
Tuning the electronic structure of Pd by the surface configuration of Al2O3 for hydrogenation reactions†
Yinglei
Liu
a,
Chicheng
Ma
a,
Jiye
Zhang
a,
Huiying
Zhou
a,
Gaowu
Qin
 ab and
Song
Li
*a
ab and
Song
Li
*a
aKey Lab for Anisotropy and Texture of Materials (MoE), School of Materials Science and Engineering, Northeastern University, Shenyang 110819, China. E-mail: lis@atm.neu.edu.cn
bInstitute of Materials Intelligent Technology, Liaoning Academy of Materials, Shenyang 110004, China
First published on 23rd November 2023
Abstract
The electronic interaction between a metal and a support modulates the electronic structures of supported metals and plays an important role in manipulating their catalytic performance. However, this interaction is mainly realized in heterogeneous catalysts composed of reducible oxides. Herein, we demonstrate the electronic interaction between γ-Al2O3 and η-Al2O3 with varying acid–base properties and supported Pd nanoparticles (NPs) of 2 nm in size. The strength and number of acid–base sites on the supports and catalysts were systemically characterized by FT-IR spectroscopy and TPD. The supported Pd NPs exhibit electron-rich surface properties by receiving electrons from the electron-donating basic sites on γ-Al2O3, which are beneficial for catalyzing the hydrogenation of nitrobenzene. In contrast, Pd NPs loaded on η-Al2O3 are electron-deficient because of the rich electron-withdrawing acid sites of η-Al2O3. As a result, Pd/η-Al2O3 exhibits higher catalytic activity in phenylacetylene hydrogenation than Pd/γ-Al2O3. Our results suggest a promising route for designing high-performance catalysts by adjusting the acid–base properties of Al2O3 supports to maneuver the electronic structures of metals.
Introduction
Supported noble metal catalysts have been extensively utilized in both fundamental research and industrial applications owing to their superior reactivity, selectivity, and thermostability.1–4 The supports play important roles in addition to adjusting the spatial distribution and geometric configuration of active metals.5,6 They participate in catalytic reactions by cooperating with metal phases through spillover or synergistic interactions.7,8 Furthermore, electronic metal–support interactions (EMSIs) occur on the interface between metals and their supports, which results in a redistribution of charges in both phases.9–11 The rearrangement of electrons on metal atoms has a profound impact on the adsorption of reactive intermediates and energy barriers in heterogeneous catalysis.12–14 Hence, strategies for modulating the electronic properties of metals by EMSIs to optimize their catalytic performance are highly desirable.In recent years, oxide supports exhibiting reducible or semiconducting behaviors have gained widespread attention in manipulating the electronic structures of metals.15–17 He and coworkers demonstrated that oxygen defects obtained on Pd/TiO2 during the high-temperature reduction enrich the electron state of metallic Pd by extracting electrons from the oxide.16 In our previous work, we reported that the electronic structure of Au NPs is regulated by tailoring the Fermi level of the spinel structured ZnFexCo2−xO4 semiconductors, and the properly charged Au NPs could effectively accelerate the oxidation of CO or benzyl alcohol.17 Therefore, designing supports with the capability of adjusting the electronic properties of noble metals to enhance the catalytic performance remains a research priority.
Al2O3 is considered one of the most commonly used support materials for loading noble metals due to its unique surface configuration and excellent chemical stability.18–20 Unlike reducible oxides and semiconducting oxides, Al2O3 acts as an inert support with non-reducing and insulating properties, which leads to the electronic interaction between metal NPs and Al2O3 not being a concern.21 In previous studies, the coordinatively unsaturated Al3+ centers and terminal hydroxyls on the surface of γ-Al2O3 have been identified as anchoring sites for metallic atoms to achieve high dispersion and thermal stability of the supported metal phase.22,23 Additionally, some researchers have focused on introducing a series of alkali metal oxides into the Al2O3 support to modulate the acid–base sites on the surface of Al2O3 or the electronic properties of supported metal particles to improve the catalytic performance.24–26 It is noteworthy that the Al2O3 surface possesses both Lewis acid and Lewis basic sites, which can capture and donate electrons, respectively, and paramorphic alumina such as γ-Al2O3 and η-Al2O3 exhibit different acid–base properties.27–29 Consequently, Al2O3 shows promise as a support for modulating the electronic properties of the catalytically active metal phase through EMSIs, which has previously been overlooked.
To this end, we synthesized γ-Al2O3 and η-Al2O3 supports with varying levels of acidity and basicity by calcining AlOOH and Al(OH)3 precursors, respectively. Pd NPs with similar particle sizes were loaded onto the supports using a deposition–precipitation method. The surface configurations of γ-Al2O3 and η-Al2O3 before and after Pd deposition, as well as the electronic properties of Pd NPs modulated by the acid–base sites on the surface of Al2O3 supports, have been fully characterized. The influence of electronic structures on the catalytic performance of Pd NPs has been elucidated in hydrogenation reactions with nitrobenzene and phenylacetylene as model substrates. Understanding the electronic modulation induced by the acid–base properties of Al2O3 supports is crucial for the rational design of supported metal catalysts and optimization of the catalytic performance of metal NPs.
Experimental
Materials
Aluminum nitrate, sodium tetrachloropalladate, ammonium hydroxide, sodium carbonate and sodium borohydride were purchased from Sinopharm. Nitrobenzene and phenylacetylene were purchased from Aladdin.Synthetic procedures of catalysts
Synthesis of Pd/Al2O3
Pd NPs were deposited onto the Al2O3 supports by a deposition–precipitation method. 100 mg of supports were completely dispersed in 30 mL of deionized water, and then the mixture was transferred to a water bath at 353 K. Subsequently, 69 μL of Na2PdCl4 aqueous solution (20 g L−1) was added. After stirring the mixture for 1 h, Na2CO3 aqueous solution (0.1 M) was added dropwise until the pH reached 8.5 to precipitate PdCl42− on the surface of Al2O3, and the mixture was then stirred for another 2 h. After that, a freshly prepared NaBH4 aqueous solution (0.1 M) was slowly added dropwise into the mixture at 303 K to reduce Pd(OH)2 with a molar ratio of NaBH4 to Pd2+ of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. After stirring for 1 h, the solid product was washed with deionized water and ethyl alcohol and then dried in a vacuum at 333 K. The two types of catalysts were denoted as Pd/η-Al2O3 and Pd/γ-Al2O3, respectively.
:1. After stirring for 1 h, the solid product was washed with deionized water and ethyl alcohol and then dried in a vacuum at 333 K. The two types of catalysts were denoted as Pd/η-Al2O3 and Pd/γ-Al2O3, respectively.
Characterization
X-ray diffraction (XRD) patterns were recorded using a Rigaku-D/max 2500 V instrument with a Cu Kα radiation source. Transmission electron microscopy (TEM) and dark-field scanning transmission electron microscopy (DF-STEM) images were acquired on a JEM-2100F or JEM-ARM200F microscope operated at 200 kV. N2 adsorption/desorption isotherms were measured using a Quantachrome evo Mikete ASAP2460 volumetric adsorption analyzer. Thermal gravimetric (TG) measurements were performed on a Setaram Setsys 18 series thermal analyzer, with the samples being heated at a ramping rate of 10 K min−1 under air. The Pd loadings of Pd/Al2O3 catalysts were measured using an Agilent 720 inductively coupled plasma optical emission spectrometer (ICP-OES). CO pulse chemisorption was measured at 323 K on a Micromeritics/AUTOCHEM II 2920 analyzer. X-ray photoelectron spectroscopy (XPS) spectra were recorded using a Thermo Fisher 250Xi spectrometer with an Al Kα X-ray radiation source. The XPS spectra were acquired by collecting 5 scans at a scanning step of 0.05 eV, and the deconvolution of XPS spectra was performed using the Casa XPS program with the Lorentzian–Gaussian function after subtraction of a Shirley background. The binding energy scale of all spectra was calibrated to the C 1s peak (248.8 eV). Temperature-programmed desorption (TPD) experiments were performed using a Quantachrome Chembet Pulsar. NH3–TPD and CO2–TPD were conducted from 308 K to 973 K at a heating rate of 10 K min−1 under a He flow. In situ FT-IR spectra were recorded on a ThermoFisher Nicolet is50 infrared spectrometer by collecting 64 scans at a resolution of 4 cm−1.General procedure for hydrogenation reactions
The hydrogenation reactions were carried out in a three-neck round bottom flask with a water bath for temperature control. For the hydrogenation of nitrobenzene or phenylacetylene, 10 mg of catalyst was placed in the reactor, followed by the addition of 50 μL of substrate and 5 mL of ethanol as a solvent. The reaction was conducted at 303 K under a H2 flow at a flow rate of 100 mL min−1. To calculate the apparent activation energy, the reaction rates were measured while maintaining the conversion below 20% to minimize the influence of diffusion. Qualitative and quantitative analyses of reactants and products were conducted on a gas chromatography (GC) apparatus equipped with a flame ionization detector and capillary columns of HP-5 and DB-WAX. The conversion, selectivity, reaction rate and apparent activation energy were calculated as follows: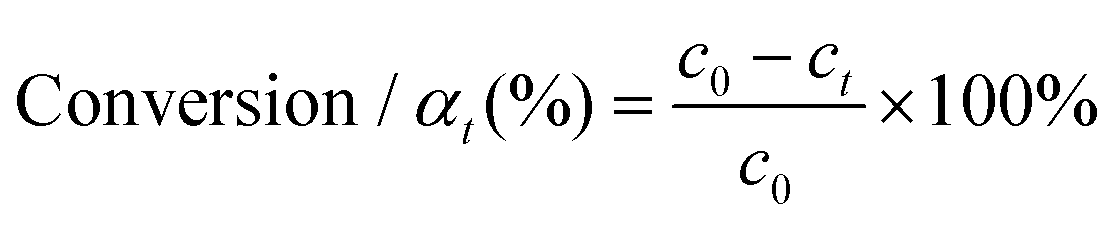

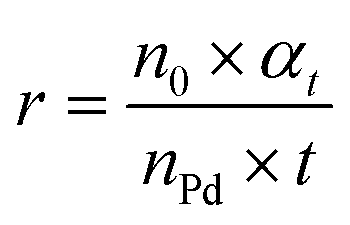
where c0 and ct are the concentrations of the reactant before and after the reaction for t h, respectively, ca is the concentration of the product after the reaction for t h, n0 is the moles of the initial reactant, and nPd is the moles of total Pd atoms.
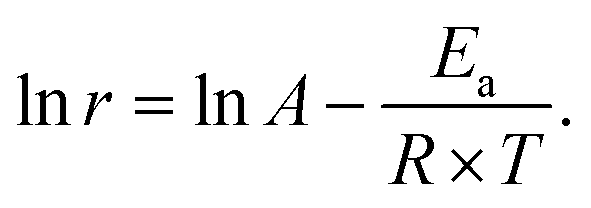
Computational method
Density functional theory (DFT) calculations were carried out in the Perdew–Burke–Ernzerhof (PBE) form of the generalized gradient approximation functional (GGA) using the Vienna ab initio simulation package (VASP). The plane wave basis set with a kinetic cutoff energy of 600 eV was applied to describe the interactions of valence electrons. The energy convergence of 1.0 × 10−5 eV per atom and the force convergence of 0.03 eV Å−1 were used for the geometric optimization. The Brillouin zone integration was set with 3 × 3 × 1 Monkhorst–Pack grids for the surface structure, and a vacuum layer of 15 Å was added along the c axis.Results and discussion
To elucidate the transformations of precursors during calcination and the crystallographic structures of both precursors and supports, XRD and TG-DTA measurements were performed. As shown in Fig. 1(a), a pure phase of Al(OH)3 was synthesized via the precipitation method, which involves adding ammonium hydroxide dropwise to an aqueous solution of aluminum nitrate until the pH reaches 11. According to the TG curve (Fig. S1(a)†), the total weight loss approaches 34.6%, which is consistent with the theoretical weight loss from Al(OH)3 to Al2O3.19 AlOOH was obtained by further dehydrating the amorphous aluminum hydroxide precipitate in a hot alkali solution. For AlOOH, the weight loss of 12.5% below 400 K is attributed to the loss of physical adsorption of water. Another weight loss of 15% is observed with an increase in temperature from 410 to 1073 K, which results from the dehydration process that converts AlOOH into Al2O3 (Fig. S1(b)†).30 After calcination, the XRD patterns of the two supports exhibit highly similar diffraction peaks. The phases of Al2O3 are distinguished by comparing the full width at half maximum of individual diffraction peaks and the relative intensities of diffraction peaks (Fig. 1(b)).19,31,32 The subtle differences in the XRD patterns of γ-Al2O3 and η-Al2O3 are as follows: (a) a shift towards lower angles for the (220) and (311) reflections in γ-Al2O3 and slight splitting or the appearance of an obvious shoulder on the (400) and (440) reflections of γ-Al2O3, due to its more pronounced tetragonal deformation compared to η-Al2O3, (b) the stronger (222) reflection in γ-Al2O3 because of its fairly well-ordered oxygen sublattice, and (c) the absence of a sharp (111) reflection but a stronger and sharper (220) reflection due to a better ordered tetrahedral Al sublattice in γ-Al2O3.27,28,31,32 Through the analysis, Al(OH)3 and AlOOH were converted to η-Al2O3 and γ-Al2O3, respectively. The results of 27Al MAS NMR show that the fraction of cations in tetrahedral positions is higher in η-Al2O3 than that in γ-Al2O3 (Fig. S2†). According to previous studies, it has been suggested that the (110) or (100) face preferentially terminates the crystallites of γ-Al2O3, while the (111) face terminates those of η-Al2O3.27,28 In Table S1,† the possible configuration and net charge of Al2O3 indicate that γ-Al2O3 may have a greater ability to donate electrons and η-Al2O3 tends to capture electrons.28,29 Therefore, the proposed crystal structures and electron transfer of Pd/Al2O3 are shown in Fig. 1(c).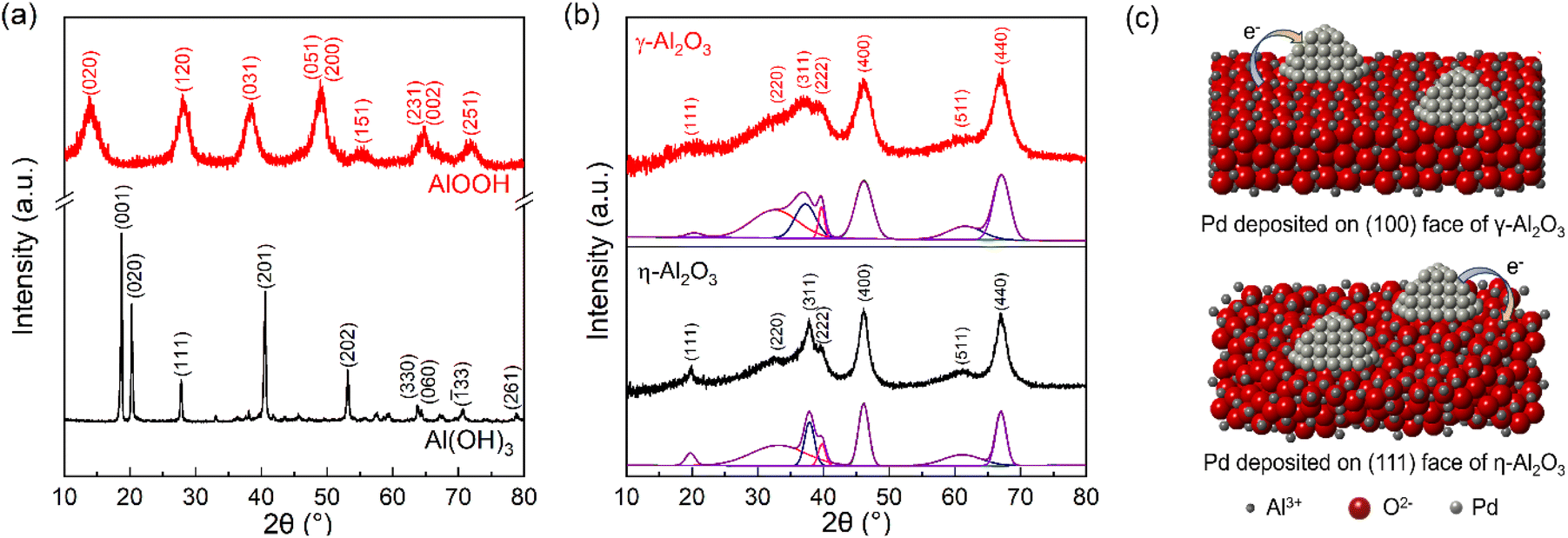 | ||
| Fig. 1 XRD patterns of (a) precursors and (b) supports. (c) The proposed crystal structures and electron transfer of Pd/γ-Al2O3 and Pd/η-Al2O3. | ||
Pd NPs were deposited on γ-Al2O3 and η-Al2O3 supports by the deposition–precipitation method, with Pd loadings of 0.474 wt% and 0.466 wt% (as determined by ICP), respectively. The specific surface areas and pore size distributions of the supports, as well as the morphology characteristics of the catalysts, are shown in Fig. S3† and Fig. 2. Due to the high specific surface areas and suitable pore sizes of γ-Al2O3 and η-Al2O3, disorderly-shaped Pd NPs are uniformly distributed over the surfaces of supports without obvious agglomeration, and there is little difference in the average particle sizes of Pd on γ-Al2O3 and η-Al2O3. The HR-TEM images indicate that the lattice spacing of Pd NPs is 2.25 Å, which can be attributed to the (111) plane of Pd. The Pd dispersions on γ-Al2O3 and η-Al2O3 are 58.8% and 64.1%, respectively, based on a combination of the CO pulse chemisorption results and the ratios of linear-adsorbed CO to bridge-adsorbed CO as determined by the in situ FT-IR spectra of CO adsorption in Fig. 4(d).
 | ||
| Fig. 2 DF-STEM images, HR-TEM images and Pd size histograms of Pd/γ-Al2O3 (a, b and c) and Pd/η-Al2O3 (d, e and f). | ||
To understand the influence of the surface configurations of the two supports on the electronic structures of Pd NPs, the acid–base properties of the supports and catalysts were studied by in situ FT-IR spectroscopy and TPD. In Fig. 3(a), the bands at approximately 1580 cm−1 and 1595 cm−1 are attributed to the physical adsorption of pyridine and the H-bonds formed between the pyridine and OH groups of Al2O3, respectively. Other bands in the FT-IR spectra are assigned to the C–C ring vibration of pyridine that is adsorbed on the Lewis acid sites.29,33 As previously reported, the bands in the region of 1600–1630 cm−1 are highly sensitive to the strength of Lewis acid sites.33 Compared to γ-Al2O3, a band at 1625 cm−1 is observed on η-Al2O3, revealing the presence of stronger Lewis acid sites.29,33 As shown in Fig. S4(a) and (b),† the overlapping NH3–TPD curves are fitted into three subpeaks using the Gaussian fitting method, including the physisorption of NH3 (red-filled), weak acid sites (blue-filled) and strong acid sites (green-filled).34 The NH3–TPD profiles indicate that the η-Al2O3 support has a greater number of strong acid sites, which is consistent with the results of FT-IR spectra (Fig. 3(c) and Table S2†). Loading Pd NPs onto the supports results in a substantial decrease in the number of strong acid sites on η-Al2O3 (59 μmol g−1), whereas that on γ-Al2O3 is reduced by only 5 μmol g−1. It is well known that the strong acid sites produced by coordinatively unsaturated Al3+ centers act as Lewis acid sites and can accept electrons.28 Therefore, more Pd NPs are loaded onto the Lewis acid sites of η-Al2O3, which transfer more electrons to the coordinatively unsaturated Al3+ centers, resulting in Pd NPs with electron-deficient surface properties.
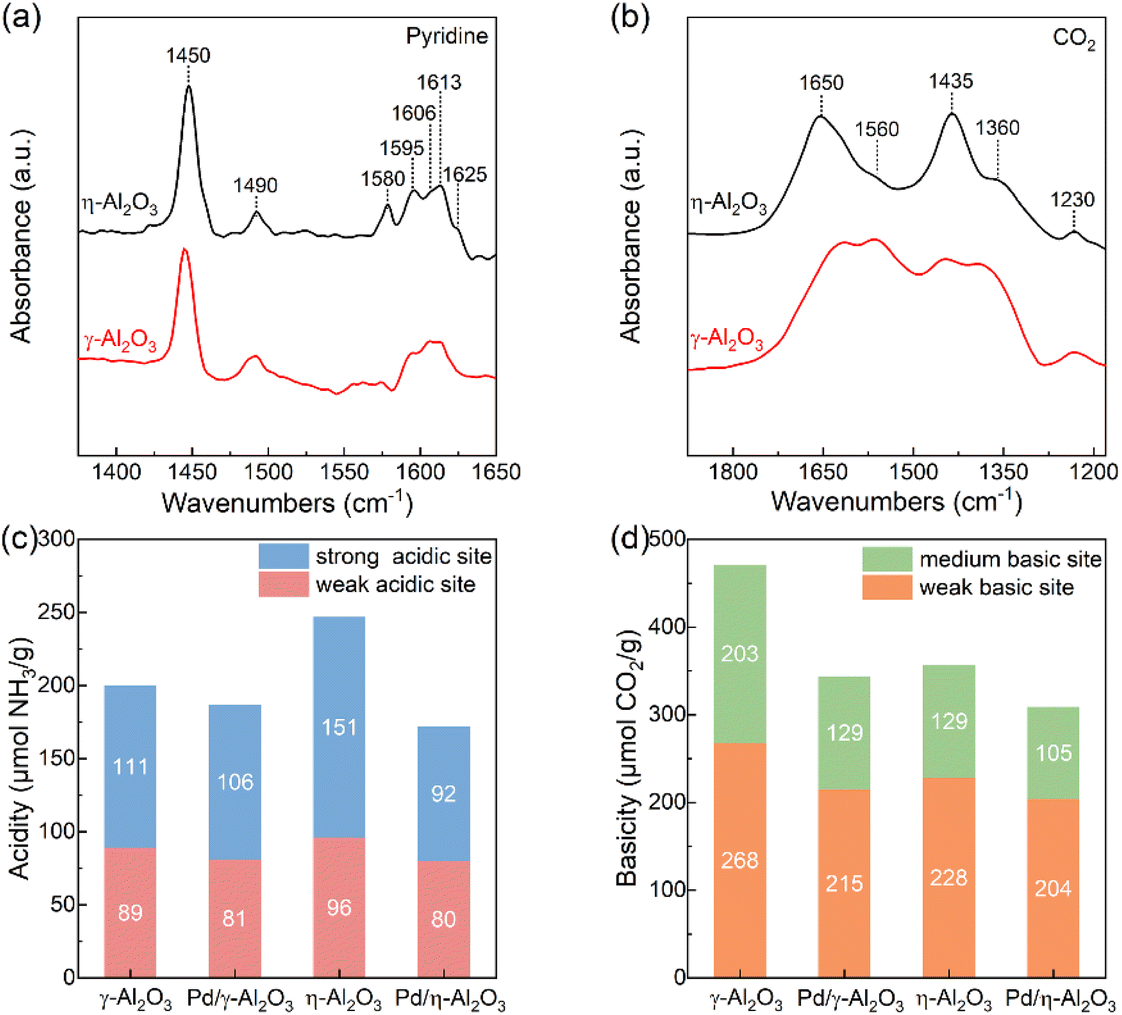 | ||
| Fig. 3 (a) FT-IR spectra of pyridine adsorption on supports. (b) FT-IR spectra of CO2 adsorption on supports. (c) Acidity of supports and catalysts. (d) Basicity of supports and catalysts. | ||
As shown in Fig. 3(b), the vibration bands of bicarbonate and carbonate were detected on γ-Al2O3 and η-Al2O3 by in situ FT-IR analysis of chemisorbed CO2. The formation of bicarbonates involves basic hydroxyl groups, which show a C–OH bending mode at 1230 cm−1 as well as symmetric and asymmetric O–C–O stretching vibrations at 1435 cm−1 and 1650 cm−1, respectively. Carbonate species form on the basic O2− sites, exhibiting a symmetric O–C–O stretching vibration at 1360 cm−1 and an asymmetric O–C–O stretching vibration at 1560 cm−1.33,35 The stronger vibration bands of carbonates on γ-Al2O3 indicate the presence of more basic O2− sites on its surface compared to η-Al2O3. In Fig. S4(c) and (d),† the CO2–TPD curves are fitted with two subpeaks, which correspond to the weak basic sites produced by the surface OH groups (red-filled) and the medium basic sites ascribed to the lattice oxygen groups (blue-filled).36 The results of CO2–TPD are the same as those of in situ FT-IR spectroscopy, indicating that the γ-Al2O3 support possesses more lattice oxygen groups, which serve as Lewis basic sites (Fig. 3(d) and Table S2†). Palladium particles deposited on the supports decrease the number of Lewis basic sites, and the reductions of γ-Al2O3 and η-Al2O3 are 74 μmol g−1 and 24 μmol g−1, respectively. Due to the negative charge on the lattice oxygen groups, more basic sites on the γ-Al2O3 support inject electrons into Pd NPs, which endow the Pd NPs with electron-rich properties (Table S1†).28 The difference in acid–base properties between γ-Al2O3 and η-Al2O3 supports may be attributed to the preferentially exposed (110) and (100) faces on γ-Al2O3, as well as the (111) face on η-Al2O3.27,28 More Lewis acid sites on η-Al2O3 and more Lewis basic sites on γ-Al2O3 act as electron acceptors and electron donors, respectively, modulating the electronic structures of Pd NPs. The OH groups function as H-bond acceptors or donors, and the similar OH groups present in both Pd/γ-Al2O3 and Pd/η-Al2O3 eliminate their influence on the catalytic performance during hydrogenation reactions (Fig. S5†).
X-ray photoelectron spectroscopy analysis was performed to identify the chemical states of aluminum, oxygen and palladium and further elucidate the interaction between Pd NPs and Al2O3 supports. As shown in Fig. 4(a), the binding energy of Al 2p at approximately 74.4 eV corresponds to the Al–O bonds present in both γ-Al2O3 and η-Al2O3.37,38 After Pd NPs are deposited on Al2O3, there are negative shifts in the Al–O peaks for both Pd/γ-Al2O3 and Pd/η-Al2O3 with offsets of 0.02 eV and 0.13 eV, respectively. More coordinatively unsaturated Al3+ centers on the surface of the η-Al2O3 support capture electrons from Pd NPs, leading to a more negative shift in binding energy, which is consistent with the results of TPD measurements. The existence of Al–OH is also observed on Al2O3 supports with a binding energy of 76.5 eV.37,38 In Fig. 4(b), the peaks at approximately 531.0 eV and 532.3 eV are attributed to the lattice oxygen and surface hydroxyl groups, respectively.39,40 Compared with the supports, Pd/γ-Al2O3 shows a positive shift of 0.11 eV from the lattice oxygen, while Pd/η-Al2O3 exhibits a positive shift of 0.02 eV. The lattice oxygen peak on Pd/γ-Al2O3 shifts more positively due to more electrons transferring from the medium basic sites on the surface of γ-Al2O3 to Pd NPs. The proportions of OH groups between the supports and catalysts in the Al 2p and O 1 s spectra change in keeping with the results of the TPD profiles and the FT-IR spectra of OH (Table S3†). From the Pd 3d XPS spectra (Fig. 4(c)), the peaks at approximately 335.3 eV and 337.1 eV are ascribed to the metallic state of Pd0 and the oxidation state of Pd2+, respectively.41 In comparison with Pd/η-Al2O3 (335.46 eV), a negative shift of 0.22 eV is observed in the Pd 3d core level spectrum of Pd/γ-Al2O3 (335.24 eV). The electronic properties of Pd NPs are further illustrated by the shift in the CO stretching frequency in the in situ FT-IR spectra of CO adsorption, as shown in Fig. 4(d). The red shift of Pd0–CO peaks for Pd/γ-Al2O3 is attributed to a stronger back-donation of electrons from Pd NPs to the 2π* anti-bonding orbital of CO, which weakens the CO bond.42 Differential charge density calculations were performed to elucidate the electron redistribution behaviors at the interface between Pd and Al2O3. As shown in Fig. 4(e) and (f), strong electron coupling occurs at the interface between Pd clusters and the Al2O3 support. For the model of a Pd cluster formed by 12 atoms, the Bader charge analysis indicates that 0.5989 e is transferred from the γ-Al2O3 support to the Pd cluster, while 0.5123 e is transferred from the Pd cluster to η-Al2O3. The above results indicate that the γ-Al2O3 support prefers to donate electrons to the Pd NPs, leading to electron-rich Pd/γ-Al2O3. Conversely, the η-Al2O3 support tends to capture electrons from the Pd NPs, resulting in Pd/η-Al2O3 with electron-deficient properties. These effects arise from the electronic metal–support interaction between the Pd NPs and acid–base sites on the surface of Al2O3.
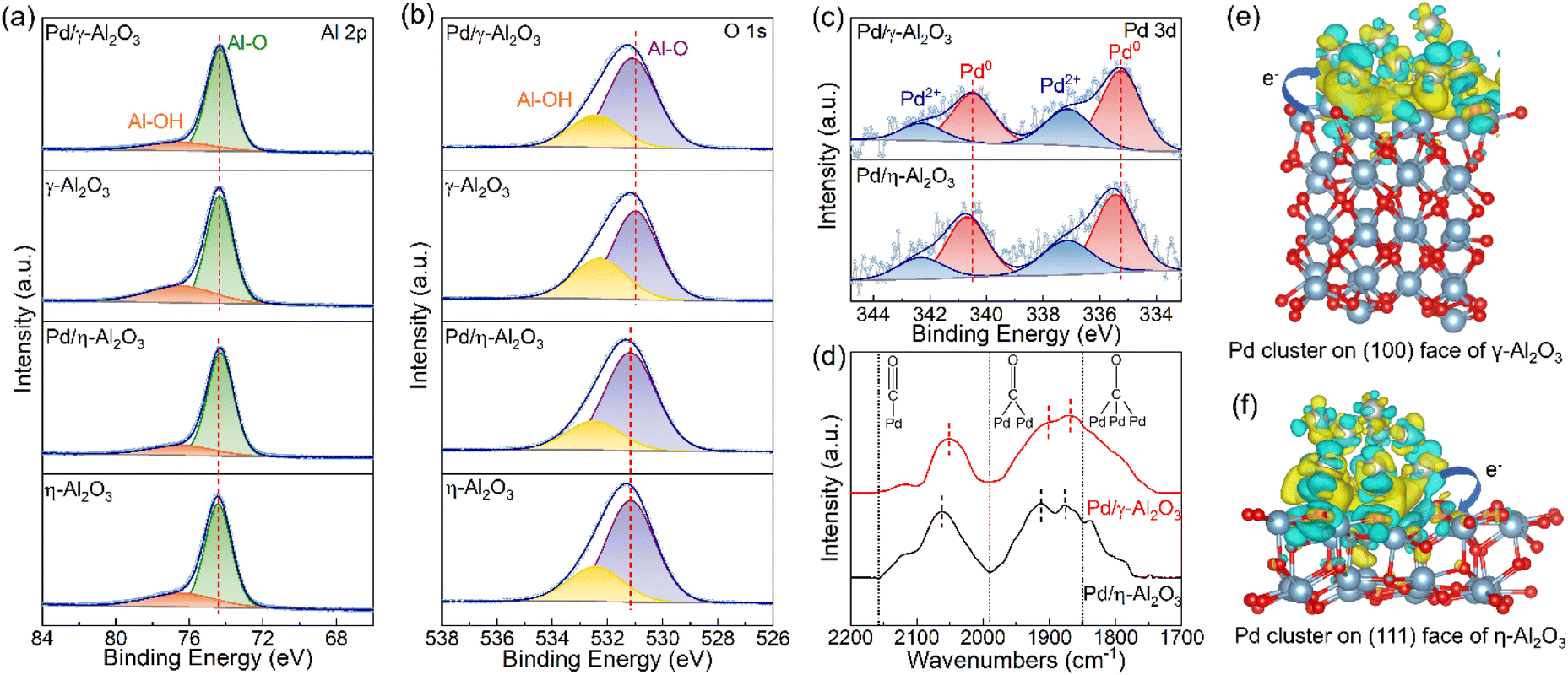 | ||
| Fig. 4 XPS spectra of the supports and Pd-supported catalysts: (a) Al 2p, (b) O 1s and (c) Pd 3d. (d) In situ FT-IR spectra of CO adsorption on Pd/γ-Al2O3 and Pd/η-Al2O3. Different charge densities on (e) Pd/γ-Al2O3 and (f) Pd/η-Al2O3 (the yellow and cyan colors represent the electron accumulation and deletion, respectively, and the gray, red, and blue spheres represent Pd, O, and Al atoms, respectively). | ||
To clarify the effect of the electronic structures of Pd NPs on their catalytic performance, the hydrogenation of nitrobenzene as a significant industrial reaction was performed. As shown in Fig. 5(a), Pd/γ-Al2O3 and Pd/η-Al2O3 could smoothly catalyze the reaction to achieve full conversion with reaction times of 40 min and 110 min, respectively. Pd/γ-Al2O3 exhibits a reaction rate of 2419 h−1, which is triple that of Pd/η-Al2O3 (795 h−1). The faster kinetic reaction process on Pd/γ-Al2O3 is due to the lower apparent activation energy (Fig. 5(b)). A previous study on the substrate reactivity of nitrobenzene derivatives indicated that the hydrogenation of nitrobenzene exhibits a strong nucleophilic character, and the rate-determining step of the reaction is the attack of nucleophilic hydride (H−) produced by hydrogen dissociation on the Pd surface toward the nitro groups in nitrobenzene.43 This suggests that Pd/γ-Al2O3 with electron-rich surface properties is beneficial for producing H− as the nucleophile, which reduces the energy barrier and promotes the reaction rate. In Fig. S6,† the selectivity of aniline increases as nitrobenzene conversion increases due to further hydrogenation of the intermediate product of nitrosobenzene. Less nitrosobenzene is produced in the hydrogenation of nitrobenzene catalyzed by Pd/γ-Al2O3, indicating that electron-rich Pd/γ-Al2O3 is more effective in the hydrogenation of nitrosobenzene and inhibits its desorption. Although the electron-deficient Pd/η-Al2O3 favors the adsorption of nucleophilic nitrobenzene, the enhanced activity of Pd/γ-Al2O3 reveals that accelerating the rate-determining step of the reaction plays an important role compared to the adsorption strength of substrates.14 In comparison with previous Pd-based catalysts, Pd/γ-Al2O3 is one of the most active catalysts for nitrobenzene hydrogenation under mild conditions (Fig. 5(c)).44–49 The recycling stability of Pd/γ-Al2O3 in the hydrogenation of nitrobenzene shows that the catalyst has no obvious loss of catalytic performance in five cycles (Fig. S7†). The morphology and electronic structure characterization studies of the used catalysts indicate that both catalysts exhibit good stability during the hydrogenation reaction (Fig. S8 and S9†).
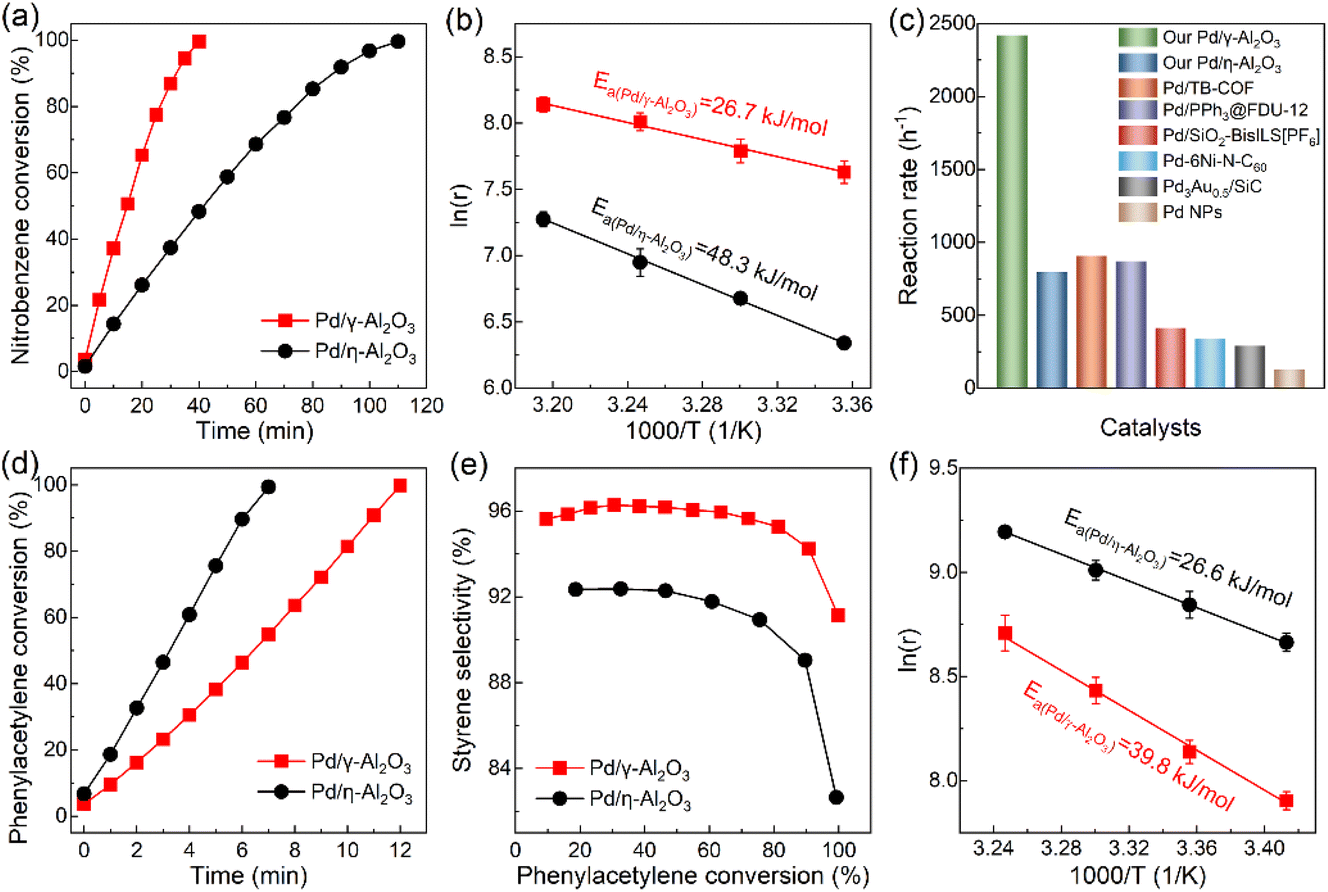 | ||
| Fig. 5 Catalytic performance of catalysts for nitrobenzene hydrogenation (a and b) and phenylacetylene hydrogenation (d, e and f). (c) Performance comparison of nitrobenzene hydrogenation under 1 bar H2. | ||
In addition to nitrobenzene hydrogenation, the catalytic performance of palladium for phenylacetylene hydrogenation is also closely related to the electronic structure of palladium. As shown in Fig. 5(d), the activity of Pd/η-Al2O3 (8116 h−1) is higher than that of Pd/γ-Al2O3 (4071 h−1). The electron-deficient Pd/η-Al2O3 strengthens the adsorption of phenylacetylene and the Pd–H bond, leading to an increased coverage of phenylacetylene and hydrogen on the Pd surface and a reduction in the energy barrier for the hydrogenation of terminal carbon (Fig. 5(f)).45,50 In Fig. 5(e), a comparison of the styrene selectivity of the two catalysts based on the same phenylacetylene conversion level clearly shows that Pd/γ-Al2O3 affords higher selectivity to styrene than Pd/η-Al2O3, which is derived from the weakened adsorption of styrene on the electron-rich Pd/γ-Al2O3 hindering the complete hydrogenation of phenylacetylene to ethylbenzene.45 Pd/η-Al2O3 exhibits good recycling stability during the hydrogenation of phenylacetylene over five cycles (Fig. S10†). Consequently, nitrobenzene hydrogenation and phenylacetylene hydrogenation serve as probe reactions to indicate that the entirely different electronic structures of Pd/γ-Al2O3 and Pd/η-Al2O3 can effectively modulate the adsorption strength of substrates and energy barriers, optimizing the catalytic performance.
Conclusions
In conclusion, modulating the electronic structure of Pd NPs by tuning the surface configuration of Al2O3 supports successfully promotes their catalytic activity and product selectivity. The γ-Al2O3 and η-Al2O3 supports prepared using AlOOH and Al(OH)3 precursors exhibit more basicity and more acidity, respectively. The medium basic sites of the lattice oxygen groups on the γ-Al2O3 support have an electron-donating effect, which endows the Pd NPs with an electron-rich surface. In contrast, Pd NPs deposited on η-Al2O3 inject electrons into the electron-withdrawing sites of coordinatively unsaturated Al3+ centers and form electron-deficient Pd/η-Al2O3. In the hydrogenation of nitrobenzene, electron-rich Pd/γ-Al2O3 facilitates the rate-determining step of the nucleophilic addition of hydride (H−) toward nitrobenzene, resulting in higher catalytic activity and aniline selectivity compared to Pd/η-Al2O3. In addition, electron-deficient Pd/η-Al2O3 accelerates the hydrogenation of phenylacetylene and strengthens the adsorption of both phenylacetylene and styrene, leading to higher activity but lower selectivity for styrene in comparison with Pd/γ-Al2O3. Herein, we revealed the importance of the acid–base sites of Al2O3 in modulating the surface electronic structures of metal nanoparticles to promote their catalytic activity and product selectivity, which provides a feasible approach for designing the electronic properties of supported metal catalysts.Author contributions
Y. Liu: conceptualization, investigation, validation, visualization, and writing – original draft. C. Ma: validation. J. Zhang: validation. H. Zhou: investigation. G. Qin: funding acquisition. S. Li: funding acquisition, conceptualization, supervision, and writing – review & editing.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was funded by the National Key Research and Development Program of China (2021YFB3801600), the National Natural Science Foundation of China (52331001), and the Fundamental Research Funds for the Central Universities (N2202012).References
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS.
- T. W. van Deelen, C. Hernández Mejía and K. P. de Jong, Nat. Catal., 2019, 2, 955–970 CrossRef CAS.
- Y. Lou, J. Xu, Y. Zhang, C. Pan, Y. Dong and Y. Zhu, Mater. Today Nano, 2020, 12, 100093 CrossRef.
- C. Dong, Y. Li, D. Cheng, M. Zhang, J. Liu, Y.-G. Wang, D. Xiao and D. Ma, ACS Catal., 2020, 10, 11011–11045 CrossRef CAS.
- Y. Rao, Y. Wu, X. Dai, Y. Zhang, G. Qin, W. Qi and S. Li, Small, 2022, 18, 2204611 CrossRef CAS.
- H. Tang, Y. Su, B. Zhang, A. F. Lee, M. A. Isaacs, K. Wilson, L. Li, Y. Ren, J. Huang, M. Haruta, B. Qiao, X. Liu, C. Jin, D. Su, J. Wang and T. Zhang, Sci. Adv., 2017, 3, e1700231 CrossRef PubMed.
- M. Xiong, Z. Gao and Y. Qin, ACS Catal., 2021, 11, 3159–3172 CrossRef CAS.
- Y. G. Wang, Y. Yoon, V. A. Glezakou, J. Li and R. Rousseau, J. Am. Chem. Soc., 2013, 135, 10673–10683 CrossRef CAS.
- T. Binninger, T. J. Schmidt and D. Kramer, Phys. Rev. B, 2017, 96, 165405 CrossRef.
- G. Pacchioni and H.-J. Freund, Chem. Soc. Rev., 2018, 47, 8474–8502 RSC.
- M. J. Zachman, V. Fung, F. Polo-Garzon, S. Cao, J. Moon, Z. Huang, D. Jiang, Z. Wu and M. Chi, Nat. Commun., 2022, 13, 3253 CrossRef CAS.
- S. Naya, M. Teranishi, R. Aoki and H. Tada, J. Phys. Chem. C, 2016, 120, 12440–12445 CrossRef CAS.
- L. Zhang, F. Mao, L. R. Zheng, H. F. Wang, X. H. Yang and H. G. Yang, ACS Catal., 2018, 8, 11035–11041 CrossRef CAS.
- Z. He, B. Dong, W. Wang, G. Yang, Y. Cao, H. Wang, Y. Yang, Q. Wang, F. Peng and H. Yu, ACS Catal., 2019, 9, 2893–2901 CrossRef CAS.
- Y. Lykhach, S. M. Kozlov, T. Skála, A. Tovt, V. Stetsovych, N. Tsud, F. Dvořák, V. Johánek, A. Neitzel, J. Mysliveček, S. Fabris, V. Matolín, K. M. Neyman and J. Libuda, Nat. Mater., 2016, 15, 284–288 CrossRef CAS PubMed.
- Y. Li, C. Zhang, J. Ma, M. Chen, H. Deng, H. He, Y. Li, H. He, J. Ma, M. Chen, C. Zhang, J. Ma, M. Chen, H. Deng and H. He, Appl. Catal., B, 2017, 217, 560–569 CrossRef CAS.
- Y. Liu, H. Chen, C. Xu, Y. Sun, S. Li, M. Jiang and G. Qin, Small, 2019, 15, 1901789 CrossRef PubMed.
- K. Murata, Y. Mahara, J. Ohyama, Y. Yamamoto, S. Arai and A. Satsuma, Angew. Chem., Int. Ed., 2017, 56, 15993–15997 CrossRef CAS PubMed.
- C. Dai, Y. Li, C. Ning, W. Zhang, X. Wang and C. Zhang, Appl. Catal., A, 2017, 545, 97–103 CrossRef CAS.
- Y. Liu, C. Liu, H. Zhou, G. Qin and S. Li, Colloids Surf., A, 2023, 667, 131392 CrossRef CAS.
- G. Pacchioni, Phys. Chem. Chem. Phys., 2013, 15, 1737–1757 RSC.
- C. Xu, Y. Liu, B. Singh, S. Yi, G. Qin and S. Li, Surf. Interfaces, 2022, 33, 102276 CrossRef CAS.
- F. Wang, J. Ma, S. Xin, Q. Wang, J. Xu, C. Zhang, H. He and X. Cheng Zeng, Nat. Commun., 2020, 11, 529 CrossRef CAS PubMed.
- X. Sun, F. Li, J. Shi, Y. Zheng, H. Su, L. Sun, S. Peng and C. Qi, Appl. Surf. Sci., 2019, 487, 625–633 CrossRef CAS.
- L. Yang, Z. Pan, D. Wang, S. Wang, X. Wang, H. Ma, H. Liu, C. Wang, W. Qu and Z. Tian, ACS Appl. Mater. Interfaces, 2021, 13, 28064–28071 CrossRef CAS.
- Y. Jing, G. Wang, K. W. Ting, Z. Maeno, K. Oshima, S. Satokawa, S. Nagaoka, K. Shimizu and T. Toyao, J. Catal., 2021, 400, 387–396 CrossRef CAS.
- H. Knözinger, P. Ratnasamy, H. Knazdtgelit and P. Ratnasamy, Catal. Rev., 1978, 17, 31–70 CrossRef.
- H. Knozinger, in Studies in Surface Science and Catalysis, 1985, vol. 20, pp. 111–125 Search PubMed.
- P. O. Scokart and P. G. Rouxhet, J. Colloid Interface Sci., 1982, 86, 96–104 CrossRef CAS.
- J. Huang, Y. Wang, J. Zheng, W.-L. Dai and K. Fan, Appl. Catal., B, 2011, 103, 343–350 CrossRef CAS.
- R.-S. Zhou and R. L. Snyder, Acta Crystallogr., Sect. B: Struct. Sci., 1991, 47, 617–630 CrossRef.
- B. C. Lippens and J. H. de Boer, Acta Crystallogr., 1964, 17, 1312–1321 CrossRef CAS.
- S. G. Scire, C. Crisafulli, R. Maggiore and S. Minico, Appl. Surf. Sci., 1998, 136, 311–320 CrossRef CAS.
- F. Wang, J. Ma, G. He, M. Chen, C. Zhang and H. He, ACS Catal., 2018, 8, 2670–2682 CrossRef CAS.
- M. A. Al-Daous, A. A. Manda and H. Hattori, J. Mol. Catal. A: Chem., 2012, 363–364, 512–520 CrossRef CAS.
- N. T. Tran, P. S. Kumar, Q. Van Le, N. Van Cuong, P. T. T. Phuong, A. A. Jalil, G. Sharma, A. Kumar, A. Sharma, B. V. Ayodele, S. Z. Abidin and D.-V. N. Vo, Top. Catal., 2021, 64, 338–347 CrossRef CAS.
- K. H. Yoon, H. Kim, Y.-E. E. Koo Lee, N. K. Shrestha and M. M. Sung, RSC Adv., 2017, 7, 5601–5609 RSC.
- C. Wu, X. Pan, F. Lin, G. Chen, L. Xu, Y. Zeng, Y. He and D. Sun, Polymers, 2022, 14, 3281 CrossRef CAS.
- Y. Fan, F. Wang, R. Li, C. Liu and Q. Fu, ACS Catal., 2023, 13, 2277–2285 CrossRef CAS.
- J. Castillo-Saenz, N. Nedev, B. Valdez-Salas, M. Curiel-Alvarez, M. I. Mendivil-Palma, N. Hernandez-Como, M. Martinez-Puente, D. Mateos, O. Perez-Landeros and E. Martinez-Guerra, Coatings, 2021, 11, 1266 CrossRef CAS.
- M. Guo, S. Jayakumar, M. Luo, X. Kong, C. Li, H. Li, J. Chen and Q. Yang, Nat. Commun., 2022, 13, 1770 CrossRef CAS PubMed.
- Y. Cao, Z. Sui, Y. Zhu, X. Zhou and D. Chen, ACS Catal., 2017, 7, 7835–7846 CrossRef CAS.
- P. Lu and N. Toshima, Bull. Chem. Soc. Jpn., 2000, 73, 751–758 CrossRef CAS.
- C. Li, X. Ren, M. Guo, W. Li, H. Li and Q. Yang, Catal. Sci. Technol., 2021, 11, 3873–3879 RSC.
- M. Guo, H. Li, Y. Ren, X. Ren, Q. Yang and C. Li, ACS Catal., 2018, 8, 6476–6485 CrossRef CAS.
- J. Li, X.-Y. Shi, Y.-Y. Bi, J.-F. Wei and Z.-G. Chen, ACS Catal., 2011, 1, 657–664 CrossRef CAS.
- Y. Qu, T. Chen and G. Wang, Appl. Surf. Sci., 2019, 465, 888–894 CrossRef CAS.
- C.-H. Hao, X.-N. Guo, M. Sankar, H. Yang, B. Ma, Y.-F. Zhang, X.-L. Tong, G.-Q. Jin and X.-Y. Guo, ACS Appl. Mater. Interfaces, 2018, 10, 23029–23036 CrossRef CAS PubMed.
- F. A. Harraz, S. E. El-Hout, H. M. Killa and I. A. Ibrahim, J. Catal., 2012, 286, 184–192 CrossRef CAS.
- J. Audevard, J. Navarro-Ruiz, V. Bernardin, Y. Tison, A. Corrias, I. Del Rosal, A. Favre-Réguillon, R. Philippe, I. C. Gerber and P. Serp, ChemCatChem, 2023, 15, e202300036 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr05258c |
| This journal is © The Royal Society of Chemistry 2024 |