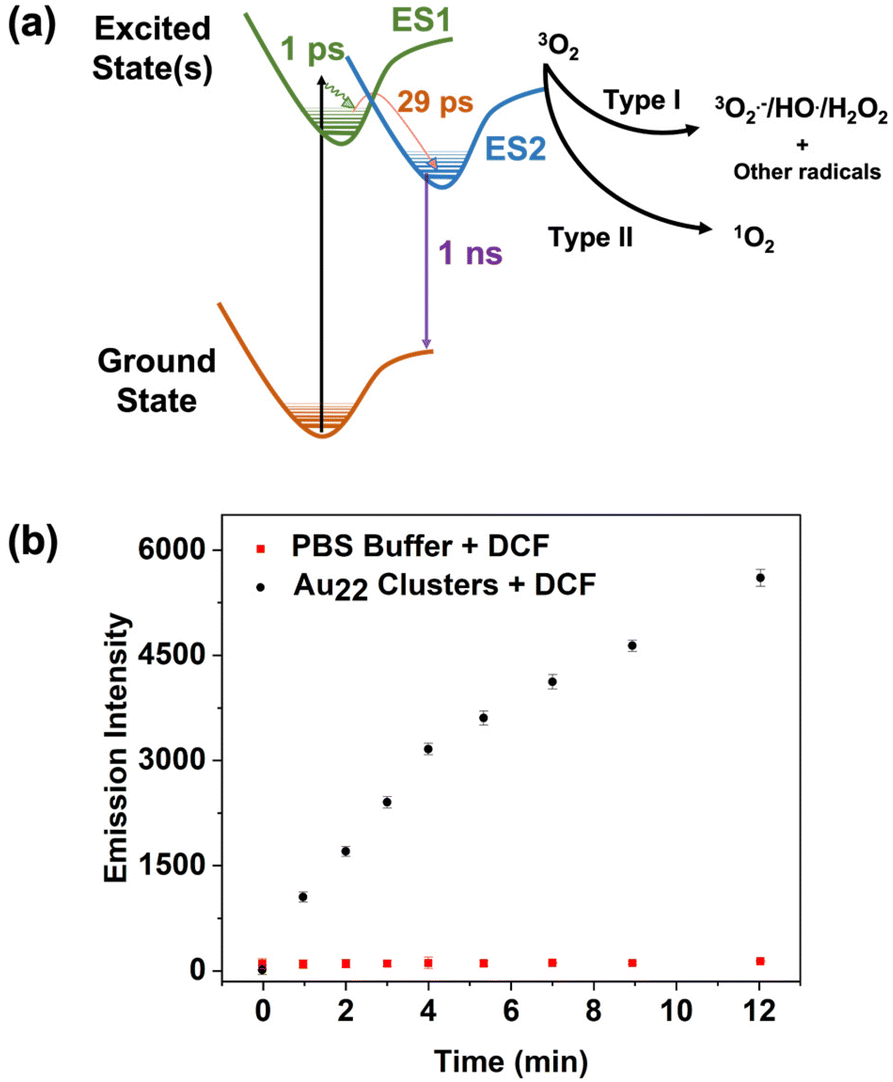 | ||
| Fig. 4 (a) Illustration of the electronic excitation and excited state dynamics involved in the relaxation of Au22(Lys–Cys–Lys)16 clusters and ROS generation pathways. (b) Fluorescent intensity of DCF probe at 525 nm with Xe lamp irradiation at the presence of Au22(Lys–Cys–Lys)16 nanoclusters and PBS blank at 488 nm excitation wavelength. | ||
The efficacy in excited state reactivity and generation of reactive oxygen species of metal NCs is highly dependent to their exact composition.37–39 The excited states of metal clusters have been shown to be reactive with different substrates. In the case of photoinduced therapies such as photodynamic therapy (PDT), the substrate is usually oxygen, and the products are ROS.39–43
In this work, two chemical probes were used to study ROS generation of Au22(Lys–Cys–Lys)16. The production of 1O2 was assessed using 9,10-anthracenediyl-bis(methylene) dimalonic acid (ABDA), which is water-soluble and specific for 1O2 detection.44 While the positive control (with methylene blue as a sensitizer) showed the expected absorbance reduction as a result of 1O2 generation,45 no specific change was observed when using Au22(Lys–Cys–Lys)16 clusters (ESI 13 and 14†). 2,7-Dichlorodihydrofluorescein (DCFH) was used to assess type-I ROS generation of the clusters. DCFH is sensitive to radicals such as H2O2, HO˙, and ROO˙.46,47 Non-fluorescent DCFH can be oxidized by ROS and form dichlorofluorescein (DCF), which is emissive at 525 nm. For this experiment, 250 μL of DCFH ethanol solution (1 mM) was added to the 1 mL sodium hydroxide solution (0.01 mM) which was subsequently diluted with 5 mL of PBS buffer in dark to hydrolyze for 30 minutes. 200 μL of the Au22(Lys–Cys–Lys)16 clusters were then added to 1 mL of DCFH solution and irradiated with a Xe lamp (100 mW cm−2) for 1 min intervals. The fluorescence spectra were measured at 488 nm of excitation wavelength. An increase in the fluorescent maxima of DCF probe observed over 12 min of irradiation, suggesting that the excited state of Au22(Lys–Cys–Lys)16 clusters is reactive, and type-I ROS has been generated (Fig. 4). The excited state reactivity of Au22(Lys–Cys–Lys)16 clusters makes them great candidates for use in photocatalysis. In addition, the ROS generation capability of Au22(Lys–Cys–Lys)16 clusters indicates their potential to function as photosensitizers in PDT.
Thiol protected nanoclusters have been studied for their excited state photocatalytic activity.48,49 The long-lived excited states observed in the present work are reactive towards molecular oxygen for the generation of ROS. While it is commonly reported that triplet excited states are necessary for generation of ROS,50,51 this is not strictly true. The reactivity of singlet excited states for the efficient formation of both type I and type II ROS are well documented and determination of the spin of the reactive excited states is beyond the scope of the present work.52–54
In conclusion, Norrish type I photochemistry followed by a new accelerated size-focusing procedure was used to isolate atomically precise Au22(Lys–Cys–Lys)16 clusters. Fluorescence EEM spectroscopy followed by PARAFAC analysis are powerful techniques in monitoring the formation of emissive intermediates during the synthesis of Au22(Lys–Cys–Lys)16 clusters and assessing purity of the final product. The assignment of the clusters as Au22(Lys–Cys–Lys)16 is supported by ESI-MS spectroscopy. The size-focusing step used in this work is a simple and useful synthetic practice for isolation/purification of new atomically precise metal clusters. The excited state reactivity of optically pure Au22(Lys–Cys–Lys)16 clusters and their photocatalytic ability towards ROS generation illustrates the utility of this material for biomedical applications and photocatalysis.
Conflicts of interest
There are no conflics to declare.Acknowledgements
The research presented herein was supposed by the Natural Science and Engineering Research Council (NSERC) through the Discovery Grant program award number RGPIN-2016-07050, New Frontiers in Research Fund (NFRFT-2020-00573), as well as Canadian Foundation for Innovation under the John R. Evans Leaders Fund program award number CFI-36423. We would also like to thank iGM Resins for the generous donation of the photoinitiator Omnirad 2959. The computational work was done at the CSC computing center at University of Jyväskylä, supported by the Academy of Finland and by the Excellence Funding from JYU Rector.References
- Metal Nanoclusters in Catalysis and Materials Science, ed. B. Corain, G. Schmid and N. Toshima, Elsevier, Amsterdam, 2008, p. v Search PubMed.
- A. Corma, Nat. Nanotechnol., 2014, 9, 412–413 CrossRef CAS PubMed.
- Y. Zhu, H. Qian, A. Das and R. Jin, Chin. J. Catal., 2011, 32, 1149–1155 CrossRef CAS.
- Z. Wu, D.-E. Jiang, A. K. P. Mann, D. R. Mullins, Z.-A. Qiao, L. F. Allard, C. Zeng, R. Jin and S. H. Overbury, J. Am. Chem. Soc., 2014, 136, 6111–6122 CrossRef CAS PubMed.
- Y.-C. Shiang, C.-C. Huang, W.-Y. Chen, P.-C. Chen and H.-T. Chang, J. Mater. Chem., 2012, 22, 12972–12982 RSC.
- Y. Zhang, C. Zhang, C. Xu, X. Wang, C. Liu, G. I. N. Waterhouse, Y. Wang and H. Yin, Talanta, 2019, 200, 432–442 CrossRef CAS.
- D. Su, L. Gao, F. Gao, X. Zhang and X. Gao, Chem. Sci., 2020, 11, 5614–5629 RSC.
- J. Li, J.-J. Zhu and K. Xu, TrAC, Trends Anal. Chem., 2014, 58, 90–98 CrossRef CAS.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- C. M. Aikens, J. Phys. Chem. Lett., 2011, 2, 99–104 CrossRef CAS PubMed.
- P. R. Nimmala and A. Dass, J. Am. Chem. Soc., 2014, 136, 17016–17023 CrossRef CAS PubMed.
- Y. Negishi, S. Hashimoto, A. Ebina, K. Hamada, S. Hossain and T. Kawawaki, Nanoscale, 2020, 12, 8017–8039 RSC.
- D. Li, B. Kumari, X. Zhang, C. Wang, X. Mei and V. M. Rotello, Adv. Colloid Interface Sci., 2020, 276, 102090 CrossRef CAS.
- Y. Niihori, Y. Kikuchi, D. Shima, C. Uchida, S. Sharma, S. Hossain, W. Kurashige and Y. Negishi, Ind. Eng. Chem. Res., 2017, 56, 1029–1035 CrossRef CAS.
- S. Knoppe and P. Vogt, Anal. Chem., 2019, 91, 1603–1609 CrossRef CAS PubMed.
- H. Ramsay, D. Simon, E. Steele, A. Hebert, R. D. Oleschuk and K. G. Stamplecoskie, RSC Adv., 2018, 8, 42080–42086 RSC.
- G. Yousefalizadeh and K. G. Stamplecoskie, J. Photochem. Photobiol., A, 2018, 353, 251–254 CrossRef CAS.
- H. S. Ramsay, M. M. Silverman, D. Simon, R. D. Oleschuk and K. G. Stamplecoskie, Nanoscale, 2019, 11, 20522–20526 RSC.
- N. Goswami, K. Zheng and J. Xie, Nanoscale, 2014, 6, 13328–13347 RSC.
- X. Wang, Y. Wang, H. He, X. Ma, Q. Chen, S. Zhang, B. Ge, S. Wang, W. M. Nau and F. Huang, ACS Appl. Mater. Interfaces, 2017, 9, 17799–17806 CrossRef CAS.
- N. L. P. Andrews, T. Ferguson, A. M. M. Rangaswamy, A. R. Bernicky, N. Henning, A. Dudelzak, O. Reich, J. A. Barnes and H. P. Loock, Anal. Chem., 2017, 89, 8554–8564 CrossRef CAS.
- N. L. P. Andrews, J. Z. Fan, H. Omrani, A. Dudelzak and H. P. Loock, Tribol. Int., 2016, 94, 279–287 CrossRef CAS.
- K. R. Murphy, C. A. Stedmon, D. Graeber and R. Bro, Anal. Methods, 2013, 5, 6557–6566 RSC.
- Y. Chen, J. W. Y. Lam, R. T. K. Kwok, B. Liu and B. Z. Tang, Mater. Horiz., 2019, 6, 428–433 RSC.
- G. Yousefalizadeh and K. G. Stamplecoskie, J. Phys. Chem. A, 2018, 122, 7014–7022 CrossRef CAS PubMed.
- R. Jin, H. Qian, Z. Wu, Y. Zhu, M. Zhu, A. Mohanty and N. Garg, J. Phys. Chem. Lett., 2010, 1, 2903–2910 CrossRef CAS.
- J. Enkovaara, C. Rostgaard, J. J. Mortensen, J. Chen, M. Dułak, L. Ferrighi, J. Gavnholt, C. Glinsvad, V. Haikola, H. A. Hansen, H. H. Kristoffersen, M. Kuisma, A. H. Larsen, L. Lehtovaara, M. Ljungberg, O. Lopez-Acevedo, P. G. Moses, J. Ojanen, T. Olsen, V. Petzold, N. A. Romero, J. Stausholm-Møller, M. Strange, G. A. Tritsaris, M. Vanin, M. Walter, B. Hammer, H. Häkkinen, G. K. Madsen, R. M. Nieminen, J. K. Norskov, M. Puska, T. T. Rantala, J. Schiotz, K. S. Thygesen and K. W. Jacobsen, J. Phys.: Condens.Matter, 2010, 22, 253202 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- Q. Li, S. Yang, T. Chen, S. Jin, J. Chai, H. Zhang and M. Zhu, Nanoscale, 2020, 12, 23694–23699 RSC.
- W. Han, E. Wang and W. W. Xu, Phys. Chem. Chem. Phys., 2022, 24, 15920–15924 RSC.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- T. Ressler, J. Synchrotron Radiat., 1998, 5, 118–122 CrossRef CAS PubMed.
- A. L. Ankudinov, B. Ravel, J. J. Rehr and S. D. Conradson, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 7565–7576 CrossRef CAS.
- D. M. Chevrier, M. A. MacDonald, A. Chatt, P. Zhang, Z. Wu and R. Jin, J. Phys. Chem. C, 2012, 116, 26947–26947 CrossRef CAS.
- Y. Yu, Z. Luo, D. M. Chevrier, D. T. Leong, P. Zhang, D.-E. Jiang and J. Xie, J. Am. Chem. Soc., 2014, 136, 1246–1249 CrossRef CAS PubMed.
- T. D. Green and K. L. Knappenberger, Nanoscale, 2012, 4, 4111–4118 RSC.
- C. Zeng, C. Liu, Y. Pei and R. Jin, ACS Nano, 2013, 7, 6138–6145 CrossRef CAS PubMed.
- Y.-S. Chen, H. Choi and P. V. Kamat, J. Am. Chem. Soc., 2013, 135, 8822–8825 CrossRef CAS.
- V. Poderys, G. Jarockyte, S. Bagdonas, V. Karabanovas and R. Rotomskis, J. Photochem. Photobiol., B, 2020, 204, 111802 CrossRef CAS PubMed.
- R. Ho-Wu, S. H. Yau and T. Goodson, J. Phys. Chem. B, 2017, 121, 10073–10080 CrossRef CAS PubMed.
- L. Tang, X. D. Zeng, H. Zhou, C. H. Gui, Q. L. Luo, W. Y. Zhou, J. Wu, Q. Q. Li, Y. Li and Y. L. Xiao, Chem. Res. Chin. Univ., 2021, 37, 934–942 CrossRef CAS.
- D. Yang, G. X. Yang, S. L. Gai, F. He, G. H. An, Y. L. Dai, R. C. Lv and P. P. Yang, Nanoscale, 2015, 7, 19568–19578 RSC.
- A. McLean, R. F. Wang, Y. Huo, A. Cooke, T. Hopkins, N. Potter, Q. Li, J. Isaac, J. Haidar, R. C. Jin and R. Kopelman, ACS Appl. Nano Mater., 2020, 3, 1420–1430 CrossRef CAS.
- T. Entradas, S. Waldron and M. Volk, J. Photochem. Photobiol., B, 2020, 204, 111787 CrossRef CAS.
- L. V. Lutkus, S. S. Rickenbach and T. M. McCormick, J. Photochem. Photobiol., A, 2019, 378, 131–135 CrossRef CAS.
- S. Hui, Q. Liu, Z. Huang, J. Yang, Y. Liu and S. Jiang, Bioconjugate Chem., 2020, 31, 2439–2445 CrossRef CAS.
- A. Gomes, E. Fernandes and J. L. Lima, J. Biochem. Biophys. Methods, 2005, 65, 45–80 CrossRef CAS.
- K. G. Stamplecoskie and P. V. Kamat, J. Am. Chem. Soc., 2014, 136, 11093–11099 CrossRef CAS PubMed.
- X. Wang, R. Liu, L. Tian, J. Bao, C. Zhao, F. Niu, D. Cheng, Z. Lu and K. Hu, J. Phys. Chem. C, 2022, 126, 18374–18382 CrossRef CAS.
- H. Fakhouri, M. P. Bakulić, I. Zhang, H. Yuan, D. Bain, F. Rondepierre, P.-F. Brevet, Ž. S. Maršić, R. Antoine, V. Bonačić-Koutecký and D. Maysinger, Commun. Chem., 2023, 6, 97 CrossRef CAS.
- R. Ho-Wu, S. H. Yau and T. Goodson, J. Phys. Chem. B, 2017, 121, 10073–10080 CrossRef CAS PubMed.
- G. A. Zalesskaya and A. V. Kuchinsky, Spectrochim. Acta, Part A, 2010, 75, 406–410 CrossRef CAS.
- W. R. Ware, J. Phys. Chem., 1962, 66, 455–458 CrossRef CAS.
- A. Moss, Y. Jang, J. Arvidson, V. N. Nesterov, F. D'Souza and H. Wang, Chem. Sci., 2022, 13, 9880–9890 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr04793h |
| This journal is © The Royal Society of Chemistry 2024 |